

Abstract
Objective: To explore the association between APOE*4 and pathologically confirmed cases of the Lewy body (LB) variant of Alzheimer disease (AD). Methods: With use of α-synuclein (AS) immunohistochemistry, LBs were detected in 74 of 131 (56.5%) of the AD + LB cases; the remaining 57 cases (43.5%) did not have LBs. Results: There were no differences in gender or age between Caucasian subjects with AD + LB or AD alone or control subjects. The APOE*4 allele frequency was highest in the AD + LB group (47.3%; 95% CI = 37.8 to 57.0%), intermediate in the AD-alone group (35.1%; 95% CI = 25.3 to 46.3%), and lowest in the control group (14.2%; 95% CI = 10.5 to 18.9%). With use of logistic regression analysis, the odds of having AD + LB vs AD alone were 2.1-fold (95% CI = 1.0 to 4.5, p = 0.055) greater in persons with an APOE*4 allele than in those without an APOE*4 allele. Conclusion: The APOE*4 allele is associated with the presence of concomitant Lewy bodies in Alzheimer disease.
There is a strong association between APOE*4 and Alzheimer disease (AD), but the role of APOE in the susceptibility to develop the Lewy body (LB) variant of AD (AD + LB) is uncertain. Studies that investigate APOE*4 as a risk factor for AD + LB have had conflicting results. Some studies suggest that APOE*4 allele frequency is similar in AD + LB and in AD alone.1,2 Others report that the APOE*4 allele frequency in AD + LB either falls between that of AD-alone cases and normal control subjects3,4 or is higher than that of AD-alone cases.5 Conversely, another recent study reported an increased frequency of the APOE*3/*4 genotype only in males with AD + LB.6 Interestingly, the APOE*4 allele is associated with increased neocortical LBs in patients with Parkinson disease (PD) with dementia.7,8
Unfortunately, because of the varying clinical and neuropathologic classification of AD + LB and PD with dementia, comparisons between studies are difficult. Furthermore, none of the published studies investigating the association between APOE and AD + LB has used the most sensitive methods for LB detection (i.e., α-synuclein [AS] immunostaining and amygdala sampling). With use of these more sensitive methods of LB detection, the current study investigates the relationship between the APOE*4 allele and LBs in neuropathologic AD.
Methods
Study population
All assessments were conducted after obtaining written informed consent from subjects according to procedures approved by the University of Pittsburgh Institutional Review Board. Only subjects with APOE genotype available from a blood sample or postmortem tissue were considered for inclusion in the study.
The study consists of 133 patients who were clinically diagnosed as having “probable AD” using the National Institute of Neurological and Communication Disorders and Stroke/Alzheimer's Disease and Related Disorders Association (NINCDS-ADRDA) criteria9 at the Pittsburgh Alzheimer's Disease Research Center from 1995 to 2001. Brains were examined as previously described.10 All 133 patients met the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) neuropathologic criteria for the diagnosis of “definite AD.”11 One hundred thirty-one cases were classified as Braak stage III or above, thus fulfilling the National Institute on Aging and Reagan Institute (NIA-RI) criteria for “intermediate or high” likelihood that dementia is due to AD. Ninety-six cases were classified as Braak stage V or VI, thus fulfilling the NIA-RI criteria for “high” likelihood that dementia is due to AD.
The control group included 197 participants in the Monogahela Valley Independent Elders Survey (MoVIES). MoVIES was a prospective community study of cognitive impairment and dementia in the semirural mid-Monongahela Valley region of southwestern Pennsylvania. Sampling, recruitment, and clinical dementia assessment for MoVIES have been described previously.12 The 197 control subjects provided blood samples for APOE genotyping between 1994 and 1997 and were not demented on the date that blood was drawn.
We excluded non-Caucasian subjects from both the case and the control groups because the association between APOE genotyping and AD differs across ethnic groups.13 We did not have sufficient non-Caucasian subjects to analyze results from these ethnic groups separately.
Clinical assessments
Each subject underwent an extensive neuropsychiatric evaluation. This assessment included a medical history and physical examination, a neurologic history and examination, and behavioral and neuropsychological evaluations. Subjects with objective evidence of progressive alterations in functional status secondary to cognitive deficits were diagnosed with dementia. The diagnosis of dementia also required that, in the absence of reversible causes of cognitive impairment, neuropsychological testing reveal impairments in two or more cognitive domains. Once a subject was diagnosed as having dementia, we applied the NINCDS-ADRDA criteria for AD and excluded subjects with other potential causes of dementia. At least two experienced clinicians agreed on a consensus diagnosis for each subject.
Neuropathologic assessments
The pathologic diagnoses for this study were based on an examination of the following brain areas: frontal cortex, anterior cingulate gyrus, insular cortex, amygdala, hippocampus, entorhinal and trans-entorhinal cortices, parietal cortex, superior and middle temporal cortices, thalamus, caudate nucleus, putamen, globus pallidus, substantia nigra, and locus ceruleus. Each case was examined for the presence of senile plaques and neurofibrillary tangles using modified Bielschowsky stain. The neuropathologic diagnosis of AD was based on CERAD and NIA-RI neuropathologic criteria.11,14
AS immunostaining with protease pretreatment
The paraffin blocks were cut at 6 μm. Our study identified LBs using monoclonal antibodies against AS.10 The deparaffinized sections of the appropriate regions were digested for 1 minute with a 100 mg of type XXIV protease (Sigma-Tau Pharmaceutical, Gaithersburg, MD) in 300 mL of distilled water at 37 °C; next, the primary antibody was diluted in a common antibody dilutor (1:1,200 for LB509; Zymed, San Francisco, CA). The staining was visualized using a commercially available kit (LSAB-2 kit; Dako, Carpinteria, CA) containing diaminobenzidine. Then the tissue was counterstained with Mayer hematoxylin. All round, homogeneously AS-positive structures were considered LBs, including intracytoplasmatic, intraneuritic, and extracellular LBs.
AS immunostains were performed on all paraffin blocks. As previously reported,10 all AD + LB cases had LBs in the amygdala, but in some cases, there were no LBs detected in other areas of the brain or brainstem.10 We evaluated the extent of LB formation using a semiquantitative LB scoring system (LB score) based on the International Consensus Criteria for the diagnosis of dementia with LBs (DLB).15 Our assessments included a determination of the presence or absence of LBs and an assessment of the severity (semiquantitative) of LB pathology in paraffin blocks from the following regions: frontal, parietal, temporal, anterior cingulate gyrus, and trans-entorhinal cortices.16 We then counted LBs in a designated area of the AS-stained section.15 Each area was given a score of 0, 1, or 2. A score of 0 corresponded to an absence of LBs, a score of 1 to the presence of one to five LBs, and a score of 2 to more than five LBs in the area of evaluation as specified by the International Consensus Criteria for the diagnosis of DLB.15 Scores from these five regions were then summed. Cases without LBs were assigned a score of “negative,” whereas a total LB score of 0 indicated the presence of minimal neocortical LBs (with LBs restricted to the amygdala), and a total LB score of “10” indicated the presence of widespread neocortical LBs. As LB pathology in the amygdala was not specifically addressed in the International Consensus Criteria, these cases were grouped with the brainstem-predominant cases. Cases with a total LB score of 0 were classified as brainstem/amygdala, cases with a total LB score of 1 to 6 were classified as limbic, and cases with a total LB score of 7 to 10 were classified as neocortical.
APOE genotyping
DNA was isolated with the QIAmp kit from Qiagen (Chatsworth, CA). Genotyping was performed using an established PCR protocol that was described previously.17
Statistical analysis
Neuropathologic AD cases were divided into two categories: AD alone and AD + LB. The latter cases were defined by the presence of any number of AS-immunoreactive LBs in any neocortical, amygdala, or brainstem section. Subsequent analyses examined the relationship of the APOE genotypes to our semiquantitative measure of cortical LB density. Continuous variables were compared using t tests for two-group comparisons and analysis of variance for three-group comparisons. Categorical variables were compared using χ2 statistics. APOE allele frequencies were calculated by the allele-counting method. The relationship between the APOE*4 allele frequency and the presence of concomitant LB pathology was explored using logistic regression analyses. Effect modification was explored by stratifying by gender. This decision was influenced by the results of previous studies, which observed a stronger APOE*4–AD association in women than men.13 Separate analyses were conducted for all cases with Braak stage IIIC or higher (intermediate or high pathologic criteria for AD) and cases with only Braak stage VC or higher (high pathologic criteria for AD).
Results
Classification of AD and AD + LB
After applying intermediate or high pathologic criteria for AD criteria (Braak stage IIIC or higher), the study sample consisted of 131 Caucasian cases of clinically diagnosed and neuropathologically confirmed AD and 197 nondemented Caucasian control subjects. All 131 AD cases satisfied NIA-RI neuropathologic criteria for having either an intermediate or a high likelihood that dementia was due to AD. AS immunohistochemistry detected LBs in 74 of 131 (56.5%) of the cases (AD + LB), whereas 57 cases (43.5%) did not have LBs (AD alone).
Clinical and demographic characteristics
Table 1 shows both the demographic characteristics of the control subjects and patients with AD (AD alone and AD + LB) and the clinical characteristics of the AD cases. The proportion of men and women and the age at study enrollment were similar in all three groups. Furthermore, comparison between cases with AD (without and without LBs) showed similar education level, age at onset, age at death, and duration of illness.
Table 1.
Demographic and clinical characteristics of control, AD-alone, and AD + LB cases
| Controls, n = 197 | AD alone, n = 57 | AD + LB, n = 74 | Test statistic, df | p Value | |
|---|---|---|---|---|---|
| Sex | |||||
| Female | |||||
| n | 104 | 38 | 42 | χ2 (2) = 3.47 | 0.18 |
| % | 52.8 | 66.7 | 56.8 | ||
| Male | |||||
| n | 93 | 19 | 32 | ||
| % | 47.2 | 33.3 | 43.2 | ||
| Age at entry, mean ± SD; y | 73.1 ± 5.0 | 72.4 ± 9.8 | 72.6 ± 7.4 | ANOVA F = 0.40 | 0.67 |
| Age at onset, mean ± SD; y | N/A | 67.7 ± 10.7 | 68.2 ± 7.8 | t = −0.33 | 0.74 |
| Age at death, mean ± SD; y | N/A | 79.2 ± 9.5 | 79.2 ± 6.9 | t = 0 | 0.99 |
| Duration, mean ± SD; y | N/A | 11.5 ± 5.0 | 11.0 ± 4.5 | t = 0.64 | 0.52 |
| Education | |||||
| <HS | |||||
| n | — | 12 | 21 | χ2 (2) = 1.56 | 0.46 |
| % | — | 21.1 | 28.4 | ||
| HS | |||||
| n | — | 21 | 29 | ||
| % | — | 36.8 | 39.2 | ||
| >HS | |||||
| n | — | 24 | 24 | ||
| % | — | 42.1 | 32.4 |
AD = Alzheimer disease; LB = Lewy body; ANOVA = analysis of variance; HS = high school.
These results did not change when the analyses were repeated on cases with only high pathologic criteria for AD. Therefore, subsequent analyses include all cases with intermediate or high pathologic criteria for AD.
APOE allele frequencies
The APOE allele frequencies (see table 2) within the control group, group with AD alone, and group with AD + LB were each in Hardy–Weinberg equilibrium (p > 0.9 all groups). The distributions of APOE allele frequency demonstrated that APOE*4 allele frequencies were highest in the AD + LB group (47.3%; 95% CI = 37.8 to 57.0%), intermediate in the AD-alone group (35.1%; 95% CI = 25.3 to 46.3%), and lowest in the control group (14.2%; 95% CI = 10.5 to 18.9%).
Table 2.
APOE genotype and allele frequencies of control, AD-alone, and AD + LB cases: APOE genotype distribution* and APOE allele frequencies†
| Controls, n = 197 | AD alone, n = 57 | AD + LB, n = 74 | |
|---|---|---|---|
| APOE genotype | |||
| 22 | |||
| n | 0 | 0 | 0 |
| % | — | — | — |
| 23 | |||
| n | 26 | 1 | 1 |
| % | 13.2 | 1.8 | 1.4 |
| 24 | |||
| n | 3 | 1 | 2 |
| % | 1.5 | 1.8 | 2.7 |
| 33 | |||
| n | 118 | 21 | 16 |
| % | 59.9 | 36.8 | 21.6 |
| 34 | |||
| n | 47 | 29 | 42 |
| % | 23.9 | 50.9 | 56.8 |
| 44 | |||
| n | 3 | 5 | 13 |
| % | 1.5 | 8.8 | 17.6 |
| APOE allele frequency | |||
| 2 | |||
| n | 29 | 2 | 3 |
| % | 7.4 | 1.8 | 2.0 |
| 3 | |||
| n | 309 | 72 | 75 |
| % | 78.4 | 63.2 | 50.7 |
| 4 | |||
| n | 56 | 40 | 70 |
| % | 14.2 | 35.1 | 47.3 |
Comparisons of APOE genotypes between all three groups: overall χ2 = 73.91, df = 8, p < 0.001; comparison between AD alone vs AD + LB: χ2 = 4.68, df = 3, p = 0.20 (APOE*2 individuals within each group were collapsed into one cell owing to small numbers).
Comparisons of APOE*4 allele frequencies between AD alone vs AD + LB: χ2 = 3.94, df = 1, p = 0.05; AD + LB vs controls: χ2 = 66, df = 1, p < 0.0001.
AD = Alzheimer disease; LB = Lewy body.
The APOE*4 allele frequency in the AD + LB cases was higher than that found in AD-alone cases (χ2 = 3.94, df = 1, p = 0.05) as well as that found in control subjects (χ2 =66, df = 1, p < 0.001).
Logistic regression analyses
Logistic regression analysis showed that the odds of having AD + LB vs being a control were 9.1-fold (95% CI = 4.9 to 17.0, p < 0.0001) greater in persons with an APOE*4 allele than in persons without an APOE*4 allele. The odds of having AD + LB vs AD alone were 2.1-fold (95% CI = 1.0 to 4.5, p = 0.055) greater in persons with an APOE*4 allele than in those without an APOE*4 allele.
In exploratory analyses, the odds of having AD + LB vs AD alone were 2.8-fold (95% CI = 0.9 to 9.3, p = 0.08) greater in men with an APOE*4 allele than in men without an APOE*4 allele. In women, the odds were 2.0-fold (95% CI = 0.7 to 5.5, p = 0.2) greater in persons with an APOE*4 allele than in those without an APOE*4 allele. Although neither odds ratio was significant, there was a trend toward significance in the men.
APOE by age at onset
Within both AD groups (with and without LBs), individuals with one or two APOE*4 alleles had a younger age at onset than those with no APOE*4 alleles, but these differences were not significant.
APOE allele frequencies by total LB score
The extent of LB formation in AD + LB cases ranged from those with numerous neocortical and nigral LBs to cases with LBs only in the amygdala. Of the AD + LB cases, 31 of 74 (41.9%) received total LB scores of 7 to 10, whereas the remaining cases (43/74 = 58.1%) received total LB scores that were <7 (figure). With use of our modified consensus neuropathologic classifications (where brainstem/amygdala predominant was defined as total LB score of 0, limbic was defined as a total LB score of 1 to 6, and neocortical was defined as a total LB score of 7 to 10), AD + LB cases were further divided according to the degree of neocortical AS-immunoreactive LB accumulation. There was no significant association between APOE*4 allele frequency and extent of neocortical LB involvement.
Figure.
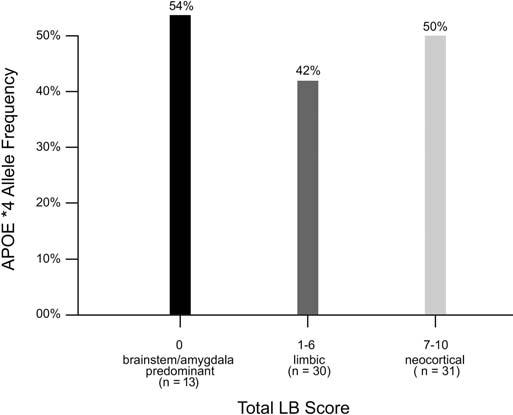
Distribution of APOE*4 allele frequencies (%) by total Lewy body (LB) score classifications in cases with Alzheimer disease + LBs. The x axis indicates the number of subjects in the three total LB score categories (brainstem/amygdala predominant, limbic, and neocortical), and the y axis indicates APOE*4 allele frequencies.
Braak stage by APOE genotype
Comparison of Braak stage by APOE genotype demonstrated that the APOE*4 allele frequencies were lower in AD-alone cases with Braak stages III or IV (25%) than in AD-alone cases with Braak stages V and VI (38%). However, there was no clear association between Braak stages and APOE allele frequencies in cases with AD + LB. In fact, AD + LB cases with Braak stages III or IV had similar APOE*4 allele frequencies to those with Braak stages V or VI (45 vs 48%).
Discussion
We confirm that there is a genetic association between the APOE*4 allele and LBs in AD. Four prior studies reported an overrepresentation of APOE*4 alleles in cases with AD + LB.2,6,18 We also confirmed reports that men with APOE*4 have higher odds of having AD/LB than men without an APOE*4 allele.6 However, the reasons accounting for these gender differences remain unclear.
Fifty-six percent of cases of neuropathologic AD in our sample had concomitant LB pathology. Other studies have reported a prevalence of DLB in sporadic AD cases that ranges from 7 to 30%. However, they did not use systematic AS immunohistochemistry for LB detection.19-25 Two studies that also used the LB509 antibody reported that approximately 50% of cases with familial AD and Down syndrome with concomitant AD have coexistent LBs.26,27 Therefore, we anticipate that our observed LB prevalence is comparable with that observed in other AD research centers.
The presence of an APOE*4 allele may accelerate the underlying pathologic process of both AD and AD + LB. Although there was no significant association between APOE*4 allele frequency and the extent of neocortical LBs, this association needs to be explored in larger autopsy samples.
The APOE*4 allele also appears to be associated with PD. Although previous studies have inconsistently reported this association, a recent large family-based association study showed that, independently of cognitive impairment, the APOE*4 allele is associated with both an increased risk of PD and a decreased age at onset of PD.28 However, the mechanism by which an APOE*4 allele may influence the development of LBs in AD and PD remains unclear.
AD and PD both involve the accumulation of insoluble protein deposits: β-amyloid and tau in AD and AS in PD. Recently, several studies have suggested that the pathologic cascades that lead to the accumulation of these different proteins may sometimes overlap or operate synergistically.29 A study utilizing a doubly transgenic (tg) mouse model showed that mice expressing both human α-synuclein (hSYN) and human β-amyloid derived from amyloid precursor protein (hAPP) developed earlier motor deficits and more severe learning deficits than singly tg hAPP mice.30 Doubly tg hSYN/hAPP mice also developed fibrillar intraneuronal inclusions of synuclein more similar to humans LBs than the amorphous accumulations of synuclein found in singly tg hSYN mice. Therefore, the development of one insoluble protein aggregate may precipitate the development of another protein aggregate. The presence of an APOE*4 allele may facilitate the development of these insoluble proteins.
On the other hand, APOE*4 may play a direct role in neurofibrillary tangle formation.31 It is plausible that an APOE*4 allele influences the formation of intraneuronal protein aggregation. APOE*4 may stress the brain by causing synaptic disruption.32 In response, the brain may attempt to restore neuronal integrity by increasing the production of tau or AS. Alternatively, the degradation process may decrease, resulting in protein accumulation and aggregation. Despite such hypotheses, the role of APOE*4 in the development of LBs remains unclear. Therefore, additional investigations focused on the pathogenesis of intraneuronal protein aggregation in AD and AD + LB are necessary.
Acknowledgment
The authors thank Mary Ganguli, MD, MPH (professor of psychiatry and epidemiology, Division of Geriatrics and Neuropsychiatry, Department of Psychiatry, University of Pittsburgh School of Medicine, and Department of Epidemiology, University of Pittsburgh Graduate School of Public Health; Dr. Ganguli is supported by NIA AG07562), for contribution of the MoVIES data; Thomas Montine, MD, PhD (Alvord Professor, Department of Pathology, University of Washington), for suggestions regarding the manuscript; Andrew David, BA (Department of Psychiatry and Behavioral Sciences, University of Washington), for editorial assistance; and Kate Simpson, MPH (Department of Psychiatry and Behavioral Sciences, University of Washington), for statistical assistance.
Footnotes
Supported in part by research grant AG05133 from the National Institute on Aging and the Alzheimer's Disease Research Center at the University of Pittsburgh (R.L.H., S.T.D., O.L.L., E.K.L.-Z., M.I.K.), by National Institute of Neurological Disorders and Stroke grant RO1 NS48595 (R.L.H., D.W.T., J.B.L.), by National Institute on Aging grant AG13672 (M.I.K., E.K.L.-Z.), and the Veteran Affairs VISN-20 Mental Illness Research, Education, and Clinical Center and Parkinson's Disease Research, Education, and Clinical Center (D.W.T., J.B.L.).
References
- 1.Gearing M, Schneider J, Rebeck G, Hyman B, Mirra S. Alzheimer's disease with and without coexisting Parkinson's disease changes: apolipoprotein E genotype and neuropathologic correlates. Neurology. 1995;45:1985–1990. doi: 10.1212/wnl.45.11.1985. [DOI] [PubMed] [Google Scholar]
- 2.Galasko D, Saitoh T, Xia Y, et al. The apolipoprotein E allele epsilon 4 is overrepresented in patients with the Lewy body variant of Alzheimer's disease. Neurology. 1994;44:1950–1951. doi: 10.1212/wnl.44.10.1950. [DOI] [PubMed] [Google Scholar]
- 3.Hardy J, Crook R, Prihar G, Roberts G, Raghavan R, Perry R. Senile dementia of the Lewy body type has an apolipoprotein E epsilon 4 allele frequency intermediate between controls and Alzheimer's disease. Neurosci Lett. 1994;182:1–2. doi: 10.1016/0304-3940(94)90190-2. [DOI] [PubMed] [Google Scholar]
- 4.Lamb H, Christie J, Singleton AB, et al. Apolipoprotein E and alpha-1 antichymotrypsin polymorphism genotyping in Alzheimer's disease and in dementia with Lewy bodies. Distinctions between diseases. Neurology. 1998;50:388–391. doi: 10.1212/wnl.50.2.388. [DOI] [PubMed] [Google Scholar]
- 5.Martinoli MG, Trojanowski JQ, Schmidt ML, et al. Association of apolipoprotein epsilon 4 allele and neuropathologic findings in patients with dementia. Acta Neuropathol (Berl) 1995;90:239–243. doi: 10.1007/BF00296506. [DOI] [PubMed] [Google Scholar]
- 6.Rosenberg CK, Cummings TJ, Saunders AM, Widico C, McIntyre LM, Hulette CM. Dementia with Lewy bodies and Alzheimer's disease. Acta Neuropathol (Berl) 2001;102:621–626. doi: 10.1007/s004010100415. [DOI] [PubMed] [Google Scholar]
- 7.Mattila P, Koskela T, Roytta M, et al. Apolipoprotein E epsilon4 allele frequency is increased in Parkinson's disease only with co-existing Alzheimer pathology. Acta Neuropathol (Berl) 1998;96:417–420. doi: 10.1007/s004010050913. [DOI] [PubMed] [Google Scholar]
- 8.Wakabayashi K, Kakita A, Hayashi S, et al. Apolipoprotein E epsilon4 allele and progression of cortical Lewy body pathology in Parkinson's disease. Acta Neuropathol (Berl) 1998;95:450–454. doi: 10.1007/s004010050824. [DOI] [PubMed] [Google Scholar]
- 9.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan E. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 10.Hamilton RL. Lewy bodies in Alzheimer's disease: a neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol. 2000;10:378–384. doi: 10.1111/j.1750-3639.2000.tb00269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mirra S, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 12.Ganguli M, Belle S, Ratcliff G, et al. Sensitivity and specificity for dementia of population-based criteria for cognitive impairment: the MoVIES project. J Gerontol. 1993;48:M152–M161. doi: 10.1093/geronj/48.4.m152. [DOI] [PubMed] [Google Scholar]
- 13.Farrer L, Cupples L, Haines J, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997;278:1349–1356. [PubMed] [Google Scholar]
- 14.National Institute on Aging and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's D Consensus Recommendations for the Postmortem Diagnosis of Alzheimer's Disease. Neurobiol Aging. 1997;18:S1–S2. [PubMed] [Google Scholar]
- 15.McKeith I, Galasko D, Kosaka K, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the Consortium on DLB International Workshop. Neurology. 1996;47:1113–1124. doi: 10.1212/wnl.47.5.1113. [DOI] [PubMed] [Google Scholar]
- 16.Wilson R, Luedecking-Zimmer E, Kamboh M, et al. Association of the ApoE4 allele with Lewy bodies in Alzheimer's disease. J Neuropathol Exp Neurol. 2002;61:455. [Google Scholar]
- 17.Kamboh MI, Aston CE, Hamman RF. The relationship of APOE polymorphism and cholesterol levels in normoglycemic and diabetic subjects in a biethnic population from the San Luis Valley, Colorado. Atherosclerosis. 1995;112:145–159. doi: 10.1016/0021-9150(94)05409-c. [DOI] [PubMed] [Google Scholar]
- 18.Harrington CR, Louwagie J, Rossau R, et al. Influence of apolipoprotein E genotype on senile dementia of the Alzheimer and Lewy body types. Significance for etiological theories of Alzheimer's disease. Am J Pathol. 1994;145:1472–1484. [PMC free article] [PubMed] [Google Scholar]
- 19.Leverenz J, Sumi S. Parkinson's disease in patients with Alzheimer's disease. Arch Neurol. 1986;43:662–664. doi: 10.1001/archneur.1986.00520070020010. [DOI] [PubMed] [Google Scholar]
- 20.Victoroff J, Mack WJ, Lyness SA, Chui HC. Multicenter clinicopathological correlation in dementia. Am J Psychiatry. 1995;152:1476–1484. doi: 10.1176/ajp.152.10.1476. [DOI] [PubMed] [Google Scholar]
- 21.Ince PG, McArthur FK, Bjertness E, Torvik A, Candy JM, Edwardson JA. Neuropathological diagnoses in elderly patients in Oslo: Alzheimer's disease, Lewy body disease, vascular lesions. Dementia. 1995;6:162–168. doi: 10.1159/000106940. [DOI] [PubMed] [Google Scholar]
- 22.Galasko D, Katzman R, Salmon D, Hansen L. Clinical and neuropathological findings in Lewy body dementias. Brain Cogn. 1996;31:166–175. doi: 10.1006/brcg.1996.0040. [DOI] [PubMed] [Google Scholar]
- 23.Heyman A, Fillenbaum GG, Gearing M, et al. Comparison of Lewy body variant of Alzheimer's disease with pure Alzheimer's disease: Consortium to Establish a Registry for Alzheimer's Disease, part XIX. Neurology. 1999;52:1839–1844. doi: 10.1212/wnl.52.9.1839. [DOI] [PubMed] [Google Scholar]
- 24.Massoud F, Devi G, Stern Y, et al. A clinicopathological comparison of community-based and clinic-based cohorts of patients with dementia. Arch Neurol. 1999;56:1368–1373. doi: 10.1001/archneur.56.11.1368. [DOI] [PubMed] [Google Scholar]
- 25.Haroutunian V, Serby M, Purohit DP, et al. Contribution of Lewy body inclusions to dementia in patients with and without Alzheimer disease neuropathological conditions. Arch Neurol. 2000;57:1145–1150. doi: 10.1001/archneur.57.8.1145. [DOI] [PubMed] [Google Scholar]
- 26.Lippa CF, Fujiwara H, Mann DM, et al. Lewy bodies contain altered alpha-synuclein in brains of many familial Alzheimer's disease patients with mutations in presenilin and amyloid precursor protein genes. Am J Pathol. 1998;153:1365–1370. doi: 10.1016/s0002-9440(10)65722-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lippa C, Schmidt M, Lee V, Trojanowski J. Antibodies to alpha-synuclein detect Lewy bodies in many Down's syndrome brains with Alzheimer's disease. Ann Neurol. 1999;45:353–357. doi: 10.1002/1531-8249(199903)45:3<353::aid-ana11>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 28.Li YJ, Hauser MA, Scott WK, et al. Apolipoprotein E controls the risk and age at onset of Parkinson disease. Neurology. 2004;62:2005–2009. doi: 10.1212/01.wnl.0000128089.53030.ac. [DOI] [PubMed] [Google Scholar]
- 29.Kurosinski P, Guggisberg M, Gotz J. Alzheimer's and Parkinson's disease—overlapping or synergistic pathologies? Trends Mol Med. 2002;8:3–5. doi: 10.1016/s1471-4914(01)02246-8. [DOI] [PubMed] [Google Scholar]
- 30.Masliah E, Rockenstein E, Veinbergs I, et al. Beta-amyloid peptides enhance alpha-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer's disease and Parkinson's disease. Proc Natl Acad Sci USA. 2001;98:12245–12250. doi: 10.1073/pnas.211412398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strittmatter WJ, Saunders AM, Goedert M, et al. Isoform-specific interactions of apolipoprotein E with microtubule-associated protein tau: implications for Alzheimer disease. Proc Natl Acad Sci USA. 1994;91:11183–11186. doi: 10.1073/pnas.91.23.11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lauderback CM, Kanski J, Hackett JM, Maeda N, Kindy MS, Butterfield DA. Apolipoprotein E modulates Alzheimer's Abeta(1–42)-induced oxidative damage to synaptosomes in an allele-specific manner. Brain Res. 2002;924:90–97. doi: 10.1016/s0006-8993(01)03228-0. [DOI] [PubMed] [Google Scholar]