

Abstract
X-linked hypophosphatemia (XLH), a disorder characterized by hypophosphatemia, impaired skeletal mineralization, and aberrant regulation of 1, 25(OH)2D3, is caused by inactivating mutations of Phex, which results in the accumulation of putative phosphaturic factors, called phosphatonins. Matrix extracellular phosphoglycoprotein (Mepe) is a proposed candidate for phosphatonin. The authors found that Hyp mice had increased expression of the MEPE and another phosphaturic factor, Fgf23. To establish MEPE’s role in the pathogenesis of the XLH, Mepe-deficient mice were back-crossed onto the Hyp mouse homologue of XLH and phenotypes of wild-type, Mepe−/−, Hyp, and Mepe−/−/Hyp mice were examined. Transfer of Mepe deficiency onto the Phex-deficient Hyp mouse background failed to correct hypophosphatemia and aberrant serum 1,25(OH)2D3 levels. Increased Fgf23 levels in Hyp mice were not affected by superimposed Mepe deficiency. In addition, Mepe-deficient Hyp mice retained bone mineralization defects in vivo, characterized by decreased bone mineral density, reduced mineralized trabecular bone volume, lower flexural strength, and histologic evidence of osteomalacia; however, cultures of Hyp-derived bone marrow stromal cells in the absence of Mepe showed improved mineralization and normalization of osteoblast gene expression profiles observed in cells derived from Mepe-null mice. These results demonstrate that MEPE elevation in Hyp mice does not contribute to the hypophosphatemia associated with inactivating Phex mutations and is therefore not phosphatonin.
X-linked hypophosphatemia (XLH), a disorder characterized by defective calcification of cartilage and bone, impaired renal tubular reabsorption of phosphate (Pi), and aberrant regulation of 1,25(OH)2D3 production, is caused by inactivating mutations of the cell surface metalloprotease, PHEX (1). The mouse Phex cDNA sequence is highly homologous to that of humans (2), and several mouse XLH homologues have been identified with inactivating Phex mutations, including Hyp, Gy, and Ska1 mice (2,3). Phex is most highly expressed in osteoblasts, osteocytes, and odontoblasts (4–7). Current data indicate that Phex regulates the production and/or degradation of phosphaturic hormones, referred to as phosphatonins (8), and putative local inhibitors of mineralization, called minhibins (9), that may contribute to an intrinsic mineralization defect in osteoblasts that is independent of hypophosphatemia (9–13). Currently, there are several candidates for these factors.
FGF23 is a strong candidate for phosphatonin. FGF23, a phosphaturic member of the fibroblast growth factor (FGF) family (14–17), is the disease-causing gene in autosomal-dominant hypophosphatemic rickets (16–18). In addition, circulating levels of FGF23 are elevated in most patients with XLH (19–21). Moreover, Fgf23-null mice are hyperphosphatemic (22,23) and the transfer of Fgf23 deficiency onto the Hyp mouse background increases serum phosphate in Hyp mice (23). Patients with tumor-induced osteomalacia (TIO) also have increased messenger RNA expression of FGF23 in the tumors, as well as elevated circulating levels of FGF23 that decrease after tumor removal (14,19–21,24–26). Finally, patients with hypophosphatemic forms of McCune Albright syndrome have increased FGF23 transcripts in fibrous dysplastic bone lesions and elevated serum levels of FGF23 (27). FGF23, however, may not fully account for the pathogenesis of XLH. In this regard, there are discrepancies between FGF23-induced phosphaturia in vivo (14) and inconsistent effects of recombinant FGF23 to inhibit renal tubular phosphate uptake in vitro (14,28), suggesting possible intermediate steps. Moreover, the defective mineralization in XLH/Hyp is not fully explained by excess FGF23 or hypophosphatemia, because Fgf23-deficient mice paradoxically have defective mineralization (22) and correction of hypophosphatemia fails to fully cure rickets and osteomalacia in this disorder (29). Thus, additional factors may be involved in the renal and skeletal phenotype in XLH.
sFRP-4 is another candidate for phosphatonin. sFRP-4, which belongs to the family of secreted decoy receptors blocking Wnt-dependent signaling, is also increased in tumors from subjects with TIO and has phosphaturic activity when administered in vivo (30). However, elevated serum sFRP4 levels have not been reported in patients with TIO and expression of sFRP4 has not been assessed in XLH/Hyp.
Matrix extracellular phosphoglycoprotein (MEPE), also called OF45, a bone-specific extracellular matrix protein belonging to the SIBLING protein family (31), has also been implicated in the pathogenesis of XLH as a candidate for phosphatonin and minhibin. MEPE is expressed in osteoblasts and osteocytes (31–34), and its expression is temporally coordinated with PHEX (32–39). MEPE has phosphaturic activity when injected into mice (39), a function that appears to be mediated by C-terminal ASARM motif that localizes to the proximal renal tubule. The ASARM peptide also inhibit mineralization (39,40). Mepe expression is increased in bone derived from Hyp mice (35), and MEPE C-terminal ASARM peptide is increased in serum of humans with XLH and Hyp (41). In addition, recent studies indicate that MEPE binds to PHEX (40), and the proteolysis of MEPE is inhibited by PHEX (36). Moreover, TIO patients have increased MEPE expression in tumors (32). Also, Mepe transcripts are positively correlated with Fgf23 expression in Hyp bone, suggesting a functional interrelationship between MEPE and FGF23 in the pathogenesis of XLH/Hyp phenotype (42).
In this study, we examine the role of Mepe in mediating the hypophosphatemia and impaired mineralization in Hyp mice by transferring Mepe deficiency onto the Hyp mice background.
Materials and Methods
Transfer of Mepe Deficiency onto the Phex-Deficient Hyp Background
We obtained male heterozygous Mepe-deficient mice (Mepe+/−/XY) and female double heterozygous Mepe-deficient Hyp mice (Mepe+/−/HypX) from the laboratory of Thomas A. Brown (Department of Genetic Technologies, Pfizer Global Research and Development, Groton, CT) (34). The Mepe-deficient mice had been back-crossed onto a C57BL/6 background for more than nine generations and maintained on the same background. Crosses between heterozygous Mepe-deficient male mice and female double heterozygous Mepe-deficient Hyp mice produced 12 genotypes at the predicted frequency. Because we were interested only in effects of Mepe-null mice on the Hyp phenotype, we limited our investigations to 12- to 13-wk-old male and female wild-type (WT) Mepe-null (Mepe−/−), Hyp, and combined Mepe-null and Hyp mice (Mepe−/−/Hyp). We observed no sex-dependent difference in serum biochemical measurements within any group, allowing the combination of data from male and female mice in the following analysis. All mice were maintained and used in accordance with recommendations in the Guide for the Care and Use of Laboratory Animals, prepared by the Institute on Laboratory Animal Resources, National Research Council (DHHS Publication, National Institutes of Health 86-23, 1985). All mice were fed with LabDiet 5001 Rodent Diet (PMI Nutrition International, LLC, Brentwood, MO) consisting of 0.95% calcium and 0.67% phosphorus.
Genotyping
Genomic DNA tissue was extracted and purified from the tail of each mouse using a QIAGEN DNeasy Tissue Kit (QIAGEN Inc, Valencia, CA). To genotype Mepe-deficient mice, we performed PCR with the following primers specific for the native Mepe gene sequence and the inserted neomycin gene sequence: Mepe1016F (5′-CCCAAGAGCAGCAAAGGTAG-3′), Mepe1231R (5′-CCGCTGTGACATCCCTTTAT-3′), Neo50F (5′-AGAGGCTATTCGGCTATGACTG-3′), and Neo-480R (5′-ATCGCCTTCTATCGCCTTCTTGACGAGTTC-3′). Amplification products were resolved by electrophoresis on a 1.5% agarose gel and visualized by ethidium bromide staining. Because we can only genotype male Hyp mice, we only planned to use male mice in our original study design. After we found that Mepe-deficient Hyp females still had hypophosphatemia and skeletal phenotypes, we diagnosed female Hyp mice by hypophosphatemia, growth retardation, and characteristic on radiography (11).
Serum and Urine Assays
Serum calcium was measured using Calcium kit (Sigma-Aldrich, St. Louis, MO), and urine and serum phosphorus levels were measured by the phosphomolybdate–ascorbic acid method as described previously (11). Urine and serum creatinine levels were measured using the Stanbio Creatinine (Stanbio Laboratory, Boerne, TX). Serum parathyroid hormone (PTH) levels were measured by a mouse intact PTH ELISA kit (Immutopics, Carlsbad, CA). Serum 1,25-(OH)2-vitamin D3 levels were measured using Gamma-B 1,25-Dihydroxy Vitamin D kit (Immunodiagnostic Systems Limited, Boldon, UK). Serum Fgf23 levels were measured by using FGF-23 ELISA kit (Kainos Laboratories Inc, Tokyo, Japan) following the manufacturer’s protocol. Fractional excretion phosphate was calculated as described previously (39).
Bone Densitometry and Dry Ash Weight
Bone mineral density (BMD) of femurs was assessed at age 13 wk using a LUNARPIXIMUS bone densitometer (Lunar Corp, Madison, WI). Dry ash weight of femurs collected from 13-wk-old mice was measured as described previously (11).
Three-Dimensional Analysis of the Femurs by Microcomputed Tomography
High-resolution microcomputed tomograph (μCT) was used to evaluate trabecular volume fraction and microarchitecture in the distal femur (μCT40; Scanco Medical AG, Basserdorf, Switzerland). The femur was scanned in a 12.3-mm diameter sample holder at 45 kEv, with cone beam mode and a slice increment of 6 μm. Images from each group were generated at identical threshold. Morphometric parameters included the bone volume fraction (bone volume/trabecular volume, %), trabecular thickness (mm), trabecular number (mm-1), and trabecular separation (mm). Trabecular thickness, trabecular number, and trabecular separation were computed using model-independent distance transformation algorithms (43). To determine cortical thickness, cortical bone was imaged over a distance of 0.4 mm at the mid-shaft of the femurs. The three-dimensional structure was generated and morphometric analysis was conducted on a length of 0.3 mm using the “midshaft” program built into the μCT system software.
Biomechanical Testing of the Femurs
Femurs were dissected and cleaned soft tissue attachments and stored at −20°C until testing. On the day of testing, the femurs were thawed and rehydrated with PBS then photographed in the anterior and medial views to measure bone length. Bending tests were performed using a three-point fixture on the ELF 3200 (EnduraTEC Inc, Minnetonka, MN). The femurs were flexed in the anterior–posterior plane by displacing the loading point at 5 mm/min to failure by modifications of previously described techniques (44).
Histomorphometric Analysis of Nondecalcified Bone
Skeletons of mice were prelabeled with tetracycline hydrochloride (Sigma-Aldrich, St. Louis, MO) and calcine (Sigma-Aldrich) by intra-peritoneal injection at days 1 and 3 in 12-wk-old mice before collection of tibias. Tibias were removed from 13-wk-old mice, fixed in 70% ethanol, prestained in Villanueva stain, and processed for methyl methacrylate embedding. Five-micrometer sections were stained with Goldner’s stain and analyzed under transmitted light, and 10-μm Villanueva-prestained sections were evaluated under fluorescent light as previously reported (11).
Bone Marrow Harvest and Stromal Cell Culture
Bone marrow stromal cells (BMSC) were cultured as described previously from long bones isolated from 13-wk-old male animals from each group (45). Adherent cells were grown in the differentiation medium (α-MEM containing 10% FBS supplemented with 5 mmol/L β-glycerophosphate and 25 μg/ml of ascorbic acid) to induce osteoblastic differentiation. Mineralization was assessed by staining with Alizarin red-S, as described previously (11).
RNA extraction and Real-Time PCR
Total RNA was extracted from cultured BMSC with Trizol Reagent (Invitrogen, Carlsbad, CA). The real-time PCR were performed as described previously (42). Briefly, cDNA was synthesized from 2 μg of total RNA using TaqMan reverse-transcription reagent kit and random hexamers according to the manufacturer’s directions (Applied Biosystems, Branchburg, NJ). The reactions were incubated at 25°C for 10 min, at 42°C for 15 min, and at 99°C for 5 min, and then stored at −20°C until use. The primer sets for specific genes are shown in Table 1. The iCycler iQ Real-Time PCR Detection System and iQ SYBR Green Supermix (Bio-Rad, Hercules, CA) were used for real-time PCR analysis. We optimized the real-time PCR conditions of all selected genes by testing efficiency of the reactions and the optimal annealing temperature. The relative differences in expression between groups were expressed using cycle threshold values, and cycle threshold values for interested genes were first normalized with cyclophilin A in the same sample. Assuming that the cycle threshold value is reflective of the initial starting copy and that there is 100% efficacy, a difference of one cycle is equivalent to a two-fold difference in starting copy using the formula of 2(−ddCT).
Table 1.
Primers used for real-time PCR
| Gene | Accession Number | Forward Primer | Reverse Primer |
|---|---|---|---|
| Cyclophilin A | NM_008907 | CTGCACTGCCAAGACTGAAT | CCACAATGTTCATGCCTTCT |
| Fgf23 | NM_022657 | ACTTGTCGCAGAAGCATCA | GTGGGCGAACAGTGTAGAA |
| Sfrp 4 | NM_016687 | TGGCCATCGAACAGTATGAA | GCAGGCACTCCAGGGTACAG |
| Mepe | AF314964 | ATGCAGGGAGAGCTGGTTAC | TGGTTCCCTTTGGACTCTTC |
| Dmp1 | NM_016779 | AGTGAGGAGGACAGCCTGAA | GAGGCTCTCGTTGGACTCAC |
| Osteopontin | AF515708 | TCTGATGAGACCGTCACTGC | CCTCAGTCCATAAGCCAAGC |
| Osteocalcin | NM_007541 | AGCAGGAGGGCAATAAGGTA | CAAGCAGGGTTAAGCTCACA |
| AKP2 | NM_007431 | TTCCTGGCTCTGCCTTTAT | GGGAATCTGTGCAGTCTGT |
| Cathepsin B | BC006656 | GGAGATACTCCCAGGTGCAA | CTGCCATGATCTCCTTCACA |
| Phex | NM_011077 | GTGGTGGTCTGTGGAATCAG | AGCCGGCTTTCTTCCAATA |
| Osterix | AF184902 | ACTGGCTAGGTGGTGGTCAG | GGTAGGGAGCTGGGTTAAGG |
| Runx2 type II | NM_009820 | GCCTCACAAACAACCACAGA | TTAAACGCCAGAGCCTTCTT |
| RankL | NM011613 | GCAGAAGGAACTGCAACACA | TGATGGTGAGGTCCAAGGTTT |
| Osteoprotegerin | MMU94331 | GTTCCTGCACAGCTTCACAA | AAACAGCCCAGTGACCATTC |
Statistical Analyses
We evaluated differences between groups by one-way ANOVA. All values are expressed as means ± SEM. P < 0.05 was considered statistically significant. All computations were performed using the Statgraphics statistical graphic system (STSC Inc, Rockville, MD).
Results
Effects of Superimposed Mepe Deficiency on Hypophosphatemia and Abnormal Vitamin D Metabolism in Hyp Mice
Hyp mice had significantly lower serum phosphate levels (40% reduction), a two-fold increase in fractional excretion of phosphate, and inappropriately normal 1,25-(OH)2-vitamin D3 levels for the degree of hypophosphatemia compared with WT mice (Table 2). Mepe knockout mice had serum phosphate and 1,25-(OH)2-vitamin D3 levels that were not statistically different from WT mice. Combined Mepe-null and Hyp mice had persistent hypophosphatemia and increased fractional excretion of phosphate, as well as inappropriately low 1,25-(OH)2-vitamin D3 for the degree of hypophosphatemia (Table 2).
Table 2.
Serum and urine markers
| Serum Markers | WT | Hyp | Mepe−/− | Mepe−/−/Hyp | P |
|---|---|---|---|---|---|
| Phosphorus, mg/dl | 8.9 ± 0.5a (n = 12) | 5.3 ± 0.5b (n = 12) | 9.4 ± 0.3a (n = 24) | 5.8 ± 0.5b (n = 11) | 30.0001 |
| Calcium, mg/dl | 8.6 ± 0.2a (n = 12) | 8.5 ± 0.2a (n = 12) | 9.1 ± 0.1b (n = 24) | 8.4 ± 0.2a (n = 13) | 0.0050 |
| 1,25-(OH)2-VitD3, pM | 217.3 ± 34.4 (n = 8) | 194.8 ± 43.5 (n = 5) | 262.2 ± 34.4 (n = 8) | 220.9 ± 43.5 (n = 5) | 0.6435 |
| PTH, pg/ml | 24.6 ± 12.3a (n = 22) | 84.9 ± 14.0c (n = 17) | 30.4 ± 10.2ab (n = 32) | 60.5 ± 11.6bc (n = 25) | 0.0035 |
| Fgf23, pg/ml | 123.1 ± 10.6a (n = 4) | 2192.6 ± 159.2b (n = 4) | 125.3 ± 10.0a (n = 6) | 2287.2 ± 234.4b (n = 3) | <0.0001 |
| FEP, % | 9.3 ± 2.7a (n = 5) | 20.7 ± 3.2b (n = 5) | 7.6 ± 0.8a (n = 6) | 19.3 ± 2.7b (n = 6) | 0.0005 |
Data shown are mean ± SEM from 12-wk-old mice, except for Fgf23, which are from 5-mo-old mice. Values sharing the same letter superscript with each row are not significantly different at P < 0.05 by one-way ANOVA analysis. FEP indicates fractional excretion of phosphate.
Regarding other potential phosphaturic factors, we unexpectedly found that serum PTH levels were increased in Hyp mice compared with WT mice. We also found that serum Fgf23 was markedly elevated (18 fold) in Hyp mice (Table 2). Superimposed Mepe deficiency did not change the elevated Fgf23 or PTH levels in Hyp mice.
Effects of Mepe Deficiency Mice on the Skeletal Phenotype in Hyp Mice
Bone Histology
To assess the effect of superimposed Mepe deficiency on bone mineralization, we examined nondecalcified bone histology of tibial metaphyseal bone derived from WT, Hyp, Mepe−/−, and combined Mepe−/−/Hyp mice (Figure 1, A to H). Compared with normal amount of osteoid in WT littermates (Figure 1A), Hyp mouse bone exhibited a striking excess of osteoid (Figure 1B) that was caused by an increase in osteoid seam width as well as the extent of osteoid covered bone surfaces. This increase in osteoid was the result of impaired mineralization as evidenced by the marked reduction in surfaces undergoing mineralization in Hyp mice (Figure 1F), compared with the normal percentage of mineralizing surfaces and width of double labels in WT mice (Figure 1E). The hyperosteoidosis and diminished fluorescence labeling of bone in Hyp mice are classic histologic features of osteomalacia. Trabecular bone from Mepe-null mice was indistinguishable from WT littermates (Figure 1C), except that there was a trend toward increased mineral apposition as evidenced by the slightly greater distance between double fluorescence bone labels (Figure 1G). Combined Mepe−/− and Hyp mouse bone more closely resembled that of Hyp mice, with persistent evidence of osteomalacia characterized by excessive osteoid relative to mineralized bone (Figure 1D) and no fluorescence labels at the bone–osteoid interface (Figure 1H), indicating that superimposed Mepe deficiency failed to rescue the bone phenotype in Hyp mice.
Figure 1.
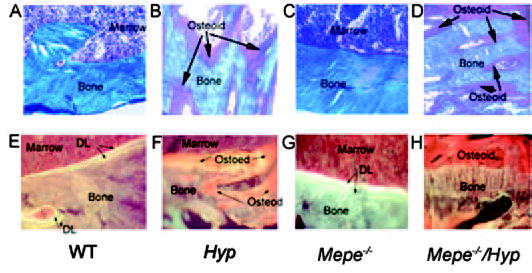
Representative nondecalcified bone sections of tibias from wild-type (WT) and mutant mice. (A to D) Goldner-stained sections are shown in the upper panel (×500). In Goldner-stained sections, mineralized bone is blue and unmineralized bone is red–brown in color. WT mice have small amounts of osteoid relative to mineralized bone (A). In contrast, Hyp bone has an increased relative osteoid volume (B). Mepe-null mice did not alter the ratio of osteoid to mineralized bone (C). Superimposed Mepe deficiency in Hyp mice (D) failed to correct the hyperosteoidosis, as evidenced by the persistent increase in relative osteoid volume in Mepe−/−/Hyp mice. (E to H) Villanueava-stained section viewed under florescent light in the lower panels (×500). Wild-type mice have two distinct double labels indicative of normal mineralization (E). Hyp mouse bone has diffuse labels consistent with impaired mineralization (F). Trabecular bone from Mepe-null mice exhibit an apparent increase in mineral apposition, as evidenced by a greater distance between double labels (G). Similar to Hyp mice, combined Mepe−/−/Hyp mice have diminished florescent labeling of bone (H), indicating that Mepe deficiency failed to correct the mineralization defect in vivo.
Bone Mineral Density
We have previously shown that BMD of femurs measured by DEXA is highly correlated with dry ash weights and both serve as surrogate markers for the degree of impaired mineralization of bone in Hyp mice (11). Consistent with the presence of osteomalacia, Hyp mice exhibited significantly lower BMD (Figure 2A) and dry ash weight (Figure 2B) of the femur compared with WT littermates. We failed to observe an increase in BMD or dry ash weight of bone in Mepe-null mice. More importantly, superimposed Mepe deficiency did not alter the diminished BMD or dry ash weight in Hyp mice. Similar to Hyp mice, BMD and dry ash weight were significantly lower in Mepe−/−/Hyp mice compared with WT littermates.
Figure 2.
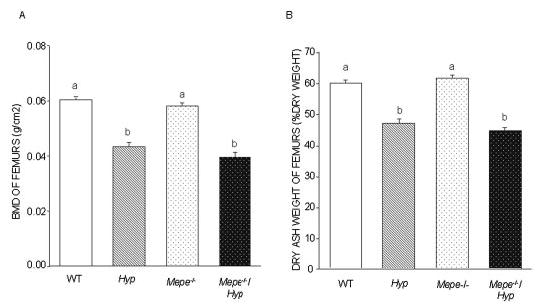
BMD and dry ash weight of femurs in WT and mutant mice. (A) BMD of the femur was assessed with the PIXImus mouse densitometer in 13-wk-old male and female WT, Hyp, Mepe−/−, and Mepe−/−/Hyp mice. (B) Dry ash weight was assessed in 13-wk-old male and female WT, Hyp, Mepe−/−, and Mepe−/−/Hyp mice. The decrease in mineralization of the skeleton in the Hyp compared with WT was confirmed by significantly reduced bone ash weight and BMD; however, Mepe−/−/Hyp exhibited no significant increment in bone ash weight and BMD compared with Hyp mice. Values sharing the same superscript are not significantly statistic different at P < 0.05.
The μCT analysis of femurs provided additional characterization of Hyp bone and the effects of superimposed Mepe deficiency (Figure 3, A through P and Table 3). Consistent with the presence of rickets and osteomalacia, the femurs of Hyp mice (Figure 3, B and F) were short, widened, and bowed compared with WT littermates (Figure 3, A and E). These changes were most pronounced in the distal femur. In addition, the growth plates were widened in Hyp compared with WT mice (Figure 3F, indicated by brackets). In addition, metaphyseal mineralized bone volume was markedly diminished in Hyp compared with WT littermates, reflecting the osteomalacia seen in histologic sections. Also, increased perpendicular lucency in the cortical bone, representing nonmineralized osteoid, is consistent with defective mineralization. In contrast, we failed to observe any significant changes in the gross appearance of bone from Mepe−/− mice (Figure 3, C and G), which was indistinguishable from WT. In addition, we did not observe any quantifiable differences in trabecular or cortical bone between Mepe-null and WT mice. Similar to the histologic and BMD results, the skeletal phenotype of combined Mepe−/−/Hyp mice (Figure 3, D and H) had the same appearance as Hyp mice, consisting of short and widened femurs width with diminished mineralized trabecular bone and increased cortical porosity consistent with persistent rickets and osteomalacia. Quantitative analysis of metaphyseal and cortical bone confirmed these impressions. In this regard, Hyp mice had significant reductions in all trabecular bone parameters and diminished cortical thickness compared with WT mice. We were unable to identify any effects of Mepe deficiency on trabecular and cortical bone measurements in 12-wk-old mice, and superimposed Mepe deficiency failed to alter the trabecular and cortical parameters in Hyp mice (Table 3).
Figure 3.
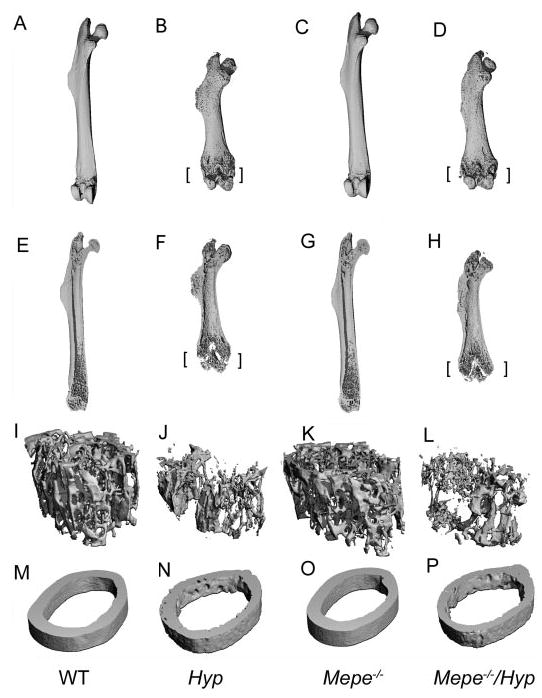
Three-dimensional structures measured by μCT of the femurs from 13-wk-old male WT and mutant mice. (A to D) Gross appearance of whole femurs. Femurs derived from Hyp mice are short, widened, and bowed, consistent with rickets/osteomalacia. Superimposed Mepe−/−/Hyp does not alter the gross appearance of the skeleton. (E to H) Sagittal views of femurs. Femurs of Hyp and Mepe−/−/Hyp mice show unmineralized growth plates and much less trabecular bones compared with WT mice. There are no significant changes between WT and Mepe knockout mice. (I to L) Three-dimensional imagine of trabecular architecture of femoral metaphysis of distal femurs. (M to P) Three-dimensional imagine of cortical bones in mid-shaft of femurs. Cortical thickness is decreased and cortical porosity is increased in both Hyp and Mepe−/−/Hyp mice, consistent with excessive unmineralized osteoid on the endosteal surfaces and Haversian channels. [brackets] represent widened growth plate.
Table 3.
Micro CT analysis of femurs in WT and mutant mice
| Group | WT | Hyp | Mepe−/− | Mepe−/−/Hyp | P |
|---|---|---|---|---|---|
| BV/TV, % | 12.91 ± 0.73a | 5.17 ± 0.81b | 10.70 ± 0.48a | 5.60 ± 0.96b | <0.0001 |
| Tb. N, mm−1 | 5.09 ± 0.14a | 3.13 ± 0.20b | 5.18 ± 0.10a | 3.79 ± 0.13c | <0.0001 |
| Tb. Th, mm | 0.042 ± 0.003a,b | 0.047 ± 0.005b | 0.036 ± 0.001a | 0.040 ± 0.006a,b | 0.0350 |
| Tb. Sp, mm | 0.193 ± 0.006a | 0.337 ± 0.024b | 0.189 ± 0.004a | 0.267 ± 0.013c | <0.0001 |
| Ct.Th, mm | 0.186 ± 0.004a | 0.117 ± 0.006b | 0.177 ± 0.002a | 0.105 ± 0.012b | <0.0001 |
Data shown are mean ± SEM from 5 male 13-wk-old mice. Values sharing the same letter superscript are not significantly different at P < 0.05 by one-way ANOVA analysis.
Biomechanical Properties
Next, we investigated the biomechanical properties of WT and mutant mice. To accomplish this, we performed biomechanical testing on femurs using a three-point bending fixture (Table 4). Compared with WT mice, the bones of Hyp mice were more pliable and could tolerate significantly less force before failure. Mepe-null mice also exhibited a decreased biomechanical strength as evidenced by a significant reduction in the maximum force and deflection at failure compared with WT mice. The reduced bone biomechanical properties were more severe in combined Mepe-null and Hyp mice than in either Mepe-null or Hyp mice alone. Taken together, these findings indicate that Mepe deficiency worsened the mechanical properties of bone.
Table 4.
Bone mechanical testing using three-point bending fixture
| Group | WT (n = 5) | Hyp (n = 5) | Mepe−/− (n = 14) | Mepe−/−/Hyp (n = 5) | P |
|---|---|---|---|---|---|
| Maximum force, N | 25.8 ± 1.2a | 8.0 ± 1.0 b | 23.1 ± 0.7c | 4.5 ± 0.9d | <0.0001 |
| Maximum deflection,* mm | 0.71 ± 0.03a | 0.96 ± 0.20a | 0.62 ± 0.04a | 1.89 ± 0.29b | <0.0001 |
| Deflection at failure, mm | 0.93 ± 0.14a | 2.08 ± 0.08b | 0.71 ± 0.06c | 2.11 ± 0.22d | <0.0001 |
| Stiffness, N/mm | 55.4 ± 4.3a | 17.6 ± 2.5b | 54.3 ± 2.5a | 6.4 ± 1.4c | <0.0001 |
Data shown are mean ± SEM from 13-wk-old male mice. Values sharing the same letter superscript with each row are not significantly different at P < 0.05 by one-way ANOVA analysis.
Maximum deflection represents the deformation corresponding to maximum force.
Effects of Mepe Deficiency on the BMSC in Hyp Mice
Although Mepe ablation did not rescue the bone phenotype in Hyp mice, we tested if Mepe deficiency altered the intrinsic mineralization defect observed in osteoblasts from Hyp mice (9). BMSC were grown in the presence of ascorbate and β-glycerophosphate to induce osteoblast differentiation and promote bone nodule formation. Consistent with an earlier report (10), BMSC derived from Hyp mice displayed impaired mineralization compared with WT mice as assessed by alizarin red staining of mineralization nodules, suggesting an intrinsic defect in mineralization in the Hyp osteoblast cells (Figure 4). Also consistent with earlier report (34), BMSC derived from Mepe-null mice had an increased mineralization potential compared with WT BMSC ex vivo (Figure 4). More importantly, BMSC derived from combined Mepe-null and Hyp mice displayed greater mineralization potential after induction of osteoblast differentiation than BMSC derived from Hyp mice (Figure 4). The improvement of the mineralization defect, however, was limited, because mineralization remained significantly less in combined Mepe−/−/Hyp-derived BMSC than in WT cultures (Figure 4B).
Figure 4.
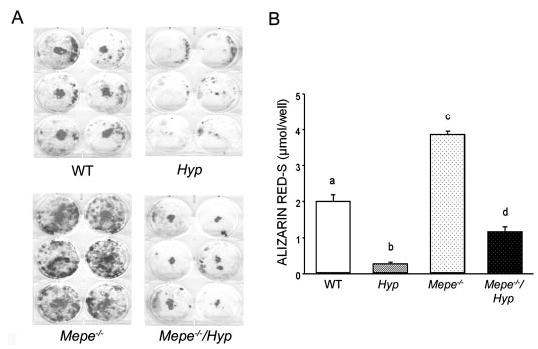
Effects of Mepe deficiency on Hyp-derived bone marrow stromal cell maturation and mineralization. (A) Alizarin red-S staining of primary bone marrow stromal cells derived from WT, Hyp, Mepe-null, and combined Mepe-null Hyp mice. Compared with WT, bone marrow stromal cells derived from Hyp mice formed less mineralization nodules, whereas the cells from Mepe−/− mice formed much more mineralized nodules. Mepe−/−/Hyp mice formed more mineralized nodules compared with cells from Hyp. (B) Quantification of mineralization. The alizarin red-S stain was extracted with 10% cetylpyridinium chloride and quantified by absorbance measurement at 562 nm as described in Materials and Methods. Numeric values represent the mean ± SEM of six wells. Bone marrow-derived mesenchymal stem cells were culture from WT and mutant mice for 14 d in the presence of ascorbic acid and β-glycerophosphate to induce osteoblast differentiation. Values sharing the same letter superscript are not significantly different at P < 0.05 by one-way ANOVA analysis.
To further explore the effects of combined Phex and Mepe deficiency on osteoblast function, we evaluated gene expression profiles of BMSC (Table 3). Except for impaired mineralization, Hyp-derived BMSC had normal levels of osteoblast transcripts, except for increased Mepe and Fgf23 expression, and a small but significant decrease in the osteoblast transcription factor Runx2-II. In contrast, Mepe deficiency, consistent with the enhanced mineralization, resulted in significant alterations in the osteoblast phenotype ex vivo that was characterized by an increase in the osteoblastic transcription factors Runx2-II and osterix, as well as osteoblastic differentiation markers alkaline phosphatase, DMP-1, and osteocalcin, consistent with increased osteoblastogenesis and/or differentiation. Expression of the osteoclastic coupling factor Rank L was decreased in BMSC from Mepe-null mice. The presence of combined Phex and Mepe deficiency in the Mepe−/−/Hyp mouse-derived BMSC reversed all of the effects of Mepe deficiency on gene expression profiles, suggesting that the lack of Phex function somehow offset the effects of Mepe deficiency on osteoblast gene expression.
Discussion
We transferred Mepe deficiency onto the Hyp mouse background to examine the role of Mepe in the pathogenesis of hypophosphatemia and defective mineralization in XLH/Hyp. We found that Mepe-deficient mice had serum phosphate levels and fractional excretion of phosphate indistinguishable from WT littermates (Table 2), and Mepe-deficient Hyp mice remained hypophosphatemic and phosphaturic, as well as had persistent inappropriately normal 1,25(OH)2D3 levels (Table 2). The inability of Mepe deficiency to rescue the renal abnormalities in Hyp mice indicates that Mepe is not the phosphaturic factor, phosphatonin.
Superimposing Mepe deficiency onto the Hyp background also did not correct the gross skeletal abnormalities associated with Phex mutations, indicating that hypophosphatemia and/or associated factors are more important than Mepe in the defective skeletal mineralization in Hyp mice. In vitro data, however, implicate a role for Mepe in regulating mineralization that may have been masked by persistent hypophosphatemia in vivo. We observed improved mineralization in Hyp-derived BMSC ex vivo in the absence of Mepe (Figure 4). In addition, Mepe deficiency was associated with increased osteoblastic markers, including DMP1, osteocalcin, Osterix, and Runx2-II, and inactivating Phex mutations counteracts the changes in osteoblastic gene expression profiles induced by Mepe deficiency (Table 5). Mepe deficiency also increases Phex expression in BMSC.
Table 5.
Real-time PCR of bone marrow stromal cells from WT and mutant mice
| Gene | WT | Hyp | Mepe−/− | Mepe−/−/Hyp | P |
|---|---|---|---|---|---|
| Phex | 0.01 ± 0.001a | Not detectable | 0.09 ± 0.017b | Not detectable | 0.0003 |
| Akp2 | 0.35 ± 0.043a | 0.39 ± 0.115a | 1.87 ± 0.593b | 0.28 ± 0.073a | 0.0165 |
| Cathepsin B | 4.89 ± 0.613a | 3.60 ± 1.043a | 3.15 ± 0.622a | 3.20 ± 0.281a | 0.3232 |
| Mepe | 0.002 ± 0.0005a | 0.008 ± 0.0028b | Not detectable | Not detectable | 0.0035 |
| Dmp1 | 0.13 ± 0.028a | 0.17 ± 0.023a | 0.32 ± 0.055b | 0.12 ± 0.017a | 0.0120 |
| Osteocalcin | 0.05 ± 0.012a | 0.06 ± 0.012a | 0.53 ± 0.067b | 0.06 ± 0.009a | <0.0001 |
| Osteoprotegerin | 0.04 ± 0.004a | 0.04 ± 0.006a | 0.02 ± 0.003a | 0.03 ± 0.005a | 0.1174 |
| Fgf23 | 0.0006 ± 0.0001a | 0.0046 ± 0.0012b | 0.0001 ± 0.00001a | 0.0064 ± 0.0020b | 0.0022 |
| sfrp4 | 0.11 ± 0.003a | 0.08 ± 0.010a | 0.09 ± 0.007a | 0.08 ± 0.013a | 0.0920 |
| RankL | 0.001 ± 0.00025a | 0.001 ± 0.00017a | 0.0001 ± 0.00002b | 0.0012 ± 0.00025a | 0.0147 |
| Runx2 II | 0.011 ± 0.0004a | 0.007 ± 0.0012b | 0.017 ± 0.0014c | 0.008 ± 0.0007ab | 0.0004 |
| Osterix | 0.004 ± 0.0007a | 0.003 ± 0.0005a | 0.014 ± 0.0037b | 0.004 ± 0.0002a | 0.0109 |
Data shown are mean ± SEM from bone marrow stromal cell cultures from WT and mutant mice at 14 d of culture duration with ascorbic acid and β-glycerophosphate (n ≥ 3). Values sharing the same letter superscript within each row are not significantly different at P < 0.05 by one-way ANOVA analysis.
Our studies differ from previous studies by Gowen et al. (34) because we failed to observe an increase in bone mass by both DEXA and μCT or to demonstrate increased bone strength by biomechanical testing (Figures 1 to 3). The reasons for these discrepancies are not clear, although notable differences exist with regard to age, techniques for assessing the skeletal phenotype, and genetic background between the two studies.
With regard to other potential phosphaturic factors, we found that sfrp-4 (30) was not different in BMSC from Hyp or Mepe-deficient mice (Table 5). Another interesting finding is the three-fold increased circulating levels of the phosphaturic hormone PTH in Hyp mice. PTH, however, is not the primary phosphaturic factor in Hyp, because parathyroidectomy does not correct the hypophosphatemia in Hyp mice (46). The mechanism of PTH elevation in Hyp remains an enigma, but Phex has been identified in the parathyroid gland (47), raising the possibility that a primary abnormality in parathyroid gland function may also exist in XLH.
Finally, we observed an 18-fold increase in serum Fgf23 levels in Hyp mice compared with WT littermates (Table 2) and an eight-fold increase in Fgf23 transcripts in BMSC from Hyp mice compared with WT mice (Table 5). Recent studies demonstrate that transfer of Fgf23 deficiency onto the Hyp background increases serum phosphorus in Hyp mice (23). Collectively, these findings implicate Fgf23, rather than Mepe, as the leading candidate for phosphatonin. The mechanisms whereby Phex deficiency results in increased Fgf23 expression are not certain, but the original notion that Fgf23 is a substrate of Phex (28) has not been confirmed (42,48). We interpret the concordant increase in serum levels and expression of Fgf23 to indicate that inactivating Phex mutations lead to the increased production of Fgf23 through intermediate steps that likely involve potential Phex substrates and/or other intermediate factors (13).
In conclusion, Mepe deficiency does not correct the hypophosphatemia in Hyp mice and therefore is not phosphatonin, but Mepe may play a role in regulating the local mineralization process in Hyp mice. It may be necessary to correct the hypophosphatemia in combined Mepe−/−/Hyp mice to uncover the in vivo function of Mepe in the pathogenesis of XLH. Regardless, the identification of the physiologically relevant Phex substrates will be necessary to fully understand the pathogenesis of XLH.
Acknowledgments
This work was supported by National Institutes of Health grant RO1 AR-45955 from the National Institutes of Arthritis and Musculoskeletal and Skin Diseases.
References
- 1.The HYP Consortium. A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. Nat Genet. 1995;11:130–136. doi: 10.1038/ng1095-130. [DOI] [PubMed] [Google Scholar]
- 2.Strom TM, Francis F, Lorenz B, Boddrich A, Econs MJ, Lehrach H, Meitinger T. Pex gene deletions in Gy and Hyp mice provide mouse models for X-linked hypophosphatemia. Hum Mol Genet. 1997;6:165–171. doi: 10.1093/hmg/6.2.165. [DOI] [PubMed] [Google Scholar]
- 3.Carpinelli MR, Wicks IP, Sims NA, O’Donnell K, Hanzinikolas K, Burt R, Foote SJ, Bahlo M, Alexander WS, Hilton DJ. An ethyl-nitrosourea-induced point mutation in phex causes exon skipping, x-linked hypophosphatemia, and rickets. Am J Pathol. 2002;161:1925–1933. doi: 10.1016/S0002-9440(10)64468-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beck L, Soumounou Y, Martel J, Krishnamurthy G, Gauthier C, Goodyer CG, Tenenhouse HS. Pex/PEX tissue distribution and evidence for a deletion in the 3′ region of the Pex gene in X-linked hypophosphatemic mice. J Clin Invest. 1997;99:1200–1209. doi: 10.1172/JCI119276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Du L, Desbarats M, Viel J, Glorieux FH, Cawthorn C, Ecarot B. cDNA cloning of the murine Pex gene implicated in X-linked hypophosphatemia and evidence for expression in bone. Genomics. 1996;36:22–28. doi: 10.1006/geno.1996.0421. [DOI] [PubMed] [Google Scholar]
- 6.Guo R, Quarles LD. Cloning and sequencing of human PEX from a bone cDNA library: Evidence for its developmental stage-specific regulation in osteoblasts. J Bone Miner Res. 1997;12:1009–1017. doi: 10.1359/jbmr.1997.12.7.1009. [DOI] [PubMed] [Google Scholar]
- 7.Ruchon AF, Marcinkiewicz M, Siegfried G, Tenenhouse HS, DesGroseillers L, Crine P, Boileau G. Pex mRNA is localized in developing mouse osteoblasts and odontoblasts. J Histochem Cytochem. 1998;46:459–468. doi: 10.1177/002215549804600405. [DOI] [PubMed] [Google Scholar]
- 8.Nesbitt T, Fujiwara I, Thomas R, Xiao ZS, Quarles LD, Drezner MK. Coordinated maturational regulation of PHEX and renal phosphate transport inhibitory activity: evidence for the pathophysiological role of PHEX in X-linked hypophosphatemia. J Bone Miner Res. 1999;14:2027–2035. doi: 10.1359/jbmr.1999.14.12.2027. [DOI] [PubMed] [Google Scholar]
- 9.Xiao ZS, Crenshaw M, Guo R, Nesbitt T, Drezner MK, Quarles LD. Intrinsic mineralization defect in Hyp mouse osteoblasts. Am J Physiol. 1998;275:E700–E708. doi: 10.1152/ajpendo.1998.275.4.E700. [DOI] [PubMed] [Google Scholar]
- 10.Miao D, Bai X, Panda D, McKee M, Karaplis A, Goltzman D. Osteomalacia in hyp mice is associated with abnormal phex expression and with altered bone matrix protein expression and deposition. Endocrinology. 2001;142:926–939. doi: 10.1210/endo.142.2.7976. [DOI] [PubMed] [Google Scholar]
- 11.Liu S, Guo R, Tu Q, Quarles LD. Overexpression of Phex in osteoblasts fails to rescue the Hyp mouse phenotype. J Biol Chem. 2002;277:3686–3697. doi: 10.1074/jbc.M107707200. [DOI] [PubMed] [Google Scholar]
- 12.Bai X, Miao D, Panda D, Grady S, McKee MD, Goltzman D, Karaplis AC. Partial rescue of the Hyp phenotype by osteoblast-targeted PHEX (phosphate-regulating gene with homologies to endopeptidases on the X chromosome) expression. Mol Endocrinol. 2002;16:2913–2925. doi: 10.1210/me.2002-0113. [DOI] [PubMed] [Google Scholar]
- 13.Quarles LD. Evidence for a bone-kidney axis regulating phosphate homeostasis. J Clin Invest. 2003;112:642–646. doi: 10.1172/JCI19687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A. 2001;98:6500–6505. doi: 10.1073/pnas.101545198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamashita T, Yoshioka M, Itoh N. Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem Biophys Res Commun. 2000;277:494–498. doi: 10.1006/bbrc.2000.3696. [DOI] [PubMed] [Google Scholar]
- 16.White KE, Carn G, Lorenz-Depiereux B, Benet-Pages A, Strom TM, Econs MJ. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int. 2001;60:2079–2086. doi: 10.1046/j.1523-1755.2001.00064.x. [DOI] [PubMed] [Google Scholar]
- 17.Saito H, Kusano K, Kinosaki M, Ito H, Hirata M, Segawa H, Miyamoto K, Fukushima N. Human fibroblast growth factor-23 mutants suppress Na+-dependent phosphate co-transport activity and 1alpha,25-dihydroxyvitamin D3 production. J Biol Chem. 2003;278:2206–2211. doi: 10.1074/jbc.M207872200. [DOI] [PubMed] [Google Scholar]
- 18.The ADHR Consortium. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet. 2000;26:345–348. doi: 10.1038/81664. [DOI] [PubMed] [Google Scholar]
- 19.Yamazaki Y, Okazaki R, Shibata M, Hasegawa Y, Satoh K, Tajima T, Takeuchi Y, Fujita T, Nakahara K, Yamashita T, Fukumoto S. Increased circulatory level of biologically active full-length FGF-23 in patients with hypophosphatemic rickets/osteomalacia. J Clin Endocrinol Metab. 2002;87:4957–4960. doi: 10.1210/jc.2002-021105. [DOI] [PubMed] [Google Scholar]
- 20.Weber TJ, Liu S, Indridason OS, Quarles LD. Serum FGF23 levels in normal and disordered phosphorus homeostasis. J Bone Miner Res. 2003;18:1227–1234. doi: 10.1359/jbmr.2003.18.7.1227. [DOI] [PubMed] [Google Scholar]
- 21.Jonsson KB, Zahradnik R, Larsson T, White KE, Sugimoto T, Imanishi Y, Yamamoto T, Hampson G, Koshiyama H, Ljunggren O, Oba K, Yang IM, Miyauchi A, Econs MJ, Lavigne J, Juppner H. Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med. 2003;348:1656–1663. doi: 10.1056/NEJMoa020881. [DOI] [PubMed] [Google Scholar]
- 22.Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K, Yamashita T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest. 2004;113:561–568. doi: 10.1172/JCI19081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sitara D, Razzaque MS, Hesse M, Yoganathan S, Taguchi T, Erben RG, H JA-P, Lanske B. Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol. 2004;23:421–432. doi: 10.1016/j.matbio.2004.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takeuchi Y, Suzuki H, Ogura S, Imai R, Yamazaki Y, Yamashita T, Miyamoto Y, Okazaki H, Nakamura K, Nakahara K, Fukumoto S, Fujita T. Venous sampling for fibroblast growth factor-23 confirms preoperative diagnosis of tumor-induced osteomalacia. J Clin Endocrinol Metab. 2004;89:3979–3982. doi: 10.1210/jc.2004-0406. [DOI] [PubMed] [Google Scholar]
- 25.De Beur SM, Finnegan RB, Vassiliadis J, Cook B, Barberio D, Estes S, Manavalan P, Petroziello J, Madden SL, Cho JY, Kumar R, Levine MA, Schiavi SC. Tumors associated with oncogenic osteomalacia express genes important in bone and mineral metabolism. J Bone Miner Res. 2002;17:1102–1110. doi: 10.1359/jbmr.2002.17.6.1102. [DOI] [PubMed] [Google Scholar]
- 26.Larsson T, Zahradnik R, Lavigne J, Ljunggren O, Juppner H, Jonsson KB. Immunohistochemical detection of FGF-23 protein in tumors that cause oncogenic osteomalacia. Eur J Endocrinol. 2003;148:269–276. doi: 10.1530/eje.0.1480269. [DOI] [PubMed] [Google Scholar]
- 27.Riminucci M, Collins MT, Fedarko NS, Cherman N, Corsi A, White KE, Waguespack S, Gupta A, Hannon T, Econs MJ, Bianco P, Gehron Robey P. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest. 2003;112:683–692. doi: 10.1172/JCI18399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bowe AE, Finnegan R, Jan de Beur SM, Cho J, Levine MA, Kumar R, Schiavi SC. FGF-23 inhibits renal tubular phosphate transport and is a PHEX substrate. Biochem Biophys Res Commun. 2001;284:977–981. doi: 10.1006/bbrc.2001.5084. [DOI] [PubMed] [Google Scholar]
- 29.Marie PJ, Travers R, Glorieux FH. Healing of bone lesions with 1,25-dihydroxyvitamin D3 in the young X-linked hypophosphatemic male mouse. Endocrinology. 1982;111:904–911. doi: 10.1210/endo-111-3-904. [DOI] [PubMed] [Google Scholar]
- 30.Berndt T, Craig TA, Bowe AE, Vassiliadis J, Reczek D, Finnegan R, Jan De Beur SM, Schiavi SC, Kumar R. Secreted frizzled-related protein 4 is a potent tumor-derived phosphaturic agent. J Clin Invest. 2003;112:785–794. doi: 10.1172/JCI18563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fisher LW, Fedarko NS. Six genes expressed in bones and teeth encode the current members of the SIBLING family of proteins. Connect Tissue Res. 2003;44(Suppl 1):33–40. [PubMed] [Google Scholar]
- 32.Rowe PS, de Zoysa PA, Dong R, Wang HR, White KE, Econs MJ, Oudet CL. MEPE, a new gene expressed in bone marrow and tumors causing osteomalacia. Genomics. 2000;67:54–68. doi: 10.1006/geno.2000.6235. [DOI] [PubMed] [Google Scholar]
- 33.Petersen DN, Tkalcevic GT, Mansolf AL, Rivera-Gonzalez R, Brown TA. Identification of osteoblast/osteocyte factor 45 (OF45), a bone-specific cDNA encoding an RGD-containing protein that is highly expressed in osteoblasts and osteocytes. J Biol Chem. 2000;275:36172–36180. doi: 10.1074/jbc.M003622200. [DOI] [PubMed] [Google Scholar]
- 34.Gowen LC, Petersen DN, Mansolf AL, Qi H, Stock JL, Tkalcevic GT, Simmons HA, Crawford DT, Chidsey-Frink KL, Ke HZ, McNeish JD, Brown TA. Targeted disruption of the osteoblast/osteocyte factor 45 gene (OF45) results in increased bone formation and bone mass. J Biol Chem. 2003;278:1998–2007. doi: 10.1074/jbc.M203250200. [DOI] [PubMed] [Google Scholar]
- 35.Argiro L, Desbarats M, Glorieux FH, Ecarot B. Mepe, the gene encoding a tumor-secreted protein in oncogenic hypophosphatemic osteomalacia, is expressed in bone. Genomics. 2001;74:342–351. doi: 10.1006/geno.2001.6553. [DOI] [PubMed] [Google Scholar]
- 36.Guo R, Rowe PS, Liu S, Simpson LG, Xiao ZS, Darryl Quarles LD. Inhibition of MEPE cleavage by Phex. Biochem Biophys Res Commun. 2002;297:38–45. doi: 10.1016/s0006-291x(02)02125-3. [DOI] [PubMed] [Google Scholar]
- 37.Nampei A, Hashimoto J, Hayashida K, Tsuboi H, Shi K, Tsuji I, Miyashita H, Yamada T, Matsukawa N, Matsumoto M, Morimoto S, Ogihara T, Ochi T, Yoshikawa H. Matrix extracellular phosphoglycoprotein (MEPE) is highly expressed in osteocytes in human bone. J Bone Miner Metab. 2004;22:176–184. doi: 10.1007/s00774-003-0468-9. [DOI] [PubMed] [Google Scholar]
- 38.Siggelkow H, Schmidt E, Hennies B, Hufner M. Evidence of downregulation of matrix extracellular phosphoglycoprotein during terminal differentiation in human osteoblasts. Bone. 2004;35:570–576. doi: 10.1016/j.bone.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 39.Rowe PS, Kumagai Y, Gutierrez G, Garrett IR, Blacher R, Rosen D, Cundy J, Navvab S, Chen D, Drezner MK, Quarles LD, Mundy GR. MEPE has the properties of an osteoblastic phosphatonin and minhibin. Bone. 2004;34:303–319. doi: 10.1016/j.bone.2003.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rowe PS, Garrett IR, Schwarz PM, Carnes DL, Lafer EM, Mundy GR, Gutierrez GE. Surface plasmon resonance (SPR) confirms that MEPE binds to PHEX via the MEPE-ASARM motif: A model for impaired mineralization in X-linked rickets (HYP) Bone. 2005;36:33–46. doi: 10.1016/j.bone.2004.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bresler D, Bruder J, Mohnike K, Fraser WD, Rowe PS. Serum MEPE-ASARM-peptides are elevated in X-linked rickets (HYP): Implications for phosphaturia and rickets. J Endocrinol. 2004;183:R1–R9. doi: 10.1677/joe.1.05989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu S, Guo R, Simpson LG, Xiao ZS, Burnham CE, Quarles LD. Regulation of fibroblastic growth factor 23 expression but not degradation by PHEX. J Biol Chem. 2003;278:37419–37426. doi: 10.1074/jbc.M304544200. [DOI] [PubMed] [Google Scholar]
- 43.Hildebrand T, Laib A, Muller R, Dequeker J, Ruegsegger P. Direct three-dimensional morphometric analysis of human cancellous bone: Microstructural data from spine, femur, iliac crest, and calcaneus. J Bone Miner Res. 1999;14:1167–1174. doi: 10.1359/jbmr.1999.14.7.1167. [DOI] [PubMed] [Google Scholar]
- 44.Silva MJ, Ulrich SR. In vitro sodium fluoride exposure decreases torsional and bending strength and increases ductility of mouse femora. J Biomech. 2000;33:231–234. doi: 10.1016/s0021-9290(99)00158-x. [DOI] [PubMed] [Google Scholar]
- 45.Xiao ZS, Quarles LD, Chen QQ, Yu YH, Qu XP, Jiang CH, Deng HW, Li YJ, Zhou HH. Effect of asymmetric dimethylarginine on osteoblastic differentiation. Kidney Int. 2001;60:1699–1704. doi: 10.1046/j.1523-1755.2001.00011.x. [DOI] [PubMed] [Google Scholar]
- 46.Meyer RA, Jr, Tenenhouse HS, Meyer MH, Klugerman AH. The renal phosphate transport defect in normal mice parabiosed to X-linked hypophosphatemic mice persists after parathyroidectomy. J Bone Miner Res. 1989;4:523–532. doi: 10.1002/jbmr.5650040411. [DOI] [PubMed] [Google Scholar]
- 47.Blydt-Hansen TD, Tenenhouse HS, Goodyer P. PHEX expression in parathyroid gland and parathyroid hormone dysregulation in X-linked hypophosphatemia. Pediatr Nephrol. 1999;13:607–611. doi: 10.1007/s004670050669. [DOI] [PubMed] [Google Scholar]
- 48.Benet-Pages A, Lorenz-Depiereux B, Zischka H, White KE, Econs MJ, Strom TM. FGF23 is processed by proprotein convertases but not by PHEX. Bone. 2004;35:455–462. doi: 10.1016/j.bone.2004.04.002. [DOI] [PubMed] [Google Scholar]