

Abstract
Sequencing of the human Y chromosome has uncovered the peculiarities of the genomic organization of a heterogametic sex chromosome of old evolutionary age, and has led to many insights into the evolutionary changes that occurred during its long history. We have studied the genomic organization of the medaka fish Y chromosome, which is one of the youngest heterogametic sex chromosomes on which molecular data are available. The Y specific and adjacent regions were sequenced and compared to the X. The male sex-determining gene, dmrt1bY, appears to be the only functional gene in the Y-specific region. The Y-specific region itself is derived from the duplication of a 43-kb fragment from linkage group 9. All other coduplicated genes except dmrt1bY degenerated. The Y-specific region has accumulated large stretches of repetitive sequences and duplicated pieces of DNA from elsewhere in the genome, thereby growing to 258 kb. Interestingly the non-recombining part of the Y did not spread out considerably from the original duplicated fragment, possibly because of a large sequence duplication bordering the Y-specific fragment. This may have conserved the more ancestral structure of the medaka Y and provides insights into some of the initial processes of Y chromosome evolution.
The Y chromosome is a peculiar constituent of the karyotype. In many cases, its presence is connected to the developmental decision of the undifferentiated gonads to become testis and of the whole organism to become a male. Like its counterpart in female heterogametic systems, the W chromosome, the Y chromosome is a constantly hemizygous chromosome and is generally small, gene-poor, and rich in repetitive sequences of all different kinds. On the contrary, the X and Z chromosomes are organized like autosomes.
It has been hypothesized that all sex chromosomes originated from a pair of autosomes (Muller 1914, 1918; Ohno 1967). Hence, in the early phase of sex chromosome evolution, both gonosomes are, like their autosomal progenitors, similar in shape (homomorphic), and only over time do they acquire different morphology and become heteromorphic. This theory is now generally accepted because of a large body of cytogenetic evidence (Graves 1998). An explanation for the tendency to become heteromorphic comes from the need for suppression of recombination at the sex-determining locus in order to keep the identity of the gonosomes as either an X or a Y. In the absence of recombination on the hemizygous chromosome, the effectiveness of selection is reduced (Rice 1996; Orr and Kim 1998). This causes degeneration of active genes present on the ancestral chromosome (Charlesworth 1978, 1991). Finally, genes that specifically benefit male fecundity (in particular, spermatogenesis genes) accumulate on the Y (Rice 1996; Charlesworth et al. 2005).
The human Y chromosome is a paradigmatic example of a Y at a very advanced stage of evolution. Besides a small (5%) pseudoautosomal region at the tip of the Y that still recombines with the X, the entire rest is male-specific. The male-specific part is a mosaic of large heterochromatic regions and euchromatic sequences. The euchromatic part contains a total of 156 transcription units. These include 78 protein-coding genes that encode altogether only 27 distinct proteins including the male-determining SRY gene (Rozen et al. 2003; Skaletsky et al. 2003). On the basis of these findings and many previous studies (for reviews, see Bachtrog and Charlesworth 2001; Lahn et al. 2001), it was possible to deduce the major steps in the evolutionary history of the human Y chromosome (Skaletsky et al. 2003).
For further understanding the evolution of Y chromosomes, it would be helpful to have similar detailed molecular information from a hemizygous sex chromosome at a very early stage of chromosome evolution (Charlesworth et al. 2005). The Y chromosome of a fish, the medaka, Oryzias latipes, appears to be a useful model for such studies (Schartl 2004; Matsuda 2005). The medaka has an XX-XY sex determination system (Aida 1921). The sex chromosomes are homomorphic (Matsuda et al. 1998), which was a first indication of their young age. The male sex-determining locus was precisely mapped to one region on the Y (Matsuda et al. 1999; Kondo et al. 2001). The gene that determines male sexual development has been isolated (Matsuda et al. 2002; Nanda et al. 2002). It encodes a putative transcription factor from the family of DM-domain proteins (dmrts). All members of this gene family have in common a highly conserved DNA-binding domain with a high similarity to proteins that are involved in sex determination in Drosophila and Caenorhabditis elegans. The prototype member, dmrt1, is the most closely related member to the invertebrate sex-determining genes (Zarkower 2001). The mammalian ortholog has been implicated as a dosage-dependent sex-determining gene in human XY male-to-female sex reversals (Raymond et al. 1999). The chicken ortholog has been mapped to the Z (Nanda et al. 1999) and is discussed as a likely candidate for the avian male-determining gene. In medaka, the male sex-determining gene actually is a duplicated version of dmrt1. The Y-specific gene was designated dmrt1bY (also known as dmY) to distinguish it from the autosomal gene dmrt1a. Interestingly, dmrt1a forms a cluster with two other dmrt genes on the autosomal linkage group (LG) 9 of medaka, a structural feature that is highly conserved from fish to mammals (Brunner et al. 2001).
Using nucleotide substitution rates for the dmrt1 genes and studying various species of the genus Oryzias for the presence or absence of the duplicated Y-specific gene, the duplication event that led to the formation of the dmrt1bY gene was calculated to have occurred ∼10 million years ago (Mya) (Kondo et al. 2004). Thus the medaka Y is—compared to the 170–210 million years (Myr) that have been estimated as the age of the mammalian Y (Graves 2002)—the youngest vertebrate Y known so far, for which the sex-determining gene has been identified.
To analyze the genomic organization of the Y-specific region in medaka, we have fully sequenced overlapping BAC clones from the Y chromosome and the corresponding region on the X, as well as the clone containing the autosomal dmrt1. We show that the Y-specific region is precisely limited by a long duplicated sequence, which appears to recombine in meiosis with a single locus on the X. The Y-specific region that does not pair with the X is only 258 kb in length. Gene prediction programs and expression studies revealed that dmrt1bY is the single functional gene in this region. In addition, sequencing of the regions flanking the Y chromosome-specific region revealed the presence of other genes that appear to be functional, indicating that degeneration of the Y chromosome has not occurred outside the Y-specific region, and that the rest of the Y chromosome still keeps its function.
Results
Cloning and sequence analysis of the sex-determining and flanking regions on the Y chromosome and the autosomal dmrt1 region
By screening a medaka male BAC library for dmrt1bY and then extending the region in both directions, we obtained clones Mn0168M02, Mn0209O12, and Mn0113N21 as the clones covering the Y chromosome-specific region with the minimum overlap (Fig. 1A). Mn0195H08, which contained BAC end markers derived from both Mn0168M02 and Mn0113N21 but did not contain dmrt1bY or share the BAC end markers of Mn0209O12, was identified to contain the X-chromosomal region corresponding to the region flanking the Y-specific region. In addition, we isolated BAC clone Mn0008J11 containing the autosomal dmrt1a gene. The sequences of these BAC clones were determined by the shotgun method. As a result, the Y-chromosomal region of 383,348 bp, the X-chromosomal region of 194,046 bp, and the autosomal dmrt1,3,2 region on linkage group (LG) 9 of 155,923 bp were obtained. Coding regions were identified by using 10 different gene/exon prediction programs and by BLAST search (http://www.ncbi.nlm.nih.gov/BLAST/) (Fig. 2; Table 1).
Figure 1.

The male sex-determining region of medaka and its position on the sex chromosome. (A) The BAC clones covering the sex-determining region of medaka were isolated by screening using dmrt1bY as the probe. The contig was extended by screening the BAC library with BAC end markers in both directions. Mn0168M02 (0168), Mn0209O12 (0209), and Mn0113N21 (0113) on the Y chromosome, as well as Mn0195H08 (0195) from the X chromosome were used for further analysis. The dmrt1bY gene (gray box) is located on clone Mn0113N21. The Olaflnk genes (white boxes) are located on Mn0113N21, Mn0168M02, and Mn0195H08. The gray circles indicate the markers that were derived from BAC end sequences of Mn0168M02 or Mn0113N21, present in Mn0195H08. The dotted line in Mn0195H08 indicates that this region does not exist in this clone, but the flanking regions are present. (B,C) The linkage map close to dmrt1bY. The sex-determining region is located on the long arm of the sex chromosome (LG1). All markers shown in the diagram except for dmrt1bY are present on both the X and Y chromosomes (B). Markers casp6 (caspase 6) and casp3B (caspase 3B) were used to screen the HNI BAC library and Mn0019I23 (0019), Mn0036H11 (0036), Mn0065F06 (0065) and Mn0046N20 (0046) were isolated (not drawn to scale) (C). End-sequencing of these clones revealed the existence of other genes (marked by triangles). Numbers indicate distances in centimorgans in sex-reversed XY female meiosis.
Figure 2.

Schematic representation of the genomic organization of the sex chromosomes and LG9. Respectively, 383 kb, 194 kb, and 156 kb of the Y chromosome, X chromosome, and LG9 were analyzed. Boxes indicate the predicted coding sequences with similarity to known gene sequences. The red, green, yellow, and blue triangles above the Y-region indicate locations of the Y-specific 551-bp (551), 363-bp (363), 338-bp (338), and 335-bp (335) repeat elements, respectively. In the Y-chromosomal region, vdp, and two copies of Olaflnk, dmrt1bY, and KIAA0914 were identified. In the X-chromosomal BAC clone, pkd2, abcg2, vdp, Olaflnk, and KIAA0914 were identified. The first two genes were only found on the X-chromosomal BAC clone, since the corresponding region on the Y chromosome was not cloned. For gene symbols, see Table 1 and text. The four subregions, the Y-specific region (Y-specific), the region on the Y with identity to LG9 (Y-core), the region on the X corresponding to the sequenced Y chromosome (X-corresponding), and the region on LG9 with similarity to the Y-specific region (LG9-core) are indicated by two-headed arrows.
Table 1.
Genes identified on the medaka sex chromosomes

Genes that were identified in an earlier study as well as those in the present study were either BLASTN- or BLASTX-searched in the database for annotation. The dmrt1bY gene was excluded from this table since this gene resulted from a recent duplication of the autosomal dmrt1a gene. The genes located on human chromosome 4 are shaded.
Sequence analysis of Mn0008J11 from LG9 revealed the presence of the KIAA0172, dmrt1a, dmrt3, and dmrt2 genes, and of a pseudogene, MHCLap (see below), confirming earlier data from cosmid sequences (Brunner et al. 2001). In the X-chromosomal BAC clone, Mn0195H08, five genes with similarity to known genes were identified, namely, polycystic kidney disease 2 (pkd2), ABC transporter G2 (abcg2), vesicle docking protein P115 (vdp), Olaflnk, and KIAA0914. In the BAC clones covering the Y-chromosomal region, five genes with similarity to known genes were identified. These five genes are vdp, two copies of Olaflnk, dmrt1bY, and KIAA0914. For clarity, we named the three Olaflnk genes OlaflnkX (for the copy on X), OlaflnkL, and OlaflnkR (for left and right on Y).
The Olaflnk gene is homologous to the HERC3 gene (HECT domain and RLD3, syn. KIAA0032) in humans and codes for a protein with the N-terminal region showing similarity to the cell cycle regulator RCC1 and a C-terminal HECT domain, which is found in some ubiquitin protein ligases. However, the function of this gene product is unknown. The OlaKIAA0914 gene is homologous to the human FAM13A1 (family with sequence similarity 13, member A1, syn. KIAA0914) gene, and has a conserved RhoGAP domain. The vdp gene is homologous to the human vesicle docking protein p115 gene, encoding a peripheral membrane protein required for transport from the ER to the Golgi apparatus. pkd2 is homologous to the human PKD2 gene, and mutations in this gene in human have been associated with human polycystic kidney disease. The gene product of human ABCG2 (ATP-binding cassette, subfamily G, member 2) belongs to the ATP-binding cassette transporter superfamily.
By comparing the nucleotide sequences of the X- and Y-regions in detail, we could define the borders of the Y-specific region (Fig. 3). The beginning of the Y-specific region lies downstream of the OlaflnkL gene, at position 89,978 of the Y sequence, where the identity to the X sequence is abruptly lost (Fig. 3A). At this side of the Y sequence, the X and Y sequences are almost identical until position 170,762 of the X sequence. That is ∼4 kb downstream of the OlaflnkX gene. Similarly, the end of the Y-specific region could also be identified at position 347,985 (Fig. 3B), upstream of the OlaflnkR gene, and the identity begins at position 143,660 of the X sequence, that is, ∼0.4 kb upstream of the OlaflnkX gene. Thus the Y-specific region (89,978–347,985 of Y) is 258,008 bp in length. The region of 27.1 kb (143,660–170,762 of X) containing the OlaflnkX gene of the X chromosome is duplicated on the Y chromosome. The identities between the X and Y sequences, in regions of 1 kb, were, on the homologous side of the borders 96.9% and 99.5%, at the beginning (88,978–89,977 of Y, corresponding to 169,742–170,762 of X) or the end (347,986–348,985 of Y, corresponding to 143,660–144,662 of X), respectively, excluding gaps. In stark contrast, the beginning (89,978–90,977) and the end (346,986–347,985) of the Y-specific region could not be aligned to the X.
Figure 3.

Identities between the X- and Y-chromosomal sequences at the borders of the Y-specific region. The alignment of the border sequences from the Y (first sequence) and X (second sequence) at the left (A) and right ends (B) of the Y-specific region are shown. Asterisks indicate identical nucleotides.
In the Y-specific region that we define as being between the two Olaflnk genes (Fig. 2), we found the dmrt1bY gene. Close to the dmrt1bY gene, four regions with similarity to other genes, KIAA0172p, MHCLbp, map1p, and dmrt3p, were identified. However, all these four genes have to be considered to be pseudogenes since continuous open reading frames could not be predicted, and all lack more than one exon. KIAA0172p consists of only the last and fourth last exons of the KIAA0172 gene, and dmrt3p consists of the first and second coding exons of dmrt3 but not the third; there is ∼440 bp of sequence inserted in the first exon, and the second exon is truncated.
Evolutionary distances were estimated for the autosomal and Y-chromosomal gene pairs of KIAA0172, dmrt3, and map1. Numbers of nonsynonymous and synonymous substitutions per site (dN and dS, respectively) for each branch after the split of the autosomal and Y-chromosomal sequences were calculated. For dmrt3, the dN/dS ratio was close to 0, indicating that this gene is under purifying selection, while that for dmrt3p was ∼1, and evolution is neutral. This is a pattern typical for pseudogenes. Conversely, both KIAA0172 and KIAA0172p had dN/dS ratios of <1. This suggests that the KIAA0172p sequence was functional until quite recently, or that this sequence still maintains some kind of function, although we could not detect signs of expression, and only two exons corresponding to the autosomal gene could be identified.
The Y-specific region obviously originated from a duplication of the autosomal LG9 region, since counterparts of KIAA0172, dmrt3, and dmrt1a are present. In this region a MHCLap-like sequence was also found. The sequence in Mn0008J11 from LG9 is a pseudogene, since a continuous coding region could not be predicted. Thus the sequence on the Y was named MHCLbp. A copy of dmrt2 was not found in the Y-specific region. Therefore, we defined the Y-core and LG9-core regions from the Y-region and LG9, respectively (Fig. 2), as being the regions that could be aligned to each other, starting between the fourth and fifth last exons of KIAA0172 and ending in the middle of the second coding exon of the dmrt3 gene (31,707–74,608 of LG9 sequence and 266,131–338,192 of Y sequence, respectively). We assume that the duplication of Olaflnk on the Y has occurred during the process of insertion of the duplicated dmrt1-containing region into the proto-Y-chromosome.
In addition, hypothetical genes or reverse-transcriptase-like genes, for example, similar to gypsy-type retrotransposons, and sequences predicted as possible coding sequences but with no similarity to known genes, were found on the X (four sequences) and Y (six sequences).
Several marker genes located on the sex chromosomes of medaka such as caspase 6 and C3-1 genes had been identified in an earlier analysis (Fig. 1B,C; Table 1; Kondo et al. 2001). To identify more genes on the sex chromosome, the BAC library was also screened for clones containing or being in the vicinity of the markers located close to the sex-determining region. From BAC end sequencing, additional sequences with high similarity to genes known from human were found (Table 1): frg1 (from clone Mn0046N20 containing caspase 3), tolloid-like 1 (tll1) (from Mn0065F06 containing caspase 3), and forkhead box K1 (Mn0019I23, the clone next to Mn0036H11 containing caspase 6). Eight of the genes on the medaka sex chromosome (LG1) are located on human chromosome 4q21–35. These are the genes closest to the Y-specific fragment. Yc-2, forkhead box K1, C3-1, C3-2, and dd048, which are not located close to the medaka sex-determining region, are located on different human chromosomes. Hence the region close to the sex-determining region has conserved synteny with human chromosome 4.
In the Y-chromosomal region, between coding exons 4 and 5 of dmrt1bY, a region similar to the human paraneoplastic antigen MA1 gene (PNMA1) was found. We named this map1p. It appears to be a corrupted version, since it has a frameshift mutation, which produces an immaturely terminated protein. A corresponding sequence was not found in the autosomal dmrt1a region. In order to define whether a functional version of map1 exists and to identify the origin of this insertion, map1p was used as a probe to screen the BAC library again. The overlapping clones Mn0030N03 and Mn0037B09 were obtained and considered to contain the functional map1 gene. From BAC end sequencing of Mn0037B09, a sequence that shows homology with the mouse and rat paraneoplastic antigen MA1 was found. This sequence of ∼800 bp had ∼88% identity at the nucleotide level to the Y-chromosomal map1p sequence. At one end of Mn0030N03, another gene, formin 2, similar to the human formin 2 gene (FMN2), was identified. By linkage analysis, both map1 and formin 2 were mapped to medaka LG19 (data not shown). This indicates that the dmrt1bY-containing Y-specific region is not only the result of a chromosomal duplication from LG9, but that there was at least another gene duplication event followed by an insertion that shaped the Y-specific region. The dN/dS ratio indicated purifying selection for map1. In the case of map1p, dS was close to 0, giving a dN/dS ratio far above 1, indicative of adaptive selection. However, since this sequence contains small deletions and multiple frameshifts occur, this gene clearly does not function as protein.
Sequence comparison of the X, Y, and LG9
Dot-plot analyses were performed to directly compare the sequences from the sex chromosomes and the LG9 sequence (Fig. 4). Comparing Y versus X (Fig. 4A), a high degree of sequence conservation at both ends of the sequence, but no similarity in the center, was observed. The region of 258 kb between the two Olaflnk genes is the Y-specific region (Fig. 2). Since there is only one copy of Olaflnk on the X (OlaflnkX), the similarity to the Y-chromosomal OlaflnkL and OlaflnkR is depicted as two parallel lines in the dot plot. An extremely high degree of conservation was observed among the Olaflnk genes (∼22.7 kb), both in the exon and intron sequences (Table 2). There were only three single nucleotide differences in the coding sequences among the three copies. The first one is at positions 75,700 and 360,786 of Y and 156,420 of X, corresponding to OlaflnkL, R, and X, respectively. This is a synonymous substitution, and is common to OlaflnkR and X, and different from OlaflnkL. The second is also synonymous and is shared between the two copies on the Y but differing from the X copy, at positions 77,798 and 362,885 of Y and 158,529 of X. The third, shared between OlaflnkL and X, and different from OlaflnkR, codes for valine for the first two genes, but leucine for OlaflnkR, at positions 84,437 and 369,527 of Y and 165,173 of X. The vdp genes on the X and Y chromosomes are 99.7% and 98.4%, and OlaKIAA0914 genes are 100% and 99.8%, identical in their coding sequences and on the total genomic sequence levels, respectively. All changes in vdp are silent substitutions. These genes could be considered to be in the pseudoautosomal region of the sex chromosomes.
Figure 4.

Comparison of the Y-chromosomal sequence to the X-chromosomal and LG9 sequences. Genes were identified in the sequenced regions of the sex chromosomes and LG9, represented by arrows along the dot plots. Sequences were compared using the Dotter software. Numbers on the right or at the bottom show the length of the sequences in base pairs. The two-headed arrows indicate the subregions (see legend for Fig. 2). The Y-chromosomal sequence region is compared against the X-chromosomal sequence region in A, and to LG9 in B. (A) The X and Y sequences are aligned at the left and right regions, but not in the middle of the Y region. The Olaflnk genes are delimiting the regions with homology. The sequence of OlaflnkR and the region to the right is aligned without gaps to the X sequence, whereas there are gaps in the alignment left to the OlaflnkL gene. There are also clusters of repetitive sequences in this region. Note that there are two stretches of sequence in this region that are inverted in the X sequence (depicted as two short lines running from left to right, upward). (B) The LG9 sequence cannot be aligned to most of the Y region, but there are some stretches of similarity between the sequences in the dmrt1bY and dmrt3p region.
Table 2.
DNA sequence identities (%) among the three Olaflnk genes
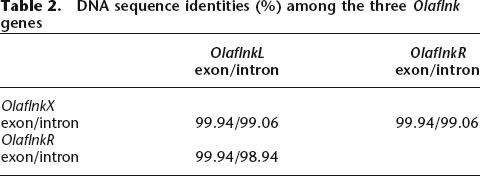
Nucleotides 85,814–179,003 of the X sequence could be aligned with the Y sequence excluding the Y-specific region. Therefore, we named this subregion of 93,190 bp as the X-corresponding region. Outside the Y-specific region, the sequences of ∼12.2 kb downstream of the OlaflnkR or OlaflnkX genes (nucleotides 371,151–383,348 of the Y sequence compared with 166,797–179,003 of the X) are highly conserved (99.90% of 12,197 bp of aligned sequence excluding gaps), with only six small gaps. The sequences of ∼58.3 kb and 63.4 kb upstream of the OlaflnkX or OlaflnkL genes, respectively (nucleotides 85,814–144,066 of the X sequence and nucleotides 1–63,392 of the Y), are of only slightly lower overall identity (99.44% of 39,023 bp of aligned sequence, excluding gaps) but with several gaps and two regions that could not be aligned. They contain two major regions of repetitive sequences only found in the Y sequence, and a short region that was inverted on the X chromosome.
That the Y-specific region originated from the autosomal dmrt1 region (LG9-core) is reflected in the short stretches of identity in the plot of Y versus LG9 (Fig. 4B), indicating the high similarity of exons. However, the sequence similarity was restricted to the exons of the dmrt1a and dmrt1bY genes. The highly repetitive satellite sequences identified in the Y-specific region (see below) were not found in the X-BAC sequence anywhere.
There was no sequence similarity detected between X and LG9 (data not shown).
Repetitive elements in the Y-specific region
Sequences were compared against themselves to identify the presence of gene duplications and satellite sequences (Fig. 5). The Y-region sequence (Fig. 5A) contains a high number of satellite sequences, indicated by the plotted signals. Here again, the two copies of Olaflnk are depicted as two parallel lines at the upper left and lower right of the dot-plot view. Satellite sequences were also found in the X sequence (Fig. 5B), but to a lower degree, and those in the middle of the sequence (Fig. 5B, box c) were also present on the Y chromosome (Fig. 5A, box b). In contrast, there are no satellite sequences in the LG9 sequence (Fig. 5C).
Figure 5.

Identification of internal repeat sequences. The Y region (A), X sequence (B), and LG9 sequence (C) were each compared against themselves, and the results are shown as dot plots. (A) The Y-region contains a high number of repetitive elements, as indicated in boxes a and b, the former being specific to the Y-specific region. The Y-specific region is indicated by a two-headed arrow. The two homologous copies of Olaflnk in the Y-region are depicted as two parallel lines. (B) The X sequence also contains repeats, as well as two stretches of sequences present in the reverse direction. The repeats present in box c, upstream of OlaflnkX, are also present in the Y-region, and are plotted in box b in A. The two-headed arrows indicate the X-corresponding region. (C) The LG9 sequence contains no clustered repetitive sequence.
In the Y-specific region, several satellite sequences are present in high numbers, and with high (almost 100%) similarity between the units. Four of the satellite sequences being 335, 338, 363, and 551 bp in length (e.g., nucleotides 94,214–94,545 [partial sequence], 131,101–131,438, 96,109–96,471, and 97,661–98,211 of the Y chromosome sequence, respectively) (Fig. 2), of 87, 78, 25, and 11 copies, respectively, are present in this region, but they were not found in the sequences of the X chromosome. These repetitive elements are plotted as black squares in the dot plot (Fig. 5A, box a). FISH analyses with all four repeats as a probe (Fig. 6) gave a strong signal on the Y chromosome and an acrocentric chromosome pair, but not with the tip of LG9, where the autosomal dmrt1a is located. This indicates that the repeats expanded after the dmrt1bY-containing duplicated fragment from LG9 inserted into the proto-Y. This process might have contributed to an enlargement of the Y-specific, nonrecombining fragment. It is possible that one or more of the repeats originated from the autosome pair giving the FISH signal. The medaka genome draft assembly database of the NIG DNA Sequencing Center (http://dolphin.lab.nig.ac.jp/medaka/), containing whole-genome shotgun sequence data from the medaka strain Hd-rR, was BLAST-searched using the four satellite sequences as query sequences. As a result, 500, 474, 500, and 97 scaffolds with high identity (E-value < e-100) to the 335-, 338-, 363-, and 551-bp satellites, respectively, were identified. This indicates that these satellite sequences- could be scattered throughout the medaka genome, but only the Y chromosome and the autosomal pair that gave the strong FISH signal contain those in high numbers or in high density.
Figure 6.
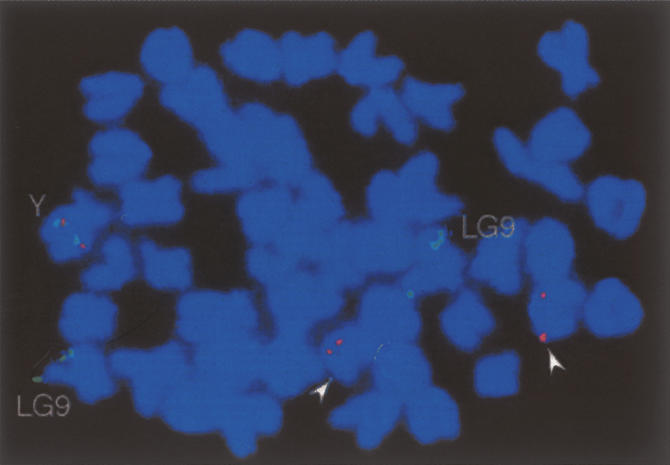
Location of repetitive elements found on the Y chromosome. A metaphase from a male fish showing the hybridization signals for dmrt1a and dmrt1bY (green signals), and the repetitive elements (335-bp, 338-bp, 363-bp, and 551-bp repeat) (red signals). These repetitive elements were initially identified on the Y chromosome in the Y-specific region. Red signals are located together with the green signal on the Y chromosome (Y), but are also detected on another pair of acrocentric chromosomes (arrowheads). The green signals at the tip of another pair of chromosomes represent the autosomal (LG9) dmrt1a gene. Consistent with the nucleotide sequence analyses, the repeats are not found on the X chromosome nor colocalize with the autosomal dmrt1a.
The sequences from the sex chromosomes as well as from LG9 were searched for repetitive elements (Table 3) using the RepeatMasker2 program and fugu as the DNA source for comparison. The total percentages of sequences that were identified as repetitive elements was not obviously different between different regions. This is in contrast to what was found in stickleback (Peichel et al. 2004), where a large difference was seen between the X and Y chromosome sequences (0.86% and 5.44%, respectively). However, when the sequences were masked using an original masking database (Keio Medaka Repeat Database 1.0), consisting of 20 Mb of medaka LG22 sequence (T. Sasaki, A. Shimizu, S.K. Ishikawa, S. Imai, S. Asakawa, Y. Murayama, M.Z. Khorasani, H. Mitani, M. Furutani-Seiki, H. Kondoh, et al., in prep.) and the publicly available RepBase of fugu and zebrafish sequences, 26.3%, 27.9%, and 44.8% of the sequences were masked from the entire LG9, X-chromosomal, and Y-chromosomal sequences, respectively. This suggests that the currently available masking database does not mask all the satellite sequences in medaka. Since in our database the repetitive elements are not classified into categories such as SINEs and LINEs, we were not able to assign the repetitive sequences into such categories. However, when we compared the corresponding sequences from the different chromosomes, the accumulation of satellite sequences on the Y chromosome was obvious. More than half (53.2%) of the Y-specific region (258 kb) was masked. The total Y region (383.3 kb) compared to the corresponding X sequence (93.2 kb) was richer in satellite sequences (44.8% vs. 24.0%). The Y-core sequence (72.1 kb) was longer, and about twice as much masked as the corresponding LG9-core sequence (42.9 kb; 45.3% vs. 20.5%).
Table 3.
Repeats in the Y-region, X, and LG9-sequences

Sequences were masked using the fugu database to identify the repeat types and the frequency.
aThe bottom row shows the percentage of masked sequence using the original database consisting of medaka, fugu, and zebrafish sequences.
The Y-core and LG9-core sequences were compared using the VISTA software. The Y-core sequence was compared without masking (Fig. 7). The long stretches of satellite sequences between KIAA0172p and MHCLbp and within dmrt1bY did not match the LG9-core sequence. These satellite sequences could be considered as those inserted after the duplication of dmrt1, and therefore explaining the expansion in length of this region. The distance between KIAA0172 and dmrt1 is 6.7 kb on the autosome, opposed to 14.9 kb on the Y. From the translation start point to the stop codon, the dmrt1a gene spans 26.6 kb, whereas dmrt1bY is 49.9 kb in length. Satellite sequences also exist in the LG9-core sequence, and in dmrt1a, but are shorter, and roughly half of those have corresponding sequences in the Y-core sequence (data not shown).
Figure 7.

Comparison of the Y-core and LG9-core sequences. The Y-core sequence (72.1 kb) (shaded yellow) and ∼4 kb each on both sides was compared to the LG9-core sequence (42.9 kb) and ∼5 kb each on both sides using VISTA. The level of conservation (vertical axis) is displayed in the coordinates of the Y-core sequence (horizontal axis). Conserved regions above the level of 70%/100 bp are shaded under the curve, with pink indicating a conserved non-coding region, purple a conserved exon, and green a pseudogene. The light blue shades label the masked sequences using our database.
Expression analysis
The expression of sequences that appear to encode transcription units was analyzed by RT-PCR (Fig. 8). The expression of dmrt1bY has been analyzed in detail earlier. The gene is expressed during embryogenesis of male embryos starting around the neurula stage (Nanda et al. 2002). In larvae the mRNA was found in the somatic cells of the developing male gonad, possibly pre-Sertoli cells (Matsuda et al. 2002). In adult fish, dmrt1bY is exclusively expressed in the Sertoli cells of the testes (Nanda et al. 2002).
Figure 8.

Expression analyses. Expression of genes in the Y-core and LG9-core sequences and Olaflnk was analyzed by RT-PCR. (A) The autosomal KIAA0172 is expressed in all tissues examined. (B) The expression of the autosomal map1 gene was detected in all tissues examined, but in slightly lower amounts in females. Using primers specific for map1p, the presence of transcripts was detected only in the testis. (C) Olaflnk was expressed in all tissues examined. The high identity of the three copies made it impossible to distinguish the transcripts from the three copies. Actin was used to calibrate for RNA amounts; the control is PCR without template.
Only the last and fourth last exons of the autosomal KIAA0172 gene exist as the Y-chromosomal copy of KIAA0172 (KIAA0172p); thus the RT-PCR products could be easily distinguished by the size difference. While the autosomal copy is ubiquitously expressed in all tested organs of males and females, no transcripts from the Y-chromosomal copy could be detected in any organ (Fig. 8A).
Primers amplifying either the autosomal or the Y-chromosomal copies of the map1 gene were designed. RT-PCR revealed a ubiquitous expression for the autosomal gene. Surprisingly, the Y-chromosomal gene was exclusively expressed in testes (Fig. 8B). As map1 on the Y chromosome (map1p) resides in intron 4 of dmrt1bY, is encoded on the same DNA strand, and exhibits the same expression pattern, the hypothesis was tested whether the RT-PCR product stems from hnRNA and not from a mature transcript. RT-PCR was performed with primers that give products spanning exon/intron borders (exon1/intron1, exon4/intron4, intron4/exon5) of dmrt1bY. Products of the expected size were obtained from testis RNA, but not from any other organ (data not shown). This shows the presence of unspliced dmrt1bY transcripts that should contain map1p in intron 4. Neither in the intron sequence between exon 4 and map1p nor upstream of this region was a promoter predicted. Thus, map1p in the Y-specific region may be not expressed in the form of a mature transcript.
The X- and both Y-chromosomal copies of Olaflnk are so similar that it was impossible to design specific primers. By RT-PCR, transcripts were found in all organs from males and females (Fig. 8C). To answer the question whether the Y-chromosomal copies are expressed, testis RNA from YY males was analyzed. Expression of Olaflnk was readily detected. The RT-PCR product was cloned, and 40 clones were sequenced. This revealed, on the basis of single nucleotide differences, that predominantly the left copy is expressed at a ratio of 39:1.
Outside of the Y-specific region, the vdp gene was checked for expression in adult tissues, using cDNA from testis of YY males and ovary, eyes, and liver of XX females. PCR products of expected sizes were obtained from all tissues tested, indicating that both the X- and Y-chromosomal copies are expressed (data not shown).
Discussion
The complete sequences of the sex-determining region of the medaka Y chromosome and the corresponding segment on the X give information that can be used to infer some of the events that shaped the structure of these sex chromosomes during their evolutionary history.
First, there is no homology of the Y-specific fragment with the X. All sequences are either derived from the piece of LG9 that was duplicated and inserted into the proto-Y or from elsewhere in the genome. This is different from the prediction of the genetic theory of sex chromosome evolution (Charlesworth 1978; Charlesworth et al. 2005), where proto-sex chromosomes arise from a stepwise allelic diversification at one locus of a chromosome pair. It is possible that the situation in medaka is an example for a single-gene sex determination as in the fly Musa domestica (Shearman 2002), where a new sex-determining gene becomes epistatic to the previous sex determiners, so that the original genes no longer control sex development. Unfortunately, we have not enough data from the other Oryzias species, which do not possess the dmrt1bY gene. Some of these species have XY-XX, others ZW-ZZ systems, but chromosome homologies have not been established (Hamaguchi et al. 2004). None of these are heteromorphic, thus they might represent young sex chromosomes as well. Environmental sex determination might be the ancestral state, but this has not been studied, except for the fact that in medaka high temperature can override the genetic system leading to fully fertile XX males (Sato et al. 2005).
The first stage in the evolution of a Y chromosome, in which a single dominant factor like dmrt1bY determines maleness, has been called “genic sex determination” (Rice 1996). Following a trajectory based on cytogenetic findings on a variety of fish Y chromosomes (Kirpichnikov 1981), the next stage is reached when the Y chromosome stops recombining with the X in a limited region (the differential segment) around the sex-determining gene. This situation has been called “semichromosomal sex determination” (Rice 1996). In the medaka Y, there is a differential segment, the Y-specific fragment (Fig. 2), which does not recombine with the X, owing to a total lack of homology. Also outside this Y-specific fragment, a region of ∼30 cM on the X (or proto-X) shows strongly reduced recombination frequencies for homologous markers on the Y (or proto-Y) (Kondo et al. 2001).
Another hallmark of Y chromosome evolution is degeneration. This has happened to a high degree in the Y-specific region. All genes that have been coduplicated with dmrt1 are inactivated by mutations. Also map1, which was inserted into intron 4 of dmrt1bY, has acquired frameshift mutations.
It is well known that duplicated genes evolve into pseudogenes (Wagner 2001; Samonte and Eichler 2002). While dmrt1bY has evolved a novel function in triggering male development, all other genes became redundant. The Y-specific segment is small, and the evolutionary factors needed to produce degeneration as a result of lack of recombination will not be very effective (Charlesworth and Charlesworth 2000; Gordo and Charlesworth 2000). Thus the degeneration seen on the medaka Y may be different from the usual Y chromosome situation. However, the strong accumulation of repeats on the Y-specific segment might be a result of absence of recombination. So far, from the study of duplicated segments in other genomes, there is no indication that repeats specifically accumulate there. On the other hand, suppressed recombination causes the accumulation of repetitive sequences (Charlesworth et al. 1986, 1994). On the neo-Y chromosome of Drosophila miranda, which was formed only 1.2 Mya, a massive accumulation of transposable elements was found (Bachtrog 2005; Steinemann and Steinemann 2005).
Another step in sex chromosome evolution is that the non-recombining part of the Y spreads out from the sex-determining genes, leading to a continuous shrinking of the pseudoautosomal region. It appears as if this has not happened to a great extent during the ∼10 million years of evolution of the medaka Y (Kondo et al. 2004), due to a peculiar genomic structure of the Y-specific fragment.
The left and right copies of Olaflnk that flank the Y-specific region are highly similar to each other (nucleotide identity in exons 99.94%, in introns 98.94%) and to the X-chromosomal copy (nucleotide identity in exons 99.94%, in introns 99.06%). In contrast, dmrt1a and dmrt1bY share only a similarity of 92.4% in exons and an even lower one (48.1%) in introns. It is reasonable to assume that the duplication of dmrt1 and the insertion event that created the two Y-chromosomal copies of Olaflnk occurred at the same time. The extreme sequence similarity of the Olaflnk copies, as opposed to the lower similarity of the dmrt1 copies, can be explained by assuming that both Y-chromosomal Olaflnk copies undergo crossing-over with the X-chromosomal copy in an alternating way during meiosis, which makes all three copies alike (Fig. 9). This may be supported by the fact that from the three nucleotide differences found between the three Olaflnk copies, the most 5′ one in the X is shared with the left copy on the Y and the most 3′ is shared with the right copy on the Y. This could be explained by intragenic crossovers of the X copy with both Y copies.
Figure 9.

Hypothetical scheme of pairing of X and Y chromosomes during meiosis. Since the X and Y chromosomes are highly homologous, the left and right borders of the Y-specific region could pair, either by the X copy of Olaflnk pairing with the L copy and the Y-specific region, while the R copy and the Y-specific region are looping out, or by OlaflnkX pairing with OlaflnkR, and OlaflnkL and the Y-specific region looping out.
An alternative explanation for the high sequence conservation of both Olaflnk copies on the Y could be X/Y crossing-over between one peculiar Y copy and the X copy, and gene conversion between the two copies on the Y chromosome.
The regions downstream of the OlaflnkR gene and upstream of the OlaflnkL gene are highly similar between both sex chromosomes. There are, however, some small islands of sequence divergence visible. Whether these differences are already sufficient to interfere with chromosome pairing and crossing-over in meiosis is not clear, but they are certainly the first sign of differentiation of the Y and X in the homologous (pseudoautosomal) regions. For instance, around the vdp gene, which obviously undergoes X/Y crossing-over (98.4% sequence identity) and which is functional on both chromosomes, there is an obvious accumulation of satellite sequences on the Y as well as a small inversion (Fig. 4A). The differences between the X and the Y away from the inserted region could be interpreted as polymorphisms within the species. This is, however, very unlikely, as we used a highly inbred strain (over 30 generations of brother–sister matings) for our analysis. In fact, using another, but similarly highly inbred strain of medaka for the complete BAC clone by clone sequencing of linkage group 22 (T. Sasaki, A. Shimizu, S.K. Ishikawa, S. Imai, S. Asakawa, Y. Murayama, M.Z. Khorasani, H. Mitani, M. Furutani-Seiki, H. Kondoh, et al., in prep.), no polymorphisms were detected between overlapping clones. This indicates that the differences outside the Y-specific region may reflect a reduced recombination and sequence diversification between the sex chromosomes. The analyses of larger parts also from the region downstream of OlaflnkR and the comparison of sequences from different Y and X chromosomes of medaka are necessary to answer this question.
It can be predicted that once one of the two copies of Olaflnk on the Y diverges in sequence or is lost, the region of restricted recombination will spread out there. Interestingly, the left copy is much more abundantly expressed than the right one, indicating that the latter could become dispensable.
The extremely small region of differentiation between the medaka X and Y is in stark contrast to other “young” Y chromosomes for which sequence information is available. In the stickleback, the Y chromosome has been estimated to be also ∼10 Myr old. Here, a considerable sequence divergence between X- and Y-chromosomal BAC clones, which are derived from regions quite distant from the sex-determining locus, is readily recognizable (Peichel et al. 2004). In the papaya plant, the sex-determining region spans ∼4–5 Mb (Liu et al. 2004). In the well-studied sex chromosomes of the plant Silene latifolia, sequence divergence of expressed genes on the X and the Y increases over a large distance (50 cM) from the pseudoautosomal region toward the sex-determining locus (Nicolas et al. 2005).
So far, dmrt1bY appears to be the only functional gene in the Y-specific region. Mutations in dmrt1bY lead to male-to-female sex reversal (Matsuda et al. 2002), showing that this gene is necessary for male sex determination in these genotypes. The question whether it is also sufficient has not been answered. The fact that it appears to be the only functional gene in the Y-specific fragment might point toward such an argument. However, transgenic experiments are necessary to conclusively answer this question.
Degeneration of genes on the Y chromosome of medaka is obviously restricted to the duplicated fragment from LG9. No mutations that would be predicted to interfere with gene function were detected in sequences outside of the Y-specific fragment, and all genes outside that are studied so far have alleles on the X and Y and appear to be functional. This is in line with the fact that YY males of medaka are fully viable (Yamamoto 1965).
Our sequence analysis also did not uncover another hallmark of Y chromosome evolution, the acquisition of male-specific genes (Vallender and Lahn 2004), for example, spermatogenesis genes. This is in agreement with the observation that XX males of medaka are not compromised in fertility (Nanda et al. 2003). Loss of essential genes and accumulation of male beneficial genes on the Y are processes possibly restricted to sex chromosomes, which are much older, like the mammalian Y. However, more “young” Y chromosomes need to be analyzed to draw conclusions on how “early” or “late” during the evolutionary history such changes occur.
Ancient sex chromosomes are typically composed of several temporal clusters, so-called evolutionary strata (Lahn and Page 1999), that represent the sequential acquisition of genes or the sequence divergence between X- and Y-linked genes. Four such strata have been identified on the human Y (Skaletsky et al. 2003; Fraser et al. 2004) and the MAT locus of the fungal pathogen Cryptococcus (Fraser et al. 2004), while the Y chromosomes of Silene have at least three. In medaka, we found only one temporal cluster represented by the degenerated duplicated fragment from the autosomal LG9.
The medaka homologs of human HERC3, FAM13A1, FRG1, TLL1, CASP3, and CASP6 were identified on the sex chromosomes. They are all located on human chromosome 4, indicating conserved synteny between a human autosome and the region in medaka, which was the target for the insertion of the duplicated dmrt1. For the stickleback sex chromosomes, a region of conserved synteny to human chromosome 15 was found near the sex-determining locus (Peichel et al. 2004). This confirms an independent origin of both X/Y sex chromosomal systems.
In summary, the origin of the male sex-determining gene in medaka from a local gene duplication event had certain consequences for the evolution of the Y chromosome. The prospective sex-determining locus was isolated from recombination with the X ab initio, because of a total lack of homologous sequences in the newly inserted fragment on the proto-Y chromosome. A large target sequence duplication (Olaflnk) that was probably generated during the process of insertion of the duplicated fragment into the proto-Y has limited the spreading out of the genetic degeneration from the Y-specific region or at least slowed down for some time. It will be interesting to compare the situation found for the medaka with the other “young” Y chromosomes from papaya, Silene, and stickleback, once the sequence information about the male sex-determining locus and their surrounding chromosomal regions will be available.
Methods
Medaka fish
All experimental animals were from an inbred line of the northern Japanese population of medaka (HNI) and from an outbred strain derived from the southern Japanese population of medaka (Carbio), which were kept at the fish facilities of the University of Tokyo and the University of Wuerzburg, respectively. To obtain YY males, at first XY females from the Carbio strain were produced by estrogen treatment of embryos (Iwamatsu 1999). These females were mated to males of HNI. The YY males were identified by PCR by checking for the presence of both Y chromosomes of Carbio and HNI. For the Carbio Y chromosome, primers DMT1k, 5′-CAACTTTGTCCAAACTCTGA-3′, and DMT1l, 5′-AACTAATTCATCCCCATTCC-3′ were used to amplify the dmrt1bY gene, and for HNI, primers Casp6-intf2, 5′-TAGCA CTTTCACATTTCCAAGC-3′, and Casp6R, 5′-CGTCTCTCGAT GAGAATAGAAACC-3′ were used to test for the presence of the Y-specific copy of the caspase 6 gene (Kondo et al. 2001).
Isolation and DNA sequencing of BAC clones
A 20-fold genome coverage BAC library from the HNI strain of medaka was screened as described previously (Kondo et al. 2002; Nanda et al. 2002). Briefly, a PCR fragment of the highly conserved DM domain of medaka dmrt4 (Kondo et al. 2002) was used as a probe for hybridization under low stringency, and several BAC clones containing dmrt genes were obtained. Of the isolated clones, clones Mn0015H17 and Mn0008J11were identified to contain dmrt1bY and dmrt1a, respectively, based on PCR amplification of the DM domain region and sequencing of the PCR products. The dmrt1bY BAC contig was extended by using PCR-amplified fragments from BAC ends for screening the HNI BAC library. Clones Mn0209O12, Mn0095J10, Mn0168M02, Mn0105G20, Mn0113N21, Mn0125L10, and Mn0195H08 were obtained. The clones were mapped in relation to each other by PCR of BAC end sequences, or by HindIII fingerprints. Mn0195H08 did not fit into the contig built up by the other clones (described in Results), and later was found to correspond to the X chromosome. Of these clones, Mn0113N21, Mn0168M02, Mn0209O12, Mn0195H08, and Mn0008J11 were selected for shotgun sequencing. Using a PCR-amplified map1p fragment as a probe, BAC clones containing map1 (Mn0030N03 and Mn0037B09) were obtained.
Using marker sequences caspase 6 (GenBank AB047772) and caspase 3B (GenBank AB032608) from the vicinity of the sex-determining region, clones Mn019I23, Mn0036H11, Mn0046N20, and Mn0065F06 were isolated, and their ends were sequenced.
Nucleotide sequences of entire BAC clones were determined by the shotgun method and assembled by Phred/Phrap/Consed, except for the satellite sequence region (Gordon et al. 1998). The nucleotide sequences of satellite sequence regions were assembled manually using GAP4. The sequence gaps were filled by primer walking.
BAC DNA sequences are deposited in GenBank under the accession numbers AP006150–AP006154.
Sequence analysis
To identify possible coding sequences, sequences were first analyzed by application of the NIX program package (http://www.hgmp.mrc.ac.uk/NIX/). Putative coding sequences, which were predicted by multiple prediction programs such as GRAIL and GenScan, were BLAST-searched for sequences with identity to known genes and were selected for annotation.
Nucleotide sequence identities were analyzed using GENETYX-MAC Version 11.1.0.
Repetitive sequences were masked using RepeatMasker2 (A.F.A. Smit, R. Hubley, and P. Green, unpubl., http://www.repeatmasker.org) against the fugu database or a generated database consisting of fugu, zebrafish, and medaka sequences. For comparison of repetitive element content, the sequences were divided into corresponding regions as follows: Y-core (266,131–338,192 bp of the Y region), Y-specific (89,978–347,985 bp of the Y region), X-corresponding (85,814–179,003 bp of Mn0195H08), and LG9-core (31,707–74,608 bp of Mn0008J11).
Dot-plot analysis was performed with the software Dotter (Sonnhammer and Durbin 1995), using the assembled BAC DNA sequences.
Molecular evolutionary analyses were conducted using MEGA version 3.1 (Kumar et al. 2004). Evolutionary distances of the autosomal and pseudogene sets were calculated by synonymous–nonsynonymous substitutions with the modified Nei-Gojobori method (Jukes-Cantor correction) included in this software. Fugu dmrt3 (AB201463), Tetraodon nigroviridis chromosome 12 sequence (SCAF14996; http://www.genoscope.cns.fr/externe/tetranew/), and zebrafish (XM_692055) sequences were used for comparison. The latter two sequences were assumed to be homologous to KIAA0172 and map1, respectively, since these sequences were identified from BLAST searches. Sequence alignments were refined manually.
FISH analysis
For FISH analysis, two BAC clones (Mn0008J11 and Mn0015H17) containing the autosomal dmrt1a and the Y-specific dmrt1bY, respectively, as well as a plasmid subclone of Mn0209O12 (1P14) containing four kinds of satellite sequences (dmrt1bY 338-bp repeat, dmrt1bY 363-bp repeat, Md0147L05 335-bp repeat, and dmrt1bY 551-bp repeat) were labeled either with digoxigenin-16-dUTP or biotin-16-dUTP. Mitotic metaphase spreads from medaka were prepared using standard methodology, and dual-color FISH was performed as described (Nanda et al. 2002). A minimum of 10 representative chromosome spreads was analyzed to evaluate the genomic locations of the probes used.
Expression analyses
Total RNA was extracted from pooled organs of several adult male or female medaka using the TRIZOL reagent (Invitrogen) according to the supplier’s recommendation. After DNase treatment, reverse transcription was done with 2 μg of total RNA using Superscript II Reverse Transcriptase (Invitrogen) and random primers. cDNA from 10 ng (actin) or 200 ng of total RNA was used for PCR with gene-specific primers (Supplemental table). For negative control, RNA that was not reverse-transcribed was used in the PCR reaction. RT-PCR products were cloned into a pCRII-TOPO vector using the TOPO TA-Cloning Kit (Invitrogen) and sequenced.
Linkage analyses
The male meiotic mapping panel consisting of 39 backcross progeny from the medaka strains HNI and AA2 was used as described previously (Naruse et al. 2000). For mapping of formin 2, primers F30N03F, 5′-TCAGAGTACCTGCTGCCAAATT-3′, and F30N03R, 5′-AATGTGCATTCATGGAGTTGG-3′; for map1, primers F37B09F, 5′-ATGGGTCTGATGCGGAGAA-3′, and F37B09R, 5′-TC TTTCGAGGCGTCTCAGAA-3′ were used for amplification. The former pair of primers amplified a 497-bp product only from HNI but not AA2, and the latter pair produced a 545-bp product from HNI and a slightly larger product from AA2. The PCR products were electrophoresed on 12% polyacrylamide gels for typing. Segregation analysis was done with MAPMAKER (Apple Macintosh version 2).
Acknowledgments
We thank G. Schneider, H. Schwind, and P. Weber, University of Wuerzburg, for breeding of the fish. This work was supported by grants supplied by the Deutsche Forschungsgemeinschaft through Graduiertenkolleg 637 “Organogenesis” and Fonds der Chemischen Industrie to M. Schartl, a grant from the Deutsche Forschungsgemeinschaft (SCHM 484/18-1) to M. Schmid, and the Ito Grant for Ichthyology, Fujiwara Natural History Foundation to M. Kondo.
Footnotes
[Supplemental material is available online at www.genome.org. The sequence data from this study have been submitted to GenBank under accession nos. AP006150–AP006154.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.5016106
References
- Aida T. On the inheritance of color in a fresh-water fish, Aplocheilus latipes Temmick and Schlegel, with special reference to sex-linked inheritance. Genetics. 1921;6:554–573. doi: 10.1093/genetics/6.6.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtrog D. Sex chromosome evolution: Molecular aspects of Y-chromosome degeneration in Drosophila. Genome Res. 2005;15:1393–1401. doi: 10.1101/gr.3543605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtrog D., Charlesworth B., Charlesworth B. Towards a complete sequence of the human Y chromosome. Genome Biol. 2001;2:reviews1016. doi: 10.1186/gb-2001-2-5-reviews1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner B., Hornung U., Shan Z., Nanda I., Kondo M., Zend-Ajusch E., Haaf T., Ropers H.H., Shima A., Schmid M., Hornung U., Shan Z., Nanda I., Kondo M., Zend-Ajusch E., Haaf T., Ropers H.H., Shima A., Schmid M., Shan Z., Nanda I., Kondo M., Zend-Ajusch E., Haaf T., Ropers H.H., Shima A., Schmid M., Nanda I., Kondo M., Zend-Ajusch E., Haaf T., Ropers H.H., Shima A., Schmid M., Kondo M., Zend-Ajusch E., Haaf T., Ropers H.H., Shima A., Schmid M., Zend-Ajusch E., Haaf T., Ropers H.H., Shima A., Schmid M., Haaf T., Ropers H.H., Shima A., Schmid M., Ropers H.H., Shima A., Schmid M., Shima A., Schmid M., Schmid M., et al. Genomic organization and expression of the doublesex-related gene cluster in vertebrates and detection of putative regulatory regions for dmrt1. Genomics. 2001;77:8–17. doi: 10.1006/geno.2001.6615. [DOI] [PubMed] [Google Scholar]
- Charlesworth B. Model for evolution of Y chromosomes and dosage compensation. Proc. Natl. Acad. Sci. 1978;75:5618–5622. doi: 10.1073/pnas.75.11.5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B. The evolution of sex chromosomes. Science. 1991;251:1030–1033. doi: 10.1126/science.1998119. [DOI] [PubMed] [Google Scholar]
- Charlesworth B., Charlesworth D., Charlesworth D. The degeneration of Y chromosomes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2000;355:1563–1572. doi: 10.1098/rstb.2000.0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B., Langley C.H., Stephan W., Langley C.H., Stephan W., Stephan W. The evolution of restricted recombination and the accumulation of repeated DNA sequences. Genetics. 1986;112:947–962. doi: 10.1093/genetics/112.4.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B., Sniegowski P., Stephan W., Sniegowski P., Stephan W., Stephan W. The evolutionary dynamics of repetitive DNA in eukaryotes. Nature. 1994;371:215–220. doi: 10.1038/371215a0. [DOI] [PubMed] [Google Scholar]
- Charlesworth D., Charlesworth B., Marais G., Charlesworth B., Marais G., Marais G. Steps in the evolution of heteromorphic sex chromosomes. Heredity. 2005;95:118–128. doi: 10.1038/sj.hdy.6800697. [DOI] [PubMed] [Google Scholar]
- Fraser J.A., Diezmann S., Subaran R.L., Allen A., Lengeler K.B., Dietrich F.S., Heitman J., Diezmann S., Subaran R.L., Allen A., Lengeler K.B., Dietrich F.S., Heitman J., Subaran R.L., Allen A., Lengeler K.B., Dietrich F.S., Heitman J., Allen A., Lengeler K.B., Dietrich F.S., Heitman J., Lengeler K.B., Dietrich F.S., Heitman J., Dietrich F.S., Heitman J., Heitman J. Convergent evolution of chromosomal sex-determining regions in the animal and fungal kingdoms. PLoS Biol. 2004;2:e384. doi: 10.1371/journal.pbio.0020384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordo I., Charlesworth B., Charlesworth B. On the speed of Muller’s ratchet. Genetics. 2000;156:2137–2140. doi: 10.1093/genetics/156.4.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D., Abajian C., Green P., Abajian C., Green P., Green P. Consed: A graphical tool for sequence finishing. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- Graves J.A. Interactions between SRY and SOX genes in mammalian sex determination. Bioessays. 1998;20:264–269. doi: 10.1002/(SICI)1521-1878(199803)20:3<264::AID-BIES10>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Graves J.A. Evolution of the testis-determining gene—The rise and fall of SRY. Novartis Found. Symp. 2002;244:86–97. [PubMed] [Google Scholar]
- Hamaguchi S., Toyazaki Y., Shinomiya A., Sakaizumi M., Toyazaki Y., Shinomiya A., Sakaizumi M., Shinomiya A., Sakaizumi M., Sakaizumi M. The XX–XY sex-determination system in Oryzias luzonensis and O. mekongensis revealed by the sex ratio of the progeny of sex-reversed fish. Zoolog. Sci. 2004;21:1015–1018. doi: 10.2108/zsj.21.1015. [DOI] [PubMed] [Google Scholar]
- Iwamatsu T. Convenient method for sex reversal in a freshwater teleost, the medaka. J. Exp. Zool. 1999;283:210–214. [Google Scholar]
- Kirpichnikov V.A. Genetic basis of fish selection. Springer-Verlag; New York: 1981. [Google Scholar]
- Kondo M., Nagao E., Mitani H., Shima A., Nagao E., Mitani H., Shima A., Mitani H., Shima A., Shima A. Differences in recombination frequencies during female and male meioses of the sex chromosomes of the medaka, Oryzias latipes. Genet. Res. 2001;78:23–30. doi: 10.1017/s0016672301005109. [DOI] [PubMed] [Google Scholar]
- Kondo M., Froschauer A., Kitano A., Nanda I., Hornung U., Volff J.N., Asakawa S., Mitani H., Naruse K., Tanaka M., Froschauer A., Kitano A., Nanda I., Hornung U., Volff J.N., Asakawa S., Mitani H., Naruse K., Tanaka M., Kitano A., Nanda I., Hornung U., Volff J.N., Asakawa S., Mitani H., Naruse K., Tanaka M., Nanda I., Hornung U., Volff J.N., Asakawa S., Mitani H., Naruse K., Tanaka M., Hornung U., Volff J.N., Asakawa S., Mitani H., Naruse K., Tanaka M., Volff J.N., Asakawa S., Mitani H., Naruse K., Tanaka M., Asakawa S., Mitani H., Naruse K., Tanaka M., Mitani H., Naruse K., Tanaka M., Naruse K., Tanaka M., Tanaka M., et al. Molecular cloning and characterization of DMRT genes from the medaka Oryzias latipes and the platyfish Xiphophorus maculatus. Gene. 2002;295:213–222. doi: 10.1016/s0378-1119(02)00692-3. [DOI] [PubMed] [Google Scholar]
- Kondo M., Nanda I., Hornung U., Schmid M., Schartl M., Nanda I., Hornung U., Schmid M., Schartl M., Hornung U., Schmid M., Schartl M., Schmid M., Schartl M., Schartl M. Evolutionary origin of the medaka Y chromosome. Curr. Biol. 2004;14:1664–1669. doi: 10.1016/j.cub.2004.09.026. [DOI] [PubMed] [Google Scholar]
- Kumar S., Tamura K., Nei M., Tamura K., Nei M., Nei M. MEGA3: Integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief. Bioinform. 2004;5:150–163. doi: 10.1093/bib/5.2.150. [DOI] [PubMed] [Google Scholar]
- Lahn B.T., Page D.C., Page D.C. Four evolutionary strata on the human X chromosome. Science. 1999;286:964–967. doi: 10.1126/science.286.5441.964. [DOI] [PubMed] [Google Scholar]
- Lahn B.T., Pearson N.M., Jegalian K., Pearson N.M., Jegalian K., Jegalian K. The human Y chromosome, in the light of evolution. Nat. Rev. Genet. 2001;2:207–216. doi: 10.1038/35056058. [DOI] [PubMed] [Google Scholar]
- Liu Z., Moore P.H., Ma H., Ackerman C.M., Ragiba M., Yu Q., Pearl H.M., Kim M.S., Charlton J.W., Stiles J.I., Moore P.H., Ma H., Ackerman C.M., Ragiba M., Yu Q., Pearl H.M., Kim M.S., Charlton J.W., Stiles J.I., Ma H., Ackerman C.M., Ragiba M., Yu Q., Pearl H.M., Kim M.S., Charlton J.W., Stiles J.I., Ackerman C.M., Ragiba M., Yu Q., Pearl H.M., Kim M.S., Charlton J.W., Stiles J.I., Ragiba M., Yu Q., Pearl H.M., Kim M.S., Charlton J.W., Stiles J.I., Yu Q., Pearl H.M., Kim M.S., Charlton J.W., Stiles J.I., Pearl H.M., Kim M.S., Charlton J.W., Stiles J.I., Kim M.S., Charlton J.W., Stiles J.I., Charlton J.W., Stiles J.I., Stiles J.I., et al. A primitive Y chromosome in papaya marks incipient sex chromosome evolution. Nature. 2004;427:348–352. doi: 10.1038/nature02228. [DOI] [PubMed] [Google Scholar]
- Matsuda M. Sex determination in the teleost medaka, Oryzias latipes. Annu. Rev. Genet. 2005;39:293–307. doi: 10.1146/annurev.genet.39.110304.095800. [DOI] [PubMed] [Google Scholar]
- Matsuda M., Matsuda C., Hamaguchi S., Sakaizumi M., Matsuda C., Hamaguchi S., Sakaizumi M., Hamaguchi S., Sakaizumi M., Sakaizumi M. Identification of the sex chromosomes of the medaka, Oryzias latipes, by fluorescence in situ hybridization. Cytogenet. Cell Genet. 1998;82:257–262. doi: 10.1159/000015113. [DOI] [PubMed] [Google Scholar]
- Matsuda M., Sotoyama S., Hamaguchi S., Sakaizumi M., Sotoyama S., Hamaguchi S., Sakaizumi M., Hamaguchi S., Sakaizumi M., Sakaizumi M. Male-specific restriction of recombination frequency in the sex chromosomes of the medaka, Oryzias latipes. Genet. Res. 1999;73:225–231. [Google Scholar]
- Matsuda M., Nagahama Y., Shinomiya A., Sato T., Matsuda C., Kobayashi T., Morrey C.E., Shibata N., Asakawa S., Shimizu N., Nagahama Y., Shinomiya A., Sato T., Matsuda C., Kobayashi T., Morrey C.E., Shibata N., Asakawa S., Shimizu N., Shinomiya A., Sato T., Matsuda C., Kobayashi T., Morrey C.E., Shibata N., Asakawa S., Shimizu N., Sato T., Matsuda C., Kobayashi T., Morrey C.E., Shibata N., Asakawa S., Shimizu N., Matsuda C., Kobayashi T., Morrey C.E., Shibata N., Asakawa S., Shimizu N., Kobayashi T., Morrey C.E., Shibata N., Asakawa S., Shimizu N., Morrey C.E., Shibata N., Asakawa S., Shimizu N., Shibata N., Asakawa S., Shimizu N., Asakawa S., Shimizu N., Shimizu N., et al. DMY is a Y-specific DM-domain gene required for male development in the medaka fish. Nature. 2002;417:559–563. doi: 10.1038/nature751. [DOI] [PubMed] [Google Scholar]
- Muller H.J. A gene for the fourth chromosome of Drosophila. J. Exp. Zool. 1914;17:325–336. [Google Scholar]
- Muller H.J. Genetic variability, twin hybrids and constant hybrids, in a case of balanced lethal factors. Genetics. 1918;3:422–499. doi: 10.1093/genetics/3.5.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanda I., Shan Z., Schartl M., Burt D.W., Koehler M., Nothwang H., Grutzner F., Paton I.R., Windsor D., Dunn I., Shan Z., Schartl M., Burt D.W., Koehler M., Nothwang H., Grutzner F., Paton I.R., Windsor D., Dunn I., Schartl M., Burt D.W., Koehler M., Nothwang H., Grutzner F., Paton I.R., Windsor D., Dunn I., Burt D.W., Koehler M., Nothwang H., Grutzner F., Paton I.R., Windsor D., Dunn I., Koehler M., Nothwang H., Grutzner F., Paton I.R., Windsor D., Dunn I., Nothwang H., Grutzner F., Paton I.R., Windsor D., Dunn I., Grutzner F., Paton I.R., Windsor D., Dunn I., Paton I.R., Windsor D., Dunn I., Windsor D., Dunn I., Dunn I., et al. 300 million years of conserved synteny between chicken Z and human chromosome 9. Nat. Genet. 1999;21:258–259. doi: 10.1038/6769. [DOI] [PubMed] [Google Scholar]
- Nanda I., Kondo M., Hornung U., Asakawa S., Winkler C., Shimizu A., Shan Z., Haaf T., Shimizu N., Shima A., Kondo M., Hornung U., Asakawa S., Winkler C., Shimizu A., Shan Z., Haaf T., Shimizu N., Shima A., Hornung U., Asakawa S., Winkler C., Shimizu A., Shan Z., Haaf T., Shimizu N., Shima A., Asakawa S., Winkler C., Shimizu A., Shan Z., Haaf T., Shimizu N., Shima A., Winkler C., Shimizu A., Shan Z., Haaf T., Shimizu N., Shima A., Shimizu A., Shan Z., Haaf T., Shimizu N., Shima A., Shan Z., Haaf T., Shimizu N., Shima A., Haaf T., Shimizu N., Shima A., Shimizu N., Shima A., Shima A., et al. A duplicated copy of DMRT1 in the sex determining region of the Y chromosome of the medaka Oryzias latipes. Proc. Natl. Acad. Sci. 2002;99:11778–11783. doi: 10.1073/pnas.182314699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanda I., Hornung U., Kondo M., Schmid M., Schartl M., Hornung U., Kondo M., Schmid M., Schartl M., Kondo M., Schmid M., Schartl M., Schmid M., Schartl M., Schartl M. Common spontaneous sex-reversed XX males of the medaka, Oryzias latipes. Genetics. 2003;163:245–251. doi: 10.1093/genetics/163.1.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naruse K., Fukamachi S., Mitani H., Kondo M., Matsuoka T., Kondo S., Hanamura N., Morita Y., Hasegawa K., Nishigaki R., Fukamachi S., Mitani H., Kondo M., Matsuoka T., Kondo S., Hanamura N., Morita Y., Hasegawa K., Nishigaki R., Mitani H., Kondo M., Matsuoka T., Kondo S., Hanamura N., Morita Y., Hasegawa K., Nishigaki R., Kondo M., Matsuoka T., Kondo S., Hanamura N., Morita Y., Hasegawa K., Nishigaki R., Matsuoka T., Kondo S., Hanamura N., Morita Y., Hasegawa K., Nishigaki R., Kondo S., Hanamura N., Morita Y., Hasegawa K., Nishigaki R., Hanamura N., Morita Y., Hasegawa K., Nishigaki R., Morita Y., Hasegawa K., Nishigaki R., Hasegawa K., Nishigaki R., Nishigaki R., et al. A detailed linkage map of medaka, Oryzias latipes: Comparative genomics and genome evolution. Genetics. 2000;154:1773–1784. doi: 10.1093/genetics/154.4.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas M., Marais G., Hykelova V., Janousek B., Laporte V., Vyskot B., Mouchiroud D., Negrutiu I., Charlesworth D., Moneger F., Marais G., Hykelova V., Janousek B., Laporte V., Vyskot B., Mouchiroud D., Negrutiu I., Charlesworth D., Moneger F., Hykelova V., Janousek B., Laporte V., Vyskot B., Mouchiroud D., Negrutiu I., Charlesworth D., Moneger F., Janousek B., Laporte V., Vyskot B., Mouchiroud D., Negrutiu I., Charlesworth D., Moneger F., Laporte V., Vyskot B., Mouchiroud D., Negrutiu I., Charlesworth D., Moneger F., Vyskot B., Mouchiroud D., Negrutiu I., Charlesworth D., Moneger F., Mouchiroud D., Negrutiu I., Charlesworth D., Moneger F., Negrutiu I., Charlesworth D., Moneger F., Charlesworth D., Moneger F., Moneger F. A gradual process of recombination restriction in the evolutionary history of the sex chromosomes in dioecious plants. PLoS Biol. 2005;3:e4. doi: 10.1371/journal.pbio.0030004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S. Springer; Berlin: 1967. Sex chromosomes and sex-linked genes. [Google Scholar]
- Orr H.A., Kim Y., Kim Y. An adaptive hypothesis for the evolution of the Y chromosome. Genetics. 1998;150:1693–1698. doi: 10.1093/genetics/150.4.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peichel C.L., Ross J.A., Matson C.K., Dickson M., Grimwood J., Schmutz J., Myers R.M., Mori S., Schluter D., Kingsley D.M., Ross J.A., Matson C.K., Dickson M., Grimwood J., Schmutz J., Myers R.M., Mori S., Schluter D., Kingsley D.M., Matson C.K., Dickson M., Grimwood J., Schmutz J., Myers R.M., Mori S., Schluter D., Kingsley D.M., Dickson M., Grimwood J., Schmutz J., Myers R.M., Mori S., Schluter D., Kingsley D.M., Grimwood J., Schmutz J., Myers R.M., Mori S., Schluter D., Kingsley D.M., Schmutz J., Myers R.M., Mori S., Schluter D., Kingsley D.M., Myers R.M., Mori S., Schluter D., Kingsley D.M., Mori S., Schluter D., Kingsley D.M., Schluter D., Kingsley D.M., Kingsley D.M. The master sex-determination locus in threespine sticklebacks is on a nascent Y chromosome. Curr. Biol. 2004;14:1416–1424. doi: 10.1016/j.cub.2004.08.030. [DOI] [PubMed] [Google Scholar]
- Raymond C.S., Parker E.D., Kettlewell J.R., Brown L.G., Page D.C., Kusz K., Jaruzelska J., Reinberg Y., Flejter W.L., Bardwell V.J., Parker E.D., Kettlewell J.R., Brown L.G., Page D.C., Kusz K., Jaruzelska J., Reinberg Y., Flejter W.L., Bardwell V.J., Kettlewell J.R., Brown L.G., Page D.C., Kusz K., Jaruzelska J., Reinberg Y., Flejter W.L., Bardwell V.J., Brown L.G., Page D.C., Kusz K., Jaruzelska J., Reinberg Y., Flejter W.L., Bardwell V.J., Page D.C., Kusz K., Jaruzelska J., Reinberg Y., Flejter W.L., Bardwell V.J., Kusz K., Jaruzelska J., Reinberg Y., Flejter W.L., Bardwell V.J., Jaruzelska J., Reinberg Y., Flejter W.L., Bardwell V.J., Reinberg Y., Flejter W.L., Bardwell V.J., Flejter W.L., Bardwell V.J., Bardwell V.J., et al. A region of human chromosome 9p required for testis development contains two genes related to known sexual regulators. Hum. Mol. Genet. 1999;8:989–996. doi: 10.1093/hmg/8.6.989. [DOI] [PubMed] [Google Scholar]
- Rice W.R. Evolution of the Y sex chromosome in animals. Bioscience. 1996;46:331–343. [Google Scholar]
- Rozen S., Skaletsky H., Marszalek J.D., Minx P.J., Cordum H.S., Waterston R.H., Wilson R.K., Page D.C., Skaletsky H., Marszalek J.D., Minx P.J., Cordum H.S., Waterston R.H., Wilson R.K., Page D.C., Marszalek J.D., Minx P.J., Cordum H.S., Waterston R.H., Wilson R.K., Page D.C., Minx P.J., Cordum H.S., Waterston R.H., Wilson R.K., Page D.C., Cordum H.S., Waterston R.H., Wilson R.K., Page D.C., Waterston R.H., Wilson R.K., Page D.C., Wilson R.K., Page D.C., Page D.C. Abundant gene conversion between arms of palindromes in human and ape Y chromosomes. Nature. 2003;423:873–876. doi: 10.1038/nature01723. [DOI] [PubMed] [Google Scholar]
- Samonte R.V., Eichler E.E., Eichler E.E. Segmental duplications and the evolution of the primate genome. Nat. Rev. Genet. 2002;3:65–72. doi: 10.1038/nrg705. [DOI] [PubMed] [Google Scholar]
- Sato T., Endo T., Yamahira K., Hamaguchi S., Sakaizumi M., Endo T., Yamahira K., Hamaguchi S., Sakaizumi M., Yamahira K., Hamaguchi S., Sakaizumi M., Hamaguchi S., Sakaizumi M., Sakaizumi M. Induction of female-to-male sex reversal by high temperature treatment in medaka Oryzias latipes. Zoolog. Sci. 2005;22:985–988. doi: 10.2108/zsj.22.985. [DOI] [PubMed] [Google Scholar]
- Schartl M. Sex chromosome evolution in non-mammalian vertebrates. Curr. Opin. Genet. Dev. 2004;14:634–641. doi: 10.1016/j.gde.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Shearman D.C. The evolution of sex determination systems in dipteran insects other than Drosophila. Genetica. 2002;116:25–43. doi: 10.1023/a:1020955507978. [DOI] [PubMed] [Google Scholar]
- Skaletsky H., Kuroda-Kawaguchi T., Minx P.J., Cordum H.S., Hillier L., Brown L.G., Repping S., Pyntikova T., Ali J., Bieri T., Kuroda-Kawaguchi T., Minx P.J., Cordum H.S., Hillier L., Brown L.G., Repping S., Pyntikova T., Ali J., Bieri T., Minx P.J., Cordum H.S., Hillier L., Brown L.G., Repping S., Pyntikova T., Ali J., Bieri T., Cordum H.S., Hillier L., Brown L.G., Repping S., Pyntikova T., Ali J., Bieri T., Hillier L., Brown L.G., Repping S., Pyntikova T., Ali J., Bieri T., Brown L.G., Repping S., Pyntikova T., Ali J., Bieri T., Repping S., Pyntikova T., Ali J., Bieri T., Pyntikova T., Ali J., Bieri T., Ali J., Bieri T., Bieri T., et al. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature. 2003;423:825–837. doi: 10.1038/nature01722. [DOI] [PubMed] [Google Scholar]
- Sonnhammer E.L., Durbin R., Durbin R. A dot-matrix program with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene. 1995;167:GC1–GC10. doi: 10.1016/0378-1119(95)00714-8. [DOI] [PubMed] [Google Scholar]
- Steinemann S., Steinemann M., Steinemann M. Y chromosomes: Born to be destroyed. Bioessays. 2005;27:1076–1083. doi: 10.1002/bies.20288. [DOI] [PubMed] [Google Scholar]
- Vallender E.J., Lahn B.T., Lahn B.T. How mammalian sex chromosomes acquired their peculiar gene content. Bioessays. 2004;26:159–169. doi: 10.1002/bies.10393. [DOI] [PubMed] [Google Scholar]
- Wagner A. Birth and death of duplicated genes in completely sequenced eukaryotes. Trends Genet. 2001;17:237–239. doi: 10.1016/s0168-9525(01)02243-0. [DOI] [PubMed] [Google Scholar]
- Yamamoto T.O. Estriol-induced XY females of the medaka (Oryzias latipes) and their progenies. Gen. Comp. Endocrinol. 1965;5:527–533. doi: 10.1016/0016-6480(65)90041-9. [DOI] [PubMed] [Google Scholar]
- Zarkower D. Establishing sexual dimorphism: conservation amidst diversity? Nat. Rev. Genet. 2001;2:175–185. doi: 10.1038/35056032. [DOI] [PubMed] [Google Scholar]