

Abstract
Calcitonin gene-related peptide (CGRP) and nitric oxide are involved in the underlying pathophysiology of migraine and other diseases involving neurogenic inflammation. We have tested the hypothesis that nitric oxide might trigger signaling mechanisms within the trigeminal ganglia neurons that would coordinately stimulate CGRP synthesis and release. Treatment of primary trigeminal ganglia cultures with nitric oxide donors caused a greater than four-fold increase in CGRP release compared with unstimulated cultures. Similarly, CGRP promoter activity was also stimulated by nitric oxide donors and overexpression of inducible nitric oxide synthase (iNOS). Cotreatment with the antimigraine drug sumatriptan greatly repressed nitric oxide stimulation of CGRP promoter activity and secretion. Somewhat surprisingly, the mechanisms of nitric oxide stimulation of CGRP secretion did not require cGMP or PI3-kinase signaling pathways, but rather, nitric oxide action required extracellular calcium and likely involves T-type calcium channels. Furthermore, nitric oxide was shown to increase expression of the active forms of the mitogen-activated protein kinases Jun amino-terminal kinase and p38 but not extracellular signal-related kinase in trigeminal neurons. In summary, our results provide new insight into the cellular mechanisms by which nitric oxide induces CGRP synthesis and secretion from trigeminal neurons.
Keywords: calcium channels, migraine, mitogen-activated protein kinases, sumatriptan
Introduction
Calcitonin gene-related peptide (CGRP) is a multifunctional neuropeptide that plays an important role in the pathology of all vascular headaches, including migraine (van Rossum et al., 1997; Edvinsson et al., 2001; Pietrobon & Striessnig, 2003). Migraine is a chronic, painful, neurovascular disorder that affects an estimated 12% of the population (Stewart et al., 1994; Ferrari, 1998). Serum levels of CGRP are elevated in patients during migraine and cluster headaches (Goadsby & Edvinsson, 1993, 1994; Fanciullacci et al., 1995). The trigeminal ganglia are the major source of both CGRP and sensory, nociceptive innervation that connect the cranial vasculature to the central nervous system (McCulloch et al., 1986). Activation of trigeminovascular afferents in the meninges releases CGRP, which causes vasodilation and neurogenic inflammation, and its efferent release contributes to pain, central sensitization, and allodynia (Buzzi et al., 1995; Buzzi, 2001; Williamson & Hargreaves, 2001). Further evidence for a role of CGRP in migraine comes from clinical studies in which the serotonergic antimigraine drug sumatriptan lowers CGRP levels, coincident with relief of headache pain (Goadsby & Edvinsson, 1991), and a novel CGRP receptor antagonist was shown to treat migraine effectively (Olesen et al., 2004). Despite the importance of CGRP in migraine pathology, the cellular mechanisms that regulate CGRP secretion in trigeminal neurons during migraine attacks are not well understood.
Release of nitric oxide and other inflammatory agents following hypercortical activity are thought to be instrumental in the pathogenesis of migraine (Bolay et al., 2002; Pietrobon & Striessnig, 2003). Nitric oxide, an important messenger involved in cerebrovascular regulation (Yun et al., 1996), is produced in a wide variety of tissues by the nitric oxide synthase isoforms nNOS, iNOS and eNOS (Faraci & Heistad, 1998). Physiological studies have demonstrated that nitric oxide derived from nitroglycerin induces a severe headache that is indistinguishable from a spontaneous migraine attack (Iversen & Olesen, 1996). Interestingly, CGRP levels in the blood plasma of patients suffering from episodic cluster headache also are increased during headaches experimentally induced by nitroglycerin (Fanciullacci et al., 1995). Thus, it seems likely that nitric oxide production and neuropeptide release are functionally linked in severe vascular headaches. However, the mechanisms by which nitric oxide controls CGRP gene expression in trigeminal neurons are not known.
The CGRP gene has been shown to be regulated by the cellular signaling cascades collectively known as the mitogen-activated protein kinases (MAPK) pathways (Thiagalingam et al., 1996; Durham & Russo, 1998, 2000). Interestingly, several agents implicated in the initiation or maintenance of migraine headache pain can directly activate MAP kinases. This family of important signal-transducing enzymes regulates the function of other proteins via a series of reversible phosphorylation events (Seger & Krebs, 1995; Cohen, 1997). It is now known that at least four distinctly regulated groups of MAP kinases are present in mammalian cells, extracellular signal-regulated kinases (ERK)-1/2, Jun amino-terminal kinases (JNK1/2/3), p38 proteins (p38α/β/γ/δ), and ERK5, that are activated by specific MAPK kinases (MAPKK; Schaeffer & Weber, 1999; Chang & Karin, 2001). Signaling through MAP kinase pathways allows a cell to respond to changes in its environmental conditions in a regulated and defined manner. In this study, the effect of nitric oxide on MAP kinase signaling was investigated.
Materials and methods
Animals and cell culture
All animal care and procedures were conducted in accordance with institutional and National Institutes of Health guidelines. Adult female Sprague-Dawley rats (Charles River, Wilmington, MA, USA) were housed in clean plastic cages on a 12-h light/dark cycle with unrestricted access to food and water. Trigeminal ganglia primary cultures were established based on our published protocol (Durham & Russo, 1999, 2003). Briefly, cells from ganglia isolated from 3-to 4-day-old Sprague-Dawley rats (killed with CO2 inhalation) were resuspended in L15 medium containing 10% fetal bovine serum, 50 mm glucose, 250 μm ascorbic acid, 8 μm glutathione, 2 mm glutamine and 10 ng/mL mouse 2.5S nerve growth factor (Alomone Laboratories, Israel) at 37 °C at ambient CO2 levels. Dissociated cells were plated on either six-well (promoter studies) or 24-well (secretion studies) tissue culture plates (Becton Dickinson, Franklin Lakes, NJ, USA), or 11-mm glass or plastic coverslips (immunostaining) coated with poly-d-lysine (relative mol. wt 30 000-70 000; Sigma, St. Louis, MO, USA). The cells from the equivalent of three ganglia were plated per -sixwell plate, one per 24-well plate, and 0.5 per 11-mm coverslip. Penicillin (100 units/mL), streptomycin (100 lg/mL) and amphotericin B (2.5 lg/mL, Sigma) were added to all media. The culture medium was changed after 24 h and every other day thereafter. Sodium nitroprusside (SNP), S-nitroso-N-acetylpenicillamine (SNAP), zaprinast, 1H-[1,2,4]oxadiazolo [4,3-a]quinoxalin-1-one (ODQ), capsazepine, thapsigargin, mibefradil and 8-bromo-cGMP were purchased from Sigma, while sumatriptan succinate (GlaxoSmithKline, UK) was obtained from the University of Iowa Pharmacy. The compounds LY 249002, N-monomethyl-l-arginine acetate (L-NMMA), N-nitro-l-arginine methyl ester hydrochloride (L-NAME) and wortmannin were purchased from Tocris (Ellisville, MO, USA). In all studies, cells were treated with equivalent amounts of vehicle.
CGRP secretion
For the CGRP secretion studies, cells maintained for 24 h were incubated in HBS (22.5 mm HEPES, 135 mm NaCl, 3.5 mm KCl, 1mm MgCl, 2.5 mm CaCl, 3.3 mm glucose, 0.1% BSA, pH 7.4) (Durham & Russo, 1999), and the amount of CGRP released from trigeminal neurons into the culture media was determined using a CGRP-specific radioimmunoassay (Bachem/Peninsula Laboratories, Inc., San Carlos, CA, USA). As a control, the basal (unstimulated) rate of CGRP secreted into the media in 1 h was determined, and this value was used to normalize for differences between dishes. Cells were pretreated with the indicated concentrations of inhibitors or appropriate vehicle for 10-15 min before addition of HBS (control), KCl, capsaicin, nitric oxide donors and/or cGMP. Treatment with KCl was used to induce a calcium influx via voltage-gated calcium channels, while capsaicin was used to cause a calcium influx via TRPV-1 channels. Each experimental condition was repeated in at least four independent experiments done in duplicate. The data were reported as mean ± SEM. Statistical analysis was performed using the Wilcoxon rank test. Results were considered significant at P < 0.05. Neuronal survival was assessed in cultures 24 h after the secretion studies by applying 1 mL of trypan blue diluted 1: 10 in PBS for 3 min, after which the cultures were washed three times with PBS and viewed via bright-field microscopy. The ratio of viable cells to total cells was determined for each secretion condition. Neuronal viability was shown to be > 90% for untreated cultures (113/120 total cells) as well as for cultures treated with SNP (105/113) and SNAP (122/134).
Immunocytochemistry
Dissociated trigeminal ganglia cells were plated at a density of 500-1000 cells on poly-d-lysine-coated 13-mm glass or plastic coverslips and incubated in complete L15 medium. Primary cultures (days 1-2) were incubated in 100% methanol for 10 min at -20 °C to fix and permeabilize the cells. For localization of CGRP, fixed cells were incubated for 30 min in PBS containing 5% donkey serum, then for 1 h with rabbit antirat CGRP polyclonal antibodies (1: 1000 dilution in PBS, Sigma), and for 1 h in Rhodamine Red-X-conjugated donkey antirabbit IgG (1: 100; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA). For 5-HT1 receptor localization, methanol-fixed cultures were blocked using 5% donkey serum and costained overnight at 4 °C with goat antirat 5-HT1B, 5-HT1D or 5-HT1F polyclonal antibodies (diluted 1: 100 in PBS, C-19, S-18, or C-19, Santa Cruz Biotechnology, Santa Cruz, CA, USA) and a β-tubulin monoclonal antibody (1: 1000 dilution in PBS, Sigma). Immunoreactive proteins were detected following a 1-h incubation with FITC-conjugated donkey antigoat IgG and/or Texas-Red-conjugated donkey antimouse IgG polyclonal antibodies (both diluted 1: 100 in PBS, Jackson ImmunoResearch Laboratories, Inc).
For the signaling pathway studies, untreated 2-day-old primary cultures or cultures treated with 1 mm SNAP, 0.6 m sorbitol or vehicle (DMSO) were briefly rinsed with PBS and incubated in 4% paraformaldehyde for 30 min and then 0.2% trition X-100 in PBS for 15 min. Stimulation with sorbitol (30 min) was used as a positive control for p38, JNK and ERK activation. For the signaling pathway studies, the cells were subcultured in L15 medium that contained 0.5% FBS for 24 h prior to addition of stimulatory agents. Cultures immunostained for the phosphorylated MAP kinase proteins were treated for 30 min. Fixed cells were incubated for 30 min in PBS containing 5% donkey serum, then for 1 h with primary antibodies and for 1 h with secondary antibodies. Cultures were also stained with antibodies directed against the phosphorylated (active) MAP kinase proteins p38 (Promega, Madison, WI, USA, 1: 8000), JNK (Promega, 1: 4000), and ERK (Promega, 1: 8000) as well as antibodies that recognize the unphosphorylated and phosphorylated forms (total protein) of p38, JNK or ERK (1 lg/mL, Santa Cruz Biotechnology). Immunoreactive proteins were detected following incubation with FITC-conjugated donkey antirabbit IgG, Texas-red-conjugated donkey antirabbit IgG, or FITC-conjugated donkey antimouse IgG polyclonal antibodies (diluted 1: 100 in PBS, Jackson ImmunoResearch Laboratories). No staining was observed in the absence of primary antibodies. The number of neuronal cells that responded to SNAP stimulation was determined by analysis of immunopositive cells from ten random fields of cells from two coverslips. The reported numbers are the average of the counts obtained by two laboratory staff who were blinded to the experimental design. Neuronal cells were identified based on their unique cell morphology and size (round cell body of 30-50 μm) from pictures obtained using phase contrast microscopy.
Transfection of trigeminal cultures
The CGRP luciferase reporter plasmids and the cytomegalovirus (CMV) β-galactosidase reporter plasmid have been described previously (Tverberg & Russo, 1993; Lanigan & Russo, 1997). The 1250-bp CGRP promoter-luciferase reporter contains sequences from the Kpn I site ()1250) to the Sau3A site (+21) in exon 1. The CMV-iNOS expression plasmid was a kind gift from D. Heistad (Gunnett et al., 2001).
Trigeminal ganglia cultures were transiently transfected 1 h after plating with Lipofectamine 2000 (Invitrogen Life Technologies) following the manufacturer's instructions. Approximately 3-5 × 104 cells (3-4 ganglia) per well were transfected 1 h after plating with 1-2 lg of CGRP-luciferase reporter, 0.5 lg gal1-luciferase reporter and/or 0.5 lg CMV-iNOS plasmid DNAs. The amount of DNA was kept constant by addition of the empty expression vector CMV-5 (Durham & Russo, 1998). The DNA and Lipofectamine 2000 reagent (1: 2 ratio) were incubated together for 20 min in L15 medium. Based on cell counts, it is estimated that 5% of the neurons are transfected by this method. The nitric oxide donor SNAP or cGMP were added to transfected cells 2 h before harvesting. In some experiments, the cells were preincubated for 30 min with 10 μm sumatriptan, prior to addition of SNAP. Cells cotransfected with CGRP and iNOS plasmid DNA were incubated overnight in serum-containing medium in the absence or presence of 10 μm sumatriptan and harvested 20 h later. Luciferase activity was measured using the Luciferase Assay System (Promega) and was reported as relative light units/20 lg protein. Protein concentrations were determined by Bradford assays (Bio-Rad Laboratories, Hercules, CA, USA). In all experiments, transfection efficiencies were normalized to CMV-β-galactosidase activity measured using Galacto-Light Plus reagents (Applied Biosystems, Foster City, CA, USA). No difference in cell viability was observed following transfection with the different plasmids or treatments as determined by trypan blue exclusion. The normalized luciferase activities are reported as means with standard errors per 20 lg protein. Each experimental condition was repeated in at least three independent experiments performed in duplicate. Statistical analyses were under-taken using the Wilcoxon rank test.
Results
Nitric oxide stimulation of CGRP secretion from cultured trigeminal ganglia neurons
In this study, primary trigeminal cultures were used to investigate the effects of nitric oxide on CGRP secretion and promoter activity. We have previously utilized cultured trigeminal ganglia neurons as a model to investigate the cellular mechanisms by which inflammatory agents and 5-HT1 receptor agonists regulate CGRP gene expression in sensory neurons (Durham et al., 1997, 2004; Durham & Russo, 1999, 2003). Under our culture conditions, > 80% of the cultured cells are neuronal, based on morphology (round cell body of 30-50 μm) and expression of neuronal-specific proteins, and > 90% of the neurons express detectable levels of CGRP. This is a much higher percentage than reported in human and rat trigeminal ganglia in situ, where 40% of the neurons are CGRP positive (Edvinsson et al., 1998). As shown in Fig. 1A, CGRP is found abundantly in the cell body and is also localized in the neuronal processes. In the absence of primary antibodies, no specific staining was observed.
Fig. 1.

Effect of nitric oxide on CGRP secretion from primary trigeminal neurons. (A) Cells from day 2 trigeminal ganglia cultures were stained with anti-CGRP antibodies and immunoreactivity detected using rhodamine-conjugated secondary antibodies. Neuronal cell bodies are indicated by arrows. Scale bar, 50 μm. (B) The relative amount of CGRP secreted in 1 h from untreated control cultures (CON), cultures treated with 60 mm KCl, 1 μm capsaicin (CAP), 1 mm SNAP or cotreated is shown. The mean basal rate of CGRP release was 112 ± 11 pg/h per well. The secretion rate for each condition was normalized to the basal rate for each well. The means and the SE from ten independent experiments are shown. (C) The relative amount of CGRP released from untreated control cultures (CON) and cultures treated with 100 μm L-NAME or L-NMMA, and KCl-depolarized trigeminal neurons in the absence or presence of L-NAME and L-NMMA. The mean basal rate of CGRP release was 99 ± 8 pg/h per well. The means and the SE from five independent experiments are shown. *P < 0.01 compared with control.
Treatment of trigeminal ganglia cultures maintained for 24 h with a depolarizing stimulus of 60 mm KCl for 1 h caused a five-fold increase in CGRP secretion as compared with the amount of CGRP released from unstimulated control cells (Fig. 1B). A similar stimulatory response was observed for 1 μm capsaicin, which is known to activate sensory C-fibers via activation of TRPV1 and increases in intracellular calcium. To determine whether nitric oxide could also stimulate CGRP secretion, trigeminal cultures were treated with SNAP, which spontaneously releases nitric oxide in aqueous solution. SNAP treatment increased CGRP release greater than four-fold as compared with prestimulated secretion rates. This level of CGRP stimulation was much lower than that seen in unstimulated (18.6 ± 1.8-fold; n = 4) or SNAP-treated cultures (16.4 ± 1.4-fold; n = 4) in response to 0.1% triton (data not shown). Cotreatment of the cultures with SNAP and either KCl or capsaicin did not result in a significant increase in the amount of secreted CGRP. This effect was not due to a depletion of CGRP in the neurons as treatment with 0.1% triton caused a much greater CGRP release (> 10-fold; n = 3, data not shown). These data demonstrate that nitric oxide can directly stimulate CGRP release from cultured trigeminal ganglia neurons.
To determine the potential contribution of nitric oxide generated by endogenous NOS activity on basal and KCl-stimulated CGRP secretion, trigeminal cultures were treated with the nonselective NOS inhibitors L-NAME and L-NMMA. Preincubation with 100 μm L-NAME or L-NmmA had no significant effect on unstimulated CGRP secretion or KCl-mediated release from trigeminal neurons as compared with control or KCl-treated secretion values (Fig. 1C). These results demonstrate that endogenous NOS activity is not required for basal or depolarization-stimulated CGRP secretion in cultured trigeminal neurons. In agreement with out finding, NOS activity was shown to have no significant role in trigeminal nerve-evoked changes in cerebral blood flow in vivo (Edvinsson et al., 1998). Furthermore, rat trigeminal ganglia neurons that innervate cerebral blood vessels were reported to express CGRP but only a small percentage contained NOS (Edvinsson et al., 2001).
cGMP and PI3-kinase independent regulation of CGRP secretion by nitric oxide
Numerous physiological studies in smooth muscle cells have shown that nitric oxide activates soluble guanylyl cyclase leading to the conversion of GTP to cGMP that can stimulate protein kinase G activity (Southam & Garthwaite, 1993). However, data from more recent studies have demonstrated that nitric oxide signaling can occur via cGMP-independent pathways. The mechanism of nitric oxide signaling in trigeminal neurons has not been investigated. To determine whether CGRP secretion involves activation of the cGMP pathway, primary trigeminal neurons were stimulated directly with cGMP or a selective inhibitor of soluble guanylyl cyclase. As seen in Fig. 2A, treatment of cultures with the nitric oxide donors SNP or SNAP caused approximately a three-fold increase in CGRP secretion. Treatment with the cell-permeable analog of cGMP, 8-bromo-cGMP (1 mm), did not stimulate CGRP release. This finding is in agreement with our promoter data, in which cGMP treatment of transfected cells 2 h before harvesting did not stimulate CGRP promoter activity in trigeminal neurons (data not shown). In addition, cotreatment with SNAP and the potent and selective inhibitor of guanylyl cyclase, ODQ (4 μm), did not repress nitric oxide stimulation. As an alternative approach, cells were preincubated with the nonselective phosphodiesterase inhibitor zaprinast, to block cGMP degradation. Exposure of cells to 100 μm zaprinast had no significant effect on basal CGRP secretion or on nitric oxide-stimulated release. Based on these data, nitric oxide-mediated stimulation of CGRP secretion in trigeminal neurons does not appear to involve activation of the cGMP signaling cascade.
Fig. 2.

Nitric oxide stimulation of CGRP secretion from trigeminal neurons is cGMP independent and does not involve PI3-kinase. (A) The relative amounts of CGRP secreted in 1 h from untreated control cells (CON) or cells treated with the SNP (10 mm), SNAP (1 mm), 8-bromo-cGMP (1 mm, cGMP), zaprinast (100 μm, ZAP), ODQ (4 μm) or cotreatment are shown. Cultures were pretreated for 15 min with zaprinast or ODQ prior to addition of NO donors. The mean basal rate of CGRP release was 108 ± 14 pg/h per dish. The means and the SE from five independent experiments are shown. *P < 0.05 compared with control. (B) The relative amounts of CGRP secreted in 1 h from untreated control cells (CON) or cells treated with 1 mm SNAP, 100 μm wortmanin (WOR), 100 μm LY 294002 (LY) or cotreatment are shown. Cultures were pretreated for 15 min with wortmanin or LY 294002 prior to addition of SNAP. The mean basal rate of CGRP release was 103 ± 7 pg/h per well. The means and the SE from four independent experiments are shown. *P < 0.05 compared with control.
It has been shown in recent studies in non-neuronal cells that nitric oxide triggers Ras activation and recruitment of an effector, phophatidylinositol 3’-kinase (PI3-kinase), which mediates an increase in secretion (Deora et al., 1998). The potential involvement of PI3-kinase in nitric oxide stimulation of CGRP was tested using two known inhibitors of PI3-kinase. Pretreatment of trigeminal cultures with the selective PI3-kinase inhibitors wortmannin or LY294002 (100 μm) had no effect on basal or nitric oxide-stimulated secretion (Fig. 2B). Thus, nitric oxide stimulation of CGRP secretion from trigeminal neurons does not require induction of the PI3-kinase pathway.
Nitric oxide signaling requires extracellular calcium and involves T-type calcium channels
Studies on nitric oxide signaling have shown that the effects of nitric oxide may involve changes in calcium levels within cells (Pinilla et al., 1998; Horn et al., 2002). We have previously reported that CGRP gene expression and secretion are differentially regulated by the amplitude and duration of a calcium signal (Durham & Russo, 1999, 2003). In this study, the potential role of calcium in nitric oxide signaling in trigeminal neurons was initially evaluated by performing the secretion experiments in the presence and absence of extracellular calcium. As seen in Fig. 3, the stimulatory effect of nitric oxide was greatly diminished when cells were stimulated in the absence of calcium in the culturing media while basal CGRP secretion was not significantly altered. This finding is in agreement with results from previous studies that demonstrated that evoked CGRP release from sensory neurons was dependent on extracellular calcium (Hingtgen & Vasko, 1994; Vasko et al., 1994). To test whether nitric oxide may also be causing release of calcium from intracellular stores, cells were treated with 10 μm thapsigargin, which irreversibly blocks endoplasmic reticulum Ca-ATPase and depletes IP3-sensitive calcium stores. Cotreatment with thapsigargin did not repress nitric oxide stimulation of CGRP. These results suggest that nitric oxide signaling is mediated via an influx of extracellular calcium. To determine the mechanism of nitric oxide-induced calcium entry, trigeminal cultures were initially cotreated with metal ions known selectively to inhibit voltage-gated calcium channels. Cotreatment with SNAP and 50 μm cadmium ions, a concentration reported to eliminate N- and L-type calcium currents but leave T-type relatively unaffected (Fox et al., 1987), did not cause inhibition of nitric oxide stimulation. However, the effect of nitric oxide was greatly reduced in cultures treated with 100 μm NiCl, which strongly reduces T-currents but leaves N- and L-type currents little changed (Fox et al., 1987; Ikeda et al., 2003). To demonstrate further that the effects of nitric oxide involve activation of the T-type channel, cultures were incubated with the T-type calcium channel antagonist mibefradil (1 μm). The concentration of mibefradil used in this study is reported to be selective for blocking T currents in neurons (Todorovic et al., 2001). As seen in Fig. 3, mibefradil treatment greatly diminished the stimulatory effect of SNAP. Taken together, these data strongly suggest that nitric oxide stimulation of CGRP secretion from trigeminal neurons involves activation of T-type channels and an increase in intracellular calcium levels.
Fig. 3.

Nitric oxide-stimulated release of CGRP is dependent on extracellular calcium and T-type calcium channel activation. The relative amounts of CGRP secreted in 1 h from untreated control cells (CON), or cells treated with 1 mm SNAP in the presence or absence of extracellular calcium, cells cotreated with 50 μm CdCl, 100 μm NiCl, 1 μm mibefradil (MIB), or 10 μm thapsigargin (THRAPS) are shown. Cultures were incubated in calcium-free media or channel inhibitors for ∼ 10 min prior to addition of SNAP. The mean basal rate of CGRP release was 76 ± 4 pg/h per dish. The means and the SE from four independent experiments are shown. *P < 0.01 compared with control; #P < 0.05 compared with SNAP.
Repression of nitric oxide stimulation of CGRP by antimigraine drug sumatriptan
Having shown that primary trigeminal neurons could be activated by nitric oxide, a signaling molecule implicated in migraine pathogenesis, we wanted to determine whether the serotonergic antimigraine drug sumatriptan succinate could repress this stimulatory effect on CGRP secretion. Sumatriptan has been shown to exhibit selectivity and potency towards the 5-HT1B, 5-HT1D and 5-HT1F receptors, which are expressed by rat trigeminal neurons (Ma, 2001; Ma et al., 2001). To determine which sumatriptan-activated 5-HT1 receptors are expressed by cultured trigeminal neurons, immunolocalization studies were performed. Day 2 cultures were stained with antibodies directed against 5-HT1 receptors (Fig. 4A). The same cultures were also stained for the cytoskeletal protein β-tubulin III, which is specifically expressed by neuronal cells (Fig. 4B). The 5-HT1B, 5-HT1D and 5-HT1F receptors were expressed primarily by medium-sized neurons (∼30-50 μm) in our cultures. We found that only a subpopulation (< 50%) of the total number of neurons in each culture expressed a specific 5-HT1 receptor. However, when antibodies directed against all three 5-HT1 receptors were used, > 90% of β-tubulin-stained cells were positively stained. The percentage of medium-sized neurons that express any one subtype of 5-HT1 receptor appears consistent with previous in situ studies in rats (Ma et al., 2001) and humans (Hou et al., 2001). Thus, the physiological effects of sumatriptan under our culture conditions are likely mediated via activation of the 5-HT1B, 5-HT1D and/or 5-HT1F receptors.
Fig. 4.
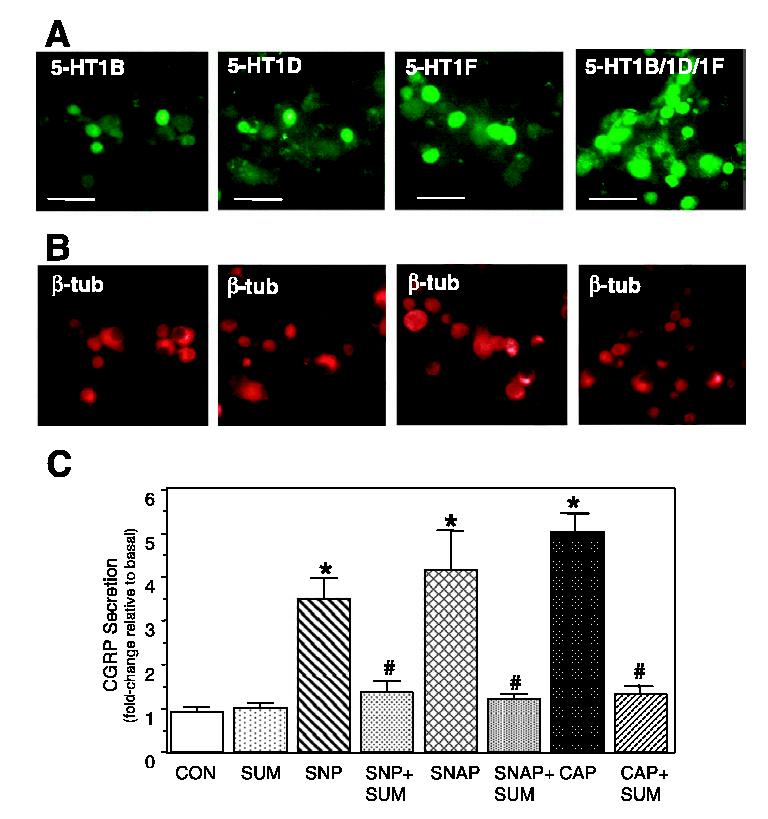
Expression of 5-HT1 receptors in cultured trigeminal neurons and sumatriptan repression of nitric oxide-stimulated CGRP secretion. Fluorescent photomicrographs of day 2 trigeminal ganglia cultures costained with 5-HT1 antibodies (A) and with β-tubulin antibodies (B). 5-HT1 receptor staining was visualized using FITC-conjugated donkey antirabbit IgG antibodies and β-tubulin using rhodamine-conjugated donkey antimouse IgG antibodies. Scale bars, 100 μm. (C) Effect of nitric oxide and sumatriptan on CGRP release from cultured trigeminal ganglia neurons. The relative amounts of CGRP secreted in 1 h from untreated control cells (CON), and cells treated with 10 mm SNP or 1 mm SNAP, 1 μm capsaicin (CAP), the antimigraine drug sumatriptan (10 μm, SUM) or cotreatment are shown. Cultures were pretreated for 30 min with sumatriptan prior to addition of NO donors or capsaicin. The mean basal rate of CGRP release was 114 ± 13 pg/h per dish. The means and the SE from five independent experiments are shown. *P < 0.01 compared with control; #P < 0.05 compared with NO donors.
In a clinical study, sumatriptan was shown to alleviate the pain associated with cluster headache, which was induced by nitric oxide following nitroglycerin administration to susceptible individuals (Fanciullacci et al., 1995). To test whether sumatriptan could repress nitric oxide-stimulated release of CGRP, trigeminal cultures were pretreated with sumatriptan for 30 min prior to stimulation with nitric oxide donors. In agreement with our previous findings (Durham & Russo, 1999), sumatriptan had no effect on basal CGRP secretion rates (Fig. 4C). However, sumatriptan repressed stimulated CGRP release mediated by SNP or SNAP to near basal levels. Similarly, treatment with sumatriptan significantly repressed capsaicin stimulation of CGRP secretion. In an earlier study, sumatriptan was shown to inhibit stimulation of CGRP by KCl depolarization and a cocktail of inflammatory agents (Durham & Russo, 1999). Taken together, these data demonstrate that sumatriptan can repress stimulation of CGRP secretion mediated by multiple external stimuli, including nitric oxide.
Nitric oxide stimulation of CGRP promoter activity and repression by sumatriptan
Having shown that nitric oxide could stimulate CGRP secretion, transient transfections of primary trigeminal ganglia cultures were used to determine whether nitric oxide could also stimulate CGRP synthesis via activation of the CGRP promoter. CGRP transcription is controlled by a cell-specific enhancer (HO) (Tverberg & Russo, 1993; Lanigan & Russo, 1997), and by hormonal stimuli via a cAMP-responsive element (CRE) (de Bustros et al., 1986) and a Ras-responsive element (RRE) (Thiagalingam et al., 1996). Trigeminal ganglia cultures were transfected with a 1.25-kb fragment that contains the cell-specific enhancer, RRE, and CRE of the rat CGRP gene. We previously have shown that this DNA fragment is active in primary trigeminal neurons (Durham & Russo, 2003). Treatment with the spontaneous nitric oxide donor SNAP resulted in a marked increase in CGRP promoter activity (∼four-fold) compared with vehicle-treated control values (Fig. 5). Cultures were also cotransfected with a constitutively active form of inducible nitric oxide synthase (iNOS, kindly provided by Dr D. Heistad, University of Iowa) to test for the effect of the endogenous production of nitric oxide. It has been reported that iNOS can produce large quantities of nitric oxide, which can promote tissue damage and inflammation. Furthermore, infusion of the nitric oxide donor glyceryl trinitrate was shown to cause an increase in iNOS expression in an animal model used to study meningeal inflammation (Reuter et al., 2001). As seen in Fig. 5, overexpression of iNOS in primary cultures caused a greater than three-fold increase in CGRP promoter activity, a level similar to that observed with SNAP. These data provide evidence that nitric oxide released from nitric oxide donors or generated by iNOS activity can directly stimulate the synthesis of CGRP via activation of the CGRP promoter in trigeminal neurons.
Fig. 5.

Sumatriptan repression of nitric oxide-stimulated CGRP promoter activity. The 1250-bp CGRP promoter fragment contains proximal cAMP- and ras-responsive regions (black box) and a distal enhancer that contains both cell-specific (grey box) and noncell-specific (striped box) elements. Trigeminal cultures transfected with the CGRP-luciferase reporter or cotransfected with the iNOS expression vector were either untreated (CON) or treated for 2 h with 1 mm SNAP and reporter activity then measured. Unstimulated or iNOS-transfected cultures were incubated overnight in 10 μm sumatriptan (SUM) or pretreated with sumatriptan 30 min prior to addition of SNAP. Mean luciferase activity (normalized to β-galactosidase activities) per 20 lg protein with SEM from five experiments is shown.
To determine whether sumatriptan could repress nitric oxide stimulation of CGRP promoter activity as observed for CGRP secretion, trigeminal cultures were treated either overnight (basal and iNOS-treated) or 30 min prior to addition of SNAP. As shown in Fig. 5, treatment with 10 μm sumatriptan did not alter basal levels CGRP promoter activity but greatly repressed nitric oxide-stimulated promoter activity in response to SNAP or iNOS overexpression. These results are in agreement with our secretion data that showed sumatriptan repression of nitric oxide-stimulated CGRP release (Fig. 4C). Thus, sumatriptan can inhibit both nitric oxide-induced CGRP synthesis via repression of promoter activity as well as CGRP secretion.
Nitric oxide signaling involves activation of JNK and p38 MAP kinases
Having previously demonstrated that activity of the CGRP promoter is stimulated by MAP kinases (Durham & Russo, 1998, 2000), we wanted to determine if nitric oxide stimulation could activate MAP kinase pathways in cultured trigeminal neurons. Nitric oxide-induced MAP kinases were identified by immunocytochemistry using phospho-specific ERK, JNK and p38 antibodies that recognize only the dual phosphorylated (activated) proteins. Trigeminal ganglia cultures were treated for 30 min with vehicle, 1 mm SNAP or 0.6 m sorbitol (positive control). Sorbitol was used as a positive control as it has been reported to stimulate ERK, JNK, and p38 kinase activity strongly (Kayali et al., 2000). In the vehicle control cultures, cytoplasmic and nuclear levels of each of the MAP kinases were barely detectable (Fig. 6B, F and J). However, treatment of cultures with nitric oxide caused a large increase in the percentage of cells exhibiting nuclear staining for active JNK and p38 (Figs 6H and L, and 7). The percentage of JNK-positive cells was similar to that following treatment with sorbitol (> 90%), whereas the percentage of p38-positive neurons (∼60%) was lower than that for sorbitol (∼85%). By contrast, nitric oxide did not cause an increase in active ERK staining compared with control values (both ∼ 20%). As a control, cultures treated with sorbitol had a marked increase in the percentage of neurons with nuclear localization of active ERK (∼85%). In addition, we stained untreated and treated trigeminal cultures with antibodies that recognize both the phosphorylated as well as the unphosphorylated forms of ERK, JNK and p38. However, the level of cytoplasmic or nuclear staining for ERK, JNK and p38 total proteins in the nitric oxide treated cultures was not increased compared with untreated cultures (data not shown). Taken together, these data demonstrate that nitric oxide treatment leads to increased phosphorylation and nuclear localization of JNK and p38, but not ERK in trigeminal ganglion neurons.
Fig. 6.
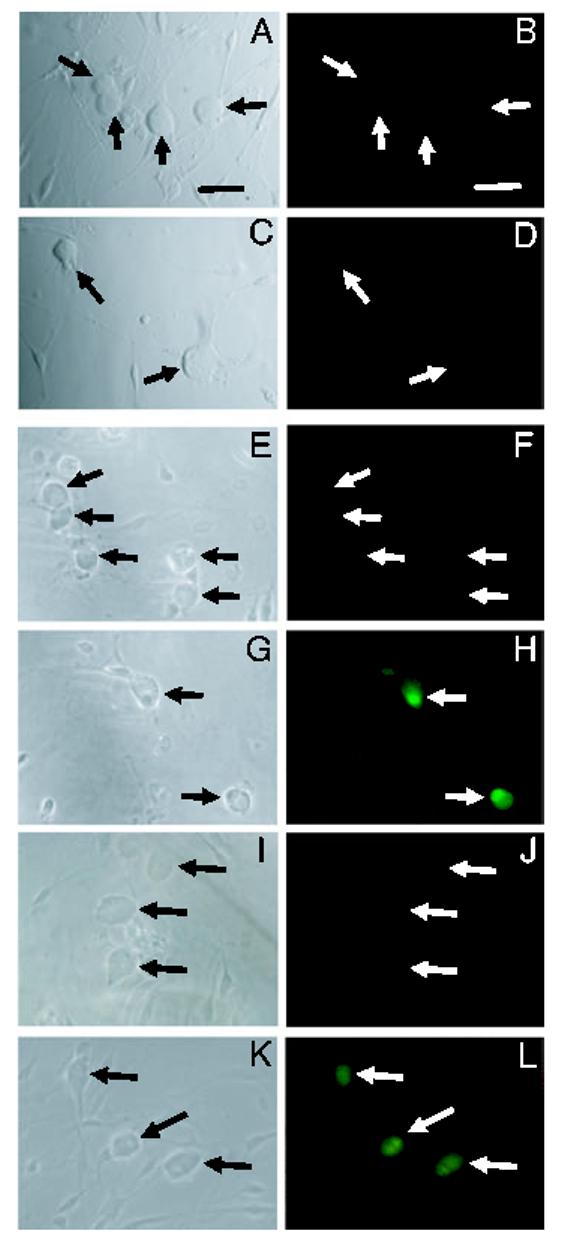
Nitric oxide stimulation of JNK and p38 MAP kinases. Trigeminal cultures were stained with antibodies directed against the active (phosphorylated) forms of ERK (A-D), JNK (E-H) or p38 (I-L). Unstimulated trigeminal cultures (A and B, E and F, I and J) or cultures treated with 1 mm SNAP (C and D, G and H, K and L) were stained with antibodies directed against the active (phosphorylated) forms of ERK (A-D), JNK (E-H) or p38 (I-L). Phase contrast microscopy (left panels) and fluorescence microscopy (right panels) photographs of the same culture are shown. Arrows indicate neuronal cell bodies. Scale bars, 50 μm.
Fig. 7.
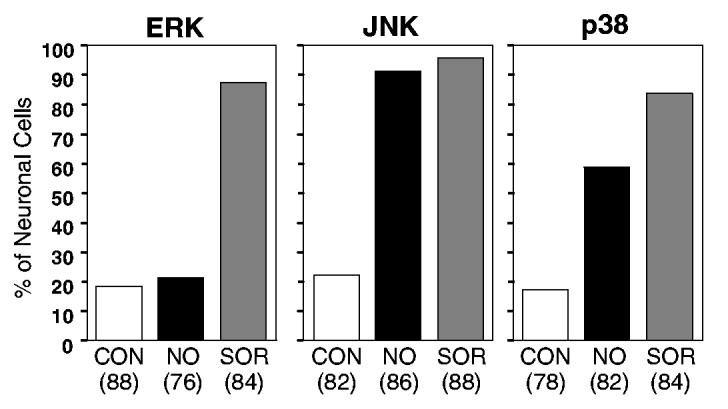
Percentage of trigeminal neurons expressing the active form of ERK, JNK or p38 in response to nitric oxide treatment. The number of neuronal cells in which nuclear staining was detected following sorbitol (SOR, positive control, grey bars) or nitric oxide (NO, black bars) treatment was compared with unstimulated cultures (CON, control, open bars). The total number of neuronal cells analysed is given in parentheses below the condition. Data from two independent experiments performed in duplicate for each condition are shown.
Discussion
In this study, primary cultures of rat trigeminal neurons were used to investigate the mechanisms by which nitric oxide regulates CGRP gene expression. We found that nitric oxide increased both the synthesis and the release of CGRP. The stimulatory effects of nitric oxide on CGRP promoter activity and secretion were repressed by the antimigraine drug sumatriptan. Unexpectedly, nitric oxide-mediated stimulation of CGRP release occurred independent of cGMP and PI3-kinase pathway activation, but appears to involve T-type calcium channels. In addition, the stimulatory effects of nitric oxide on CGRP promoter activity likely involve activation of the MAP kinases JNK and p38. Our results suggest that activation of T-type calcium channels and MAP kinase signal transduction cascades play important roles in controlling CGRP gene expression in trigeminal neurons and in mediating the pathological effects of nitric oxide.
Regulation by nitric oxide is particularly relevant to understanding the role of CGRP in migraine pathology as nitric oxide is thought to be instrumental in the pathogenesis of migraine (Bolay et al., 2002; Pietrobon & Striessnig, 2003). Migraine, a chronic disease with episodic occurrences, is one of the most underdiagnosed and undertreated of all neurological conditions (Ferrari, 1998). The wave of neuronal depolarization that spreads across the cerebral cortex causing release of protons, K+ ions and nitric oxide can sensitize and activate perivascular trigeminal nerves, leading to neurogenic inflammation within the meninges, central sensitization and allodynia. Based on results from clinical studies, nitric oxide is now thought to play an important role in the pathology of migraine and cluster headaches (Olesen & Jansen-Olesen, 2000). Intravenous infusions of glyceryl trinitrate, an exogenous nitric oxide donor, have been reported to produce a delayed headache in migraine and cluster headache patients that was indistinguishable from a spontaneous attack (Olesen et al., 1993; Iversen & Olesen, 1996; Bank, 2001). In addition, NOS inhibitors have been shown to reduce the severity of migraine headaches in spontaneous attacks (Olesen & Jansen-Olesen, 2000). Results from animal studies have provided evidence for the involvement of nitric oxide in sensitization and/or activation of the trigeminovascular system and release of CGRP from dural afferents (Messlinger et al., 2000; Strecker et al., 2002). However, the cellular mechanisms of the nitric oxide effects on CGRP were not investigated.
In this study, we demonstrated that nitric oxide stimulation of CGRP secretion was dependent on extracellular calcium and likely involves activation of T-type calcium channels, and does not require release of intracellular calcium from thapsigargin-sensitive pools. Our findings differ from data reported for secretion from striatal neurons in which nitric oxide caused increases in intracellular calcium via release from mitochondria (Horn et al., 2002). Interestingly, T-type (low-voltage activated) calcium channels are abundantly expressed by sensory neurons (Cardenas et al., 1995) and have been shown to play a role in nociceptive transmission and sensitization of spinal dorsal horn neurons in vivo (Todorovic et al., 2001). Furthermore, it has been shown that hyperalgesia in animals involves calcium-dependent signal transduction pathways (Kawamata & Omote, 1996). The finding that nitric oxide can stimulate CGRP secretion via activation of T-type calcium channels is particularly significant as T-type channels facilitate activity- and calcium-dependent long-term potentiation at synapses from nociceptive neurons (Ikeda et al., 2003). Thus, T-type channels may be performing a similar function in trigeminal neurons during migraine pathology. Based on our data, we have identified a novel nitric oxide signaling mechanism in sensory neurons that involves T-type calcium channels and likely occurs independent of the cGMP and PI3-kinase pathways.
Traditionally, nitric oxide signaling is reported to involve activation of guanylate cyclase and subsequent increases in intracellular cGMP levels that mediate the cellular response (Yun et al., 1996; Faraci & Heistad, 1998). However, most studies to date that have focused on the signaling cascades activated by nitric oxide in non-neuronal cells. In agreement with our findings, other investigators have also reported that nitric oxide-mediated stimulation of secretion can occur via cGMP-independent mechanisms (Pinilla et al., 1998, 1999; Garry et al., 2000).
The recent report that a nonpeptide CGRP receptor antagonist is effective as an acute treatment for migraine (Olesen et al., 2004) supports a critical role of CGRP in migraine pathology. Furthermore, results from a study by Juhasz et al. (2003) demonstrated that plasma CGRP levels correlated with the timing and severity of nitric oxide-induced migraine attacks. Although several studies have focused on nitric oxide stimulation of CGRP secretion (Messlinger et al., 2000; Strecker et al., 2002), the effects of nitric oxide on the CGRP promoter have not been investigated. In our study, we found that nitric oxide released from exogenous donors or generated by iNOS could stimulate CGRP promoter activity in trigeminal neurons. In agreement, CGRP mRNA was shown to be increased by nitric oxide derived from nitroglycerin in lumbar dorsal root ganglia neurons (Du et al., 2004). Our data demonstrate that CGRP synthesis and release can be stimulated by nitric oxide. This may be important in understanding how the pain of migraine can persist for up to 72 h if untreated, as an increase in CGRP synthesis would be necessary to sustain the elevated levels of CGRP. It will be of therapeutic interest to identify the nitric oxide-responsive regulatory sequences within the CGRP promoter and to determine the signaling pathways involved. Towards this end, we used immunocytochemistry to demonstrate that nitric oxide could induce the JNK and p38 MAP kinase pathways but not ERK in trigeminal neurons. We have previously shown that the CGRP gene is MAP kinase-responsive (Durham & Russo, 2003). Thus, the stimulatory effects of nitric oxide on CGRP promoter activity will likely involve activation of the JNK and p38 MAP kinase signal transduction cascades.
We have shown here that the antimigraine drug sumatriptan, which has affinity for 5-HT1B, 5-HT1D and 5-HT1F receptors, can repress both the synthesis and the release of CGRP from trigeminal neurons in response to nitric oxide treatment. These inhibitory effects likely involve activation of multiple 5-HT1 receptors as we found that 5-HT1B, 5-HT1D and 5-HT1F receptors were expressed on cultured trigeminal neurons. The rationale for the sumatriptan treatment was based on clinical evidence that sumatriptan decreased headaches induced by intravenous administration of nitroglycerin as a source of exogenous nitric oxide (Iversen & Olesen, 1996). Sumatriptan, the most-studied of the serotonergic drugs now known collectively as the triptans, has been shown to be an effective therapy for the acute treatment of migraine and other types of vascular headaches (Longmore et al., 1999). Based on our previous studies, we propose that the inhibitory effects of sumatriptan on nitric oxide-stimulated secretion and promoter activity are mediated by a sustained increase in intracellular calcium in response to 5-HT1 receptor activation and induction of specific phosphatases (Durham et al., 1997; Durham & Russo, 2003). In particular, we have shown that MAP kinase phosphatase-1 expression is increased in response to 5-HT1 receptor activation. The effectiveness of sumatriptan in aborting migraine attacks and preventing nitric oxide-induced headaches may be due in part to its ability to repress the production and release of CGRP from trigeminal neurons.
In conclusion, we have shown that CGRP promoter activity and as well as its release from trigeminal neurons is increased by nitric oxide. Our data suggest that nitric oxide induction of select MAP kinase pathways and T-type calcium channels are involved in coordinating CGRP synthesis and secretion. The antimigraine drug sumatriptan was found to suppress nitric oxide stimulation of CGRP promoter activity and release, presumably via activation of 5-HT1 receptors. Results from our study provide new insights into the cellular mechanisms by which nitric oxide, an inflammatory mediator implicated in migraine pathology, and sumatriptan, an effective antimigraine drug, function to regulate CGRP gene expression in trigeminal neurons.
Acknowledgements
We would like to thank Drs Donald D. Heistad and Frank Faraci for providing reagents and Ms T. Fielder for her technical assistance. This work was supported by NIH grants NS044033-01 and DE015385, and a grant from the National Headache Foundation to P.L.D. and NIH grants DE016511 and HL14388 to A.F.R.
Footnotes
- CGRP
- calcitonin gene-related peptide
- ERK
- extracellular signal-regulated kinases
- iNOS
- inducible nitric oxide synthase
- JNK
- Jun amino-terminal kinases
- L-NAME
- N-nitro-l-arginine methyl ester hydrochloride
- L-NMMA
- N-monomethyl-l-arginine acetate
- MAPK
- mitogen-activated protein kinases
- ODQ
- 1H-[1,2,4]oxadiazolo [4,3-a]quinoxalin-1-one
- PI3-kinase
- phophatidylinositol 3’-kinase
- SNAP
- S-nitroso-N-acetylpenicillamine
- SNP
- sodium nitroprusside.
References
- Bank J. Migraine with aura after administration of sublingual nitroglycerin tablets. Headache. 2001;41:84–87. doi: 10.1046/j.1526-4610.2001.111006084.x. [DOI] [PubMed] [Google Scholar]
- Bolay H, Reuter U, Dunn A, Huang Z, Boas D, Moskowitz M. Intrinsic brain activity trigger trigeminal meningeal afferents in a migraine model. Nat. Med. 2002;8:136–142. doi: 10.1038/nm0202-136. [DOI] [PubMed] [Google Scholar]
- de Bustros A, Baylin S, Levine M, Nelkin B. Cyclic AMP and phorbol esters separately induce growth inhibition, calcitonin secretion, and calcitonin gene transcription in cultured human medullary thyroid carcinoma. J. Biol. Chem. 1986;261:8036–8041. [PubMed] [Google Scholar]
- Buzzi M. Trigeminal pain pathway: peripheral and central activation as experimental models of migraine. Funct. Neurol. 2001;16:77–81. [PubMed] [Google Scholar]
- Buzzi M, Bonamini M, Moskowitz M. Neurogenic model of migraine. Cephalalgia. 1995;15:277–280. doi: 10.1046/j.1468-2982.1995.1504277.x. [DOI] [PubMed] [Google Scholar]
- Cardenas C, Del Mar L, Scroggs R. Variation in serotonergic inhibition of calcium channel currents in four types of rat sensory neurons differentiated by membrane properties. J. Neurophysiol. 1995;74:1870–1879. doi: 10.1152/jn.1995.74.5.1870. [DOI] [PubMed] [Google Scholar]
- Chang L, Karin M. Mammalian MAP kinase signaling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- Cohen P. The search for physiological substrates of MAP and SAP kinases in mammalian cells. Trends Cell Biol. 1997;7:353–361. doi: 10.1016/S0962-8924(97)01105-7. [DOI] [PubMed] [Google Scholar]
- Deora A, Win T, Vanhasesebroeck B, Lander H. A Redox-triggered Ras-effector interaction: recruitment of phosphtidylinositol 3’-kinase to Ras by redox stress. J. Biol. Chem. 1998;273:29923–29928. doi: 10.1074/jbc.273.45.29923. [DOI] [PubMed] [Google Scholar]
- Du Y, Peng J, Huang Z, Jiang D, Deng H, Li Y. Delayed cardioprotection afforded by nitroglycerin is mediated by alpha-CGRP via activation of inducible nitric oxide synthase. Int. J. Cardiol. 2004;93:49–54. doi: 10.1016/s0167-5273(03)00123-2. [DOI] [PubMed] [Google Scholar]
- Durham P, Dong P, Belasco K, Kasperski J, Gierasch W, Edvinsson L, Heistad D, Faraci F, Russo A. Neuronal expression and regulation of CGRP promoter activity following viral gene transfer into cultured trigeminal ganglia neurons. Brain Res. 2004;997:103–110. doi: 10.1016/j.brainres.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Durham P, Russo A. Serotonergic repression of mitogen-activated protein kinase control of the calcitonin gene-related peptide enhancer. Mol. Endocrinol. 1998;12:1002–1009. doi: 10.1210/mend.12.7.0135. [DOI] [PubMed] [Google Scholar]
- Durham P, Russo A. Regulation of calcitonin gene-related peptide secretion by a serotonergic antimigraine drug. J. Neurosci. 1999;19:3423–3429. doi: 10.1523/JNEUROSCI.19-09-03423.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham P, Russo A. Differential regulation of mitogen-activated protein kinase-responsive genes by the duration of a calcium signal. Mol. Endocrinol. 2000;14:1570–1582. doi: 10.1210/mend.14.10.0529. [DOI] [PubMed] [Google Scholar]
- Durham P, Russo A. Stimulation of the calcitonin gene-related peptide enhancer by mitogen-activated protein kinases and repression by an antimigraine drug in trigeminal ganglia neurons. J. Neurosci. 2003;23:807–815. doi: 10.1523/JNEUROSCI.23-03-00807.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham P, Sharma R, Russo A. Repression of the calcitonin gene-related peptide promoter by 5-HT1 receptor activation. J. Neurosci. 1997;17:9545–9553. doi: 10.1523/JNEUROSCI.17-24-09545.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvinsson L, Elsas T, Suzuki N, Shimizu T, Lee T. Origin and co-localization of nitric oxide synthase, CGRP, PACAP, and VIP in the cerebral circulation of the rat. Microsc. Res. Tech. 2001;53:221–228. doi: 10.1002/jemt.1086. [DOI] [PubMed] [Google Scholar]
- Edvinsson L, Mulder H, Goadsby P, Uddman R. Calcitonin gene-related peptide and nitric oxide in the trigeminal ganglion: cerebral vasodilation from trigeminal nerve stimulation involves mainly calcitonin gene-related peptide. J. Auton. Nerv. Syst. 1998;70:15–22. doi: 10.1016/s0165-1838(98)00033-2. [DOI] [PubMed] [Google Scholar]
- Fanciullacci M, Alessandri M, Figini M, Geppetti P, Michelacci S. Increase in plasma calcitonin gene-related peptide from the extracerebral circulation during nitroglycerin-induced cluster headache attack. Pain. 1995;60:119–123. doi: 10.1016/0304-3959(94)00097-X. [DOI] [PubMed] [Google Scholar]
- Faraci F, Heistad D. Regulation of the cerebral circulation: role of endothelium and potassium channels. Physiol. Rev. 1998;78:53–97. doi: 10.1152/physrev.1998.78.1.53. [DOI] [PubMed] [Google Scholar]
- Ferrari MD. Migraine. Lancet. 1998;351:1043–1051. doi: 10.1016/S0140-6736(97)11370-8. [DOI] [PubMed] [Google Scholar]
- Fox A, Nowycky M, Tsien R. Kinetic and pharmacological properties distinguishing three types of calcium currents in chick sensory neurons. J. Physiol. 1987;394:149–172. doi: 10.1113/jphysiol.1987.sp016864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garry M, Walton L, Davis M. Capsaicin-evoked release of immunoreactive calcitonin gene-related peptide from the spinal cord is mediated by nitric oxide by not by cyclic GMP. Brain Res. 2000;861:208–219. doi: 10.1016/s0006-8993(99)02448-8. [DOI] [PubMed] [Google Scholar]
- Goadsby P, Edvinsson L. Sumatriptan reverses the changes in calcitonin gene-related peptide seen in the headache phase of migraine. Cephalalgia. 1991;11:3–4. [Google Scholar]
- Goadsby P, Edvinsson L. The trigeminovascular system and migraine: studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann. Neurol. 1993;33:48–56. doi: 10.1002/ana.410330109. [DOI] [PubMed] [Google Scholar]
- Goadsby P, Edvinsson L. Human in vivo evidence for trigeminovascular activation in cluster headache. Neuropeptide changes and effects of acute attacks therapies. Brain. 1994;117:427–434. doi: 10.1093/brain/117.3.427. [DOI] [PubMed] [Google Scholar]
- Gunnett C, Lund D, Chu Y, Brooks R, II, Faraci F, Heistad D. NO-dependent vasorelaxation is impaired after gene transfer of inducible NO-synthase. Arterioscler. Thromb. Vasc. Biol. 2001;21:1281–1287. doi: 10.1161/hq0801.093509. [DOI] [PubMed] [Google Scholar]
- Hingtgen CM, Vasko MR. The phosphatase inhibitor, okadaic acid, increases peptide release from rat sensory neurons in culture. Neurosci. Lett. 1994;178:135–138. doi: 10.1016/0304-3940(94)90308-5. [DOI] [PubMed] [Google Scholar]
- Horn T, Wolf G, Duffy S, Weiss S, Keilhoff G, MacVicar B. Nitric oxide promotes intracellular calcium release from mitochandria in striatal neurons. FASEB J. 2002;16:1611–1622. doi: 10.1096/fj.02-0126com. [DOI] [PubMed] [Google Scholar]
- Hou M, Kanje M, Longmore J, Tajti J, Uddman R, Edvinsson L. 5-HT1B and 5-HT1D receptors in the human trigeminal ganglion: colocalization with calcitonin gene-related peptide, substance P and nitric oxide synthase. Brain Res. 2001;909:112–120. doi: 10.1016/s0006-8993(01)02645-2. [DOI] [PubMed] [Google Scholar]
- Ikeda H, Heinke B, Ruscheweyh R, Sandkuhler J. Synaptic plasticity in spinal lamina I projection neurons that mediate hyperalgesia. Science. 2003;299:1237–1240. doi: 10.1126/science.1080659. [DOI] [PubMed] [Google Scholar]
- Iversen H, Olesen J. Headache induced by a nitric oxide donor (nitroglycerin) responds to sumatriptan. A human model for development of migraine drugs. Cephalalgia. 1996;16:412–418. doi: 10.1046/j.1468-2982.1996.1606412.x. [DOI] [PubMed] [Google Scholar]
- Juhasz G, Zsombok T, Modos E, Olajos S, Jakab B, Nemeth J, Szolcsanyi J, Vitrai J, Bagdy G. NO-induced migraine attack: strong increase in plasma calcitonin gene-related peptide (CGRP) concentration and negative correlation with platelet serotonin release. Pain. 2003;106:461–470. doi: 10.1016/j.pain.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Kawamata M, Omote K. Involvement of increased excitatory amino acids and intracellular Ca2+ concentration in the spinal dorsal horn in an animal model of neuropathic pain. Pain. 1996;68:85–96. doi: 10.1016/S0304-3959(96)03222-8. [DOI] [PubMed] [Google Scholar]
- Kayali A, Austin D, Webster N. Stimulation of MAPK cascades by insulin and osmotic shock: lack of an involvement of p38 mitogen-activated protein kinase in glucose transport in 3T3-L1 adipocytes. Diabetes. 2000;49:1783–1793. doi: 10.2337/diabetes.49.11.1783. [DOI] [PubMed] [Google Scholar]
- Lanigan T, Russo A. Binding of upstream stimulatory factor and a cell-specific activator to the calcitonin/calcitonin gene-related peptide enhancer. J. Biol. Chem. 1997;272:18316–18324. doi: 10.1074/jbc.272.29.18316. [DOI] [PubMed] [Google Scholar]
- Longmore J, Dowson A, Hill R. Advances in migraine therapy-5-HT receptor subtype-specific agonist drugs. Curr. Opinion CPNS Invest. Drugs. 1999;1:39–53. [Google Scholar]
- Ma Q. Co-localization of 5-HT (1B/1D/1F) receptors and glutamate in trigeminal ganglia in rats. Neuroreport. 2001;12:1589–1591. doi: 10.1097/00001756-200106130-00015. [DOI] [PubMed] [Google Scholar]
- Ma Q, Hill R, Sirinathsinghji D. Colocalization of CGRP with 5-HT1B/1D receptors and substance P in trigeminal ganglion neurons in rats. Eur. J Neurosci. 2001;13:2099–2104. doi: 10.1046/j.0953-816x.2001.01586.x. [DOI] [PubMed] [Google Scholar]
- McCulloch J, Uddman R, Kingman T, Edvinsson L. Calcitonin gene-related peptide: functional role in cerebrovascular regulation. Proc. Natl Acad. Sci. USA. 1986;83:5731–5735. doi: 10.1073/pnas.83.15.5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messlinger K, Suzuki A, Pawlak M, Zehnter A, Schmidt R. Involvement of nitric oxide in the modulation of dural arterial blood flow in the rat. Br. J. Pharmacol. 2000;129:1397–1404. doi: 10.1038/sj.bjp.0703220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesen J, Diener H, Husstedt I, Goadsby P, Hall D, Meier U, Pollentier S, Lesko L. Calcitonin gene-related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. N. Eng J. Med. 2004;350:1104–1110. doi: 10.1056/NEJMoa030505. [DOI] [PubMed] [Google Scholar]
- Olesen J, Iversen H, Thomsen L. Nitric oxide supersensitivity: a possible molecular mechanism of migraine pain. Neuroreport. 1993;4:1027–1030. doi: 10.1097/00001756-199308000-00008. [DOI] [PubMed] [Google Scholar]
- Olesen J, Jansen-Olesen I. Nitric oxide mechanisms in migraine. Pathol. Biol. (Paris) 2000;48:648–657. [PubMed] [Google Scholar]
- Pietrobon D, Striessnig J. Neurobiology of migraine. Nat. Rev. Neurosci. 2003;4:386–398. doi: 10.1038/nrn1102. [DOI] [PubMed] [Google Scholar]
- Pinilla L, Gonzalez D, Tena-Sempere M, Aguilar E. Nitric oxide (NO) stimulates gonadotropin secretion in vitro through a calcium-dependent, cGMP-independent mechanism. Neuroendocrinology. 1998;68:180–186. doi: 10.1159/000054364. [DOI] [PubMed] [Google Scholar]
- Pinilla L, Tena-Sempere M, Aguilar E. Nitric oxide stimulates growth hormone secretion in vitro through a calcium- and cyclic guanosine monophosphate-independent mechanism. Horm. Res. 1999;51:242–247. doi: 10.1159/000023378. [DOI] [PubMed] [Google Scholar]
- Reuter U, Bolay H, Jansen-Olesen I, Chiarugi A, Sanchez del Rio M, Letourneau R, Theoharides T, Waeber C, Moskowitz M. Delayed inflammation in rat meninges: implications for migraine pathophysiology. Brain. 2001;124:2490–2502. doi: 10.1093/brain/124.12.2490. [DOI] [PubMed] [Google Scholar]
- van Rossum D, Hanisch U, Quirion R. Neuroanatomical localization, pharmacological characterization and functions of CGRP, related peptides and their receptors. Neurosci. Biobehav. Rev. 1997;21:649–678. doi: 10.1016/s0149-7634(96)00023-1. [DOI] [PubMed] [Google Scholar]
- Schaeffer H, Weber M. Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol. Cell Biol. 1999;19:2435–2444. doi: 10.1128/mcb.19.4.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seger R, Krebs E. The MAPK signaling cascade. FASEB J. 1995;9:726–735. [PubMed] [Google Scholar]
- Southam E, Garthwaite J. The nitric oxide-cyclic GMP signaling system in rat brain. Neuropharmacology. 1993;32:1267–1277. doi: 10.1016/0028-3908(93)90021-t. [DOI] [PubMed] [Google Scholar]
- Stewart W, Schechter A, Rasmussen B. Migraine heterogeneity: disability, pain, intensity, and attack frequency and duration. Neurology. 1994;44:S24–S39. [PubMed] [Google Scholar]
- Strecker T, Dux M, Messlinger K. Nitric oxide releases calcitonin-gene related peptide from rat dura mater encephali promoting increases in meningeal blood flow. J. Vasc. Res. 2002;39:489–496. doi: 10.1159/000067206. [DOI] [PubMed] [Google Scholar]
- Thiagalingam A, deBustros A, Borges M, Jasti R, Compton D, Diamond L, Mabry M, Ball D, Baylin S, Nelkin B. RREB-1, a novel zinc finger protein, is involved in the differentiation response to ras in human medullary thyroid carcinomas. Mol. Cell Biol. 1996;16:5335–5345. doi: 10.1128/mcb.16.10.5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorovic S, Jevtovic-Todorovic V, Meyenburg A, Mennerick S, Perez-Reyes E, Romano C, Olney J, Zorumski C. Redox modulation of T-type calcium channels in rat peripheral nociceptors. Neuron. 2001;31:75–85. doi: 10.1016/s0896-6273(01)00338-5. [DOI] [PubMed] [Google Scholar]
- Tverberg L, Russo A. Regulation of the calcitonin/calcitonin gene-related peptide gene by cell-specific synergy between helix-loop-helix and octamer-binding transcription factors. J. Biol. Chem. 1993;268:15965–15973. [PubMed] [Google Scholar]
- Vasko MR, Campbell WB, Waite KJ. Prostaglandin E2 enhances bradykinin-stimulated release of neuropeptides from rat sensory neurons in culture. J. Neurosci. 1994;14:4987–4997. doi: 10.1523/JNEUROSCI.14-08-04987.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson D, Hargreaves R. Neurogenic inflammation in the context of migraine. Microsc. Res. Tech. 2001;53:167–178. doi: 10.1002/jemt.1081. [DOI] [PubMed] [Google Scholar]
- Yun H-Y, Dawson V, Dawson T. Neurobiology of nitric oxide. Crit. Rev. Neurobiol. 1996;10:291–316. doi: 10.1615/critrevneurobiol.v10.i3-4.20. [DOI] [PubMed] [Google Scholar]