

Abstract
Ca2+ sparks in heart muscle are activated on depolarization by the influx of Ca2+ through dihydropyridine receptors in the sarcolemmal (SL) and transverse tubule (TT) membranes. The cardiac action potential is thus able to synchronize the [Ca2+]i transient as Ca2+ release is activated throughout the cell. Increases in the amount of Ca2+ within the sarcoplasmic reticulum (SR) underlie augmented Ca2+ release globally and an increase in the sensitivity of the ryanodine receptors (RyRs) to be triggered by the local [Ca2+]i. In a similar manner, phosphorylation of the RyRs by protein kinase A (PKA) increases the sensitivity of the RyRs to be activated by local [Ca2+]i. Heart failure and other cardiac diseases are associated with changes in SR Ca2+ content, phosphorylation state of the RyRs, [Ca2+]i signaling defects and arrhythmias. Additional changes in transverse tubules and nearby junctional SR may contribute to alterations in local Ca2+ signaling. Here we briefly discuss how TT organization can influence Ca2+ signaling and how changes in SR Ca2+ release triggering can influence excitation–contraction (EC) coupling. High speed imaging methods are used in combination with single cell patch clamp experiments to investigate how abnormal Ca2+ signaling may be regulated in health and disease. Three issues are examined in this presentation: (1) normal Ca2+-induced Ca2+ release and Ca2+ sparks, (2) abnormal SR Ca2+ release in disease, and (3) the triggering and propagation of waves of elevated [Ca2+]i.
Keywords: Ca2+ sparks, Ca2+ waves, ryanodine receptors, transverse tubules
INTRODUCTION
Mammalian heart muscle cells distinguish themselves from reptilian, bird, and fish cardiac ventricular myocytes by the presence of an extensive transverse tubule (TT) system as shown in Figure 1. This system of tubules permits each of the larger heart cells of the mammalian ventricular myocardium to nearly synchronously trigger the release of Ca2+ from the sarcoplasmic reticulum (SR) in response to its depolarization.
FIGURE 1.

Transverse tubules (TTs) in rat heart cells. (A) A wide field transmitted light image of a rat ventricular myocyte. (B) A confocal fluorescence image of the TTs in a living rat ventricular myocyte stained with the membrane marker di-8 ANEPPS imaged with 488 nm excitation. The myocyte was exposed to 10 μM dye for 10 min. The striation or TT separation along the long axis of the cell is 1.8 μm.
The propagated action potential (AP) originates in the sino-atrial (SA) node, is conducted through the atria, the atrio-ventricular (AV) node, and the His-Purkinje fiber system before it reaches the septum and the endocardial surfaces of the ventricles. This conducted AP then propagates from cell to cell within the myocardium moving from endocardium to epicardium. In this manner, cardiac myocytes are sequentially activated as the wave of depolarization spreads. The TT system is largely responsible for the synchrony of local Ca2+ release within each cell. It would appear to be advantageous for each cell to generate force in an organized manner so that one region of a cell does not serve to stretch another region of the cell as would tend to occur if the Ca2+ release and contractions were asynchronous. The cellular contractions within the entire heart are coordinated so that the contraction of the myocardium in mammals squeezes blood out of the heart as the intraventricular pressure increases and volume is reduced. Here we briefly examine the TT system in heart with respect to its role in Ca2+ signaling in mammalian heart muscle. In addition, we discuss issues related to TT development, de-differentiation, and disease. Recent work has made the local signaling within the ventricular myocyte of extraordinary interest because it underlies normal excitation–contraction (EC) coupling.1,2 When abnormalities develop in this process, local Ca2+ overload may occur and this may lead to abnormal Ca2+ release, which can underlie contractile dysfunction and arrhythmogenesis.
TRANSVERSE TUBULES
TTs are physical invaginations (100 to 300 nm in diameter3) that occur at regular intervals (every 1.2 microns along the Z-line4) and run deep into the ventricular cell as shown in Figure 1. One TT is separated from the next TT along the long axis of the cell by junctional SR (jSR) and a nearly 2 micron long mitochondrion, as diagrammed in Figure 2. With a roughly 2 micron Z-line spacing on the long axis of the cell, there is virtually no region inside a ventricular myocyte more than about 1.2 microns from a TT or SL membrane. This is illustrated in Figure 3. At every cross-section of the ventricular myocyte along its long axis at a Z-line there is a grid of TTs that conducts the cardiac AP into the depths of the cell. In this manner, the TTs form a network of membranes within the heart cell, and this extension of the surface sarcolemmal (SL) membrane contains many channels and transporters5 (see Figs. 3 and 4). This includes all major channel types in heart: L-type Ca2+ channels (also known as dihydropyridine receptors, or DHPRs), Na+ channels, and diverse K+ channels. Critical transporters include the Na+/Ca2+ exchanger (NCX), multiple isoforms of the Na+, K+ ATPase, transporters that regulate pH, and those that move chloride. Of particular importance is the spatial organization of these proteins along the TT, because it affects local Ca2+ signaling which underlies EC coupling.6–9 Because of the critical roles played by the TTs and their array of local proteins, the TTs have become an area of considerable research interest.
FIGURE 2.
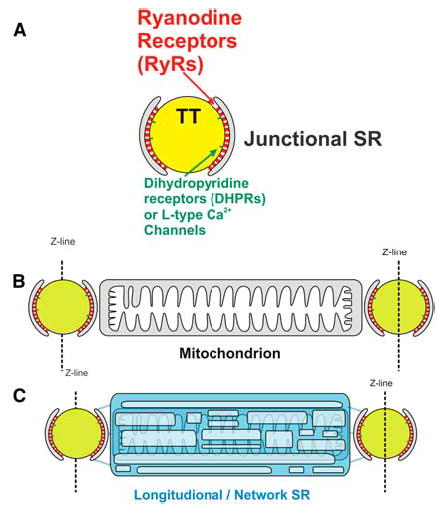
Transverse tubules (TTs) and other related structures in the sarcomere. (A) The TT, schematic diagram of the TT, ryanodine receptor (RyR) cluster, and junctional sarcoplasmic reticulum (jSR). (B) The intermyofibrillar mitochondrion. Relationship between TT structures and the intermyo-fibrillar mitochondrion. (C) The reticular longitudinal sarcoplasmic reticulum (SR). The longitudinal SR wraps around the mitochondrion and is linked to the jSR through diffusion-restricted connections [in color in Annals Online].
FIGURE 3.
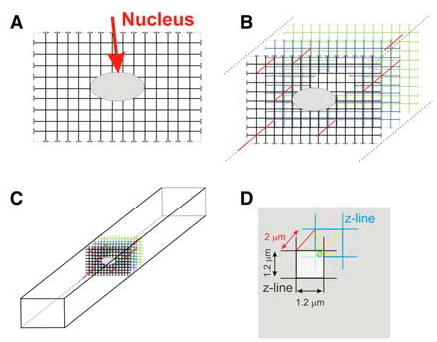
The transverse tubule (TT) array in a mammalian ventricular myocyte. (A) Diagram of a fully populated TT system (array) at the level of the nucleus (arrow). The TTs flare open at the sarcolemmal surface and do not penetrate the nucleus. The TTs are approximately 1.2 microns apart. (B) An array of TTs are found at every Z-line and are sparsely connected to the neighboring Z-line array by longitudinal tubules (lines at 45 degrees, shown in red in Annals Online). There are three Z-line arrays shown in this diagram. (C) Diagram of the placement of the array of TTs in an intact cell. (D) Diagram of a TT elementary unit. An elementary unit of TTs is composed of a square (1.2 microns on an edge) in successive Z-line arrays. The small sphere (shown in green online) represents the most distant location in the elementary unit from a TT, and it is approximately 1.17 microns from the nearest TT, assuming the distance between Z-lines is 2 microns. Yellow lines (seen online) connect opposite corners and go through the center of the small sphere [in color in Annals Online].
FIGURE 4.
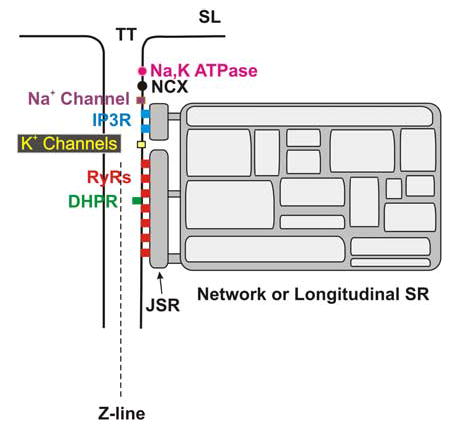
Organization of key channel and transporter proteins and sarcoplasmic reticulum (SR) structures near the transverse tubule (TT). The IP3 receptors (IP3Rs) are separated from the ryanodine receptors (RyRs) in this diagram because of recent immunofluorescence data. However, both IP3Rs and RyRs are thought to be SR structures. The key proteins are labeled in the figure. The extensive array of cytoskeletal proteins, signaling receptors, adaptor proteins, and contractile filaments are not shown. SL, sacrolemmal; NCX, Na+/Ca2+ exchanger; DHPR, dihydropyridine receptors; JSR, junctional sarcoplasmic reticulum [in color in Annals Online].
Soeller and Cannell10 have examined the organization of TTs in rat ventricular myocytes and have revealed the grid-like structure noted above. These investigators have also shown clearly that there are occasional missing elements of the grid as well as longitudinal or axial tubules that connect the TT grid at one Z-line with that at another Z-line (see, for example, solid lines at 45 degrees in Fig. 3). A similar pattern was also seen in the TT distribution of the Na+/Ca2+ exchanger in rat ventricular myocytes4 and can be observed in the TT image shown in Figure 1. In contrast, atrial myocytes have few TTs,11–13 but they can have some which show a sparse distribution and, in many cases, a more prominent longitudinal component.
Sarcoplasmic Reticulum
The SR is an organelle that consists of multiple interconnected Ca2+-containing components. Each component is a membrane-enclosed volume that is Ca2+-rich and is connected to the others.14 The jSR is joined to the network SR (or longitudinal SR) through diffusion-limiting connections. The jSR is found close to the Z-line and the TTs and has a very small volume, while the longitudinal SR spans the distance across the sarcomere. Some evidence suggests that each SR component within a cell may be directly or indirectly connected to all of the other SR components within the cell. SR Ca2+-release channels (ryanodine receptors, or RyRs) span the approximately 15 nm gap between jSR and the TT membrane and are found primarily at the SR-TT junction. At these junctions there are clusters of RyRs (between 10 and 300).15 The RyRs are sensitive to local [Ca2+]i and are activated to release Ca2+ from the SR when there is an elevation of [Ca2+]i in the gap or “subspace” between the TT membrane and the SR. When activated by [Ca2+]i a RyR opens as an SR Ca2+-release channel and floods the subspace with Ca2+ from the jSR. Elevation of subspace [Ca2+] from Ca2+ entry across the TT membrane (mainly by DHPRs facing the subspace) or across the jSR membrane by RyRs will tend to activate all RyRs facing the subspace. This regenerative RyR activation and consequent Ca2+ efflux from the SR produces a Ca2+ spark. The large efflux of Ca2+ from the small jSR volume is possible because of the SR “lumenal” buffer, calsequestrin, and the extensive network of the longitudinal SR. Termination of the Ca2+ spark is thought to occur largely due to depletion of Ca2+ from the SR, thereby decreasing RyR sensitivity to [Ca2+]i. The termination may also depend on a cooperative interaction between RyRs within the cluster.16
Ryanodine Receptors
RyRs are large homotetrameric proteins of 2 mega-Daltons. The bulk of the protein is in the cytoplasmic space and spans the gap or subspace between the jSR and the TT membrane.17,18 The RyRs form a cluster of interacting channels15,19,20 that are triggered by the Ca2+ influx into the subspace through the L-type Ca2+ channels and other channels and transporters.16 The RyRs are phosphorylated by protein kinase A (PKA) and by calmodulin-dependent protein kinase II (CaMKII),20–23 and this phosphorylation modifies the sensitivity of the RyR to [Ca2+].23 Phosphatases de-phosphorylate the RyR and provide balanced modulation of this important protein.24 There is significant evidence that indicates RyRs are regulated by the SR lumenal Ca2+. Higher [Ca2+]lumen leads to the increased sensitivity of RyRs25 so that RyRs are more readily activated by [Ca2+]subspace. The SR Ca2+ ATPase (SERCA) transports Ca2+ from the cytoplasm into the SR so that the RyRs can release it. While SERCA is clearly localized primarily to the Z-line region based on immunofluorescence data, SERCA is also found on the network SR. In a similar manner, RyRs, which are primarily found at the Z-line, are also seen between the Z-lines. This raises a question not yet fully addressed: Where exactly are RyRs that are not at the SR-TT junctions and not part of the junctional clusters?
The mitochondria span the region from one jSR to the next and are surrounded by the longitudinal SR (see Fig. 2). This intimate connection does not make it easy to separate—based on function or by experimental intervention—RyRs that may be found along the longitudinal SR from those that are on the mitochondria. Thus the question raised above remains unanswered. A second and equally important question is this: If RyRs are found elsewhere, then what do they do there?
IP3 Receptors
IP3 receptors (IP3Rs) are found in mammalian ventricular myocytes at the Z-lines.5 They appear to co-localize with NCX and Na+, K+ ATPase proteins but not with RyRs and DHPRs. The RyRs and DHPRs are co-localized and also found along the Z-line.5,26 This observation made by several groups suggests that there may be close but distinct groups of channels and transporters that form separate but interacting elements. Since both IP3Rs and RyRs are thought to reside in the SR, it is not clear in which functional parts of the SR each receptor or set of receptors is found. Atrial myocytes have more IP3Rs than ventricular myocytes, and it has been suggested that activation of IP3Rs may contribute to the Ca2+ signal by influencing RyRs.27,28 Mohler et al.5 also show that the IP3Rs are found at sites that contain the Na+/Ca2+ exchanger. It is still not clear, however, how the IP3Rs are activated and what they may do in ventricular myocytes. Since IP3Rs are “Ca2+ release channels” that are activated by IP3 and Ca2+, along with other possible agonists, the question that remains is this: What is their function in ventricular myocytes? It is tempting to speculate that they may influence local [Ca2+]i and hence influence EC coupling at nearby RyRs, as Zima & Blatter27 suggested for atrial myocytes; however, as yet there is no compelling evidence. Figure 4 shows how IP3Rs may be located close to RyRs as suggested by the current information available.
Proteins Located On or Near The Transverse Tubules
The TTs and Z-line region have many proteins including many cytoskeletal proteins, the adaptor protein ankyrin B5, transporters (e.g., NCX and the various isoforms of the Na+, K+ ATPase [Na+ pump]), SL channels including DHPRs, Na+ channels, diverse K+ channels, the Ca2+ release channels RyR and IP3R, and the SR Ca2+ ATPase (SERCA), among others. There are several specific questions that are relevant for this examination of Ca2+ signaling along the TT. Each of these questions centers on the spatial organization of the channels, transporters, and other proteins and the special role(s) they may play.
The presence of the cardiac isoform of the Na+ channel (Nav 1.5) on the SL and TT membranes underlies the rapid upstroke of the conducted cardiac action potential. Analysis of membrane currents suggest that about 300 channels per square micron may exist29 in ventricular myocytes. Tetrodotoxin (TTX) sensitivity suggests that the cardiac isoform is the dominant isoform of the Na+ channel found in ventricular myocytes,30 but some immunofluorescence findings raise the possibility that there may be neuronal isoforms of the TTX-sensitive INa in ventricular myocytes.30 It is not clear what role, if any, would be played by these other Na+ channel isoforms in heart cells, and further identification and functional investigations must still be carried out.
A question that has been raised by diverse groups is whether the Na+ channels may contribute to local Ca2+ signaling. There is some evidence that Na+ flux through Na+ channels affects Na+/Ca2+ exchanger and Ca2+ signaling.31 Immunofluorescence imaging at high resolution suggests that while the Na+ channels and Na+/Ca2+ exchanger may be close to each other, they are not co-localized with the RyRs at the jSR.26,32 This observation is consistent with investigations of other Z-line proteins.5 That leaves open the question of how Na+ influx via Na+ channels can influence local Ca2+ signaling as suggested by recent physiological studies.31
DEVELOPMENT, DE-DIFFERENTIATION, AND DISEASE AND THE TT SYSTEM: SR CA2+ OVERLOAD
All investigations to date show that “control” or “wild type” ventricular myocytes have [Ca2+]i transients that are virtually synchronously triggered upon depolarization everywhere within the cell.33 Figure 5 shows the measurement of SR Ca2+ release under normal conditions using confocal imaging in linescan mode. However, when TTs are lost (or become dysfunctional in some disease states), Ca2+ release dyssynchrony may develop. Similarly, during cardiac development or while cardiac myocytes are in culture, Ca2+-induced Ca2+-release (CICR) may spread throughout the cell as a propagating wave of elevated [Ca2+]i,34 depending on diffusion distances, cell morphology, and electrical behavior. Dyssynchrony has been shown to occur in myocytes in primary culture,35 in disease,36–38 and under conditions where the triggering Ca2+ is reduced.39 Dyssynchrony is often characterized by a propagation delay of the triggered release of Ca2+ and may contribute to dysfunction of cellular contraction35,38 and/or [Ca2+]i transient “alternans.”39 Figure 5E shows that the half-time of the increase in [Ca2+]i goes up as the square of distance that Ca2+ must diffuse. Electrical activation of EC coupling is rapid, but Ca2+ diffusion-dependent activation of Ca2+ release is slow. This raises a clear question: How do Ca2+ sparks get triggered when the local [Ca2+]i is not elevated the way it is when DHPR activate the apposed RyR cluster in the jSR? Under these conditions, a propagating “wave” can develop from a site of normal activation to the untriggered SR. For this “orphaned SR” to be triggered, it must be in a state of local Ca2+ overload with high EC coupling gain. This is clear since isolated Ca2+ sparks do not normally trigger other Ca2+ sparks,40–43 even when there is modest elevation of the SR Ca2+ load.44 Thus, as TTs de-differentiate (as may occur in primary culture), the dyssynchrony that develops is associated with local SR Ca2+ overload. Some have speculated that similar conditions develop in diseases such as heart failure.36,37 This alteration may not be an aberration but instead may reflect a cellular adaptation to a loss of TTs. Consistent with this, EC coupling resulting from propagation of Ca2+ release is a common finding in atrial myocytes,11–13 cells which often lack TTs. Because of the smaller cross-section of these cells, negative functional consequences of this release are minimal.
FIGURE 5.
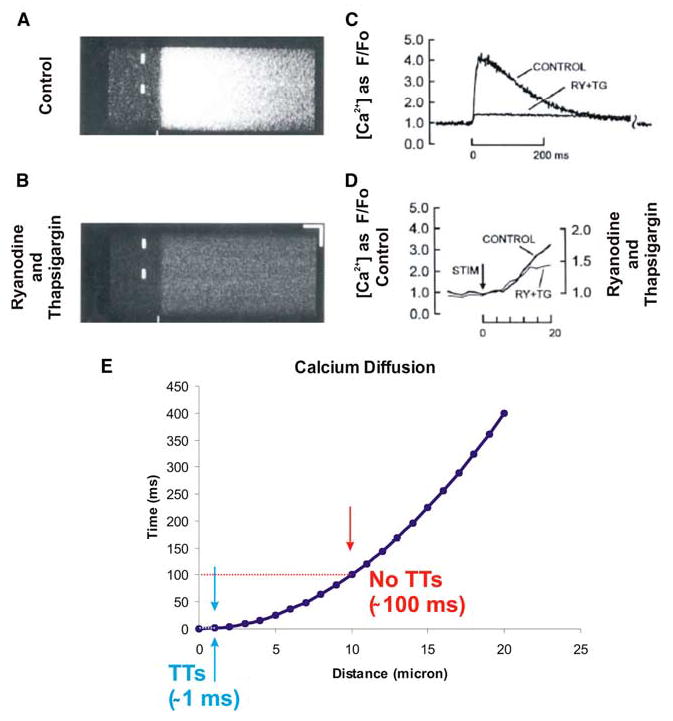
Calcium profile across the diameter of the cell during excitation–contraction (EC) coupling. (A) A [Ca2+]i transient shown using a linescan image of a heart cell taken across the diameter of the cell. Note that there is virtually synchronous activation of Ca2+ release at all points. (B) Same as A but after the application of ryanodine (RY) and thapsigargin (TG) to completely block sarcoplasmic reticulum (SR) Ca2+ release and re-uptake respectively. (C) The [Ca2+]i transients from A and B. (D) Time-course of the [Ca2+]i transient due to ICa alone and ICa plus triggered release are nearly identical during the early phase. (E) Effect of diffusion distance and diffusion half-time. (A–C modified from Cheng et al.33 Used with permission.) TT, transverse tubule; STIM, stimulus [in color in Annals Online.]
MODELING LOCAL AND GLOBAL CA2+ SIGNALING
The great stability of Ca2+-induced Ca2+-release in heart arises largely because of two factors: (1) the relative insensitivity of RyRs to be triggered by [Ca2+]i changes, and (2) local Ca2+ triggering.45 This investigation proposed that local Ca2+ signaling solved the problem posed a few years earlier:46 How can myocytes have both apparent high gain and EC coupling stability? Additional modeling investigations have supported the earlier work and explored diverse local control models.47 Importantly, the discovery of Ca2+ sparks43 provided experimental support for the local signaling and accounted for the transition to the increased sensitivity of the SR Ca2+ release process that underlies propagating waves of elevated Ca2+ and arrhythmogenesis. Figure 6 shows a sequence of images that reveals how such a propagating wave of elevated [Ca2+]i is initiated by Ca2+ sparks. This imaging is obtained at high resolution with a high-speed Zeiss confocal microscope, the LSM 5 Live. While normal modeling of Ca2+ sparks and SR Ca2+ release provides stability,16 when SR Ca2+ content becomes elevated, the “gain” of the SR Ca2+ release mechanism increases and the process becomes unstable.
FIGURE 6.
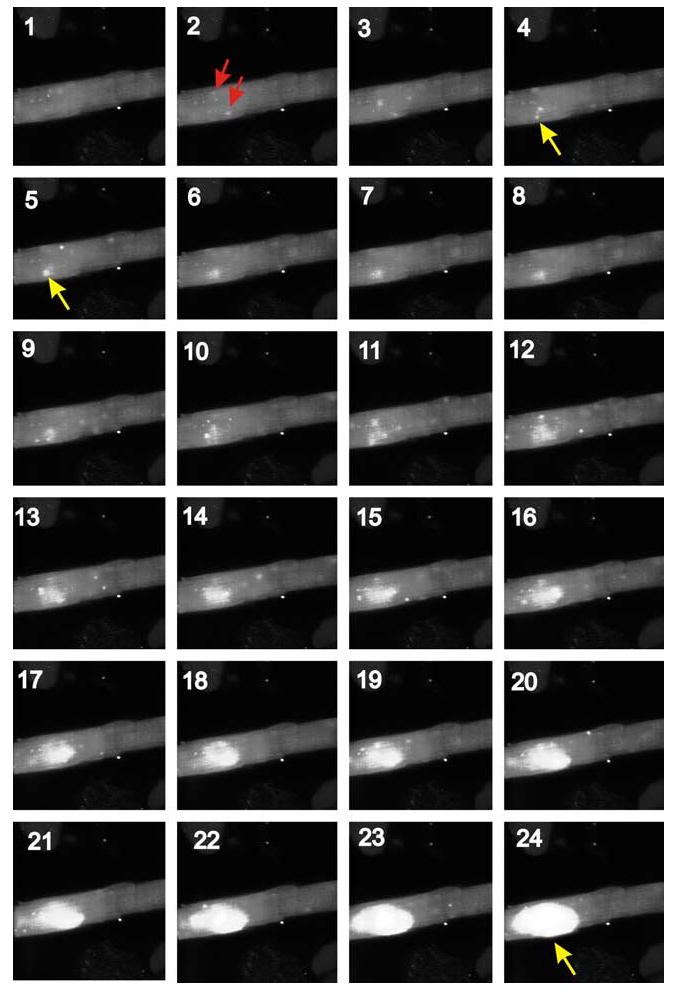
Images of the initiation of a propagated wave of elevated [Ca2+]i in a rat ventricular myocyte. Frames are numbered sequentially 1–24. Ca2+ sparks can be seen in virtually every frame. In frame 2, two of several Ca2+ sparks are identified with arrows. In Frames 4 and 5, the arrows (shown in yellow in Annals Online) mark a Ca2+ spark that initiates the wave of propagating elevated [Ca2+]i. It is clear in the frames (starting in frame 4) that there is a local instability over a region of about 8 microns, where sparks activate neighboring regions and then form an increasing cloud of activating and spreading elevated [Ca2+]i that by frame 24 has started to exit the region. This wave propagates throughout the cell in frames that follow these first 24. The cell was loaded with the Ca2+ sensitive indicator fluo-4 and was exposed to a normal extracellular solution with [Ca2+]o = 3 mM. Sequential high-resolution images acquired at 30 ms intervals using the very fast confocal microscope, the Zeiss LSM 5 Live [in color in Annals Online].
MAJOR QUESTIONS UNDER INVESTIGATION RELATED TO TTS AND SR CA2+ SIGNALING
One of the primary questions that remains open is this: Why does SERCA expression decrease in many cardiac pathologies? The cellular and molecular signals that underlie this change and the decrease in average SR Ca2+ content have not yet been identified. Further, if only SERCA were to decrease, then the SR Ca2+ content would also decline. But it is also clear that the SR Ca2+ content is not always uniformly decreased within the cell. Despite the decrease in global averaged [Ca2+]i during EC coupling, cells retain the ability to develop Ca2+-dependent (or Ca2+-triggered) arrhythmias. This raises the second major question: What underlies the development of the local SR Ca2+ overload (even when global SR Ca2+ may be depleted)? A related question is whether the non-uniform decreases in SR Ca2+ are related to the decrease in SERCA. One possible answer suggested by circumstantial evidence is that there is local Ca2+ overload in some regions. Could the local increase of Ca2+ within the SR provide a signal that leads to the decrease in SERCA globally? This local Ca2+ overload is partly a consequence of the “orphaned” SR48 that can develop as TTs become disorganized with respect to the Z-lines and SR. This hypothesis continues to be examined by several groups. A third major question focuses on the RyRs in heart. How does modulation of the RyRs by phosphorylation and by other interacting proteins influence CICR, EC coupling, and SR Ca2+ overload? Direct evidence obtained in simple systems (e.g., isolated channels) is very provocative, and circumstantial evidence suggests that these modulations of the RyRs are important in intact cells.
SUMMARY
Ca2+ signaling in mammalian ventricular myocytes depends on local signaling between DHPRs and RyR clusters to produce a high-gain Ca2+ signaling system with great stability. Disruption of the TT organization or weak Ca2+ triggering underlies an increase in SR Ca2+ content (and thus in EC coupling gain) and produces instability. Continuing investigations of cellular control of SERCA and the organization of channels, transporters, cytoskeletal proteins, adaptor proteins, intracellular organelles, and signaling cascades should provide improved understanding of Ca2+ signaling in mammalian ventricular myocytes.
Acknowledgments
This work has been supported by the National Heart, Lung, and Blood Institute; the National Science Foundation; the American Heart Association; and the Whitaker Foundation.
References
- 1.Wang SQ, Song LS, Lakatta EG, et al. Ca2+ signaling between single L-type Ca2+ channels and ryanodine receptors in heart cells. Nature. 2001;410:592–596. doi: 10.1038/35069083. [DOI] [PubMed] [Google Scholar]
- 2.Song LS, Wang SQ, Xiao RP, et al. β-adrenergic stimulation synchronizes intracellular Ca2+ release during excitation-contraction coupling in cardiac myocytes. Circ Res. 2001;88:794–801. doi: 10.1161/hh0801.090461. [DOI] [PubMed] [Google Scholar]
- 3.Bers, D.M. 2001. Excitation-Contraction Coupling and Cardiac Contractile Force. 2nd ed. Kluwer. Boston.
- 4.Kieval RS, Bloch RJ, Lindenmayer GE, et al. Immunofluorescence localization of the Na-Ca exchanger in heart cells. Am J Physiol. 1992;263:C545–C550. doi: 10.1152/ajpcell.1992.263.2.C545. [DOI] [PubMed] [Google Scholar]
- 5.Mohler PJ, Schott JJ, Gramolini AO, et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634–639. doi: 10.1038/nature01335. [DOI] [PubMed] [Google Scholar]
- 6.Wang SQ, Wei C, Zhao G, et al. Imaging microdomain Ca2+ in muscle cells. Circ Res. 2004;94:1011–1022. doi: 10.1161/01.RES.0000125883.68447.A1. [DOI] [PubMed] [Google Scholar]
- 7.Wehrens XH, Lehnart SE, Huang F, et al. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell. 2003;113:829–840. doi: 10.1016/s0092-8674(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 8.Marks AR, Marx SO, Reiken S. Regulation of ryanodine receptors via macromolecular complexes. A novel role for leucine/isoleucine zippers. Trends Cardiovasc Med. 2002;12:166–170. doi: 10.1016/s1050-1738(02)00156-1. [DOI] [PubMed] [Google Scholar]
- 9.Marx SO, Kurokawa J, Reiken S, et al. Requirement of a macromolecular signaling complex for β adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science. 2002;295:496–499. doi: 10.1126/science.1066843. [DOI] [PubMed] [Google Scholar]
- 10.Soeller C, Cannell MB. Examination of the transverse tubular system in living cardiac rat myocytes by 2-photon microscopy and digital image-processing techniques. Circ Res. 1999;84:266–275. doi: 10.1161/01.res.84.3.266. [DOI] [PubMed] [Google Scholar]
- 11.Blatter LA, Kockskamper J, Sheehan KA, et al. Local calcium gradients during excitation-contraction coupling and alternans in atrial myocytes. J Physiol. 2003;546:19–31. doi: 10.1113/jphysiol.2002.025239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kockskamper J, Sheehan KA, Bare DJ, et al. Activation and propagation of Ca2+ release during excitation-contraction coupling in atrial myocytes. Biophys J. 2001;81:2590–2605. doi: 10.1016/S0006-3495(01)75903-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kirk MM, Izu LT, Chen-Izu Y, et al. Role of the transverse-axial tubule system in generating calcium sparks and calcium transients in rat atrial myocytes. J Physiol. 2003;547:441–451. doi: 10.1113/jphysiol.2002.034355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brochet DX, Yang D, Di Maio A, et al. Ca2+ blinks: rapid nanoscopic store calcium signaling. Proc Natl Acad Sci USA. 2005;102:3099–3104. doi: 10.1073/pnas.0500059102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Franzini-Armstrong C, Protasi F, Ramesh V. Shape, size, and distribution of Ca2+ release units and couplons in skeletal and cardiac muscles. Biophys J. 1999;77:1528–1539. doi: 10.1016/S0006-3495(99)77000-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sobie EA, Dilly KW, dos Santos CJ, et al. Termination of cardiac Ca2+ sparks: an investigative mathematical model of calcium-induced calcium release. Biophys J. 2002;83:59–78. doi: 10.1016/s0006-3495(02)75149-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wagenknecht T, Samso M. Three-dimensional reconstruction of ryanodine receptors. Front Biosci. 2002;7:d1464–d1474. doi: 10.2741/A853. [DOI] [PubMed] [Google Scholar]
- 18.Sharma MR, Penczek P, Grassucci R, et al. Cryoelectron microscopy and image analysis of the cardiac ryanodine receptor. J Biol Chem. 1998;273:18429–18434. doi: 10.1074/jbc.273.29.18429. [DOI] [PubMed] [Google Scholar]
- 19.Franzini-Armstrong C, Protasi F, Ramesh V. Comparative ultrastructure of Ca2+ release units in skeletal and cardiac muscle. Ann NY Acad Sci. 1998;853:20–30. doi: 10.1111/j.1749-6632.1998.tb08253.x. [DOI] [PubMed] [Google Scholar]
- 20.Marx SO, Gaburjakova J, Gaburjakova M, et al. Coupled gating between cardiac calcium release channels (ryanodine receptors) Circ Res. 2001;88:1151–1158. doi: 10.1161/hh1101.091268. [DOI] [PubMed] [Google Scholar]
- 21.Marx SO, Reiken S, Hisamatsu Y, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 22.Wehrens XH, Lehnart SE, Reiken SR, et al. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res. 2004;94:e61–e70. doi: 10.1161/01.RES.0000125626.33738.E2. [DOI] [PubMed] [Google Scholar]
- 23.Valdivia HH, Kaplan JH, Ellis-Davies GCR, et al. Rapid adaptation of cardiac ryanodine receptors: modulation by Mg2+ and phosphorylation. Science. 1995;267:1997–2000. doi: 10.1126/science.7701323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du Bell WH, Lederer WJ, Rogers TB. Dynamic modulation of excitation-contraction coupling by protein phosphatases in rat ventricular myocytes. J Physiol (London) 1996;493:793–800. doi: 10.1113/jphysiol.1996.sp021423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gyorke I, Gyorke S. Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophys J. 1998;75:2801–2810. doi: 10.1016/S0006-3495(98)77723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scriven DR, Dan P, Moore ED. Distribution of proteins implicated in excitation-contraction coupling in rat ventricular myocytes. Biophys J. 2000;79:2682–2691. doi: 10.1016/S0006-3495(00)76506-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zima AV, Blatter LA. Inositol-1,4,5-trisphosphate-dependent Ca2+ signalling in cat atrial excitation-contraction coupling and arrhythmias. J Physiol. 2004;555:607–615. doi: 10.1113/jphysiol.2003.058529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mackenzie L, Bootman MD, Laine M, et al. The role of inositol 1,4,5-trisphosphate receptors in Ca2+ signalling and the generation of arrhythmias in rat atrial myocytes. J Physiol. 2002;541:395–409. doi: 10.1113/jphysiol.2001.013411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Makielski JC, Sheets MF, Hanck DA, et al. Sodium current in voltage clamped internally perfused canine cardiac Purkinje cells. Biophys J. 1987;52:1–11. doi: 10.1016/S0006-3495(87)83182-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maier SK, Westenbroek RE, Yamanushi TT, et al. An unexpected requirement for brain-type sodium channels for control of heart rate in the mouse sinoatrial node. Proc Natl Acad Sci USA. 2003;100:3507–3512. doi: 10.1073/pnas.2627986100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Su Z, Sugishita K, Ritter M, et al. The sodium pump modulates the influence of INa on [Ca2+]i transients in mouse ventricular myocytes. Biophys J. 2001;80:1230–1237. doi: 10.1016/S0006-3495(01)76099-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scriven DR, Klimek A, Lee KL, et al. The molecular architecture of calcium microdomains in rat cardiomyocytes. Ann NY Acad Sci. 2002;976:488–499. doi: 10.1111/j.1749-6632.2002.tb04783.x. [DOI] [PubMed] [Google Scholar]
- 33.Cheng H, Cannell MB, Lederer WJ. Propagation of excitation-contraction coupling into ventricular myocytes. Pfluegers Arch Eur J Physiol. 1994;428:415–417. doi: 10.1007/BF00724526. [DOI] [PubMed] [Google Scholar]
- 34.Lipp P, Niggli E. Modulation of Ca2+ release in cultured neonatal rat cardiac myocytes: insight from subcellular release patterns revealed by confocal microscopy. Circ Res. 1994;74:979–990. doi: 10.1161/01.res.74.5.979. [DOI] [PubMed] [Google Scholar]
- 35.Louch WE, Bito V, Heinzel FR, et al. Reduced synchrony of Ca2+ release with loss of T-tubules-a comparison to Ca2+ release in human failing cardiomyocytes. Cardiovasc Res. 2004;62:63–73. doi: 10.1016/j.cardiores.2003.12.031. [DOI] [PubMed] [Google Scholar]
- 36.Hatem S. Does the loss of transverse tubules contribute to dyssynchronous Ca2+ release during heart failure? Cardiovasc Res. 2004;62:1–3. doi: 10.1016/j.cardiores.2004.01.037. [DOI] [PubMed] [Google Scholar]
- 37.Gómez AM, Valdivia HH, Cheng H, et al. Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science. 1997;276:800–806. doi: 10.1126/science.276.5313.800. [DOI] [PubMed] [Google Scholar]
- 38.Litwin SE, Zhang D, Bridge JH. Dyssynchronous Ca2+ sparks in myocytes from infarcted hearts. Circ Res. 2000;87:1040–1047. doi: 10.1161/01.res.87.11.1040. [DOI] [PubMed] [Google Scholar]
- 39.Diaz ME, O’Neill SC, Eisner DA. Sarcoplasmic reticulum calcium content fluctuation is the key to cardiac alternans. Circ Res. 2004;94:650–656. doi: 10.1161/01.RES.0000119923.64774.72. [DOI] [PubMed] [Google Scholar]
- 40.Santana LF, Cheng H, Gómez AM, et al. Relation between the sarcolemmal Ca2+ current and Ca2+ sparks and local control theories for cardiac excitation-contraction coupling. Circ Res. 1996;78:166–171. doi: 10.1161/01.res.78.1.166. [DOI] [PubMed] [Google Scholar]
- 41.Cannell MB, Cheng H, Lederer WJ. The control of calcium release in heart muscle. Science. 1995;268:1045–1050. doi: 10.1126/science.7754384. [DOI] [PubMed] [Google Scholar]
- 42.Cheng H, Cannell MB, Lederer WJ. Partial inhibition of Ca2+ current by methoxyverapamil (D600) reveals spatial nonuniformities in [Ca2+]i during excitation-contraction coupling in cardiac myocytes. Circ Res. 1995;76:236–241. doi: 10.1161/01.res.76.2.236. [DOI] [PubMed] [Google Scholar]
- 43.Cheng H, Lederer WJ, Cannell MB. Calcium sparks: Elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- 44.Santana LF, Kranias EG, Lederer WJ. Calcium sparks and excitation-contraction coupling in phospholamban-deficient mouse ventricular myocytes. J Physiol (Lond) 1997;503:21–29. doi: 10.1111/j.1469-7793.1997.021bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Niggli E, Lederer WJ. Voltage-independent calcium release in heart muscle. Science. 1990;250:565–568. doi: 10.1126/science.2173135. [DOI] [PubMed] [Google Scholar]
- 46.Cannell MB, Berlin JR, Lederer WJ. Effect of membrane potential changes on the calcium transient in single rat cardiac muscle cells. Science. 1987;238:1419–1423. doi: 10.1126/science.2446391. [DOI] [PubMed] [Google Scholar]
- 47.Stern MD. Theory of excitation-contraction coupling in cardiac muscle. Biophys J. 1992;63:497–517. doi: 10.1016/S0006-3495(92)81615-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gomez AM, Guatimosim S, Dilly KW, et al. Heart failure after myocardial infarction: altered excitation-contraction coupling. Circulation. 2001;104:688–693. doi: 10.1161/hc3201.092285. [DOI] [PubMed] [Google Scholar]