

Summary
Glucocorticoids are potent immunosuppressive drugs that are generally withheld from cancer patients receiving immunotherapy. We sought to test the hypothesis that glucocorticoids might interfere with the function of cells after adoptive transfer. We gave dexamethasone, a potent synthetic glucocorticoid, to B16 melanoma–bearing mice receiving the adoptive cell transfer (ACT) of pmel-1 T-cell receptor transgenic CD8+ cells. Dexamethasone caused a profound lymphodepletion but, surprisingly, did not alter the antitumor efficacy of ACT-based regimens whether given before, during, or after ACT. Although dexamethasone radically decreased the number of native CD8+ splenocytes in recipient mice, it did not affect the numbers of CD8+ pmel-1 cells derived from ACT in these mice. In vitro proliferation assays revealed acute inhibition of naive pmel-1 CD8+ cells by dexamethasone without significant effect on activated cells. In vitro interferon (IFN)-γ release from activated pmel-1 CD8+ cells showed partial inhibition by dexamethasone, but this effect was relatively minor when compared with the near-complete inhibition of naive cells. Thus, glucocorticoids had a profound inhibitory effect on naive CD8+ T cells but had little impact on the proliferation and function of activated CD8+ pmel-1 T cells. Finally, because glucocorticoids had no effect on tumor regression in this model, it may be possible to use glucocorticoids in some patients receiving ACT-based immunotherapy regimens.
Keywords: immunotherapy, glucocorticoid, T cell
Glucocorticoids are potent immunosuppressive drugs that are used to treat diverse clinical conditions, but they are generally withheld from cancer patients receiving immunotherapy because of concerns that they may inhibit immune-mediated tumor regression. Cancer patients may require glucocorticoids not only for the treatment of concomitant diseases such as asthma and arthritis but for palliation of pain from bone metastases or cerebral edema from brain metastases. In addition, glucocorticoids are highly effective in treating autoimmune side effects of immunotherapy, including gastritis, ileitis, colitis, and hypophysitis, caused by anticytotoxic T-lymphocyte–associated antigen 4 (CTLA-4) therapy.1
The immunosuppressive effects of glucocorticoids include inhibition of inflammatory cytokines, upregulation of regulatory cytokines, inhibition of dendritic cells, and induction of T-cell apoptosis.2 Perhaps the strongest evidence that glucocorticoids inhibit T-cell responses, and the most compelling reason why they are withheld in patients receiving immunotherapy, is their ability to prevent and treat organ rejection, a T-cell–mediated event. There is, however, no direct evidence that glucocorticoids have any effect on adoptive cell transfer (ACT) therapy.
To determine the effect of glucocorticoids on ACT, the pmel-1 model of cell transfer tumor immunotherapy, consisting of 1) adoptively transferred T-cell receptor (TCR) transgenic CD8+ T cells reactive against the tumor/self-antigen gp100, 2) antigen-specific vaccination, and 3) administration of exogenous interleukin (IL)-2 to induce regression of B16 melanoma, was used.3–5 Dexamethasone, a potent synthetic glucocorticoid with high affinity for the glucocorticoid receptor and relatively low binding to plasma proteins, was given. Because lymphodepletion with radiation or chemotherapy before cell transfer improves ACT therapy, we explored the effects of glucocorticoids given for lymphodepletion before cell transfer as well as the effects when given after cell transfer.6–9
MATERIALS AND METHODS
Mice and Tumor Lines
The pmel-1 TCR transgenic (Tg) mice (now available from Jackson Laboratories, Bar Harbor, ME)5 were crossed with homozygous Thy1.1 mice (Jackson Laboratories) to generate pmel-1/Thy1.1 double-Tg mice. Expression of the transgenes was confirmed by polymerase chain reaction (PCR) analysis for the pmel-1 α- and β-TCR chains, and fluorescence-activated cell sorting (FACS) analysis was used to confirm the presence of the Thy1.1 congenic marker. The mice were housed at the National Institutes of Health (NIH), and procedures were performed according to the guidelines of the NIH Animal Care and Use Committee. B16 (H-2b), a spontaneous murine melanoma expressing gp100, was maintained in culture in complete media (CM) composed of RPMI 1640 with 10% heat-inactivated fetal bovine serum (Biofluids, Rockville, MD).5
In Vitro Activation, Cytokine Release, and Proliferation Assays
The pmel-1 and pmel-1/Thy1.1 splenocytes were isolated as described,5 and used immediately (naive) or cultured (activated) in the presence of 1 μM hgp10025–3310 and CM containing 60 IU/mL recombinant human IL-2 (rhIL-2; Chiron, Emeryville, CA) for 6 to 8 days. When naive cells were used, depletion of non-CD8+ cells using magnetic cell sorting negative-selection columns (Miltenyi Biotec, Auburn, CA) was performed. For in vitro coculture experiments, 104 pmel-1 cells were cocultured in CM, with or without dexamethasone (Sigma, St. Louis, MO), at concentrations indicated, with 105 irradiated (30 Gy) C57BL/6 target splenocytes pulsed with hgp10025–33 or NP366–374 peptides. Interferon (IFN)-γ release assays were performed as previously described.5 For proliferation assays, cells were pulsed with 1 μCi H3-thymidine after 6 hours of coculture, incubated 18 hours, and then harvested using a semiautomated sample harvester, and counts per minute were determined with a β-scintillation counter. Cytokine release and proliferation assays were performed in triplicate, and values are presented as mean ± standard deviation (SD).
Adoptive Cell Transfer, Vaccination, Cytokine and Dexamethasone Administration
Female C57BL/6 (Jackson Laboratories) mice 6 to 8 weeks old were injected subcutaneously with 5 × 105 B16F10 melanoma cells. Mice (n = 5–10) were treated 7 to 10 days later with intravenous adoptive transfer of 3 × 106 activated pmel-1 or pmel-1/Thy1.1 splenocytes. Indicated mice were vaccinated with intravenous administration of 2 × 107 plaque-forming units of recombinant fowlpox virus-encoding hgp100 (rFPhgp100; Therion Biologics, Cambridge, MA) as described previously3,5,10 and 600,000 IU rhIL-2 intraperitoneally twice daily for a total of 6 doses. In some experiments, lymphodepletion was accomplished by administration of 5 Gy whole-body irradiation before cell transfer. When given to induce lymphodepletion before cell transfer, dexamethasone was administered intraperitoneally at a dose of 10 mg/kg on days −6, −4, and −2 before transfer. Dexamethasone regimens of 10 mg/kg or 1 mg/kg administered intraperitoneally on days 2, 4, 6, 8, and 10 were used to model a course of dexamethasone after cell transfer. Dexamethasone at a dose of 10 mg/kg from days −1 to 17 and days 2 to 17 was used to treat mice throughout the period of antitumor activity. Tumor size was determined in a blinded fashion using calipers. The products of the perpendicular diameters are presented as mean ± standard error of the mean (SEM). Analysis of variance (ANOVA) was used to determine the significance of differences in tumor measurements between groups.
Enumeration of Splenocytes and Adoptively Transferred Cells
Enumeration of thymocytes was accomplished by harvesting thymuses and forming a single cell suspension by passage through a 100-μm cell strainer (BD Falcon, Bedford, MA). Cells were counted using trypan blue exclusion. Evaluation of splenocytes after cell transfer was accomplished by harvesting 2 pooled spleens from each group. To enumerate pmel-1 CD8+ splenocytes, cells were stained with monoclonal antibodies against CD8a and Thy1.1 and flow cytometry was performed as described below. The absolute number of pmel-1 cells was calculated by multiplying the splenocyte count by the percentage of total cells that were CD8+ and Thy1.1+.
Flow Cytometry
Cells were labeled with monoclonal antibodies (BD Biosciences, San Diego, CA) against Vβ13-fluorescein isothiocyanate (FITC), Thy1.1-phycoerythrin (PE), and CD8− antigen-presenting cells (APCs) or CD8− cychrome. Samples were analyzed using FACScalibur and CELLquest software (BD Biosciences).
RESULTS
Nonmyeloablative Lymphodepletion With Radiation Enhances pmel-1 CD8+ T-Cell Immunotherapy
To determine the effect of lymphodepletion on ACT, pmel-1 CD8+ T cells were transferred into irradiated and nonirradiated hosts and response was assessed with serial tumor measurements. The pmel-1 CD8+ T cells were activated in vitro with hgp10025–33 and IL-2 before transfer. As is required with this model, gp100-encoding fowlpox viral vaccine and IL-2 were also administered.5 Nonmyeloablative lymphodepletion with radiation before cell transfer resulted in significantly decreased tumor growth compared with treatment without lymphodepletion (P = 0.048; Fig. 1). In experiments in which only the tumor was shielded from radiation, tumor regression was similar, ruling out a direct antitumor effect of the radiation (unpublished data). Therefore, lymphodepletion with radiation before adoptive transfer improved tumor treatment. This finding led us to speculate that other agents that induce lymphodepletion may improve subsequent cell transfer therapy.
FIGURE 1.
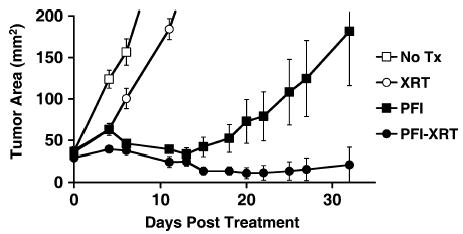
Lymphodepletion with total-body radiation augmented tumor regression mediated by adoptively transferred antigen-specific CD8+ T cells. Mice bearing large unmanipulated B16 melanoma tumors were treated with adoptive transfer of activated pmel-1 cells, vaccination with rFPgp10025–33, and IL-2. Either no lymphodepletion (PFI) or lymphodepletion with 5 Gy total-body irradiation (PFI-XRT) was administered before cell transfer. Radiation alone (XRT) had a negligible effect on tumor growth.
Dexamethasone Induces Lymphodepletion but Does Not Enhance Tumor Regression by Subsequently Transferred pmel-1 CD8+ T Cells
Having observed improved treatment with lymphodepleting radiation given before cell transfer, we hypothesized that lymphodepletion with dexamethasone would similarly enhance tumor treatment. The proapoptotic effects of glucocorticoids on immune cells are well known.11 To evaluate the lymphodepleting effects of dexamethasone, mice were treated with high-dose dexamethasone or phosphate-buffered saline (PBS). Spleens were harvested, and viable splenocytes were enumerated. Dexamethasone-treated mice had a mean of 2.83 ± 0.8 × 105 splenocytes, whereas controls had a mean of 8.7 ± 0.7 × 107 splenocytes. We concluded that dexamethasone does indeed cause lymphodepletion and sought to determine if this lymphodepletion would translate into the improved treatment seen with radiation-induced lymphodepletion.
To evaluate the effect of dexamethasone lymphodepletion on subsequent pmel-1 cell transfer therapy, tumor-bearing mice were treated with dexamethasone or PBS, followed by transfer of activated pmel-1 cells, vaccination, and IL-2. Dexamethasone was administered at a dose of 10 mg/kg on days −6, −4, and −2 before cell transfer. Tumor growth was similar in both groups (P = 0.665; Fig. 2). Despite inducing lymphodepletion, dexamethasone did not affect treatment with pmel-1 CD8+ cell transfer.
FIGURE 2.
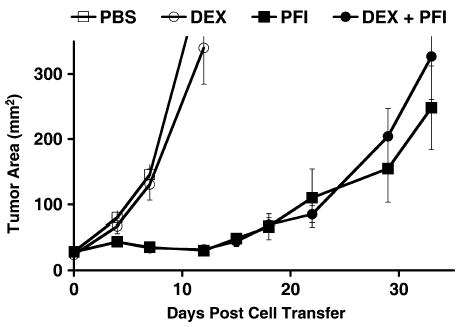
Lymphodepletion with dexamethasone did not affect subsequent ACT immunotherapy. Tumor-bearing mice were treated with PBS or dexamethasone (DEX), followed by adoptive transfer of activated pmel-1 cells, vaccination with rFPgp10025–33, and IL-2 (PFI and DEX + PFI, respectively). Tumor treatment was not affected.
Effect of a 9-Day Course of Dexamethasone After Cell Transfer on Tumor Treatment by pmel-1 CD8+ T Cells
Dexamethasone induced lymphodepletion without causing a net enhancement or impairment of tumor regression mediated by pmel-1 cells. This finding prompted us to explore the impact of a course of glucocorticoids given after cell transfer, a situation relevant in clinical practice, where patients may require steroids to treat cerebral edema, airway edema, allergic reactions, or other concomitant diseases or complications that arise after ACT. To permit assessment of a positive or negative effect of dexamethasone on therapy, a suboptimal number of activated pmel-1 cells were given to provide a “window” whereby improved treatment would decrease the slope of the tumor growth curve and impaired treatments would increase it. Tumor-bearing mice were treated with cell transfer and administered a course of dexamethasone at a dose of 10 mg/kg or 1 mg/kg every other day for 9 days starting on day 2 or no dexamethasone. Tumor growth in mice treated with cell transfer and dexamethasone was not significantly different than in mice receiving cell transfer only (P = 0.22; Fig. 3C). The thymuses of mice from each group were also harvested on day 12, and CD4+CD8+ cells were enumerated. The number of double-positive thymocytes in dexamethasone-treated mice was markedly diminished, confirming its biologic activity (see Fig. 3A). To assess the effect of dexamethasone alone on tumor growth, tumor-bearing mice received dexamethasone or PBS. Dexamethasone alone did not significantly affect tumor growth (P = 0.20; see Fig. 3B).
FIGURE 3.
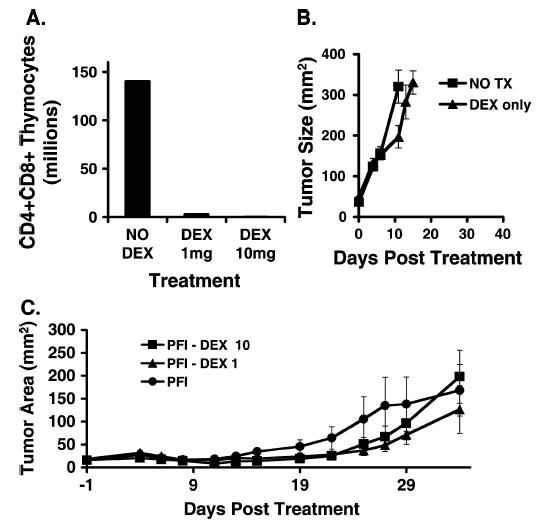
A course of dexamethasone did not augment but also did not inhibit immunotherapy with ACT. A, Dexamethasone depleted double-positive thymocytes. After a course of dexamethasone, thymocytes were enumerated and FACS analysis was used to determine the frequency of CD4+CD8+ thymocytes. Double-positive thymocyte number was reduced in the groups treated with dexamethasone at a dose of 1 mg/kg (DEX, 1 mg) and at a dose of 10 mg/kg (DEX, 10 mg) compared with mice receiving no dexamethasone (NO DEX). B, Dexamethasone alone (DEX only) had minimal effect on the growth of untreated tumors (NO TX). C, Tumor-bearing mice were treated with adoptive transfer of activated pmel-1 cells, vaccination with rFPgp10025–33, and IL-2 (PFI). Groups were treated with a course of dexamethasone at a dose of 10 mg/kg (PFI-DEX 10) or at a dose of 1 mg/kg on days 2, 4, 6, 8, and 10 after cell transfer (PFI-DEX 1). Tumor treatment was not affected by dexamethasone.
Effect of Dexamethasone on pmel-1 CD8+ T-Cell Number
We next sought to determine if a 9-day course of dexamethasone diminished the expansion of adoptively transferred pmel-1 cells in treated mice. We compared the number of pmel-1 splenocytes on day 5 after cell transfer in mice treated with and without dexamethasone. Mice in the experiment shown in Figure 3C were killed on day 5 after transfer. The number of total splenocytes in dexamethasone-treated mice was reduced, but the number of pmel-1 splenocytes was essentially the same between groups (Fig. 4). Dexamethasone seemed to impair native splenocytes selectively, while sparing transferred pmel-1 cells.
FIGURE 4.
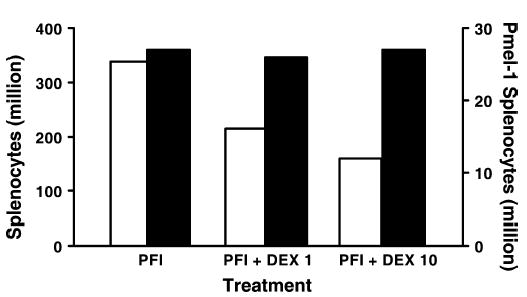
Tumor-bearing mice were treated with adoptive transfer of activated pmel-1 cells expressing the congenic marker Thy1.1, vaccination with rFPgp10025–33, and IL-2 (PFI). A course of dexamethasone at a dose of 1 mg/kg (PFI+DEX 1) or at a dose of 10 mg/kg (PFI+DEX 10) was administered to the indicated groups. Five days after cell transfer, splenocytes were enumerated and FACS analysis was performed to determine the frequency of Thy1.1+ cells. Overall splenocyte numbers (open bars) were diminished in mice receiving a course of dexamethasone. Pmel-1 numbers (solid bars) were not diminished by dexamethasone.
Effect of Dexamethasone on Proliferation of Activated and Naive pmel-1 In Vitro
We observed different responses to dexamethasone between native splenocytes and transferred pmel-1 cells and speculated that this difference may be attributable to the relative resistance of the activated pmel-1 cells to suppression by dexamethasone. To determine if the proliferation of activated and naive cells is affected differently by dexamethasone, coculture experiments were performed comparing H3-thymidine incorporation in the presence of titrated concentrations of dexamethasone. Pmel-1 splenocytes were isolated and activated in vitro in the presence of hgp10025–33 peptide and IL-2. After 6 to 8 days of activation under these conditions, cell cultures consisted of highly purified activated (expressing high levels of CD25, CD44, and CD69) pmel-1 CD8+ cells.5 Naive pmel-1 CD8+ splenocytes were freshly isolated and enriched using negative selection magnetic bead columns.
H3-thymidine incorporation by naive pmel-1 decreased with increasing concentrations of dexamethasone, whereas H3-thymidine incorporation by activated cells was not significantly affected by dexamethasone (Fig. 5). This lack of effect on activated cells may explain, at least in part, the lack of inhibition of tumor treatment, seen with a course of dexamethasone.
FIGURE 5.
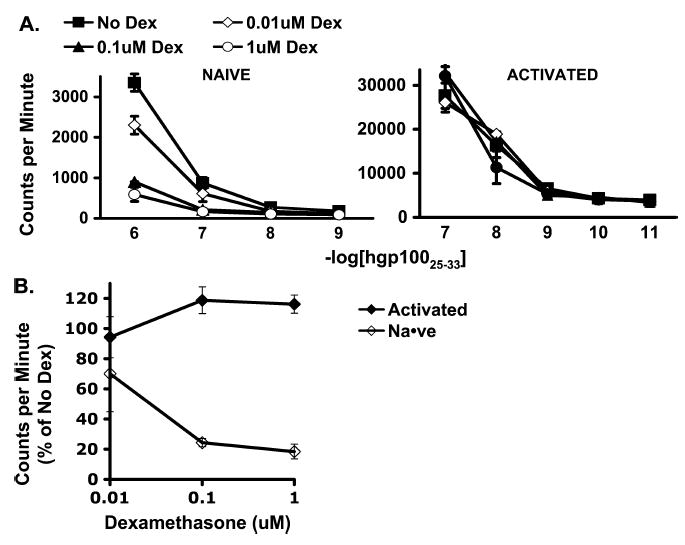
Dexamethasone impaired proliferation of naive but not activated antigen-specific CD8+ T cells. A, H3-thymidine incorporation by naive and activated pmel-1 CD8+ T cells cocultured with hgp100 peptide–pulsed irradiated splenocytes. H3-thymidine incorporation by naive cells but not by activated cells was diminished. B, H3-thymidine incorporation by naive and activated pmel-1 CD8+ T cells cocultured with irradiated splenocytes pulsed with hgp100 at 1 × 10−7 M in titrated concentrations of dexamethasone relative to H3-thymidine incorporation in the absence of dexamethasone. Proliferation of naive cells was impaired, whereas proliferation of activated cells was not affected.
Effect of Dexamethasone on Function of Activated and Naive pmel-1 In Vitro
To determine if the function of activated and naive cells was affected differently by dexamethasone, coculture experiments were performed to compare IFNγ release by naive and activated pmel-1 cells in the presence or absence of dexamethasone. Activated and naive cells were isolated as described previously. On coculture with hgp100-pulsed irradiated splenocytes, IFNγ release by naive pmel-1 CD8+ cells was markedly decreased in the presence of dexamethasone (Fig. 6A). Activated cell IFNγ production was inhibited by dexamethasone, but this inhibition was relatively low compared with that seen with naive cells (see Fig. 6A).
FIGURE 6.
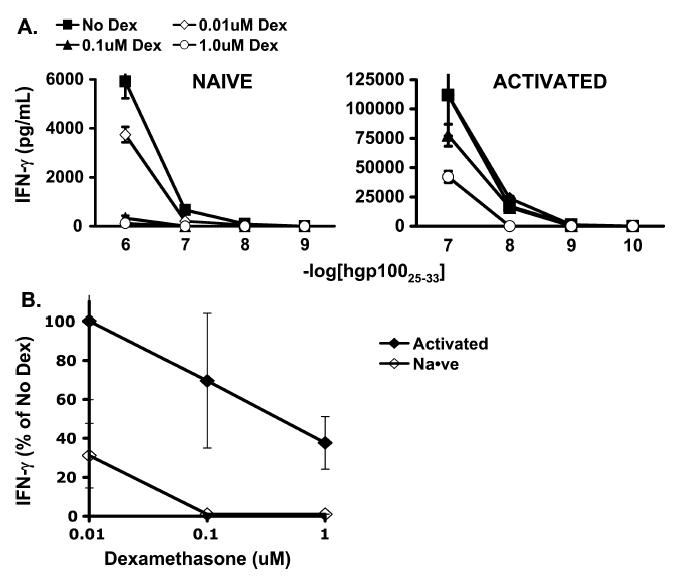
Function of naive and activated antigen-specific CD8+ T cells was impaired by dexamethasone, but naive cells were more significantly suppressed. A, IFNγ release by naive and activated pmel-1 CD8+ T cells cocultured with hgp100 peptide–pulsed irradiated splenocytes. IFNγ release by naive and activated cells was diminished. B, IFNγ release by naive and activated pmel-1 CD8+ T cells cocultured with irradiated splenocytes pulsed with hgp100 at 1 × 10−7 M in titrated concentrations of dexamethasone relative to H3-thymidine incorporation in the absence of dexamethasone. Naive cell function was more impaired than activated cell function.
In Vivo Effect of Administration of Dexamethasone During the Entire Period of Antitumor Activity by Transferred pmel-1 CD8+ Cells
Because we noted some inhibition of activated pmel-1 function with dexamethasone in vitro, we considered the possibility that the reason we had not seen impairment in tumor treatment in vivo was failure to expose the transferred cells to dexamethasone for an adequate period. We sought to determine if administration of dexamethasone throughout the entire period of antitumor activity by the transferred pmel-1 cells impaired their ability to cause tumor regression. To establish the length of time during which pmel-1 cells have a significant antitumor effect, anti-CD8a antibodies were given to pmel-1–treated mice at intervals up to 16 days after cell transfer. At time points before 16 days, anti-CD8a antibody treatment significantly impaired therapy; however, at the day 16 time point, no significant difference in treatment was observed (data not shown). We concluded that the therapeutic effect by the transferred pmel-1 cells was completed by day 16.
To test the effect of dexamethasone given during this period of antitumor effect by transferred cells, high-dose dexamethasone was started the day before cell transfer or 2 days after cell transfer and continued for 17 days. The tumor growth curves for the groups receiving dexamethasone or no dexamethasone were not significantly different (Fig. 7) as confirmed by ANOVA (P > 0.05). We tested several additional dexamethasone dosing regimens, including daily from days 2 to 6; daily from days 7 to 17; twice daily for 6 doses; and a single dose on day 0, 1, 2, 3, or 4 (data not shown). Despite our consistent observations that high-dose steroids depleted double-positive thymocytes and host splenocytes, we saw no effect on tumor treatment with any of these regimens, and therefore concluded that dexamethasone neither enhanced nor hindered cell transfer tumor therapy.
FIGURE 7.
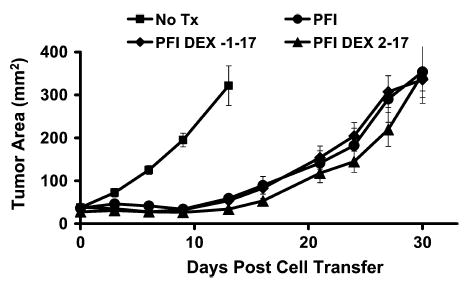
Dexamethasone given throughout the antitumor activity of transferred cells did not impair tumor treatment. Tumor-bearing mice were treated with adoptive transfer of activated pmel-1 cells, vaccination with rFPgp10025–33, and IL-2 (PFI). Groups were then given no additional therapy (PFI), high-dose dexamethasone daily from the day before transfer through day 17 after transfer (PFI DEX 1–17), or high-dose dexamethasone daily from the second day after transfer through day 17 after transfer (PFI DEX 2–17). Treatment with dexamethasone did not significantly affect tumor regression mediated by adoptively transferred cells.
DISCUSSION
Nonmyeloablative lymphodepletion with cyclophosphamide and fludarabine before ACT enhances the effectiveness of transferred tumor infiltrating lymphocytes (TILs) in patients with metastatic melanoma.9 This preparative regimen induces thrombocytopenia, anemia, and immunodeficiency, however. We therefore sought a way to induce lymphopenia and enhance cell transfer efficacy without the unintended effects of lymphodepleting chemotherapy. To determine if lymphodepletion in mice results in augmentation of ACT immunotherapy of tumors, the pmel-1 model was used. In this model, cytotoxic chemotherapy alone induced regression of B16 melanoma, thereby confounding assessment of tumor response to subsequent cell transfer treatments (unpublished data). Therefore, sublethal total-body radiation with 5 Gy was used to induce lymphodepletion because it had minimal effect on tumor growth. Lymphodepletion with total-body irradiation resulted in improved tumor treatment (see Fig. 1). We then sought to determine whether lymphodepletion with dexamethasone, a potent synthetic glucocorticoid known to induce apoptosis of lymphocytes, was possible. High-dose dexamethasone induced lymphodepletion in the spleens of treated mice; however, this lymphodepletion did not lead to improved tumor treatment with subsequent ACT (see Fig. 2).
There are several possible explanations for failure of lymphodepletion with dexamethasone to improve subsequent ACT. Lymphodepletion with chemotherapy or radiation augments ACT by inducing homeostatic cytokines, depleting regulatory and competing cell populations and enhancing APC function.12,13 By contrast, dexamethasone suppresses proinflammatory cytokines, promotes anti-inflammatory cytokines, may preferentially spare regulatory cells, and inhibits dendritic cell activation.14–23 There may also be differences in how chemotherapy, radiation, and dexamethasone deplete cells outside the spleen. In addition, the splenocyte populations in dexamethasone-treated mice began to replete earlier than in radiation-treated mice (data not shown). Thus, lymphodepletion with dexamethasone may not be as durable as with chemotherapy or radiation, and this lack of durability may contribute to its failure to augment ACT.
Dexamethasone given before ACT did not augment therapy; however, it did not impair therapy either. This finding is encouraging because glucocorticoids are valuable and may even be lifesaving in the treatment of certain medical conditions, but they are rarely administered to immunotherapy patients because of concerns that they may inhibit immune-based treatments. Even topical and inhaled glucocorticoids are discontinued when patients are enrolled in immunotherapy trials, and all patients are counseled to avoid taking these medications. Nevertheless, circumstances arise in which patients with no previous indication for treatment with steroids require a course, or even long-term treatment, with glucocorticoids. For example, cancer patients may develop brain metastases with cerebral edema that is responsive to glucocorticoid treatment. Glucocorticoids may also be required to treat allergic reactions to drugs or radiographic contrast agents. The latter is of particular concern because patients treated with IL-2 are at increased risk of hypersensitivity to radiographic contrast material, and computed tomography is routinely used to evaluate response to immunotherapy.24 It was therefore valuable to know if the immunosuppressive effects of glucocorticoids impaired immunotherapy.
To address this issue we treated mice receiving ACT immunotherapy with glucocorticoids. We found no impairment in tumor-specific CD8+ T-cell–mediated tumor regression. Furthermore, there was no in vivo reduction in the number of antigen-specific CD8+ T cells. In vitro experiments also showed no impairment of activated CD8+ proliferation but some impairment of activated CD8+ function. The impairment in function was relatively minor compared with that seen with naive cells and did not lead to decreased tumor treatment in vivo.
We were surprised by these findings; however, glucocorticoids have diverse dynamic biologic effects involving different components of the immune system, and their in vivo effects are sometimes unpredictable. In addition to immunosuppressive effects, including suppression of proinflammatory cytokines, promotion of anti-inflammatory cytokines, inhibition of dendritic cells, suppression of natural killer cells, promotion of T-regulatory cells, and induction of T-cell apoptosis, glucocorticoids have been reported to have immune-enhancing effects, which may explain why they do not inhibit tumor treatment with ACT. One of these effects is inhibition of activation-induced cell death associated with TCR signaling.11,25–30 This phenomenon was initially described in T-cell hybridomas, where activation in the absence of dexamethasone or exposure to dexamethasone in the absence of activation led to apoptosis. Activation in the presence of dexamethasone attenuated activation-induced cell death, however.31 The mechanism of this inhibition may be through repression of Fas ligand expression, which is thought to be integral in causing activation-induced cell death.32,33 The pmel-1 CD8+ T cells become highly activated (expressing high levels of CD25 and CD44 and decreased levels of CD62L) by TCR signaling from in vitro stimulation with hgp100 peptide and subsequent in vivo stimulation from vaccination with fowlpox virus-encoding hgp10025–33.5 These activated CD8+ T cells, in the presence of specific TCR signaling, were uninhibited in their proliferation in vitro, and their numbers were not reduced by dexamethasone in vivo.
In addition to TCR signaling, these lymphocytes were exposed in vitro and in vivo to high doses of IL-2, a cytokine reported to induce resistance to glucocorticoids. Some clinical evidence for this comes from study of steroid-resistant asthmatics, in whom glucocorticoids do not inhibit T-cell proliferation or reduce proinflammatory cytokines.34 The airways of these patients have elevated expression of IL-2 and IL-4.34,35 In addition, rescue of T helper subsets and protection of T-cell hybridomas and T lymphocytes from glucocorticoid-induced apoptosis have been reported.36–38 This protective effect may be mediated through STAT5 repression of glucocorticoid-dependent transcription.39 Regardless of the mechanism, the phenomenon of IL-2–mediated protection of lymphocytes from glucocorticoid-induced apoptosis has been documented by several investigators and may explain why we did not see impaired treatment in mice treated with dexamethasone and IL-2.
Signaling through several receptors may also have a role in resistance of activated T cells to glucocorticoid-induced apoptosis. GITR is a transmembrane receptor homologous to 4-1BB, CD27, and OX-40 of the tumor necrosis factor (TNF)/nerve growth factor receptor (NGFR) family that is induced by glucocorticoids.40–42 Overexpression in T-cell hybridomas was reported to inhibit CD3-mediated apoptosis.41 Nur77 is a nuclear orphan receptor, and its induction is required for activation-induced cell death of T-cell hybridomas.43,44 It has been reported to antagonize transcription effects mediated by the glucocorticoid receptor.45 Finally, glucocorticoids enhance expression of IL-7 receptor-α, the receptor for the important homeostatic cytokine IL-7.46 Signaling through these receptors may, in part, explain the absence of glucocorticoid-mediated inhibition of ACT therapy.
Although glucocorticoids mediate immune-enhancing and immunosuppressive effects, their overall effect is immunosuppressive in clinical situations, such as the treatment of asthma, arthritis, and organ transplant rejection. Organ transplant rejection parallels tumor treatment in that it is primarily a cell-mediated event, which has been shown to be dependent on either CD4+ and/or CD8+ T cells in various models.47–49 We have demonstrated that in a CD8-dependent tumor treatment model, glucocorticoids do not prevent tumor rejection. Why are glucocorticoids effective in preventing graft rejection but not tumor rejection? There are some important differences between the tumor treatment model and models of transplant rejection:
ACT involves transfer of a massive number of in vitro–activated antigen-specific cells to mediate rejection, whereas transplant rejection involves conversion of a smaller number of naive cells from the native repertoire into activated cells capable of graft rejection.
Transferred cells are driven by exogenous administration of IL-2 without which they do not mediate tumor regression5; however, no IL-2 is given to patients receiving organ transplants.
Antigen-specific vaccination is given with cell transfer and is required for effective treatment, whereas no vaccine is administered with organ transplantation.5
ACT in this model is mediated primarily by CD8+ T cells, whereas transplant rejection may be mediated by CD4+ T cells, CD8+ T cells, or both.47–49 Although transplanted allograft rejection is cell mediated, there are distinct differences between graft rejection and tumor immunotherapy that limit the applicability of lessons learned in transplantation to immunotherapy.
There are 2 situations in which glucocorticoids have been given to patients treated with immunotherapy for cancer. Anti-CTLA-4 therapy, given for treatment of melanoma and kidney cancer, induces autoimmunity, which is associated with tumor response. Glucocorticoids are effective in treating the autoimmune side effects of this therapy, which include colitis and hypophysitis.1 Interestingly, we have observed ongoing tumor regression after the initiation of steroid therapy. Glucocorticoids have also been used to treat graft-versus-host disease (GVHD) associated with allogeneic stem cell transplantation. Despite administration of glucocorticoids, ongoing responses are not uncommon. Furthermore, glucocorticoids are only approximately 50% effective in gaining durable control of GVHD, a T-cell–mediated event.50,51 The impact of steroid administration on patients receiving ACT is unknown, but the data presented here indicate that glucocorticoids may not interfere with the antitumor response of immunotherapy.
References
- 1.Phan GQ, Yang JC, Sherry RM, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci USA. 2003;100:8372–8377. doi: 10.1073/pnas.1533209100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Franchimont D, Kino T, Galon J, et al. Glucocorticoids and inflammation revisited: the state of the art. NIH Clinical Staff Conference. Neuroimmunomodulation. 2003;10:247–260. doi: 10.1159/000069969. [DOI] [PubMed] [Google Scholar]
- 3.Klebanoff CA, Finkelstein SE, Surman DR, et al. IL-15 enhances the in vivo antitumor activity of tumor-reactive CD8+ T cells. Proc Natl Acad Sci USA. 2004;101:1969–1974. doi: 10.1073/pnas.0307298101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palmer DC, Balasubramaniam S, Hanada K, et al. Vaccine-stimulated, adoptively transferred CD8+ T cells traffic indiscriminately and ubiquitously while mediating specific tumor destruction. J Immunol. 2004;173:7209–7216. doi: 10.4049/jimmunol.173.12.7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Overwijk WW, Theoret MR, Finkelstein SE, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dudley ME, Rosenberg SA. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat Rev Cancer. 2003;3:666–675. doi: 10.1038/nrc1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenberg SA, Dudley ME. Cancer regression in patients with metastatic melanoma after the transfer of autologous antitumor lymphocytes. Proc Natl Acad Sci USA. 2004;101(Suppl 2):14639–14645. doi: 10.1073/pnas.0405730101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosenberg SA, Yang JC, Robbins PF, et al. Cell transfer therapy for cancer: lessons from sequential treatments of a patient with metastatic melanoma. J Immunother. 2003;26:385–393. doi: 10.1097/00002371-200309000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Overwijk WW, Tsung A, Irvine KR, et al. gp100/pmel 17 is a murine tumor rejection antigen: induction of “self”-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J Exp Med. 1998;188:277–286. doi: 10.1084/jem.188.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ashwell JD, Lu FW, Vacchio MS. Glucocorticoids in T cell development and function. Annu Rev Immunol. 2000;18:309–345. doi: 10.1146/annurev.immunol.18.1.309. [DOI] [PubMed] [Google Scholar]
- 12.Klebanoff CA, Khong HT, Antony PA, et al. Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol. 2005;26:111–117. doi: 10.1016/j.it.2004.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wrzesinski C, Restifo NP. Less is more: lymphodepletion followed by hematopoietic stem cell transplant augments adoptive T-cell-based antitumor immunotherapy. Curr Opin Immunol. 2005;17:195–201. doi: 10.1016/j.coi.2005.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y, Louboutin JP, Zhu J, et al. Preterminal host dendritic cells in irradiated mice prime CD8+ T cell-mediated acute graft-versus-host disease. J Clin Invest. 2002;109:1335–1344. doi: 10.1172/JCI14989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rea D, van Kooten C, van Meijgaarden KE, et al. Glucocorticoids transform CD40-triggering of dendritic cells into an alternative activation pathway resulting in antigen-presenting cells that secrete IL-10. Blood. 2000;95:3162–3167. [PubMed] [Google Scholar]
- 16.Piemonti L, Monti P, Allavena P, et al. Glucocorticoids affect human dendritic cell differentiation and maturation. J Immunol. 1999;162:6473–6481. [PubMed] [Google Scholar]
- 17.Nelson DJ, McWilliam AS, Haining S, et al. Modulation of airway intraepithelial dendritic cells following exposure to steroids. Am J Respir Crit Care Med. 1995;151:475–481. doi: 10.1164/ajrccm.151.2.7842209. [DOI] [PubMed] [Google Scholar]
- 18.Chen X, Murakami T, Oppenheim JJ, et al. Differential response of murine CD4+CD25+ and CD4+CD25− T cells to dexamethasone-induced cell death. Eur J Immunol. 2004;34:859–869. doi: 10.1002/eji.200324506. [DOI] [PubMed] [Google Scholar]
- 19.Franchimont D, Louis E, Dewe W, et al. Effects of dexamethasone on the profile of cytokine secretion in human whole blood cell cultures. Regul Pept. 1998;73:59–65. doi: 10.1016/s0167-0115(97)01063-x. [DOI] [PubMed] [Google Scholar]
- 20.Almawi WY, Hess DA, Rieder MJ. Multiplicity of glucocorticoid action in inhibiting allograft rejection. Cell Transplant. 1998;7:511–523. doi: 10.1177/096368979800700602. [DOI] [PubMed] [Google Scholar]
- 21.Almawi WY, Melemedjian OK. Molecular mechanisms of glucocorticoid antiproliferative effects: antagonism of transcription factor activity by glucocorticoid receptor. J Leukoc Biol. 2002;71:9–15. [PubMed] [Google Scholar]
- 22.Barrat FJ, Cua DJ, Boonstra A, et al. In vitro generation of interleukin 10-producing regulatory CD4(+) T cells is induced by immunosuppressive drugs and inhibited by T helper type 1 (Th1)- and Th2-inducing cytokines. J Exp Med. 2002;195:603–616. doi: 10.1084/jem.20011629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Franchimont D. Overview of the actions of glucocorticoids on the immune response: a good model to characterize new pathways of immunosuppression for new treatment strategies. Ann NY Acad Sci. 2004;1024:124–137. doi: 10.1196/annals.1321.009. [DOI] [PubMed] [Google Scholar]
- 24.Zukiwski AA, David CL, Coan J, et al. Increased incidence of hypersensitivity to iodine-containing radiographic contrast media after interleukin-2 administration. Cancer. 1990;65:1521–1524. doi: 10.1002/1097-0142(19900401)65:7<1521::aid-cncr2820650712>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 25.Iwata M, Hanaoka S, Sato K. Rescue of thymocytes and T cell hybridomas from glucocorticoid-induced apoptosis by stimulation via the T cell receptor/CD3 complex: a possible in vitro model for positive selection of the T cell repertoire. Eur J Immunol. 1991;21:643–648. doi: 10.1002/eji.1830210316. [DOI] [PubMed] [Google Scholar]
- 26.Iwata M, Ohoka Y, Kuwata T, et al. Regulation of T cell apoptosis via T cell receptors and steroid receptors. Stem Cells. 1996;14:632–641. doi: 10.1002/stem.140632. [DOI] [PubMed] [Google Scholar]
- 27.Jamieson CA, Yamamoto KR. Crosstalk pathway for inhibition of glucocorticoid-induced apoptosis by T cell receptor signaling. Proc Natl Acad Sci USA. 2000;97:7319–7324. doi: 10.1073/pnas.97.13.7319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Riccardi C, Zollo O, Nocentini G, et al. Glucocorticoid hormones in the regulation of cell death. Therapie. 2000;55:165–169. [PubMed] [Google Scholar]
- 29.Ayroldi E, Migliorati G, Bruscoli S, et al. Modulation of T-cell activation by the glucocorticoid-induced leucine zipper factor via inhibition of nuclear factor kappaB. Blood. 2001;98:743–753. doi: 10.1182/blood.v98.3.743. [DOI] [PubMed] [Google Scholar]
- 30.Ko M, Jang J, Ahn J, et al. T cell receptor signaling inhibits glucocorticoid-induced apoptosis by repressing the SRG3 expression via Ras activation. J Biol Chem. 2004;279:21903–21915. doi: 10.1074/jbc.M402144200. [DOI] [PubMed] [Google Scholar]
- 31.Zacharchuk CM, Mercep M, Chakraborti PK, et al. Programmed T lymphocyte death. Cell activation- and steroid-induced pathways are mutually antagonistic. J Immunol. 1990;145:4037–4045. [PubMed] [Google Scholar]
- 32.Yang Y, Mercep M, Ware CF, et al. Fas and activation-induced Fas ligand mediate apoptosis of T cell hybridomas: inhibition of Fas ligand expression by retinoic acid and glucocorticoids. J Exp Med. 1995;181:1673–1682. doi: 10.1084/jem.181.5.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kasibhatla S, Genestier L, Green DR. Regulation of Fasligand expression during activation-induced cell death in T lymphocytes via nuclear factor kappaB. J Biol Chem. 1999;274:987–992. doi: 10.1074/jbc.274.2.987. [DOI] [PubMed] [Google Scholar]
- 34.Leung DY, Martin RJ, Szefler SJ, et al. Dysregulation of interleukin 4, interleukin 5, and interferon gamma gene expression in steroid-resistant asthma. J Exp Med. 1995;181:33–40. doi: 10.1084/jem.181.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sher ER, Leung DY, Surs W, et al. Steroid-resistant asthma. Cellular mechanisms contributing to inadequate response to glucocorticoid therapy. J Clin Invest. 1994;93:33–39. doi: 10.1172/JCI116963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fernandez-Ruiz E, Rebollo A, Nieto MA, et al. IL-2 protects T cell hybrids from the cytolytic effect of glucocorticoids. Synergistic effect of IL-2 and dexamethasone in the induction of high-affinity IL-2 receptors. J Immunol. 1989;143:4146–4151. [PubMed] [Google Scholar]
- 37.Nieto MA, Lopez-Rivas A. IL-2 protects T lymphocytes from glucocorticoid-induced DNA fragmentation and cell death. J Immunol. 1989;143:4166–4170. [PubMed] [Google Scholar]
- 38.Zubiaga AM, Munoz E, Huber BT. IL-4 and IL-2 selectively rescue Th cell subsets from glucocorticoid-induced apoptosis. J Immunol. 1992;149:107–112. [PubMed] [Google Scholar]
- 39.Biola A, Lefebvre P, Perrin-Wolff M, et al. Interleukin-2 inhibits glucocorticoid receptor transcriptional activity through a mechanism involving STAT5 (signal transducer and activator of transcription 5) but not AP-1. Mol Endocrinol. 2001;15:1062–1076. doi: 10.1210/mend.15.7.0657. [DOI] [PubMed] [Google Scholar]
- 40.Gurney AL, Marsters SA, Huang RM, et al. Identification of a new member of the tumor necrosis factor family and its receptor, a human ortholog of mouse GITR. Curr Biol. 1999;9:215–218. doi: 10.1016/s0960-9822(99)80093-1. [DOI] [PubMed] [Google Scholar]
- 41.Nocentini G, Giunchi L, Ronchetti S, et al. A new member of the tumor necrosis factor/nerve growth factor receptor family inhibits T cell receptor-induced apoptosis. Proc Natl Acad Sci USA. 1997;94:6216–6221. doi: 10.1073/pnas.94.12.6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kwon B, Yu KY, Ni J, et al. Identification of a novel activation-inducible protein of the tumor necrosis factor receptor superfamily and its ligand. J Biol Chem. 1999;274:6056–6061. doi: 10.1074/jbc.274.10.6056. [DOI] [PubMed] [Google Scholar]
- 43.Liu ZG, Smith SW, McLaughlin KA, et al. Apoptotic signals delivered through the T-cell receptor of a T-cell hybrid require the immediate-early gene Nur77. Nature. 1994;367:281–284. doi: 10.1038/367281a0. [DOI] [PubMed] [Google Scholar]
- 44.Woronicz JD, Calnan B, Ngo V, et al. Requirement for the orphan steroid receptor Nur77 in apoptosis of T-cell hybridomas. Nature. 1994;367:277–281. doi: 10.1038/367277a0. [DOI] [PubMed] [Google Scholar]
- 45.Philips A, Maira M, Mullick A, et al. Antagonism between Nur77 and glucocorticoid receptor for control of transcription. Mol Cell Biol. 1997;17:5952–5959. doi: 10.1128/mcb.17.10.5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Franchimont D, Galon J, Vacchio MS, et al. Positive effects of glucocorticoids on T cell function by up-regulation of IL-7 receptor alpha. J Immunol. 2002;168:2212–2218. doi: 10.4049/jimmunol.168.5.2212. [DOI] [PubMed] [Google Scholar]
- 47.Bumgardner GL, Gao D, Li J, et al. Rejection responses to allogeneic hepatocytes by reconstituted SCID mice, CD4, KO, and CD8 KO mice. Transplantation. 2000;70:1771–1780. doi: 10.1097/00007890-200012270-00017. [DOI] [PubMed] [Google Scholar]
- 48.Bumgardner GL, Li J, Prologo JD, et al. Patterns of immune responses evoked by allogeneic hepatocytes: evidence for independent co-dominant roles for CD4+ and CD8+ T-cell responses in acute rejection. Transplantation. 1999;68:555–562. doi: 10.1097/00007890-199908270-00019. [DOI] [PubMed] [Google Scholar]
- 49.He G, Hart J, Kim OS, et al. The role of CD8 and CD4 T cells in intestinal allograft rejection: a comparison of monoclonal antibody-treated and knockout mice. Transplantation. 1999;67:131–137. doi: 10.1097/00007890-199901150-00022. [DOI] [PubMed] [Google Scholar]
- 50.Srinivasan R, Chakrabarti S, Walsh T, et al. Improved survival in steroid-refractory acute graft versus host disease after non-myeloablative allogeneic transplantation using a daclizumab-based strategy with comprehensive infection prophylaxis. Br J Haematol. 2004;124:777–786. doi: 10.1111/j.1365-2141.2004.04856.x. [DOI] [PubMed] [Google Scholar]
- 51.Weisdorf D, Haake R, Blazar B, et al. Treatment of moderate/severe acute graft-versus-host disease after allogeneic bone marrow transplantation: an analysis of clinical risk features and outcome. Blood. 1990;75:1024–1030. [PubMed] [Google Scholar]