

Abstract
Cholecystokinin (CCK) has been identified as a pronociceptive endogenous peptide which also possesses antiopioid actions. CCK may be upregulated in conditions of chronic pain or during sustained morphine administration resulting in attenuation of opioid-mediated pain relief. These complex interactions between opioids and endogenous CCK receptor systems have suggested the need for a new paradigm in drug design for some states of chronic pain. In these circumstances the rational design of potential drugs for the treatment of these conditions must be based on one ligand for multiple targets. We have designed a single peptide which can interact with δ and μ opioid receptors as agonists and with CCK receptors as antagonists. The ligands were designed based on a model of overlapping pharmacophores of opioid and CCK peptide ligands, which incorporates opioid pharmacophores at the N-terminal and CCK tetrapeptide pharmacophores at the C-terminal of the designed ligands. We measured binding and activities of our bifunctional peptides at opioid and CCK receptors. Compound 11 (Tyr-d-Ala-Gly-d-Trp-NMeNle-Asp-Phe-NH2) demonstrated opioid agonist properties at δ and μ receptors (IC50 = 63 ± 27 nM and 150 ± 65 nM, respectively in MVD and GPI tissue assays) and high binding affinity at CCK-1 and CCK-2 receptors (Ki = 320 and 1.5 nM, respectively). Compound 9 (Tyr-d-Nle-Gly-Trp-Nle-Asp-Phe-NH2) displayed potent agonist activity at δ and μ receptors (IC50 = 23 ±10 nM and 210 ± 52 nM, respectively in MVD and GPI tissue assays), with a balanced binding affinity for CCK-1 and CCK-2 receptors (Ki = 9.6 and 15 nM, respectively). These results provide evidence supporting the concept that opioid and CCK receptors have overlapping pharmacophores required for binding affinity and biological activity and that designing overlapping pharmacophores of two peptides into a single peptide is a valid drug design approach.
Introduction
Neuropathic pain is an abnormal pain that can result from injuries to nerves1 and is difficult to treat. While opioid alkaloids have been shown to be efficacious, the use of opioids for the treatment of this chronic pain state remains problematic due to the side-effects associated with the doses necessary to achieve sufficient pain relief in patients.2 In addition, the use of opioids for the treatment of chronic pain states, including cancer pain, is sometimes limited by the development of time-dependent reduction in pain relief (i.e., analgesic tolerance).
While mechanisms of neuropathic pain are not well understood, numerous studies indicate significant neuroplasticity which include increased activities of neuromodulators and neurotransmitters including CCK3–5 in the central nervous system.6,7 Upregulation of CCK has also been observed following sustained administration of morphine to animals, raising the possibility that pain-relieving actions of the opiate may be limited by the pronociceptive and antiopiate actions of this peptide.3,8 CCK-8 has long been considered as an “antiopioid” peptide because it diminishes opioid induced antinociception.9 Additionally, CCK-8 has been shown to have direct pronociceptive actions.10 CCK-8 is a linear octapeptide found in the periphery as a hormone and which functions as a neurotransmitter in the central nervous system.11,12 The main subtypes of receptors for CCK have been classified as the CCK-1 and CCK-2 and are both distributed in the central nervous system of humans.13–15 In rodents, CCK-1 receptors are found predominantly in the peripheral tissues while the CCK-2 receptor is localized predominately in the central nervous system (CNS).16,17 Additionally, CCK and endogenous opioids and their corresponding receptors have overlapping distribution in the central nervous system.18
We, therefore, hypothesized that effective treatment of chronic pain states such as neuropathic pain and improved antinociceptive actions would be associated with compounds which could act as agonists at opioid receptors while acting to counteract the upregulation/activity of endogenous CCK (i.e., CCK antagonists). Herein we report the design and structure activity relationships of a single peptide acting as an antagonist at CCK-1 and CCK-2 receptors as well as agonists at δ and μ opioid receptors.
Results
Design
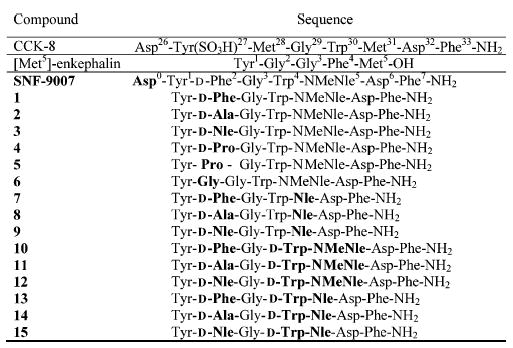
Our design of peptides that simultaneously interact with δ (and/or μ) opioid receptors as agonist, and at CCK-1 and CCK-2 receptors as antagonists, take advantage of the similarities between the pharmacophores of peptidic opioid and CCK ligands.19,20 In addition, these peptides also are designed to have similar affinities for CCK-1 and CCK-2 receptors. This balanced property addresses the issue of varying distributions of CCK-1 and CCK-2 receptors in the CNS of animals and humans, which is an important concern for in vivo studies. Previous studies in our laboratory have developed 3-D models of the bioactive conformations of ligands for both the δ opioid receptor21–23 and CCK receptors.24 From these reported 3-D models, we observed that there are topographical similarities between the opioid pharmacophore and CCK pharmacophore, particularly in the orientation of the aromatic residues.25,26 Incidentally, previous studies for structure–activity relationships of CCK receptor ligands in our laboratory resulted in the discovery of SNF-9007,27 which has a potent and very selective agonist activity at the CCK-2 receptor with a weak affinity but moderate agonist activity at the δ opioid receptor. As seen in Figure 1, SNF-9007, our model lead compound, the opioid pharmacophores at the N-terminus overlaps with the CCK pharmacophore at the C-terminal end. Thus, we redesigned SNF-9007 from a highly selective CCK-2 agonist to a CCK antagonist with balanced (similar) binding affinity for the CCK-1 and CCK-2 receptors, and for stronger affinity for both μ and δ opioid receptors. For our first modification, the N-terminal Asp of SNF-9007 was removed, since it is known that the free N-terminal tyrosine is essential for high potency of peptidic ligands at opioid receptors (Table 1). It is known that Asp0 can be removed without greatly affecting binding affinity and potency at CCK receptors, particularly at the CCK-2 receptors where CCK-4 (C-terminal tetrapeptide) remains potent.28 To explore the optimal binding at opioid receptors, as well as CCK receptors, a variety of amino acids were substituted at position 2 (analogues 1–6, Table 1). Our second goal was to obtain a balanced affinity for CCK-1 and CCK-2 receptors resulting in analogues 7–9 (Table 1). As mentioned earlier, the lead compound SNF-9007 is a highly selective CCK-2 receptor peptide, in part due to the Nα-methylated Nle5(NMeNle) which significantly reduced binding at the CCK-1 receptor. Thus, to improve the balanced binding affinity between CCK-1 and CCK-2 receptors, NMeNle was substituted with Nle, resulting in our second set of analogues. Our third goal was to improve the antagonist properties at CCK-1 and CCK-2 receptors while maintaining agonist activity at μ and δ opioid receptors. We focused on Trp4, which is a residue that has been demonstrated to have antagonist activity at the CCK receptors when modified. It is known that l-Trp can be substituted with d-Trp in peptides, peptoids, and nonpeptide analogues for CCK, we replaced l-Trp with d-Trp in the NMeNle series (analogues 10–12, Table 1).29 Finally, in our last set of analogues we sought to have the simultaneous effect of having antagonist properties at CCK-1 and CCK-2 receptors with a balanced binding affinity between CCK-1 and CCK-2 receptors while maintaining potent agonist activity. l-Trp was subsequently substituted with d-Trp4 along with Nle5 (i.e. a double substitution) (analogues 13–15, Table 1).
Figure 1.
Structure of peptides synthesized in this study.
Table 1.
Binding Affinities and Functional Activities of the CCK/Opioid Peptides at the Mu and Delta Opioid Receptors
| binding affinity
|
[35S]GTP-γ-S binding
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| hDORb, [3H]DPDPEc |
rMORb, [3H]DAMGOd |
hDORb |
rMORb |
|||||||
| peptidea | Log IC50e | Ki, nMf | Log IC50e | Ki, nMf | Log EC50g | EC50/nM | Emaxh | Log EC50g | EC50/nM | Emaxh |
| 1 | −7.73 ± 0.05 | 1.0 | −6.61 ± 0.10 | 100 | −8.60 ± 0.47 | 2.5 | 84 ± 9.0 | −7.65 ± 0.10 | 22 | 300 ± 9.0 |
| 2 | −8.28 ± 0.17 | 2.2 | −7.05 ± 0.17 | 39 | −6.94 ± 0.08 | 120 | 270 ± 7.2 | −7.75 ± 0.09 | 18 | 73 ± 2.1 |
| 3 | −8.33 ± 0.13 | 2.0 | −7.23 ± 0.04 | 25 | −7.82 ± 0.07 | 15 | 150 ± 3.6 | −6.96 ± 0.29 | 110 | 70 ± 8.3 |
| 4 | −6.70 ± 0.45 | 85 | −5.22 ± 0.19 | 2,600 | −6.05 ± 0.24 | 890 | 110 ± 8.5 | −6.05 ± 0.45 | 900 | 210 ± 39 |
| 5 | −5.47 ± 0.15 | 1500 | n.c. | - | n.s. | - | - | n.s. | - | - |
| 6 | −6.70 ± 0.22 | 85 | −6.26 ± 0.05 | 240 | −7.12 ± 0.32 | 76 | 68 ± 11 | −8.70 ± 0.26 | 2.0 | 100 ± 5.8 |
| 7 | −9.37 ± 0.13 | 0.20 | −7.10 ± 0.29 | 34 | −9.05 ± 0.11 | 0.90 | 170 ± 5.5 | −8.84 ± 0.41 | 1.4 | 180 ± 24 |
| 8 | −9.28 ± 0.16 | 0.22 | −7.74±0.11 | 7.8 | −8.23±0.17 | 5.9 | 74 ± 3.3 | −7.93±0.18 | 12 | 86 ± 4.5 |
| 9 | −8.22 ± 0.34 | 2.6 | −7.22±0.23 | 26 | −8.35±0.23 | 4.5 | 81 ± 5.1 | −9.34±0.46 | 0.46 | 88 ± 12 |
| 10 | −9.00 ± 0.17 | 0.42 | −7.86±0.07 | 5.9 | −7.54±0.34 | 29 | 14 ± 1.6 | −6.28±0.18 | 530 | 50 ± 0.18 |
| 11 | −8.46 ± 0.18 | 1.5 | −7.30±0.09 | 22 | −7.64±0.31 | 23 | 56 ± 5.2 | −7.42±0.20 | 38 | 44 ± 2.7 |
| 12 | −6.14 ± 0.11 | 310 | −5.54±0.09 | 1200 | n.s. | - | - | −6.60±0.29 | 250 | 47 ± 4.7 |
| 13 | −6.40 ± 0.17 | 170 | −6.15±0.17 | 300 | −6.89±0.14 | 130 | 170 ± 8.1 | −7.34±0.26 | 45 | 160 ± 12 |
| 14 | −5.70 ± 0.17 | 860 | −5.89±0.14 | 590 | −6.89±0.29 | 130 | 91 ± 8.7 | −6.45±0.35 | 350 | 110 ± 11 |
| 15 | −7.32 ± 0.16 | 20 | −6.20±0.16 | 270 | −8.39±0.19 | 4.1 | 75 ± 4.2 | −7.70±0.18 | 20 | 110 ± 6.5 |
| SNF-9007 | −6.26 ± 0.16 | 250 | −4.94 ± 0.19 | 5,200 | −6.98 ± 11 | 110 | 80 ± 2.8 | −7.09 ± 0.19 | 81 | 91 ± 5.5 |
| DPDPE | n.d. | - | n.d. | - | −8.8±0.25 | 1.6 | 69 ± 4.2 | n.d. | - | - |
| DAMGO | n.d. | - | n.d. | - | n.d. | - | - | −7.44±0.19 | 37 | 150 ± 9.4 |
Refer to Figure 1 for the structure of the peptides.
Carried out using membrane preparations from transfected HN9.10 cells that constitutively expressed the respective opioid receptor type.
Kd = 0.50 ± 0.1 nM.
Kd = 0.85 ± 0.2 nM.
Logarithmic values determined from the nonlinear regression analysis of data collected from at least 2 independent experiments.
Calculated using the Cheng and Prusoff equation to correct for the concentration of the radioligand used in the assay.
Logarithmic values determined from the nonlinear regression analysis of data collected from at least two independent experiments. The EC50 value is converted from the respective log EC50.
[35S]GTP-γ-S bound/basal [35S]GTP-γ-S bound] × 100 ± standard error. n.c.: incomplete competition of the radioligand at the highest test concentration of the peptide. An IC50 value cannot be determined. n.s.: nonsaturable dose–response curve. In these cases, stimulation of [35S]GTP-γ-S binding was observed only at the highest test dose(s) of the peptide (1 × 10−5 and 1 × 10−4 M). An EC50 or Emax value cannot be determined. n.d.: not determined.
Binding and Functional Activity Data
As shown previously, SNF-9007 has moderate binding affinity at the δ and μ opioid receptors (IC50 = 29 and 702 nM, respectively).27 At the cloned opioid receptors (Table 1), the binding at δ and μ receptors was poor (Ki = 250 and 5200 nM, respectively), but when the N-terminal Asp was initially removed resulting in [desAsp0] SNF-9007 (1, Table 1), the binding affinity was strong at 1 nM at the hDOR (human delta opioid receptor) to 100 nM at the rMOR (rat mu opioid receptor), respectively. In the MVD and GPI assays and [35S]GTP-γ-S-binding assay, the opioid agonist activity of 1 had a corresponding increase in agonist activity at opioid receptors (Table 2). As expected for the CCK-1 and CCK-2 receptors, the removal of Asp resulted to a high binding affinity at the CCK-2 receptor (Ki = 15 nM), while the binding affinity at the CCK-1 receptor was weak (in the high μM range) (Table 3). Compound 1 was a potent agonist at the CCK-2 receptors (Ki = 16. nM), while at the CCK-1 receptors, there was no response at 10 μM.
Table 2.
In Vitro Functional Activity of CCK/Opioid Peptides at the MVD and GPI
| opioid, IC50/nMa |
||
|---|---|---|
| no. | MVD (δ) | GPI (μ) |
| 1 | 65 ± 9 | 100 ± 23 |
| 2 | 66 ± 10 | 470 ± 60 |
| 3 | 9 ± 1 | 48 ± 6 |
| 4 | 890 ± 310 | 13% at 1 μM |
| 5 | 5% at 1 μM | 1.2% at 1 μM |
| 6 | 220 ± 16 | 4200 ± 720 |
| 7 | 12 ± 2 | 420 ± 68 |
| 8 | 45 ± 10 | 160 ± 28 |
| 9 | 23 ± 10 | 210 ± 52 |
| 10 | 24 ± 5 | 71 ± 5 |
| 11 | 63 ± 27 | 150 ± 65 |
| 12 | 700 ± 100 | 8.8% at 1 μM |
| 13 | 170 ± 20 | 2700 ± 180 |
| 14 | 1300 ± 190 | 2300 ± 330 |
| 15 | 310 ± 70 | 1000 ± 250 |
| SNF-9007 | 29 ± 10 | 220 ± 81 |
| Biphalin | 2.7 ± 1.5 | 8.8 ± 0.3 |
Concentration at 50% inhibition of muscle concentration at electrically stimulated isolated tissue.
Table 3.
Binding Affinities and Functional Activities of the CCK/Opioid Peptides at the CCK-1 and CCK-2 Receptors
| binding affinity, [125I]CCK-8(SO3)b |
PI assay
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| hCCK-1c |
hCCK-2c |
hCCK-1d |
hCCK-2d |
|||||||
| peptidea | Log IC50e | Ki, nMf | Log IC50e | Ki, nMf | Log EC50g | EC50, nM | Emaxh | Log EC50g | EC50, nM | Emaxh |
| 1 | −5.44 ± 0.17 | 3600 | −7.82 ± 0.12 | 15 | n.s. | - | - | −7.80 ± 0.17 | 16 | 11 ± 0.5 |
| 2 | −5.29 ± 0.28 | 5100 | −8.79 ± 0.17 | 1.6 | n.s. | - | - | −8.34 ± 0.22 | 4.6 | 22 ± 0.4 |
| 3 | n.c. | - | −8.42 ± 0.11 | 3.8 | n.s. | - | - | −7.72 ± 0.21 | 19 | 12 ± 0.8 |
| 4 | −4.98 ± 0.19 | >10000 | −8.57 ± 0.08 | 2.7 | n.s. | - | - | −7.44 ± 0.24 | 36 | 15 ± 1.0 |
| 5 | n.c. | - | −9.66 ± 0.25 | 0.22 | n.s. | - | - | n.s. | - | - |
| 6 | n.c. | - | −8.32 ± 0.10 | 4.8 | n.r. | - | - | −7.50 ± 0.14 | 32 | 15 ± 0.6 |
| 7 | −7.01 ± 0.19 | 98 | −7.59 ± 0.15 | 26 | −6.53 ± 0.3 | 300 | 7.1 ± 1 | −6.97 ± 0.20 | 110 | 9.5 ± 0.6 |
| 8 | −5.24 ± 0.24 | 5700 | −6.84 ± 0.27 | 150 | n.s. | - | - | −8.10 ± 0.50 | 8.0 | 13 ± 1.5 |
| 9 | −8.02 ± 0.10 | 9.6 | −7.82 ± 0.07 | 15 | −5.60 ± 0.3 | 2,500 | 12 ± 1.4 | −5.72 ± 0.19 | 1900 | 11 ± 0.9 |
| 10 | −5.96 ± 0.16 | 1100 | −8.96 ± 0.07 | 1.1 | n.r. | - | - | −8.56 ± 0.34 | 2.8 | 14 ± 1.3 |
| 11 | −6.50 ± 0.20 | 320 | −8.81 ± 0.06 | 1.5 | n.s. | - | - | −8.18 ± 0.13 | 6.6 | 13 ± 0.5 |
| 12 | −5.04 ± 0.17 | 910 | −5.96 ± 0.18 | 1100 | n.r. | - | - | n.r. | - | - |
| 13 | −5.71 ± 0.21 | 2000 | −5.76 ± 0.06 | 1700 | n.s. | - | - | n.s. | - | - |
| 14 | −5.19 ± 0.17 | 6500 | −5.57 ± 0.12 | 2700 | n.s. | - | - | −6.49 ± 0.15 | 330 | 16 ± 0.9 |
| 15 | n.c. | - | −5.66 ± 0.16 | 2200 | n.s. | - | - | n.r. | - | - |
| SNF-9007 | −5.5 ± 0.17 | 3200 | −8.67 ± 0.08 | 2.1 | n.r. | - | - | −4.71 ± 1.39 | >10000 | 14 ± 9.5 |
| CCK-8(SO3) | n.d. | - | n.d. | - | −7.55 ± 0.15 | 28 | 59 ± 3.0 | −7.57 ± 0.24 | 27 | 16 ± 1.1 |
Refer to Figure 1 for the structure of the peptides.
Kd = 1.9 ± 0.2 nM for hCCK-1; Kd = 1.3 ± 0.4 nM for hCCK-2.
Carried out using whole cell lysate preparations from transfected HEK 293 cells that constitutively expressed the respective receptor type.
Carried out on hCCK-1 or hCCK-2 expressing cells cultured in 24-well titer plates.
Logarithmic values determined from the nonlinear regression analysis of data collected from at least two independent experiments.
Antilogarthmic value of the respective IC50.
Logarithmic values determined from the nonlinear regression analysis of data collected from at least two independent experiments. The EC50 value is converted from the respective log EC50.
Saturable, maximum level of [3H]inositol phosphates produced in the cells upon incubation with the peptide, expressed as a ratio ± standard error of the amount of [3H]inositol phosphates produced in the presence of peptide over the basal level of [3H]inositol phosphates in untreated cells. n.c.: incomplete competition of the radioligand at the highest test concentration of the peptide. An IC50 value cannot be determined. n.s.: nonsaturable dose–response curve. In these cases, stimulation of [3H]phosphotidylinositol hydrolysis was observed only at the highest test dose(s) of the peptide (1 × 10−5 and 1 × 10−4 M). An EC50 or Emax value cannot be determined. n.r.: no stimulatory (or inhibitory) response at the highest test concentration of the peptide. n.d.: not determined.
Since removal of Asp0 improved the binding affinity at the opioid receptors and was well tolerated at the CCK receptors, structural modifications of 1 were further explored by substituting position 2 with various d-amino acids (d-Ala 2, d-Nle 3, d-Pro 4) as well as Pro 5 and Gly 6 to determine which residue would have an optimal effect for CCK antagonist activity while maintaining potent opioid agonist activity. Analogues 2 and 3 generally retained good binding affinity at both opioid receptors (2–40 nM), with moderate preference for the δ over the μ opioid receptor (ca. 10–20 μ/δ ) (Table 1). The binding affinities at δ opioid receptors were poor when Pro 5 and Gly 6 when placed at position 2 (Ki = 1.5 μM and 2.0 μM, respectively). However, substitution with d-Nle in 3 had the best binding affinity for the hDOR (Ki = 1.6 nM) (Table 1).
In the MVD and GPI tissue opioid agonist assays (Table 2), analogues 2 and 3 maintained or slightly decreased agonist activity at both δ and μ opioid receptors. The importance of a d-amino amino acid in position 2 was further supported when Gly was placed in position 2 which resulted in a significant decrease in activity compared to 1. Substitution of d-Pro 4 or Pro 5 resulted in a significant loss in potency all of the opioid agonist assays, suggesting that the secondary amide of Pro introduced an unfavorable turn or constraint in the peptide backbone for these enkephalin-derived peptide ligands.
When various amino acids were substituted at the position 2, the binding affinities at the CCK-2 receptors increased (Ki = 1.6–4.8 nM) when compared to 1. The binding at the CCK-1 receptor, however, remained in the micromolar range. Unlike in the opioid receptors, substitution with d-Pro 4 and Gly 6 did not decrease the binding at the CCK-2 receptor (Ki = 2.7 and 4.8 nM, respectively). Replacement with Pro 5, surprisingly, resulted in a significant increase in binding at the CCK-2 receptors (Ki = 0.22 nM) with no agonist activity at CCK-2, making 5 a very selective antagonist. The substitutions at position 2, except for Pro in 5, produced corresponding potent agonist activities at the CCK-2 receptors, while not producing any activation of CCK-1 receptors.
We hypothesized that the added constraint produced by Nα-methylation at position 5 induces a conformation that was less favorable for binding at the CCK-1 receptor. Thus, we modified the CCK-2 selective lead compound SNF-9007 (Table 3, IC50 = 3200 nM at CCK-1 receptor and 2.1 nM at CCK-2 receptor) by substituting NMeNle5 with Nle 7. The binding affinity at the CCK-1 receptor increased significantly (Ki = 9.6–5700 nM, Table 3), which resulted in a more balanced selectivity between the two receptors while maintaining the high binding affinity at the CCK-2 receptor (Ki = 26–150 nM). When position 2 was substituted with d-Phe in 7 and d-Nle in 9, the balanced CCK-1 and CCK-2 binding affinity was maintained, with a nearly 1:1 ratio for 9 (Ki = 9.6 and 15 nM for CCK-1 and CCK-2, respectively). When position 2 was substituted with a less bulky and hydrophobic residue d-Ala 8, the binding affinity at CCK-1 was very low at 5.7 μM. From this it can be suggested that position 2 does play a role in CCK-1 and CCK-2 receptor selectivity. To our knowledge, the [d-Nle2] analogue 9 shows the best binding affinity (Ki = 9.6 nM) at the CCK-1 receptor for a nonsulfated peptide analogue of CCK.
Coincidentally and surprisingly, substitution of NMeNle5 with Nle resulted in analogues with poor activity at both CCK receptor subtypes in the CCK functional assay. Relevant to the binding affinity of these peptides, analogues 7–9 (Table 3) were acting as antagonists, or possibly as partial agonists. From these it can be suggested that position 2 has a role in the bioactivity of CCK receptors.
At the δ and μ opioid receptors, 7–9 showed increased binding affinity at the δ and μ receptors compared to the NMeNle counterparts (Ki = 0.20–0.2.6 nM and 7.8–34 nM, respectively, Table 1). In the MVD and GPI assays (Table 2), substitution with Nle from NMeNle5 resulted in analogues with slightly improved agonist activity at the opioid receptors when compared to SNF-9007, while in the [35S]GTP-γ-S-binding opioid agonist assay, 7–9 displayed significant increased potencies when compared to the NMeNle counterparts.
l-Trp4 was substituted with d-Trp in the CCK/opioid analogues to introduce antagonist activity at CCK receptors while retaining agonist potency at the opioid receptors. Introducing a d-Trp substitution was expected to lead to analogues with antagonist activity at the CCK receptors. Analogues [d-Phe2, d-Trp4] 10 and [d-Ala2, d-Trp4] 11 both had binding affinities of Ki = 1–1.5 nM. Furthermore, the binding affinities of analogues 10 and 11 at the CCK-1 receptors increased significantly (Ki = 110 and 320, respectively) when compared to their l-Trp counterparts (1 and 2), resulting in an unexpected increased balanced selectivity between CCK-1 and CCK-2. Surprisingly, [d-Nle2, d-Trp4] 12 resulted in a significant loss in binding affinity at CCK-2 receptors (Ki = 1100 nM). As for the functional activity at the CCK receptors, 10 and 11 had good activity at the CCK-2 receptors (EC50 = 2.8 and 6.6 nM, respectively) while the activity at the CCK-1 receptors remained very poor or with no response at all.
In the set of peptides with NMeNle5 and various d-amino acids at position 2, the agonist activity at MVD and GPI tissue assays (Table 2) was good for analogues 10 (IC50 = 24 and 71 nM, respectively) and 11 (IC50 = 63 and 150 nM, respectively) while poor for analogue 12 (IC50 = 7000 and 8.8% at 1 μM, respectively). In comparison to SNF-9007, 10 is 2.5-fold more potent at both the MVD and the GPI tissue assays. In the competitive binding assays for δ and μ opioid receptors involving the substitution of l-Trp4 with d-Trp4 (Table 1), the binding affinity of analogues 10 and 11 increased by 2 to 10-fold compared the l-Trp4 counterparts. These analogues were moderately selective for the δ opioid receptor. These same d-Trp4 analogues had moderate activities at the δ and μ opioid receptors. The high to moderate binding affinity and bioactivity for the opioid receptors are surprising since it was found that d-aromatic amino acid residues in this position resulted in inactive peptides.19,30
Two strategies were combined to obtain the desired effect of increased binding at opioid receptors and at CCK receptors. Since substitution with d-Trp4 successfully resulted in an antagonist effect at the CCK-1 receptor, and substitution with Nle5 resulted in a balanced binding affinity between CCK-1 and CCK-2, a double substitution was attempted to obtain additive result. [desAsp0]SNF-9007 was modified to include d-Trp4 and Nle5 substitution. Analogues 13–15 showed significant decreases in opioid activity at the δ and μ opioid receptors (Table 1). The binding affinities at CCK-1 and CCK-2 also were poor, in the micromolar range (Table 3). The corresponding functional activities at these CCK receptors also were poor. These results showed that double substitution of d-Trp4 and Nle5 was not tolerated at opioid or CCK receptors and did not have an additive effect when compared to a single substitution of d-Trp4 or Nle.5
Taken together, the initial approach to truncate the N-terminal Asp of SNF-9007 increased the binding and activity at the opioid receptors without changing the profile at the CCK receptors. In our attempt for a balanced binding at CCK-1 and CCK-2 receptors, we replaced NMeNle5 with Nle5 which converted a highly CCK-2 selective SNF-9007 into more balanced ligands, particularly compound 9, for CCK-1 and CCK-2 receptors. In our attempt to make a potent CCK antagonist, we replaced Trp4 with d-Trp4. The binding affinity increased at CCK-1 receptors with no CCK-1 agonist properties. At the CCK-2 receptors the binding remained potent with high CCK-2 agonist properties for compounds 10 and 11, while compound 12 resulted in lower binding with much less CCK-2 agonist properties. In our final goal of making a balanced CCK-1 and CCK-2 antagonist with potent opioid agonist properties, simultaneous substitution with d-Trp4 and Nle5 was made, resulting in a significant drop yet balanced binding affinities at both CCK-1 and CCK-2 receptors with significantly less CCK agonist properties for compounds 13, 14, and 15. The binding and agonist opioid properties of compounds 13 and 14 dropped significantly while compound 15 remained potent. Our modifications to make a balanced CCK-1 and CCK-2 antagonists did not have an additive effect, but compounds 9 and 11 are new leads toward achieving our goals.
Discussion
Our data show that analogue 9 (Nle5) has very good binding affinity for the CCK-1 receptor. This finding is notable, as it is thought that the sulfated tyrosine is necessary for high binding affinity at the CCK-1 receptor. When compared to other SNF analogues,31 9 has the highest binding affinity (Ki = 9.6 nM) for a nonsulfated analogue of CCK at the CCK-1 receptor. The potency at the CCK functional PI assay was fairly low at 2 μM, which suggests that 9 is an antagonist.
The Peptide Backbone at Positions 4 and 5 Is Important for Selectivity at Opioid and CCK Receptors
From the perspective of the opioid receptors, these structure–function activity studies have provided interesting new insights into the stereochemical requirements of opioids at the μ and δ opioid receptors. Particularly noteworthy is the relatively high binding affinity of 10 where position 4 has a d-Trp residue. Schiller has reported that substitution of d-Phe4 or d-Trp4 in [Leu5]-enkephalin analogues resulted in analogues with low activity.19,32 Also, in the β-MePhe4 analogues of DPDPE, the (2S,3S) and (2S,3R) isomers have superior potency compared to the (2R) isomers.33 This observation is qualitatively consistent with the [d-Trp4, Nle5] analogues. Because analogues 10 and 11 [d-Trp4,N-MeNle5] had high potency at the opioid receptors, it can be suggested that the N-methylation at position 5 allows analogues with d-Trp to be potent at μ and δ opioid receptors.
The initial hypothesis was that the selectivity for CCK-2 was due to the added constraint introduced to the backbone by N-methylation. On the basis of the results, however, substitution from l-Trp to d-Trp (e.g. 10 and 11) increased the binding affinity at the CCK-1 receptor, while retaining the high affinity at CCK-2 receptor. This suggests that both [d-Trp4, NMeNle5] and [Trp4,Nle5] analogues have a common structure that is favorable to CCK-1 receptors. These positions in CCK have been explored by stabilizing the cis-amide bond between positions 4 and 5, which resulted in low binding affinities to both CCK receptors.34
Similarities in Opioid and CCK SAR Support the Overlapping Pharmacophore Hypothesis
From the perspective of overlapping pharmacophores between opioid and CCK ligands, these structure–function–activity results further show that there are common requirements for opioid and CCK ligands binding to their respective receptors, in particular the peptide backbone at position 4 and 5. As previously reported, substitution of d-Phe or d-Trp in positions 4 of [Leu5]-enkephalin resulted in very poor activity.19,30,32 Much like the enkephalin peptide, substitution of d-Trp in Ac-CCK-7 resulted in low binding affinities at CCK-1 receptors. In the CCK/opioid analogues, substitution of d-Trp in position 4 resulted in loss of binding affinity at both CCK and opioid receptors only if position 5 was Nle. But this substitution of d-Trp4 retained high binding affinity at the CCK and opioid receptors when position 5 is a N-methylated Nle. Because CCK/opioid ligands had the same structure–activity relationship at the CCK and opioid receptors, it can be inferred that CCK and opioids have similar conformations along positions 4 and 5 which are favorable for binding. These observations further support the hypothesis that CCK and opioid have overlapping pharmacophores. No particular conclusion can be suggested as to whether the backbone is of primary importance or the way the backbone orients the side-chain group has more effect on the binding affinity since all of peptides were linear and thus had considerable flexibility of possible structures without a high energy penalty.
Because positions 4 and 5 had a tremendous effect to the binding affinity at CCK and opioid receptors, it might be interesting to determine by computation methods structures of the backbones of d-Trp, N-MeNle and Trp, Nle. This could be important in the design of biologically active peptides (agonists or antagonists) especially when the structure–activity/selectivity relationships require a d-amino acid and/or N-methylated residue when applied generally. These linear analogues have already provided interesting insights into the nature of these peptides. Despite the introduction of local constraints on the peptide backbones, these linear peptides are still flexible along psi, phi, and chi space.
Conclusions
A series of linear peptides were designed and synthesized to interact with CCK receptors as antagonists and opioid receptors as agonists. The design of the linear peptides was based on our hypothesis that peptide opioid and CCK ligands have overlapping pharmacophore groups. These compounds were tested for binding and functional activity in human CCK-1 and CCK-2 receptors as well as human delta opioid receptors and rat mu opioid receptors. These compounds were also tested in vitro for opioid agonist activities in MVD and GPI.
Substitution of Nle5 produced a more balanced activity between CCK-1 and CCK-2 receptors as seen in compound 9. Also, substitution of d-Trp4 when position 5 is NMeNle showed antagonist properties at CCK receptors while maintaining the opioid agonist properties as seen in compound 11. These structure–activity relationships support the hypothesis that peptide opioid and CCK ligands have overlapping pharmacophores.
Experimental Section
Chemicals and Materials
Rink Amide AM resin (200–400 mesh, 0.6–0.7 mmol/gram substitution) was purchased from Novabiochem (San Diego, CA). Nα-Fmoc-Phe-OH, Nα-Fmoc-Asp-(O-tBu)-OH, Nα-Fmoc-Trp(Nin-Boc), Nα-Fmoc-Pro-OH, Nα-Fmoc-Gly-OH, Nα-Fmoc-Tyr(tBu)-OH, Nα-Fmoc-d-Trp(Boc) were from American Peptide Co. (Sunnyvale, CA); Nα-Fmoc-d-Ala-OH, Nα-Fmoc-d-Pro-OH, Nα-Fmoc-d-Phe-OH, Nα-Fmoc-d-Val-OH, and Nα-Fmoc-d-Nle-OH were purchased from Novabiochem; Nα-Fmoc-NMeNle-OH was from ChemImpex (Woodale, NJ); HBTU and HOBt were purchased from Peptide International (Louisville, KY). PyBrop was purchased from Advanced ChemTech (Louisville, KY). Peptide synthesis solvents were reagent grade, except for the HPLC grade CH3CN, were acquired from commercial sources and used without further purification unless otherwise noted. The purification of the crude peptides was achieved using a Hewlett-Packard 1100 series Liquid Chromatograph (Agilent-Technologies) with a C18-bonded silica column semipreparative column (Vydac 218TP1010, 300 Å, 1.0 × 25 cm, Separations Group, Hesperia, CA). The separations were monitored at 220 and 280 nm with a Hewlett-Packard 1100 series multiple variable wavelength UV detector and were integrated with a Hewlett-Packard 3396 series III integrator. Purity of the isolated peptides was assessed with analytical RP-HPLC in two different gradient systems as detected at 214, 230, 254, 280 nm. The structures of the pure peptides were confirmed by ESI-MS (Finnigan, Thermoelectron, LCQ classic) and high-resolution fast atom bombardment FAB-MS (JEOL HX110 sector instrument) or MALDI-TOF, where were performed at the University of Arizona Mass Spectrometry and Protein Sequencing Facility. TLC was done on Analtech (Newark, NJ) silica gel F254 plates (250 μM layer thickness) using the following systems: (A) 1-butanol/water/acetic acid (4:1:1); (B) ethyl acetate/1-butanol/water/acetic acid (5:3:1:1); and (C) chloroform/methanol/acetic acid (7:1:2). The peptides were detected on the TLC plates using iodine vapor. Myo-[2-3H(N)]-inositol; [tyrosyl-3,5-3H(N)] d-Ala2-MePhe4-glyol5-enkephalin (DAMGO); [tyrosyl-2,6-35H(N)]-(2-d-penicil-lamine, 5-d-penicillamine)enkephalin (DPDPE); Bolton-Hunter radioiodinated cholecystokinin octapeptide (CCK-8(s)); and [35S]-guanosine 5’-(γ-thio) triphosphate were purchased from Perkin-Elmer. Cholecystokinin (CCK-8(s)) was purchased from American Peptide Company Inc. (Sunnyvale, CA). Bovine serum albumin (BSA), protease inhibitors, Tris, and other buffer reagents were obtained from Sigma (St. Louis, MO). Culture medium (MEM, DMEM, IMDM), penicillin/streptomycin, and fetal calf serum (FCS) were purchased from Gibco (Grand Island, NY).
Peptide Synthesis
The peptides were synthesized on 0.5 g of Rink Amide AM resin using Nα-Fmoc chemistry. The resin were swollen in the reaction vessel with DMF overnight. Initially, the resin was deprotected with 20% (v/v) piperidine in DMF solution (3 × 20 min). The first amino acid, Nα-Fmoc-Phe-OH, was coupled to the resin. The following amino acids were coupled sequentially to the growing peptide chain with Nα-Fmoc-Asp(O-tBu)-OH, Nα-Fmoc-Nα-MeNle-OH, Nα-Fmoc-Nle-OH, Nα-Fmoc-Trp(Boc)-OH, Nα-Fmoc-d-Trp(Boc)-OH, Nα-Fmoc-Gly-OH, Nα-Fmoc-d-Ala-OH, Nα-Fmoc-d-Phe-OH, Nα-Fmoc-d-Nle-OH, Nα-Fmoc-Pro-OH, Nα-Fmoc-d-Pro-OH, Nα–Fmoc-d-Val-OH, or Nα-Fmoc-Tyr(tBu)-OH using standard solid-phase methods. Each coupling reaction was achieved using 3-fold excesses (relative to resin substitution) of amino acid, HBTU, and HOBt in the presence of 6-fold excess DIEA. The coupling reactions were run for 1 h. The completeness of the coupling reaction was monitored by a negative Kaiser test,35 which is indicated by a clear solution and clear resins. In the cases coupling of Nα-Fmoc-Trp(Boc)-OH following the NαMeNle, 4-fold excesses of amino acids and coupling agents each were used along with 8-fold excess of diisopropylethyamine. The coupling reaction was run for 2 h. In addition to the Kaiser test,35 the chloranil test36 and the TNBS test37 were used to monitor the reaction. The Nα-Fmoc protecting group on the amino acid was removed with piperidine (20% in DMF, 1 × 3 min, 1 × 25 min). The coupling and deprotection steps were each followed by washes with DMF (3 × 1 min) and DCM (3 × 1 min). Completion of the Nα-Fmoc deprotection was assessed by a positive Kaiser test, which is indicated by a deep blue or purple solution and dark blue resins following the test. After the deprotection of the final Nα-terminal Fmoc, the resins were washed with DMF. After a final thorough wash with DCM, removing the DMF, the peptidyl resin was dried with vacuum and ambient air. The dried peptidyl resins were transferred to a bromosilicate scintillation vial. The peptide was cleaved from the resin, and the other side chain protecting groups were removed by incubation in a cleavage cocktail (10 mL/grams) with 95% TFA, 2.5% TIS, 2.5% water for 90 min. The resins were filtered through a cotton plugged glass pipet. The resins were then washed with additional TFA (~2 mL) for 5 min. The TFA-peptide solution was transferred to a 15 mL propylethylene conical centrifuge tube (Falcon). The volume of TFA of the filtrate was reduced with a gentle flow of inert gas to about 1.5 mL. The peptide was precipitated upon slow addition of cold diethyl ether (12 mL). The precipitate was isolated by centrifugation using a benchtop centrifuge. The organic solvent was decanted and discarded. The peptide pellets were triturated with cold diethyl ether (12 mL). The pellet was dried over air, yielding 55–90% of the crude peptides.
HPLC Purification
The crude peptides were purified by using a C18 semipreparative column (VYDAC 218TP1010, 10 mm × 250 mm). The crude peptides was loaded into the column at a concentration of 10 mg/mL. For 100 mg of dried crude peptide, the peptide was “wetted” with CH3CN (~1 mL). Aqueous 0.1% TFA was added slowly until peptides precipitated (~6 mL). CH3-CN was added until the crude peptides were dissolved (~2 mL). Iterative addition of aqueous 0.1% TFA and CH3CN (or ethanol or methanol) was done until peptide was fully dissolved, without exceeding 20% aqueous CH3CN. The solution was allowed to sit at room-temperature overnight, or until the side product peak is no longer detected by HPLC, to decarboxylate the intermediate carbamic acid on the Trp. The dissolved peptide was filtered through a 0.45 μm cellulose acetate filter (Aerodisk). For the C18 semipreparatative sized column, the maximum loading capacity of 10 mg/mL was injected. In general, the gradient used was 20 to 60% CH3CN in aqueous 0.1% TFA in 30 min at a flow rate of 3 mL/min. Fractions corresponding to the major peak were collected, combined, reduced, and lyophilized to yield the final peptides as pure solids. Analytical results for HR-MS, HPLC, and TLC are summarized in the Supporting Information. All peptides were at least 99% pure.
Cell Lines
The cDNA for the human DOR was a gift from Dr. Brigitte Kieffer (IGBMC, Illkirch, France). The cDNA for the rat MOR was a gift from Dr. Huda Akil (University of Michigan). The cDNA for the human CCK-1 and CCK-2 was a gift from Dr. Alan Kopin (Tufts University, MA). Stable expression of the human CCK-1 (CMV Tag2) and CCK-2 (pCDNA HisXpress) receptors in HEK 293 cells was produced by transfecting the cells with the respective cDNA by calcium phosphate precipitation followed by clonal selection in neomycin. Stable expression of the rat MOR (pCDNA3) and the human DOR (pCDNA3) in the neuroblastoma cell line, HN9.10, were achieved with the same method. Expression of the respective receptors was initially verified and the level of expression periodically monitored by radioligand saturation analysis (see below). All cells were maintained at a 37 °C, 95% air/5% CO2, humidified atmosphere in a Forma Scientific (Marietta, OH) incubator in DMEM with 10% BSA and 100 U mL penicillin/100 μg mL streptomycin.
Radioligand Binding Analysis
For opioid receptors, crude membranes were prepared as previously described38 from the transfected cells that express the MOR (μ opioid receptor) or the DOR (δ opioid receptor). The protein concentration of the membrane preparations was determined by the Lowry method, and the membranes were stored at −80 °C until use. Membranes were resuspended in assay buffer (50 mM Tris, pH 7.4, containing 50 μg/mL bacitracin, 30 μM bestatin, 10 μM captopril, 100 μM PMSF, 1 mg/mL BSA). For saturation analysis, six concentrations of [3H]-DAMGO (0.02–6 nM, 47.2 Ci/mmol), or six concentrations of [3H]-DPDPE (0.1 nM–10 nM, 44 Ci/mmol), were each mixed with 200 μg of membranes from MOR or DOR expressing cells, respectively, in a final volume of 1 mL. For competition analysis, 10 concentrations of a test compound were each incubated with 50 μg of membranes from MOR or DOR expressing cells and the Kd concentration of [3H]DAMGO (1.0 nM, 50 Ci/mmol), or of [3H]-DPDPE (1.0 nM, 44 Ci/mmol), respectively. Naloxone at 10 μM was used to define the nonspecific binding of the radioligands in all assays. All samples were carried out in duplicate. The samples were incubated in a shaking water bath at 25 °C for 3 h, followed by rapid filtration through Whatman Grade GF/B filter paper (Gaithersburg, MD) presoaked in 1% polyethyleneimine, washed four times each with 2 mL of cold saline, and the radioactivity was determined by liquid scintillation counting (Beckman LS5000 TD).
For CCK receptors, all assays were performed using whole cells preparations expressing either the human CCK-1 or CCK-2. The assay buffer used was low glucose DMEM, pH 7.4, in the presence of 0.5 mg/mL BSA and protease inhibitors, as above. For saturation analysis, 12 concentrations (12.5 pM – 4 nM) of unlabeled CCK-8(s)/[125I]CCK-8 (labeled-to-unlabeled ratios: 1:25 to 1:65) were incubated with 200 000 cells/assay tube in a final volume of 1 mL. For [125I]CCK-8 (2200 Ci/mmol)/ligand competition analysis, 10 different concentrations (10−13 M to 10−4 M) of each test peptide was mixed with [125I]CCK-8 (1.0–1.5 pM) and 200 000 cells/assay tube in a final volume of 1 mL in assay buffer. Nonspecific binding was defined as that of [125I]CCK-8 bound in the presence of 1 μM unlabeled CCK-8. All samples were done in duplicates. Incubations were carried out at 30 °C for 60 min in a shaking water bath and the reaction was terminated by rapid filtration through Whatman GF/B filter paper (presoaked in polyethyleneimine) and washed four times with 2 mL of ice-cold saline. The radioactivity was determined by gamma counting (Packard Cobra II Gamma Counter). Data were analyzed by nonlinear regression analysis using Graph-Pad Prism4 (Graph Pad, San Diego, CA). The Ki value(s) for each ligand was calculated from the IC50 value(s) based on the Cheng and Prusoff equation from at least three independent experiments.
GPI and MVD in Vitro Bioassays
The in vitro tissue bioassays were performed as described previously.39 IC50 values represent the mean of no less than four tissues. IC50 estimates, relative potency estimates, and their associated standard errors were determined by fitting the data to the Hill equation by a computerized nonlinear least-squares method. In the MVD assay, male ICR mice under ether anesthesia were sacrificed by cervical dislocation and the vasa deferentia was removed. The tissue were tied to a gold chain with suture silk and mounted between platinum wire electrodes in 20 mL organ baths at a tension of 0.5 g and bathed in oxygenated (95% O2, 5% CO2) magnesium free Krebs buffer at 37 °C. They were stimulated electrically (0.1 Hz, single pulses, 2.0 ms duration) at supramaximal voltage. Following an equilibrium period, compounds were added to the bath cumulatively in volumes of 14–16 mL until maximum inhibition is reached. Response to an IC50 dose of DPDPE (10 nM) were measured to determine tissue integrity before compound testing begins.
In the GPI bioassay, male Hartley guinea pigs under anesthesia were sacrificed by decapitation, and a nonterminal portion of the ileum was removed. The LMMP were carefully separated from the circular muscle and were cut into strips. The tissues were tied to a gold chain with suture silk and mounted between platinum wire electrodes in 20 mL baths at a tension of 1 g containing 37 °C oxygenated (95% O2, 5% CO2) Krebs buffer (118 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.19 mM KH2PO4, 1.18 mM MgSO4, 25 mM NaHCO3, and 11.48 mM glucose) and allowed to equilibrate for 15 min. The tissues were stimulated electrically (0.1 Hz, 0.4 ms duration) at supramaximal voltage. Following equilibration, the compound was added to the baths in 15–60 μL aliquots until maximum inhibition was observed. Percent inhibition was calculated by using the average contraction height for 1 min preceding the addition of the compound divided by the contraction height 3 min after exposure to the dose of the compound. Response to an IC50 dose of PL-017 (10 nM) were measured to determine tissue integrity before compound testing begins.
Functional Assays for CCK. Phoshatidylinositol Hydrolysis Assay for the CCK Receptors
The HEK cells were seeded at 50 000 cells per well 2 days before the experiment. The next day cells were then incubated overnight with 2 μCi/mL [3H] inositol and 6% FCS. The cells were washed with fresh media and incubated with various concentrations of a test drug, in duplicates, in separate wells for 60 min at 37°C in the tissue culture incubator. The method used to measure the accumulation of [3H]inositol phosphates was according to that described,40 with two additional washes with 5 mL of 5 mM sodium tetraborate/60 mM sodium formate before the elution of [3H]inositol phosphates. Radioactivity was determined by liquid scintillation counting (Beckman LS5000 TD). Basal activity was defined as the amount of [3H]inositol phosphates detected in the absence of test drug. The effect of test drug at each concentration on [3H]inositol phosphates production was expressed as a ratio of radioactivity over basal activity. Data were expressed as mean ± SEM. from at least three independent assays and analyzed by nonlinear least squares analaysis using GraphPad Prism4.
[35S]GTPγS Binding Assay for the Opioid Receptors
The method was carried out according to that previously described.41 Membrane preparation (10 μg) to a final volume of 1 mL incubation mix (50 mM HEPES, pH 7.4, 1 mM EDTA, 5 mM MgCl2, 30 μm GDP, 1 mM dithiothreitol, 100 mM NaCl, 0.1 mM phenylmethylsulfonyl fluoride (PMSF), 0.1% BSA, 0.1 nM [35S]GTPγS) was added along with various concentrations, in duplicate or triplicate, of the test drug and incubated for 90 min at 30 °C in a shaking water bath. Reactions were terminated by rapid filtration through Whatman GF/B filters (presoaked in water), followed by four washes with 4 mL of ice-cold wash buffer (50 mM Tris, 5 mM MgCl2, 100 mM NaCl, pH 7.4). The radioactivity was determined by liquid scintillation counting as above. Basal level of [35S]GTPγS binding was defined as the amount bound in the absence of any test drug. Nonspecific binding was determined in the presence of 10 μM unlabeled GTPγS. Total binding was defined as the amount of radioactivity bound in the presence of test drug. The effect of the drug at each concentration on [35S]GTPγS binding was calculated as a percentage by the following equation: [total bound – basal]/[basal – nonspecific] × 100. Data were expressed as mean ± SEM from three independent experiments and analyzed by nonlinear regression analysis using GraphPad Prism4.
Supplementary Material
Acknowledgments
This research was supported in part by grants from the USPHS, National Institute of Drug Abuse, DA12394 and DA06284. The opinions expressed are those of the authors and not necessarily those of NIDA. Authors are thankful to Ms. Margie Colie for her assistance with the manuscript.
Footnotes
Supporting Information Available: HR-MS, TLC, HPLC data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Wiesenfeld-Hallin Z, Xu XJ, Hokfelt T. The role of spinal cholecystokinin in chronic pain states. Pharmacol Toxicol. 2002;91:398–403. doi: 10.1034/j.1600-0773.2002.910619.x. [DOI] [PubMed] [Google Scholar]
- 2.Ossipov MH, Lai J, King T, Vanderah TW, Porreca F. Underlying mechanisms of pronociceptive consequences of prolonged morphine exposure. Biopolymers. 2005;80:319–324. doi: 10.1002/bip.20254. [DOI] [PubMed] [Google Scholar]
- 3.Stanfa L, Dickenson A, Xu XJ, Wiesenfeld-Hallin Z. Cholecystokinin and morphine analgesia: variations on a theme. Trends Pharm Sci. 1994;15:65–66. doi: 10.1016/0165-6147(94)90279-8. [DOI] [PubMed] [Google Scholar]
- 4.Wiesenfeld-Hallin Z, Xu XJ. The role of cholecystokinin in nociception, neuropathic pain and opiate tolerance. Regul Pept. 1996;65:23–28. doi: 10.1016/0167-0115(96)00068-7. [DOI] [PubMed] [Google Scholar]
- 5.Wiesenfeld-Hallin Z, de Arauja Lucas G, Alster P, Xu XJ, Hokfelt T. Cholecystokinin/opioid interactions. Brain Res. 1999;848:78–89. doi: 10.1016/s0006-8993(99)01978-2. [DOI] [PubMed] [Google Scholar]
- 6.Vanderah TW, Ossipov MH, Lai J, Malan TP, Jr, Porreca F. Mechanisms of opioid-induced pain and antinociceptive tolerance: descending facilitation and spinal dynorphin. Pain. 2001;92:5–9. doi: 10.1016/s0304-3959(01)00311-6. [DOI] [PubMed] [Google Scholar]
- 7.Heinricher MM, Neubert MJ. Neural basis for the hyperalgesic action of cholecystokinin in the rostral ventromedial medulla. J Neurophysiol. 2004;92:1982–1989. doi: 10.1152/jn.00411.2004. [DOI] [PubMed] [Google Scholar]
- 8.Zhou Y, Sun YH, Zhang ZW, Han JS. Increased release of immunoreactive cholecystokinin octapeptide by morphine and potentiation of mu-opioid analgesia by CCKB receptor antagonist L-365, 260 in rat spinal cord. Eur J Pharmacol. 1993;234:147–154. doi: 10.1016/0014-2999(93)90948-h. [DOI] [PubMed] [Google Scholar]
- 9.Faris PL, Komisaruk BR, Watkins LR, Mayer DJ. Evidence for the neuropeptide cholecystokinin as an antagonist of opiate analgesia. Science. 1983;219:310–312. doi: 10.1126/science.6294831. [DOI] [PubMed] [Google Scholar]
- 10.Watkins LR, Kinscheck IB, Mayer DJ. Potentiation of morphine analgesia by the cholecystokinin antagonist proglumide. Brain Res. 1985;327:169–180. doi: 10.1016/0006-8993(85)91511-2. [DOI] [PubMed] [Google Scholar]
- 11.Noble F, Roques BP. CCK–B receptor: chemistry, molecular biology, biochemistry and pharmacology. Prog Neurobiol. 1999;58:349–379. doi: 10.1016/s0301-0082(98)00090-2. [DOI] [PubMed] [Google Scholar]
- 12.Noble F, Wank SA, Crawley JN, Bradwejn J. Seroogy KBet alInternational Union of Pharmacology, XXI. Structure, Distribution, and Functions of Cholecystokinin Receptors. Pharmacol Rev. 1999;51:745–781. [PubMed] [Google Scholar]
- 13.Innis RB, Snyder SH. Distinct cholecystokinin receptors in brain and pancreas. Proc Natl Acad Sci USA. 1980;77:6917–6921. doi: 10.1073/pnas.77.11.6917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moran TH, Robinson PH, Goldrich MS, McHugh PR. Two brain cholecystokinin receptors: implications for behavioral actions. Brain Res. 1986;362:175–179. doi: 10.1016/0006-8993(86)91413-7. [DOI] [PubMed] [Google Scholar]
- 15.Baber NS, Dourish CT, Hill DR. The role of CCK caerulein, and CCK antagonists in nociception. Pain. 1989;39:307–328. doi: 10.1016/0304-3959(89)90045-6. [DOI] [PubMed] [Google Scholar]
- 16.Ghilardi JR, Allen CJ, Vigna SR, McVey DC, Mantyh PW. Trigeminal and dorsal root ganglion neurons express CCK receptor binding sites in the rat, rabbit, and monkey: possible site of opiate-CCK analgesic interactions. J Neurosci. 1992;12:4854–4866. doi: 10.1523/JNEUROSCI.12-12-04854.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hill DR, Shaw TM, Woodruff GN. Binding sites for 125I-cholecystokinin in primate spinal cord are of the CCK-A subclass. Neurosci Lett. 1988;89:133–139. doi: 10.1016/0304-3940(88)90369-2. [DOI] [PubMed] [Google Scholar]
- 18.Stengaard-Pedersen K, Larsson LI. Localization and opiate receptor binding of enkephalin, CCK and ACTH/beta-endorphin in the rat central nervous system. Peptides. 1981;2:3–19. doi: 10.1016/0196-9781(81)90050-4. [DOI] [PubMed] [Google Scholar]
- 19.Schiller PW, Yam CF, Prosmanne J. Synthesis, opiate receptor affinity, and conformational parameters of [4-tryptophan]enkephalin analogues. J Med Chem. 1978;21:1110–1116. doi: 10.1021/jm00209a004. [DOI] [PubMed] [Google Scholar]
- 20.Pincus MR, Carty RP, Chen J, Lubowsky J, Avitable M, et al. On the biologically active structures of cholecystokinin, little gastrin, and enkephalin in the gastrointestinal system. Proc Natl Acad Sci USA. 1987;84:4821–4825. doi: 10.1073/pnas.84.14.4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Collins N, Flippen-Anderson JL, Hasseth RC, Deschamps JR, George C, et al. Conformational determinants of agonist versus antagonists properties of [d-Pen2, d-Pen5]enkephalin (DPDPE) analogues at opioid receptors. Comparison of X-ray crystallographic structure, solution 1H NMR data, and molecular dynamic simulations of [l-Ala3]DPDPE and [d-Ala3]DPDPE. J Am Chem Soc. 1996;118:2143–2152. [Google Scholar]
- 22.Nikiforovich GV, Hruby VJ, Prakash O, Gehrig CA. Topographical requirements for delta-selective opioid peptides. Biopolymers. 1991;31:941–955. doi: 10.1002/bip.360310804. [DOI] [PubMed] [Google Scholar]
- 23.Shenderovich MD, Liao S, Qian X, Hruby VJ. A three-dimensional model of the delta-opioid pharmacophore: comparative molecular modeling of peptide and nonpeptide ligands. Biopolymers. 2000;53:565–580. doi: 10.1002/(SICI)1097-0282(200006)53:7<565::AID-BIP4>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 24.Nikiforovich GV, Hruby VJ. Models for the A- and B-receptor-bound conformations of CCK-8. Biochem Biophys Res Commun. 1993;194:9–16. doi: 10.1006/bbrc.1993.1777. [DOI] [PubMed] [Google Scholar]
- 25.Hruby, V. J.; Fang, S. N.; Kramer, T. H.; Davis, P.; Parkhurst, D. et al. Analogs of cholecystokinin26–33 selective for B-type CCK receptors possess delta opioid receptor agonist activity in vitro and in vivo: evidence for similarities in CCK–B and delta.-opioid receptor requirements. In Peptides: Chemistry, Structure and Biology, Proceedings of the 13th American Peptide Sympposium; Hodges, R., Smith, A. J., Eds.; ESCOM Science Publishers B.V.: Leiden, The Netherlands, 1994; pp 669–671.
- 26.Nikiforovich GV, Kolodziej SA, Nock B, Bernad N, Martinez J, et al. Conformationally readdressed CCK–B/delta-opioid peptide ligands. Biopolymers. 1995;36:439–452. doi: 10.1002/bip.360360407. [DOI] [PubMed] [Google Scholar]
- 27.Slaninova J, Knapp RJ, Wu JJ, Fang SN, Kramer T, et al. Opioid receptor binding properties of analgesic analogues of cholecystokinin octapeptide. Eur J Pharmacol. 1991;200:195–198. doi: 10.1016/0014-2999(91)90688-m. [DOI] [PubMed] [Google Scholar]
- 28.Takeda Y, Hoshino M, Yanaihara N, Yanaihara C, Isobe J, et al. Comparison of CCK-8 receptors in the pancreas and brain of rats using CCK-8 analogues. Jpn J Pharmacol. 1989;49:471–481. doi: 10.1254/jjp.49.471. [DOI] [PubMed] [Google Scholar]
- 29.Hughes J, Boden P, Costall B, Domeney A, Kelly E, et al. Development of a class of selective cholecystokinin type B receptor antagonist having potent anxiolytic activity. Proc Natl Acad Sci USA. 1991;87:6728–6732. doi: 10.1073/pnas.87.17.6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beddell CR, Clark RB, Hardy GW, Lowe LA, Ubatuba FB, et al. Structural requirements for opioid activity of analogues of the enkephalins. Proc R Soc London –Ser B: Biol Sci. 1977;198:249–265. doi: 10.1098/rspb.1977.0096. [DOI] [PubMed] [Google Scholar]
- 31.Hruby VJ, Fang S, Knapp R, Kazmierski W, Lui GK, et al. Cholecystokinin analogs with high affinity and selectivity for brain membrane receptors. Int J Pept Protein Res. 1990;35:566–573. doi: 10.1111/j.1399-3011.1990.tb00263.x. [DOI] [PubMed] [Google Scholar]
- 32.Schiller PW, Natarajan S, Bodanszky M. Determination of the intramolecular tyrosine-tryptophan distance in a 7-peptide related to the C-terminal sequence of cholecystokinin. Int J Pept Protein Res. 1978;12:139–142. doi: 10.1111/j.1399-3011.1978.tb02877.x. [DOI] [PubMed] [Google Scholar]
- 33.Hruby VJ, Toth G, Gehrig CA, Kao LF, Knapp R, et al. Topographically designed analogues of [cyclic] [d-Pen2, d-Pen5]-enkephalin. J Med Chem. 1991;34:1823–1830. doi: 10.1021/jm00110a010. [DOI] [PubMed] [Google Scholar]
- 34.Boteju LW, Nikiforovich GV, Haskell-Luevano C, Fang SN, Zalewska T, et al. The use of topographical constraints in receptor mapping: investigation of the topographical requirements of the tryptophan 30 residue for receptor binding of Asp-Tyr-d-Phe-Gly-Trp-(NMe)Nle-Asp-Phe-NH2 (SNF 9007), a cholecystokinin (26–33) analogue that binds to both CCK–B and delta-opioid receptors. J Med Chem. 1996;39:4120–4124. doi: 10.1021/jm960078j. [DOI] [PubMed] [Google Scholar]
- 35.Kaiser E, Colescott RL, Bossinger CD, Cook PI. Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal Biochem. 1970;34:595–598. doi: 10.1016/0003-2697(70)90146-6. [DOI] [PubMed] [Google Scholar]
- 36.Vojkovsky T. Detection of secondary amines on solid phase. Pep Res. 1995;8:236–237. [PubMed] [Google Scholar]
- 37.Hancock WS, Battersby JE. A new micro-test for the detection of incomplete coupling reactions in solid-phase peptide synthesis using 2,4,6-trinitrobenzenesulphonic acid. Anal Biochem. 1976;71:260–264. doi: 10.1016/0003-2697(76)90034-8. [DOI] [PubMed] [Google Scholar]
- 38.Lai J, Ma SW, Zhu RH, Rothman RB, Lentes KU, Porreca F. Pharmacological characterization of the cloned kappa opioid receptor at a kappa 1b subtype. NeuroReport. 1994;5(16):2161–2164. doi: 10.1097/00001756-199410270-00043. [DOI] [PubMed] [Google Scholar]
- 39.Kramer TH, Shook JE, Kazmierski W, Ayres EA, Wire WS, Hruby VJ, Burks TF. Novel peptidic mu opioid antagonists: pharmacologic characterization in vitro and in vivo. J Pharmacol Exp Ther. 1989;249:544–551. [PubMed] [Google Scholar]
- 40.Berridge MJ, Downes CP, Hanley MR. Lithium amplifies agonist-dependent phosphatidylinositol responses in brain and salivary glands. Biochem J. 1982;206(3):587–595. doi: 10.1042/bj2060587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lorenzen A, Fuss M, Vogt H, Schwabe U. Measurement of guanine nucleotide-binding protein activation by A1 adenosine receptor agonists in bovine brain membranes: stimulation of guanosine-5′-O–(3-[35S]thio)triphosphate binding. Mol Pharmacol. 1993;44(1):115–123. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.