

Abstract
Immunotherapy using adoptive cell transfer is a promising approach that can result in the regression of bulky, invasive cancer in some patients. However, currently available therapies remain less successful than desired. To study the mechanisms of action and possible improvements in cell-transfer therapies, we use a murine model system with analogous components to the treatment of patients. T cell receptor transgenic CD8+ T cells (pmel-1) specifically recognizing the melanocyte differentiation antigen gp100 are adoptively transferred into lympho-depleted mice bearing large, established, 14-day subcutaneous B16 melanoma (0.5–1 cm in diameter) on the day of treatment. Adoptive cell transfer in combination with interleukin interleukin-2 or interleukin-15 cytokine administration and vaccination using an altered form of the target antigen, gp100, can result in the complete and durable regression of large tumor burdens. Complete responders frequently develop autoimmunity with vitiligo at the former tumor site that often spreads to involve the whole coat. These findings have important implications for the design of immunotherapy trials in humans.
Keywords: IFN-γ, MHC, interleukin, melanoma, adoptive cell transfer, vaccination, active immunization, cytokine, tumor
THE PROBLEM
Metastatic melanoma is a significant public health concern in the United States with increasing incidence and mortality rates over the past several decades. The estimated lifetime risk of melanoma in the United States is approximately one in 55 males and one in 82 females [1]. Approximately 55,100 cases of invasive melanoma are estimated for 2004 [1]. It is estimated that 7910 patients with metastatic melanoma will die of their disease this year [1].
The ability to successfully and consistently treat advanced melanoma has been an elusive goal. At initial presentation to physicians, the majority of patients will have skin disease only without palpable nodes or evidence of distant metastases [2]. Most patients will undergo surgical treatment by wide local excision alone; additionally, sentinel lymph node biopsy and/or regional nodal dissection may be used. After surgical resection to render patients clinically free of disease, clinical observation, adjuvant therapy using interferon-α (IFN-α) or experimental therapies may be recommended [3]. Despite these interventions, some patients will progress to develop metastatic disease and succumb to their illness [4]. Thus, new therapies capable of treating advanced metastatic melanoma are urgently needed.
IMMUNOTHERAPY TO DESTROY BULKY, INVASIVE CANCER
A wide variety of therapies for metastatic melanoma have been attempted including surgery, radiotherapy, chemotherapy, and biological therapy. In some instances, immunotherapy can be used effectively to treat patients with metastatic disease. Complete and durable regression of stage IV melanoma has been reported using interleukin-2 (IL-2)-based immunotherapy alone [5]. At our institution, 182 patients with metastatic melanoma were treated with high-dose intravenous (i.v.) bolus IL-2 between September 1985 and November 1996. As of June 2003, 12 patients (7%) were complete responders, and 16 patients (9%) were partial responders for a total response rate of 15%. All patients who were complete responders beyond 18 months (83%) remained free of disease as of June 2003.
Although a limited number of patients can be cured of metastatic melanoma solely using high-dose IL-2, the response rate still remains low. This has led to the use of IL-2 in conjunction with other treatment modalities, including vaccines, monoclonal antibodies, and the adoptive transfer of T lymphocytes. The generation of highly active, tumor-specific lymphocytes and their administration in large numbers to patients are the basis of adoptive cell-transfer therapy [6]. Recently, our group reported that after a lympho-depleting but nonmyeloablative-conditioning regimen, the adoptive transfer of highly selected, tumor antigen-specific T cells directed against self-derived differentiation antigens in combination with IL-2, can lead to objective tumor regressions in approximately 45% of patients [7]. However, the biological mechanisms by which tumor regression is elicited have not been elucidated clearly. Thus, the development of a murine model system with analogous components to the treatment of human patients could have important implications for our understanding of current therapies and the design of future immunotherapies.
THE DEVELOPMENT OF AN ANALOGOUS MODEL TO THE HUMAN EXPERIENCE
Clinical efforts using biologic therapy are largely based on mouse models, where the prevention of tumor implantation and growth is often the measure of success. Prevention models are not generally applicable with respect to the treatment of patients, as individuals rarely present to physicians for treatment before the initial development of disease. When treatment models are used for preclinical data, treated tumors in mice are usually extremely small. In studies focusing on adoptive immunotherapy, investigators frequently report on the treatment of pulmonary “metastases” created by the i.v. injection of tumor cells, which are then treated with lymphocytes injected via the same route. Although previous studies have reported approaches that may induce complete regression of established solid tumors, these immunotherapeutic regimens have largely been directed against non-self antigens. Indeed, many of the existing tumor systems target model (foreign) antigens that have been artificially inserted into the tumor genome, whereas the majority of human tumor-associated antigens targeted in clinical efforts are nonmutated self-antigens [8].
In an effort to determine the components of successful immunotherapy in a relevant model of established cancer, we sought to treat large, established, subcutaneous B16 melanoma, a highly aggressive tumor in C57BL/6 mice [9]. B16 is poorly immunogenic [10]. This tumor expresses low levels of major histocompatibility complex (MHC) class I and no class II [11]. Of note, MHC classes I and II are inducible in B16 upon treatment with IFN-γ [11]. Analogous to the human experience, B16 melanoma naturally expresses the mouse homologue (pmel-17) of human gp100, an enzyme involved in pigment synthesis that is expressed by normal and transformed melanocytes [12, 13].
Indeed, gp100 represents an example of a family of tumor-associated, unmutated “self” antigens, which are frequently found to be the target antigens recognized by T cells that infiltrate human melanoma tumors. We described previously the cloning of the unmutated mouse (m) homologue of gp100 from B16 melanoma [14]. From this work, we identified an epitope derived from human (h) gp100, KVPRNQDWL (gp10025–33), which represented an altered form of mgp10025–33, EGSRNQDWL [15], with improved binding to the MHC [16]. It is interesting that gp100-specific, H-2Db-restricted, CD8+ T cells capable of recognizing B16 melanoma and normal melanocytes could only be elicited when the altered peptide was used [15].
To further study antitumor T cell responses, we developed a transgenic mouse strain designated “pmel-1” on a C57BL/6 background [16]. Pmel-1 express the Vα1Vβ13 T cell receptor (TCR) and specifically recognize the H-2Db-restricted mouse and human gp10025–33 epitopes similar to the previously described Clone #9 T cell upon which it was based [15, 16] (Fig. 1). Thus, the pmel-1 transgenic mouse could be used to generate tumor-reactive CD8+ T cells for adoptive cell-transfer experiments, as well as provide a platform to study mechanisms of tolerance for self-reactive T cells.
Fig. 1.
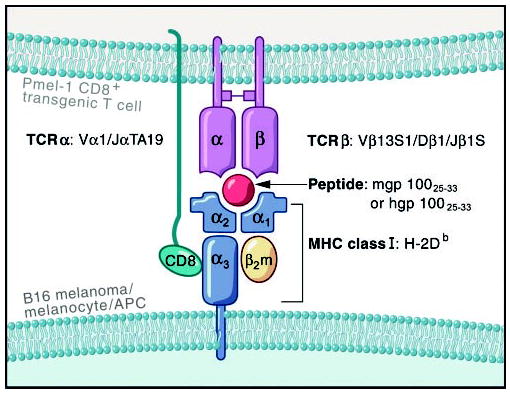
Diagram of pmel-1 CD8+ transgenic T cell interaction with B16 melanoma. T cells from transgenic mice express a TCR recognizing a H-2Db-restricted CD8+ epitope from the gp100/pmel-17 protein mouse gp10025–33 (EGSRNQDWL) or human gp10025–33 (KVPRNQDWL). APC, Antigen-presenting cell.
IMPLICATIONS FROM THE ANIMAL MODEL
Using this model system, we were able to define three elements that were all strictly necessary to induce tumor regression of large, established, poorly immunogenic, unmanipulated, solid tumors: adoptive transfer of tumor-specific T cells; T cell stimulation through antigen-specific vaccination with an altered peptide ligand, rather than the native self-peptide; and co-administration of a T cell growth and activation factor such as IL-2 or IL-15 [16, 17]. This approach can also be used to treat 7-day, established lung nodules (Fig. 2).
Fig. 2.
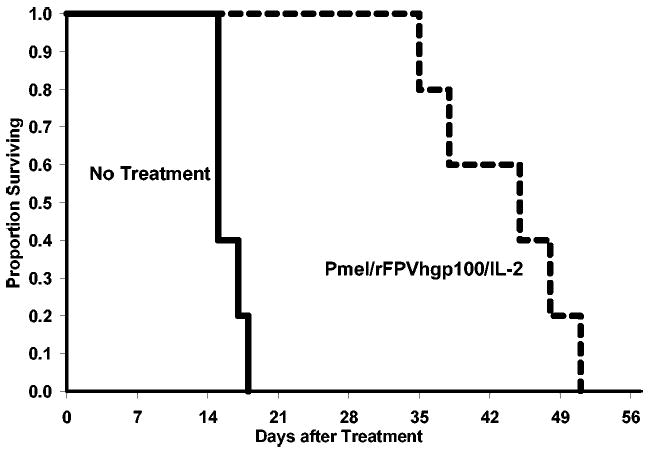
Successful treatment of lung nodules. Adoptive cell transfer of 1 × 106 fresh pmel-1 CD8+ T splenocytes was undertaken in combination with recombinant (r)hIL-2 cytokine administration at 36 μg doses intraperitoneally (i.p.), twice a day for 3 days, and a fowlpox virus encoding human gp100 (rFPVhgp100) into wild-type C57BL/6 mice 7 days after establishment of lung nodules. Kaplan-Meier survival curves illustrating significant survival benefit from with complete treatment regimen.
To further augment clinical relevance, we used this tripartite regimen of cells, vaccine, and administration of a T cell growth/activation factor against a large, established, subcutaneous (s.c.) tumor in lympho-depleted hosts. Adoptive cell transfer with the CD8+Vβ13+ pmel-1 cell was undertaken alone or in a combination with IL-2 administration with or without a fowlpox virus encoding hgp100 (rFPVhgp100; Fig. 3). Tumor regression was observed in mice receiving the complete treatment regimen consisting of adoptive transfer of pmel-1 cells, rFPVhgp100 vaccination, and IL-2 (Fig. 4A). Regimens consisting of any other combination of adoptively transferred cells, vaccine, and IL-2 did not result in dramatic tumor regression (Fig. 4A). The optimal treatment regimen led to complete regression of large tumors greater than 50 mm2 in area (Fig. 4B). The combination of pmel-1 cells, vaccine, and IL-2 prolonged the survival of mice compared with mice receiving no treatment, pmel-1 cells alone, pmel-1 cells plus vaccine, or pmel-1 cells plus IL-2. Additionally, this approach is noted to work on a large tumor burden consisting of a combination of 14-day s.c. tumor with 7-day lung nodules (data not shown).
Fig. 3.
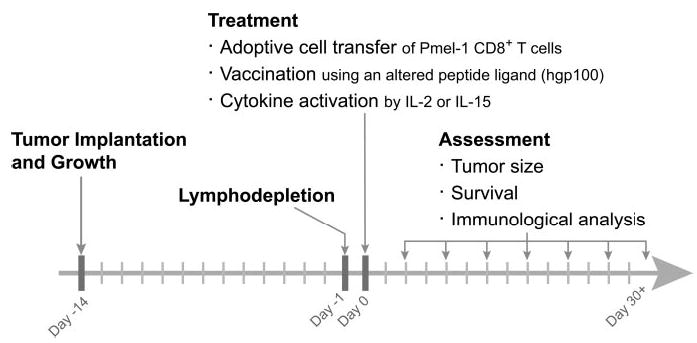
Development of an analogous model to the treatment of human patients. Schema for treatment regimen of 14-day s.c. B16 melanoma.
Fig. 4.
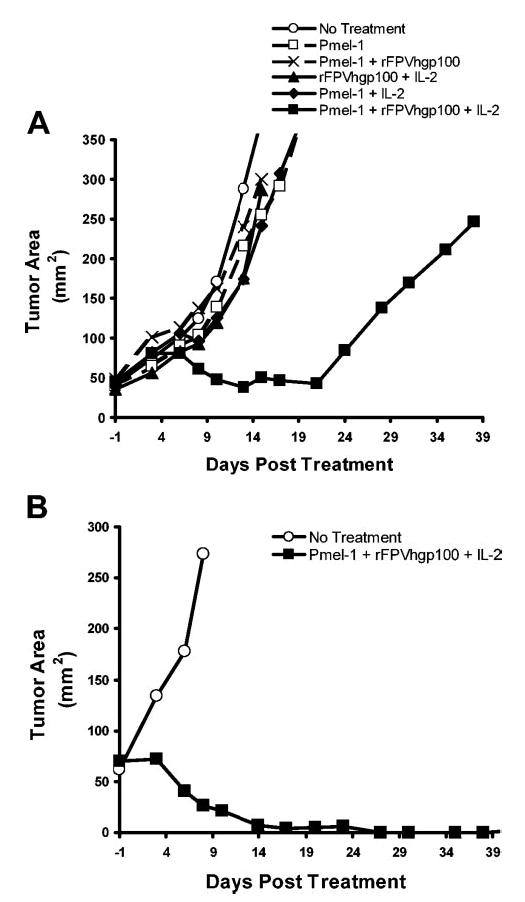
Implications from the animal model. Adoptive cell transfer of pmel-1 CD8+ T splenocytes was undertaken alone or in combination with rhIL-2 cytokine administration at 36 μg doses i.p., twice a day for 3 days, and/or a fowlpox virus encoding human gp100 (rFPVhgp100) into sublethally irradiated C57BL/6 mice bearing 14-day, established s.c. B16 melanoma. (A) Dramatic tumor regression was observed only in mice receiving the complete treatment regimen consisting of adoptive transfer of 1 × 106 fresh pmel-1 cells, rFPVhgp100 vaccine, and IL-2 (▪). (B) Complete regression of large tumor burden was achieved in mice receiving adoptive transfer of 2 × 107 fresh pmel-1 cells, rFPVhgp100 vaccine, and IL-2 (▪).
Mice with complete regression of tumor exhibited vitiligo at the previous site of tumor. Frequently, vitilgo spread randomly throughout the full coat of the mouse (Fig. 5). This phenomenon of developing vitiligo has been reported previously in melanoma patients undergoing successful immunotherapy [18, 19]. Thus, complete tumor regression seems to be correlated with the development of autoimmunity in our model.
Fig. 5.

Optimal treatment led to complete regression of large tumors and autoimmunity. Vitiligo at the former tumor site continued to spread to involve the entire coat.
FROM BEDSIDE TO BENCH AND BACK...
Immunotherapy using adoptive cell transfer is a promising approach that can result in the regression of bulky, invasive cancer in some patients. However, currently available therapies are still less successful than desired. We have developed a murine model that allows us to study the mechanisms of action in the treatment of aggressive, large, established melanoma. Recent successful manipulations in this animal model are being translated into clinical investigation. To further advance clinical therapy, intensive preclinical studies are ongoing to unravel the mechanisms of action with the hope of improving therapeutic efficacy. Research is under way investigating the use of cytokines besides IL-2, such as IL-15 [17], IL-7, and IL-21, as well as the manipulation of the host immune environment. In addition, we are actively studying the role of CD4+ T helper cells [20] and regulatory T cells during immunotherapy [21].
Acknowledgments
S. E. F. was accepted for oral presentation at the International Society for Biological Therapy of Cancer (iSBTc) and was the recipient of the 18th Annual iSBTc Presidential Award, November 1, 2003.
References
- 1.Jemal A, Tiwari RC, Murray T, Ghafoor A, Samuels A, Ward E, Feuer EJ, Thun MJ. Cancer statistics, 2004. CA Cancer J Clin. 2004;54:8–29. doi: 10.3322/canjclin.54.1.8. [DOI] [PubMed] [Google Scholar]
- 2.Balch CM, Soong SJ, Gershenwald JE, Thompson JF, Reintgen DS, Cascinelli N, Urist M, McMasters KM, Ross MI, Kirkwood JM, Atkins MB, Thompson JA, Coit DG, Byrd D, Desmond R, Zhang Y, Liu PY, Lyman GH, Morabito A. Prognostic factors analysis of 17,600 melanoma patients: Validation of the American Joint Committee on Cancer melanoma staging system. J Clin Oncol. 2001;19:3622–3634. doi: 10.1200/JCO.2001.19.16.3622. [DOI] [PubMed] [Google Scholar]
- 3.Kirkwood JM, Strawderman MH, Ernstoff MS, Smith TJ, Borden EC, Blum RH. Interferon-α-2b adjuvant therapy of high risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group trial EST 1684. J Clin Oncol. 1996;14:7–17. doi: 10.1200/JCO.1996.14.1.7. [DOI] [PubMed] [Google Scholar]
- 4.Manola J, Atkins M, Ibrahim J, Kirkwood J. Prognostic factors in metastatic melanoma: a pooled analysis of Eastern Cooperative Oncology Group trials. J Clin Oncol. 2000;18:3782–3793. doi: 10.1200/JCO.2000.18.22.3782. [DOI] [PubMed] [Google Scholar]
- 5.Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, Abrams J, Sznol M, Parkinson D, Hawkins M, Paradise C, Kunkel L, Rosenberg SA. High-dose recombinant interleukin-2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17:2105–2116. doi: 10.1200/JCO.1999.17.7.2105. [DOI] [PubMed] [Google Scholar]
- 6.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, Robinson MR, Raffeld M, Duray P, Seipp CA, Rogers-Freezer L, Morton KE, Mavroukakis SA, White DE, Rosenberg SA. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dudley ME, Rosenberg SA. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat Rev Cancer. 2003;3:666–675. doi: 10.1038/nrc1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosenberg SA. A new era for cancer immunotherapy based on the genes that encode cancer antigens. Immunity. 1999;10:281–287. doi: 10.1016/s1074-7613(00)80028-x. [DOI] [PubMed] [Google Scholar]
- 9.Poste G, Doll J, Hart IR, Fidler IJ. In vitro selection of murine B16 melanoma variants with enhanced tissue- invasive properties. Cancer Res. 1980;40:1636–1644. [PubMed] [Google Scholar]
- 10.Dranoff G, Jaffee E, Lazenby A, Golumbek P, Levitsky H, Brose K, Jackson V, Hamada H, Pardoll D, Mulligan RC. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci USA. 1993;90:3539–3543. doi: 10.1073/pnas.90.8.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seliger B, Wollscheid U, Momburg F, Blankenstein T, Huber C. Characterization of the major histocompatibility complex class I deficiencies in B16 melanoma cells. Cancer Res. 2001;61:1095–1099. [PubMed] [Google Scholar]
- 12.Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Sakaguchi K, Appella E, Yannelli JR, Adema GJ, Miki T, Rosenberg SA. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc Natl Acad Sci USA. 1994;91:6458–6462. doi: 10.1073/pnas.91.14.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kobayashi T, Urabe K, Orlow SJ, Higashi K, Imokawa G, Kwon BS, Potterf B, Hearing VJ. The Pmel 17/silver locus protein. Characterization and investigation of its melanogenic function. J Biol Chem. 1994;269:29198–29205. [PubMed] [Google Scholar]
- 14.Zhai Y, Yang JC, Spiess P, Nishimura MI, Overwijk WW, Roberts B, Restifo NP, Rosenberg SA. Cloning and characterization of the genes encoding the murine homologues of the human melanoma antigens MART1 and gp100. J Immunother. 1997;20:15–25. doi: 10.1097/00002371-199701000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Overwijk WW, Tsung A, Irvine KR, Parkhurst MR, Goletz TJ, Tsung K, Carroll MW, Liu C, Moss B, Rosenberg SA, Restifo NP. gp100/pmel 17 is a murine tumor rejection antigen: induction of “self”-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J Exp Med. 1998;188:277–286. doi: 10.1084/jem.188.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, Heimann DM, Klebanoff CA, Yu Z, Hwang LN, Feigenbaum L, Kruisbeek AM, Rosenberg SA, Restifo NP. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klebanoff CA, Finkelstein SE, Surman DR, Lichtman MK, Gattinoni L, Theoret MR, Grewal N, Spiess PJ, Antony PA, Palmer DC, Tagaya Y, Rosenberg SA, Waldmann TA, Restifo NP. IL-15 enhances the in vivo antitumor activity of tumor-reactive CD8+ T cells. Proc Natl Acad Sci USA. 2004;101:1969–1974. doi: 10.1073/pnas.0307298101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosenberg SA, White DE. Vitiligo in patients with melanoma: normal tissue antigens can be targets for cancer immunotherapy. J Immunother Emphasis Tumor Immunol. 1996;19:81–84. [PubMed] [Google Scholar]
- 19.Yee C, Thompson JA, Roche P, Byrd DR, Lee PP, Piepkorn M, Kenyon K, Davis MM, Riddell SR, Greenberg PD. Melanocyte destruction after antigen-specific immunotherapy of melanoma: direct evidence of T cell-mediated vitiligo. J Exp Med. 2000;192:1637–1644. doi: 10.1084/jem.192.11.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ho WY, Yee C, Greenberg PD. Adoptive therapy with CD8+ T cells: it may get by with a little help from its friends. J Clin Invest. 2002;110:1415–1417. doi: 10.1172/JCI17214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Antony PA, Restifo NP. Do CD4+CD25+ immunoregulatory T cells hinder tumor immunotherapy? J Immunother. 2002;25:202–206. doi: 10.1097/00002371-200205000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]