

Abstract
Platelet Endothelial Cell Adhesion Molecule (PECAM) is an adhesion and signaling molecule used for leukocyte extravasation. We have generated two strains of PECAM-deficient mouse, one in the original C57BL/6 and a second by backcrossing nice generations into the FVB/n strain. The FVB/n strain has reduced responses in models of acute inflammation. We show here that this strain is also susceptible to a chronic pneumonia which leads to pulmonary fibrosis. In contrast, PECAM-deficient C57BL/6 mice do not develop this lung disease and have normal responses in acute models of inflammation. This demonstrates that PECAM-dependent and -independent mechanisms are found in both acute and chronic inflammation. Further, the PECAM-deficient FVB/n strain has many pathologic similarities to the human disease Idiopathic Pulmonary Fibrosis, suggesting that similar molecular mechanisms may play a role in human disease.
Introduction
Leukocyte extravasation in response to inflammatory signals on the endothelial cells that line the blood vessels is one of the earliest steps in the response to a pathogen. Platelet Endothelial Cell Adhesion Molecule (PECAM) is an adhesion molecule used by leukocytes, especially monocytes and neutrophils, to exit blood vessels in this process. PECAM is a 130-kDa member of the Immunoglobulin superfamily that is expressed both on leukocytes and in the junctions between the endothelial cells that line all blood vessels. Leukocyte migration across the blood vessel wall involves interaction of PECAM on the leukocyte with the PECAM at the endothelial cell junction (Muller, 1995; Muller, 1999; Newman, 1997).
On the other hand, the immune response must be terminated after the infection is cleared to prevent further tissue damage, and PECAM has also been shown to regulate programmed cell death (apoptosis) (Bird et al., 1999; Evans et al., 2001; Gao et al., 2003; Noble et al., 1999). PECAM has two cytoplasmic Immunoreceptor Tyrosine Inhibitory Motif (ITIM) domains for intracellular signaling (Newman et al., 2001; Newton-Nash and Newman, 1999). When PECAM on live cells engages macrophage PECAM, phagocytosis is prevented. Signaling through the cytoplasmic tail and the ITIM domains is important for both preventing ingestion of live leukocytes and increasing resistance to apoptosis (Brown et al., 2002; Gao et al., 2003). Thus, PECAM function is important for both promoting and terminating inflammation.
PECAM is important for leukocyte responses to inflammation, but there are also PECAM-independent pathways. Blocking PECAM prevents extravasation of a majority of monocytes and neutrophils in vitro and in vivo. However, a minor population (∼ 20-25%) of both monocytes and neutrophils can cross by PECAM-independent mechanisms (Muller, 1999). Further, when mice genetically deficient for PECAM were first derived in the C57BL/6 strain, they were able to use PECAM-independent mechanisms to mobilize normal numbers of monocytes and neutrophils in several models of inflammation (Duncan et al., 1999). While these PECAM-deficient mice were subsequently found to have some deficiencies in their inflammatory responses in certain models (Carrithers et al., 2005; Graesser et al., 2002; Maas et al., 2005; Solowiej et al., 2003; Thompson et al., 2001), C57BL/6 mice appear to be unique in their relative insensitivity to PECAM blockade. All other mouse strains tested have significantly reduced inflammatory reactions when PECAM is genetically deficient or blocked in wild-type mice using antibodies. The other genetically PECAM-/- mouse strain, FVB/n, fails to mobilize monocytes and neutrophils in models of peritonitis and dermatitis (Schenkel et al., 2004). Studies in the literature show that inflammation can be blocked by anti-PECAM antibodies in strains like CD2F1 (Bogen et al., 1994), AKR/J (Bogen et al., 1994), AND, a T lymphocyte transgenic derived from SJL (Qing et al., 2001), and DBA1/J (Ishikaw et al., 2002). We have confirmed the result in FVB/n, SJL, and Swiss Webster (outbred) mice (Schenkel et al., 2004). In rats treated with anti-PECAM antibodies, neutrophil emigration was also blocked (Vaporciyan et al., 1993), and leukocyte adhesion to synovial blood vessels was blocked in a model of rheumatoid arthritis (Decking et al., 2001). The amount that inflammation was reduced in these in vivo studies is similar to what we have observed with human leukocytes in vitro (Muller, 1995; Muller, 1999). Thus, PECAM-dependent mechanisms are used widely in mice, rats, and humans for leukocyte trafficking.
PECAM is highly expressed on lung vasculature (Marszalek et al., 2000; Muller et al., 1989). A high proportion of PECAM-/- FVB/n mice in clean veterinary facilities spontaneously develop a chronic lung disease described in this report, while their wild-type littermates are unaffected. Further, the C57BL/6 PECAM-/- mice do not develop chronic lung disease. Thus, PECAM-independent mechanisms must play a significant role in the resistance of C57BL/6 mice to a chronic and ultimately fatal pneumonia.
Materials and methods
Mouse strains
C57BL/6 or FVB/n wild-type and PECAM knockout mice were raised and housed at Weill Medical College of Cornell University. PECAM-deficient mice in the C57BL/6 background have been previously described (Duncan et al., 1999; Schenkel et al., 2004). PECAM-deficient mice in the FVB/n background were generated by nine successive backcrosses (Schenkel et al., 2004). Mice were kept in specific pathogen-free housing. Sentinel mice were screened for antibodies to most common murine viral, bacterial, fungal, and parasitic infections. Over 3 years, only one sentinel mouse was positive for Mouse Hepatitis Virus, PECAM-deficient mice were tested but did not have detectable antibodies.
Pathologic examination
At necropsy, animals were dissected to expose the lungs. Gross observations were noted, and the lungs were removed. Lungs were fixed in 10% buffered formalin and submitted for embedding in paraffin. Sections were mounted and stained with hematoxylin and eosin stains. Blinded scoring of the specimens was done on a Zeiss microscope, and the code broken after all scoring was completed. The clinical scoring system was as follows: No pathology = 0, Multifocal inflammation = +, Inflammation/Fibrosis in 25% of lung = ++, Inflammation/Fibrosis in 25-50% of lung = ++, Inflammation/Fibrosis in >50% of lung = +++.
Detection of pathogens
Bronchoalveolar lavage fluid was collected as follows. The trachea was exposed using sterile technique and lavaged with sterile saline using an 18-gauge feeding needle attached to a 1-cm3 syringe. This fluid was cultured for aerobic bacteria. Pieces of lung tissues were also cultured for aerobic bacteria. Other pieces were stained using silver stains to identify any fungal or bacterial infections. Culture and staining were performed by veterinary services at Weill Medical College of Cornell University.
Western blot for Ym1
Lungs were prepared lavaged as described. Lavaged cells were counted and pelleted for centrifugation. Supernatants and cell pellets were lysed in lysis buffer for SDS polyacrylamide gel electrophoresis containing 2% SDS, 12% sucrose, 0.01% Bromphenol Blue in 50 mM carbonate buffer pH 8.6 ± 5% β- mercaptoethanol. Additionally, lungs were minced and lysed in a lysis buffer containing 10 mM HEPES pH 7.9, 1.5 mM MgCL2, 10 mM KCl, 0.5 mM DTT, 0.1% NP-40, and Eukaryotic Protease Inhibitor Cocktail (4-(2-aminoethyl) benzenesulfonyl fluoride, pepstatinA, E-64, bestatin, leupeptin, and aprotinin Sigma, St. Louis, MO) The lysate was centrifuged at high speed, and the insoluble pellets and soluble supernatant were diluted in lysis buffer (Neville, 1971). Proteins were separated by gel electrophoresis on 4-20% polyacrylamide gradient gels in Tris-SDS buffer. The lysate was transferred to PVDF membranes by semi-dry electrophoresis at 25 W for 1 h. The membranes were blocked with 5% bovine serum albumin for 1 h in Tris-buffered saline +0.1% Tween-20 (TBST). Rabbit anti-serum against murine Ym1 (Harbord et al., 2002) was incubated with the membranes in TBST, washed to remove unbound antibody, incubated with goat anti-rabbit secondary antiserum conjugated to horseradish peroxidase for 1 h, and washed to remove unbound antibodies. The membranes were then incubated with CDP-Star chemiluminescent substrate (Perkin Elmer, Norwalk, CT) and placed on photographic films for detection.
Immunohistochemistry
Four-micrometer paraffin sections were deparaffinzed then treated with hydrogen peroxide and then blocked with Serum Free Protein Block (DakoCytomation, Carpinteria, CA). Apoptotic cells were immunohistochemically stained with a polyclonal rabbit anti-Cleaved Caspase-3 (Asp175) antibody (Cell Signaling Technology, Beverly, MA). A pretreatment of heat-induced epitope retrieval in Target Retrieval Solution, pH 9.0 (DakoCytomation, Carpinteria, CA) was utilized. The antibody was incubated for 30 min at room temperature and then visualized using Envision + rabbit (DakoCytomation, Carpinteria, CA) followed by DAKO Liquid DAB+ (DakoCytomation, Carpinteria, CA). Cells positive for F4/80 were immunohistochemically stained with a monoclonal rat anti-mouse F4/80 (Serotec, Raleigh, NC). Prior to primary incubation, the sections were treated with proteinase K (DakoCytomation, Carpinteria, CA). The antibody was incubated for 30 min at room temperature. After incubation with primary antibody, the tissue sections were sequentially incubated with biotinylated rabbit anti-rat immunoglobulin (DakoCytomation, Carpinteria, CA) and then additionally treated with Envision + Rabbit (DakoCytomation, Carpinteria, CA). Staining was developed with Liquid DAB+ (DakoCytomation, Carpinteria, CA). YM-1 was localized with a rabbit anti-murine Ym1 (Harbord et al., 2002), and Wide Spectrum Screen Keratin (DakoCytomation, Carpinteria, CA) was used to localize cytokeratin, the antibodies were incubated for 30 min at room temperature and then additionally treated with Envision + Rabbit (DakoCytomation, Carpinteria, CA). Staining was developed with Liquid DAB+ (DakoCytomation, Carpinteria, CA).
Statistics
Survival curves between C57BL/6 and FVB/n PECAM-/- mice was done with Kaplan-Meier Survival Analysis (JMPin Software, SAS Institute, Cary, NC).
Results
The first evidence for a role of PECAM in lung inflammation came from the survival of PECAM-/- FVB/n strain mice in our breeding colony. After derivation of this strain, we initially found a surprising number of mice in respiratory distress. When we found sick mice, they were often in a single cage with a sick mother and pups or a set of siblings housed together. We found significantly higher mortality in PECAM-/- FVB/n mice compared to the PECAM-/- C57BL/6 mouse strain (P < 0.01, Kaplan-Meier Survival Analysis, Fig. 1). This disease also kills many pups, as evidenced by the sharp drop in survival by FVB/n mice at early age (Fig. 1, arrow). Hemizygous mice with one functional PECAM allele (PECAM+/-) were not affected.
Fig. 1.

Survival of PECAM-deficient mice in a breeding colony. C57BL/6 strain mice are indicated in black (n = 17), FVB/n strain in gray (n = 55). Both strains were housed in the same rooms together at Weill Medical College of Cornell University. All deaths, due to pneumonia or other disease, were included in retrospective analysis. P < 0.001 (Kaplan-Meier Survival Analysis).
Several mice were submitted for full diagnostic workup. Compared to normal lungs (Fig. 2A), two major types of pathology were noted in PECAM-/- FVB/n strain mice. In some lungs, there were numerous eosinophilic alveolar macrophages (Fig. 2B). More commonly, large regions of diffuse alveolar damage with interstitial thickening were observed in PECAM-/- FVB/n mice(Fig. 2C). In many of these lesions, type II pneumocyte hyperplasia was observed (Fig. 3), suggestive of ongoing disease, and significant numbers of alveolar macrophages were identified by the scavenger receptor F4/80 antigen (Figs. 4B-C). Fibrosis and collagen deposition were common in many of the lungs, indicative of a chronic condition (Fig. 5). Masson's Trichrome staining indicated that the collagen deposits were relatively recent because they were faintly blue, rather than the dark blue of older, more cross-linked collagen seen in human chronic lung diseases.
Fig. 2.

Gross morphology of diseased lung from PECAM-deficient FVB/n strain mice. (A) Wild-type FVB/n strain lungs. (B) PECAM-/-lung showing extensive pneumonia and fibrosis (2× magnification).
Fig. 3.

Diffuse alveolar damage in PECAM-deficient mice. (A) Diffuse alveolar damage. Near complete consolidation of the lung with large cystically dilated airspaces lined by epithelium (4×). (B) Higher power demonstrating type II pneumocyte hyperplasia in a bronchiole (*). There are alveolar macrophages with Ym1 crystals in their cytoplasm visible in airspaces on the left. The area to the right shows interstitial inflammation with fibroblast infiltration (arrows) and alveolar collapse (40×). (C) PECAM-/- FVB/n lung showing residual airspaces lined by pneumocytes marked with anti-cytokeratin. Numerous cytokeratin negative alveolar macrophages can also be seen, as well as cytokeratin negative fibroblasts in the interstitium (arrows; 10×).
Fig. 4.

Macrophages in the lung tissue stained with anti-F4/80. (A) No antibody (negative control) in a FVB/n PECAM-/- lung section. (B) Lesion with F4/80-positive cells in the midst of a region of interstitial fibrosis a FVB/n PECAM-/- mouse lung (20×). (C) Alveolar macrophages in a C57BL/6PECAM-/- lung (20×).
Fig. 5.

Collagen deposition in lung. (A) Hematoxylin and eosin stained section. (B) Trichrome stain on adjacent section showing the light blue stain characteristic of newly deposited collagen.
Another prominent feature of this disease was the appearance of Ym1 crystals within the large macrophages (Fig. 6). This has been seen in other immunocompromised mouse strains (Guo et al., 2000; Harbord et al., 2002; Jin et al., 1998). Ym1 is an antifungal β-N-acetylhexosaminidase secreted by macrophages and neutrophils (Harbord et al., 2002). Ym1 was confirmed in the large alveolar macrophages from these mice using immunohistochemistry (Fig. 6B). Ym1 is known to fluoresce and examination using FITC excitation and emission wavelengths also showed the prominent crystals within macrophages (Fig. 6D). Normal lungs also stained with Ym1, but not as prominently (Fig. 6F).
Fig. 6.

Ym1 in PECAM-/- mice. (A) Hematoxylin and eosin stained section showing pneumonic processes. (B) Immunohistochemistry on adjacent section to panel A, using an anti-Ym1 serum showing extensive localization to the large alveolar macrophages (40×). (C) Crystals in alveolar macrophages (H and E, 100×). (D) Same view as in panel C, viewed under fluorescence showing intracellular Ym1 crystals (100×). (E) Negative control (no primary antibody) in FVB/n PECAM-/- lung (20×) (B). Ym1 staining in C57BL/6 lung, localized mostly to macrophages in the subpleural space (arrows) but also found occasionally across the alveoli (20×).
Western blots showed increased levels of Ym1 protein in a diseased PECAM-/- FVB/n mother and two pups who succumbed to disease. Ym1 has a molecular weight of approximately 43 kDa. We noted that older mice in particular had a greater accumulation of macrophages and deposits of Ym1. This older mother also had reactivity to Ym1 in bronchoalveolar lavage fluids (Fig. 7). A wild-type FVB/n mouse co-housed with the sick mother did not have any sign of disease, and the expression of Ym1 was not as pronounced as in the diseased mice.
Fig. 7.
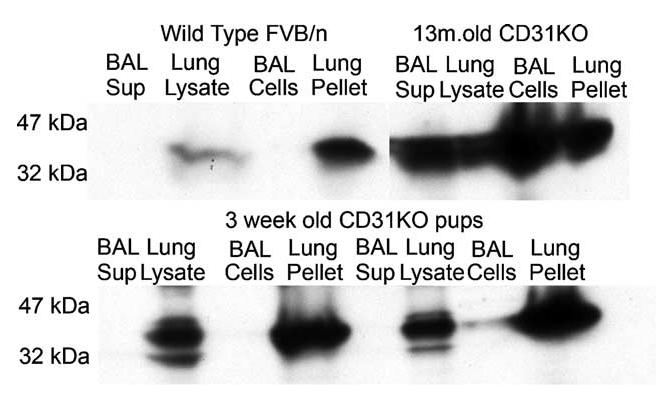
Western blot of Ym1 on lung tissues and bronchoalveolar lavage (BAL) fluids from healthy and sick mice housed together. A healthy wild-type mouse was housed for 4 weeks with a pregnant CD31-/- mouse. This surprise pregnancy resulted in two pups, who subsequently succumbed to pneumonia, along with the mother.
The incidence of this pneumonic disease is approximately 45% in aged PECAM-/- FVB/n strain mice (Table 1). Older mice (9-14 months old) sacrificed during routine culling were examined at necropsy for lung disease by blinded microscopic analysis and scored for severity of pathology and amount of fibrosis as indicated. More than half of the mice in the PECAM-/- FVB/n strain had normal lung function and structure, showing that the disease phenotype is not fully penetrant.
Table 1.
Percentages of aged mice with inflammation/pulmonary fibrosis
| Score | C57/WT | C57/CD31KO | FVB/WT | FVB/CD31KO |
|---|---|---|---|---|
| Unaffected | 93.7 | 89.7 | 89 | 56 |
| + | 6.3 | 8.8 | 11 | 23.3 |
| ++ | 0 | 0 | 0 | 16.5 |
| +++ | 0 | 1.5 | 0 | 2.8 |
| ++++ | 0 | 0 | 0 | 1.4 |
| Number of mice | 32 | 68 | 18 | 73 |
Scoring Multifocal Inflammation = + Inflammation/Fibrosis in 25% of lung = ++ Inflammation/Fibrosis in 25-50% of lung = +++ Inflammation/Fibrosis in >50% of lung = ++++ Lungs from routinely culled mice were removed at necropsy, and processe for histology. Slides were pooled and lesions were scored as indicated in a blinded manner with regard to strain.
There is no single infection clearly associated with this pneumonia. We cultured both bronchoalveolar lavage (BAL) fluid and lung tissue. However, only common bacteria can be cultured from these lungs, most often but not consistently Pasturella pneumotropica or Streptococcus acidominus (Table 2). P. pneumotropica is a common inhabitant of most mouse colonies and is not considered pathogenic in wild type mice. One PECAM-/- C57BL/6 mouse also had S. acidominus, Staphylococcus xylosus, and a Pasturella species cultured from BAL fluid. Silver stains of lung tissue to show any other organisms like fungi have been uniformly negative. Serum antibodies against most common murine pathogens like murine hepatitis virus have also been negative.
Table 2.
Bacteria Cultured From PECAM deficient mice
| Age at death (strain) | Specimen source | Aerobic culture results |
|---|---|---|
| 3 weeks (FVB/n) | Broncho-Alveolar lavage (BAL) | No growth |
| 4 weeks (FVB/n) | Lung tissue/BAL | Streptococcus acidominus, pasturella sp./No growth |
| 3 months (FVB/n) | Lung tissue | No growth |
| 10 months (FVB/n) | BAL | Pasturella sp. |
| 10 months (FVB/n) | BAL | Streptococcus sp., pasturella sp. |
| 10 months (FVB/n) | BAL | Streptococcus acidominus, pasturella sp. |
| 11 months (C57) | BAL | Streptococcus acidominus, staphylococcus xylosus, pasturella sp. |
| 11 months (FVB/n) | BAL | No growth |
| 11 months (FVB/n) | Lung tissue/BAL | Gram positive rods (Presumed: Lactobacillus) |
| 13 months (FVB/n) | BAL | Pasturella sp. |
PECAM has been shown to control apoptosis and ingestion of apoptotic cells, a process called efferocytosis (deCathelineau and Henson, 2003), so we examined the lungs for cleaved caspase-3. Many inflammatory cells in the lungs of PECAM-/- deficient FVB/n mice stained intensely (Fig. 7). The smaller cells are much more intense than the fainter and much larger macrophages. Many of the large macrophages did not stain with cleaved caspase-3 (arrows, Fig. 8). It is unclear whether the positive macrophages have ingested the smaller cells and that is why the staining is less intense, but we have only rarely observed discrete compartments of staining within the macrophages, as would be possible if an apoptotic body were ingested. The lungs from the other mouse strains had very little cleaved caspase staining (Fig. 7). This indicates that apoptosis does occur in PECAM-/- mice, but there may be a defect in clearance of apoptotic cells. Apoptotic cells are rapidly cleared from tissues under normal conditions. The accumulation of large alveolar macrophages that are positive for cleaved caspase-3 suggests that clearance may indeed be an issue.
Fig. 8.

Cleaved caspase-3 staining in PECAM-/- lung tissue. Cleaved caspase-3 was detected by immunohistochemistry. (A) No staining was detected in C57BL/6 PECAM-/- mice. (B) Extensive staining in FVB/n PECAM-/- mice. Cleaved caspase staining tended to be seen in large less intensely stained alveolar macrophages and smaller more intensely stained cells.
Discussion
Here we show that PECAM-deficient FVB/n mice are susceptible to a chronic inflammatory lung disease. We have closely characterized the penetrance of disease in different PECAM-deficient mice. The susceptibility to disease parallels the inability to mount acute inflammatory responses, suggesting that the same mechanism may play a role in a chronic inflammatory condition.
PECAM-deficient FVB/n mice have problems mobilizing leukocytes early on in an immune response (Schenkel et al., 2004). This may allow infection to develop to the point that destruction of the lung parenchyma occurs before the host can clear the infection. This would lead to the chronic inflammation and scarring we see. However, the role of infection remains unclear. We did not culture any pathogenic organisms from the lungs of affected mice. It is possible that the immune system can clear the infection and yet the chronic inflammation persists in the absence of any further infection. Alternatively or additionally, the increased sensitivity of PECAM-/- mice to apoptosis could lead to more rapid turnover of inflammatory cells, outstripping the ability of alveolar macrophages to clear them and leading to damage of parenchyma as the dead leukocytes spill their cache of hydrolytic enzymes. A third possibility is that continued cell death and efferocytosis in turn triggers TGFβ production and lung fibrosis (Huynh et al., 2002). Anyone of these mechanisms alone or together could result in the chronic disease we observe.
The role of PECAM in lung inflammation has been unclear, partially because some studies used the C57BL/6 strain of mice, which can compensate for loss or blockade of PECAM. Blockade of PECAM can reduce leukocyte responses to the lung of rats, in models of lung injury, including IgG immunecomplex deposition (Vaporciyan et al., 1993) and interleukin-1 (IL-1) instillation in PECAM-/- C57BL/6 strain mice (Albelda et al., 2004). Other studies, also using C57BL/6 mice or rats, have shown that PECAM-independent pathways are used in response to acute pneumonia caused by Escherichia coli and Streptococcus pneumoniae (Tasaka et al., 2003).
Alternative pathways for leukocyte extravasation to the lung compartment have been shown for adhesion molecules like CD44 and CD18 (β2) integrins. In CD44-deficient mice, there was actually enhanced neutrophil migration to lung upon infection with E. coli (Wang et al., 2002). Blockade of CD18 integrins inhibits migration of neutrophils into the lung in response to experimental Pseudomonas aeruginosa and E. coli/lipopolysaccharide, as well as IgG immune-complex deposition, IL-1 and phorbol myristate acetate. However, blockade of CD18 does not affect responses to S. pneumoniae, Group B Streptococcus, Staphylococcus aureus, hyperoxia, C5a, and hydrochloric acid (Doerschuk et al., 2000).
The different susceptibilities also challenge us to find the PECAM-independent pathways that confer resistance in the C57BL/6 strain. We are currently using Quantitative Trait Locus analysis to map the gene(s) that can prevent this fatal pneumonia in C57BL/6 mice. Several candidates have emerged, and we will begin testing these molecular targets in models of both acute inflammation and chronic inflammation. Once we confirm these mechanisms, these new pathways will be potential targets when addressing ways to control inflammation in lung disease.
Recent work has shown that crystals of Ym1 in Src homology 2-containing inositol-5-phosphatase-deficient (SHIP-/-) mice are a result of a subset of macrophages which have undergone “alternative activation.” These SHIP-/- macrophages are poorly responsive to inflammatory stimuli and are involved in pulmonary remodeling. Further, it appears that exposure to TGFβ in vitro causes SHIP-/- macrophages from these mice to become alternatively activated (Rauh et al., 2005). In PECAM-/- FVB/n strain mice, alternatively activated cells may also arise when TGFβ is secreted in response to some environmental insult. Future studies will address whether macrophages from PECAM-/- FVB/n mice also show similar characteristics as the macrophages from SHIP-/- mice.
Other studies have shown Ym1 crystals in other immunocompromised mouse strains like phagocyte oxidase-deficient mice (Harbord et al., 2002), as well as CD40L-deficient mice, a transgenic mouse strain expressing lung-specific surfactant apoprotein C promoter/soluble human tumor necrosis factor p75 receptor type II fusion protein, and the motheaten (Src Homology protein-tyrosine phosphatase) mouse strain (Guo et al., 2000). In contrast to these strains, PECAM-deficient mice appear to be much more susceptible to disease at a younger age, as evidenced by the loss of weanlings.
The majority of the literature on pulmonary fibrosis in mice examine fibrosis in the presence of exogenous agents like bleomycin. In the mice we report here, the disease developed spontaneously, without a clear role for an infectious agent. Additionally, the disease was not fully penetrant. The “idiopathic” nature of the disease in mice bears some pathologic similarities with human Idiopathic Pulmonary Fibrosis (IPF). In both humans and PECAM-/- mice, diseased tissue is found adjacent to healthy tissue. The role for infectious agents in IPF also is not clear. We clearly have a role for genetic susceptibility in mice, and genetic susceptibility in humans has been suggested for a number of genes involved in inflammation (Khalil and O'Connor, 2004). In humans, the disease lasts much longer, and this is likely to contribute to the extensive interstitial fibrosis (“honeycomb”) appearance of the lungs, whereas our mice are not given supplementary oxygen and instead must be humanely euthanized relatively early in the course of the disease. Further, Ym1 crystals are not found in human IPF because there is no human homolog (Boot et al., 2005), and the deposition of these crystals is only found in mice. However, we do observe the type II pneumocyte hyperplasia, interstitial fibrosis, and large-scale destruction of alveolar structure which is also a characteristic of IPF (Fig. 2). The degree of diffuse alveolar damage seems proportional in scale between mouse and man. Despite these differences, the PECAM-deficient FVB/n mouse may therefore be a model to look at early events in fibrotic disease, the role of infectious agents in initiating disease, and the molecules that contribute to susceptibility. These molecules or functionally similar ones in humans may reveal the etiology behind the “idiopathic” nature of some forms of IPF.
Acknowledgments
The authors would like to thank Dr. Randy Basaraba and Dr. Jim DeMartini for the discussion on mouse lung pathology. This project was supported by NIH Grant HL046849-16.
References
- Albelda SM, Lau KC, Chien P, Huang ZY, Arguiris E, Bohen A, Sun J, Billet JA, Christofidou-Solomidou M, Indik ZK, Schreiber AD. Role for Platelet-endothelial Cell Adhesion Molecule-1 in macrophage Fcgamma receptor function. Am. J. Respir. Cell Mol. Biol. 2004;31:246–255. doi: 10.1165/rcmb.2003-0404OC. [DOI] [PubMed] [Google Scholar]
- Bird IN, Taylor V, Newton JP, Spragg JH, Simmons DL, Salmon M, Buckley CD. Homophilic PECAM-1(CD31) interactions prevent endothelial cell apoptosis but do not support cell spreading or migration. J. Cell Sci. 1999;19891997;112(Pt 12) doi: 10.1242/jcs.112.12.1989. [DOI] [PubMed] [Google Scholar]
- Bogen S, Pak J, Garifallou M, Deng X, Muller WA. Monoclonal antibody to murine PECAM-1 [CD31] blocks acute inflammation in vivo. J. Exp. Med. 1994;179:1059–1064. doi: 10.1084/jem.179.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boot RG, Bussink AP, Aerts JM. Human acidic mammalian chitinase erroneously known as eosinophil chemotactic cytokine is not the ortholog of mouse YM1. J. Immunol. 2005;175:2041–2042. doi: 10.4049/jimmunol.175.4.2041-a. [DOI] [PubMed] [Google Scholar]
- Brown S, Heinisch I, Ross E, Shaw K, Buckley CD, Savill J. Apoptosis disables CD31-mediated cell detachment from phagocytes promoting binding and engulfment. Nature. 2002;418:200–203. doi: 10.1038/nature00811. [DOI] [PubMed] [Google Scholar]
- Carrithers M, Tandon S, Canosa S, Michaud M, Graesser D, Madri JA. Enhanced susceptibility to endotoxic shock and impaired STAT3 signaling in CD31-deficient mice. Am. J. Pathol. 2005;166:185–196. doi: 10.1016/S0002-9440(10)62243-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- deCathelineau AM, Henson PM. The final step in programmed cell death: phagocytes carry apoptotic cells to the grave. Essays Biochem. 2003;39:105–117. doi: 10.1042/bse0390105. [DOI] [PubMed] [Google Scholar]
- Decking J, Mayer A, Petrow P, Seiffge D, Karbowski A. Antibodies to PECAM-1 and glucocorticoids reduce leukocyte adhesion in adjuvant arthritis of the rat knee synovium in vivo. Inflamm. Res. 2001;50:609–615. doi: 10.1007/PL00000242. [DOI] [PubMed] [Google Scholar]
- Doerschuk CM, Tasaka S, Wang Q. CD11/CD18-dependent and -independent neutrophil emigration in the lungs: how do neutrophils know which route to take. Am. J. Respir. Cell Mol. Biol. 2000;23:133–136. doi: 10.1165/ajrcmb.23.2.f193. [DOI] [PubMed] [Google Scholar]
- Duncan GS, Andrew DP, Takimoto H, Kaufman SA, Yoshida H, Spellberg J, de la Pompa JL, Elia A, Wakeham A, Karan-Tamir B, et al. Genetic evidence for functional redundancy of Platelet/Endothelial Cell Adhesion Molecule-1 (PECAM-1): CD31-deficient mice reveal PECAM-1-dependent and PECAM-1-independent functions. J. Immunol. 1999;162:3022–3030. [PubMed] [Google Scholar]
- Evans PC, Taylor ER, Kilshaw PJ. Signaling through CD31 protects endothelial cells from apoptosis. Transplantation. 2001;71:457–460. doi: 10.1097/00007890-200102150-00020. [DOI] [PubMed] [Google Scholar]
- Gao C, Sun W, Christofidou-Solomidou M, Sawada M, Newman DK, Bergom C, Albelda SM, Matsuyama S, Newman PJ. PECAM-1 functions as a specific and potent inhibitor of mitochondrial-dependent apoptosis. Blood. 2003;102:169–179. doi: 10.1182/blood-2003-01-0003. [DOI] [PubMed] [Google Scholar]
- Graesser D, Solowiej A, Bruckner M, Osterweil E, Juedes A, Davis S, Ruddle NH, Engelhardt B, Madri JA. Altered vascular permeability and early onset of experimental autoimmune encephalomyelitis in PECAM-1-deficient mice. J. Clin. Invest. 2002;109:383–392. doi: 10.1172/JCI13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Johnson RS, Schuh JC. Biochemical characterization of endogenously formed eosinophilic crystals in the lungs of mice. J. Biol. Chem. 2000;275:8032–8037. doi: 10.1074/jbc.275.11.8032. [DOI] [PubMed] [Google Scholar]
- Harbord M, Novelli M, Canas B, Power D, Davis C, Godovac-Zimmermann J, Roes J, Segal AW. Ym1 is a neutrophil granule protein that crystallizes in p47phox-deficient mice. J. Biol. Chem. 2002;277:5468–5475. doi: 10.1074/jbc.M110635200. [DOI] [PubMed] [Google Scholar]
- Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J. Clin. Invest. 2002;109:41–50. doi: 10.1172/JCI11638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikaw J, Okada Y, Bird IN, Jasani B, Spragg JH, Yamada T. Use of anti-Platelet-Endothelial Cell Adhesion Molecule-1 antibody in the control of disease progression in established collagen-induced arthritis in DBA/1J mice. Jpn. J. Pharmacol. 2002;88:332–340. doi: 10.1254/jjp.88.332. [DOI] [PubMed] [Google Scholar]
- Jin HM, Copeland NG, Gilbert DJ, Jenkins NA, Kirkpatrick RB, Rosenberg M. Genetic characterization of the murine Ym1 gene and identification of a cluster of highly homologous genes. Genomics. 1998;54:316–322. doi: 10.1006/geno.1998.5593. [DOI] [PubMed] [Google Scholar]
- Khalil N, O'Connor R. Idiopathic pulmonary fibrosis: current understanding of the pathogenesis and the status of treatment. CMAJ. 2004;171:153–160. doi: 10.1503/cmaj.1030055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas M, Stapleton M, Bergom C, Mattson DL, Newman DK, Newman PJ. Endothelial cell PECAM-1 confers protection against endotoxic shock. Am. J. Physiol.: Heart Circ. Physiol. 2005;288:H159–H164. doi: 10.1152/ajpheart.00500.2004. [DOI] [PubMed] [Google Scholar]
- Marszalek A, Daa T, Kashima K, Nakayama I, Yokoyama S. Ultrastructural and morphometric studies related to expression of the cell adhesion molecule PECAM-1/CD31 in developing rat lung. J. Histochem. Cytochem. 2000;48:1283–1289. doi: 10.1177/002215540004800911. [DOI] [PubMed] [Google Scholar]
- Muller WA. The role of PECAM-1 [CD31] in leukocyte emigration: studies in vitro and in vivo. J. Leukocyte Biol. 1995;57:523–528. doi: 10.1002/jlb.57.4.523. [DOI] [PubMed] [Google Scholar]
- Muller WA. The role of PECAM in leukocyte emigration. In: Pearson JD, editor. Vascular Adhesion Molecules and Inflammation. Birkhauser Verlag; Basel, Switzerland: 1999. pp. 125–140. [Google Scholar]
- Muller WA, Ratti CM, McDonnell SL, Cohn ZA. A human endothelial cell-restricted, externally disposed plasmalemmal protein enriched in intercellular junctions. J. Exp. Med. 1989;170:399–414. doi: 10.1084/jem.170.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neville DM., Jr. Molecular weight determination of protein-dodecyl sulfate complexes by gel electrophoresis in a discontinuous buffer system. J. Biol. Chem. 1971;246:6328–6334. [PubMed] [Google Scholar]
- Newman PJ. The biology of PECAM-1. J. Clin. Invest. 1997;99:3–8. doi: 10.1172/JCI119129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman DK, Hamilton C, Newman PJ. Inhibition of antigen-receptor signaling by Platelet Endothelial Cell Adhesion Molecule-1 (CD31) requires functional ITIMs, SHP-2, and p56(lck) Blood. 2001;97:2351–2357. doi: 10.1182/blood.v97.8.2351. [DOI] [PubMed] [Google Scholar]
- Newton-Nash DK, Newman PJ. A new role for platelet-endothelial cell adhesion molecule-1 (CD31): inhibition of TCR-mediated signal transduction. J. Immunol. 1999;163:682–688. [PubMed] [Google Scholar]
- Noble KE, Wickremasinghe RG, DeCornet C, Panayiotidis P, Yong KL. Monocytes stimulate expression of the Bcl-2 family member, A1, in endothelial cells and confer protection against apoptosis. J. Immunol. 1999;162:1376–1383. [PubMed] [Google Scholar]
- Qing Z, Sandor M, Radvany Z, Sewell D, Falus A, Potthoff D, Muller WA, Fabry Z. Inhibition of antigen-specific T cell trafficking into the central nervous system via blocking PECAM1/CD31 molecule. J. Neuropathol. Exp. Neurol. 2001;60:798–807. doi: 10.1093/jnen/60.8.798. [DOI] [PubMed] [Google Scholar]
- Rauh MJ, Ho V, Pereira C, Sham A, Sly LM, Lam V, Huxham L, Minchinton AI, Mui A, Krystal G. SHIP represses the generation of alternatively activated macrophages. Immunity. 2005;23:361–374. doi: 10.1016/j.immuni.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Schenkel AR, Chew TW, Muller WA. Platelet Endothelial Cell Adhesion Molecule deficiency or blockade significantly reduces leukocyte emigration in a majority of mouse strains. J. Immunol. 2004;173:6403–6408. doi: 10.4049/jimmunol.173.10.6403. [DOI] [PubMed] [Google Scholar]
- Solowiej A, Biswas P, Graesser D, Madri JA. Lack of Platelet Endothelial Cell Adhesion Molecule-1 attenuates foreign body inflammation because of decreased angiogenesis. Am. J. Pathol. 2003;162:953–962. doi: 10.1016/S0002-9440(10)63890-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasaka S, Qin L, Saijo A, Albelda SM, DeLisser HM, Doerschuk CM. Platelet Endothelial Cell Adhesion Molecule-1 in neutrophil emigration during acute bacterial pneumonia in mice and rats. Am. J. Respir. Crit. Care Med. 2003;167:164–170. doi: 10.1164/rccm.2202011. [DOI] [PubMed] [Google Scholar]
- Thompson RD, Noble KE, Larbi KY, Dewar A, Duncan GS, Mak TW, Nourshargh S. Platelet-Endothelial Cell Adhesion Molecule-1 (PECAM-1)-deficient mice demonstrate a transient and cytokine-specific role for PECAM-1 in leukocyte migration through the perivascular basement membrane. Blood. 2001;97:1854–1860. doi: 10.1182/blood.v97.6.1854. [DOI] [PubMed] [Google Scholar]
- Vaporciyan AA, Delisser HM, Yan H-C, Mendiguren II, Thom SR, Jones ML, Ward PA, Albelda SM. Involvement of Platelet-Endothelial Cell Adhesion Molecule-1 in neutrophil recruitment in vivo. Science. 1993;262:1580–1582. doi: 10.1126/science.8248808. [DOI] [PubMed] [Google Scholar]
- Wang Q, Teder P, Judd NP, Noble PW, Doerschuk CM. CD44 deficiency leads to enhanced neutrophil migration and lung injury in Escherichia coli pneumonia in mice. Am. J. Pathol. 2002;161:2219–2228. doi: 10.1016/S0002-9440(10)64498-7. [DOI] [PMC free article] [PubMed] [Google Scholar]