

Abstract
The utility and advantages of the recently introduced two-dimensional quadrupole ion trap mass spectrometer in proteomics over the traditional three-dimensional ion trap mass spectrometer have not been systematically characterized. Here we rigorously compared the performance of these two platforms by using over 100,000 tandem mass spectra acquired with identical complex peptide mixtures and acquisition parameters. Specifically we compared four factors that are critical for a successful proteomic study: 1) the number of proteins identified, 2) sequence coverage or the number of peptides identified for every protein, 3) the data base matching SEQUEST Xcorr and Sp score, and 4) the quality of the fragment ion series of peptides. We found a 4–6-fold increase in the number of peptides and proteins identified on the two-dimensional ion trap mass spectrometer as a direct result of improvement in all the other parameters examined. Interestingly more than 70% of the doubly and triply charged peptides, but not the singly charged peptides, showed better quality of fragmentation spectra on the two-dimensional ion trap. These results highlight specific advantages of the two-dimensional ion trap over the conventional three-dimensional ion traps for protein identification in proteomic experiments.
Ion trap mass spectrometers are widely used in proteomic studies (1, 2). They are rugged, relatively less expensive, and provide good MS/MS1 capabilities. A number of critical technological features also complement the utility of ion traps in proteomics. For example, software control of ion trap instrument operation has facilitated data-dependent acquisition features and, along with on-line reverse phase separation, has enabled analysis of highly complex peptide mixtures from cells and tissues (3–5). Furthermore search tools such as SEQUEST automate the process of assigning uninterpreted fragmentation data to peptides from sequence data bases, making proteomic scale projects feasible (6–8).
Despite a number of technological improvements, comprehensive identification of cellular proteomes is not fully realized. This limitation is due in part to the complexity of samples in proteomic experiments where thousands of peptides elute within a narrow window of organic gradient in reverse-phase separation methods (9). In addition, conventional three-dimensional (3-D) ion traps have a slower scan rate and a limited ion capacity and trapping efficiency (10). These may contribute toward a reduced number of peptides identified as well as limited quality of fragmentation spectra. Improving the above features of the ion trap mass spectrometers is likely to enhance the identification capability. To address these issues, a new design of the ion trap mass analyzer was introduced, and this is commercially available as LTQ (10). The LTQ has a quadrupole made of four hyperbolic cross-sectional rods giving it a two-dimensional (2-D) or linear feature. The ions are trapped in an axial fashion as opposed to central trapping on 3-D ion traps. Functional enhancements of this mass analyzer include 15× higher ion capacity, 3× faster scan rate, up to 100% detection efficiency (as opposed to ~50% detection efficiency on LCQ Deca), and up to 70% trapping efficiency (as opposed to ~5% trapping efficiency on 3-D ion traps) (10). Most of other features and operations are very much like the conventional 3-D ion traps.
The utility and performance of the 2-D ion trap mass spectrometer (LTQ) in proteomic analyses compared with that of conventional 3-D ion traps have not been rigorously tested to date. Hence we analyzed identical complex peptide mixtures with identical reverse-phase organic gradients on both the 2-D ion trap (LTQ) and a 3-D ion trap (LCQ Deca). We compared over 100,000 MS/MS spectra from the two instruments with respect to the size of the proteome, sequence coverage, data base peptide match scores, and the quality of collision-induced dissociation (CID) spectra of peptides. We found a 4–6-fold increase in the number of proteins/peptides identified using the 2-D ion trap mass spectrometer. We also observed significant improvement in sequence coverage and the quality of the CID spectra of multiply charged peptides on the 2-D ion trap, which contribute to higher confidence in peptide/protein identification and in the size of the proteome defined.
EXPERIMENTAL PROCEDURES
Jurkat T leukemia cell extracts were prepared using a lysis buffer containing the non-ionic detergent maltoside (0.5% n-dodecyl-β-d-maltoside, 50 mm Tris-HCl at pH 8, 150 mm NaCl, and 1 mm EDTA). 40 μg of total protein extracts were resolved on a 10% NuPAGE gel (Invitrogen), and slices corresponding to the molecular mass range of 45–51 and 51–64 kDa were excised. Four such slices for each molecular mass range were pooled together and subjected to in-gel trypsin digestion, and the resulting tryptic peptides were extracted as described previously (11). The extracted tryptic peptide mixture was divided back into four equal samples to give identical tryptic peptide mixtures and analyzed both on LCQ Deca (a conventional 3-D ion trap) and LTQ (a 2-D ion trap) with different acquisition protocols as described below. The LCQ Deca was equipped with an in-house nanospray source, and the LTQ was equipped with a commercial nanospray source (Thermo Finnigan, San Jose, CA). Samples were loaded by means of a microautosampler (Famos, LC Packings, Sunnyvale, CA) onto an 11-cm × 100-μm fused silica capillary column packed in house with reverse-phase C18 material (5-μm, 100-Å Monitor C18 beads from Column Engineering, Ontario, Canada). The capillary column also served as the fritless emitter for nanospray (12). An organic solvent gradient was delivered through an HP1100 pump (Agilent Technologies, Palo Alto, CA) at a final flow rate of ~500 nl/min. Peptides were eluted with a linear gradient from 100% solvent A (5% acetonitrile, 0.4% acetic acid, and 0.005% heptafluorobutyric acid in water) to 10% solvent B (0.4% acetic acid and 0.005% heptafluorobutyric acid in acetonitrile) in 10 min and then to 45% solvent B in 64 min. Each full MS scan was followed by two, five, or nine MS/MS scans of the most intense full MS scan peaks (henceforth referred as “top 2,” “top 5,” and “top 9” protocols, respectively). The dynamic exclusion feature was enabled to maximize the detection of less abundant peptide ions. Sample loading, gradient delivery, and all the mass spectrometric scan functions were controlled by the Xcalibur software (Thermo Finnigan). The instrument method files and data files are available upon request. CID spectra were searched against a non-redundant human protein sequence data base from the NCI, National Institutes of Health using the SEQUEST software tool on a 16-node (32 processors) Linux cluster (6, 7). Search parameters included provisions for both unmodified and oxidized methionine (+16 Da) at a peptide mass tolerance of ±1.5 Da and full trypsin specificity allowing one missed cleavage for peptides (the SEQUEST search parameter file is available upon request). SEQUEST peptide matches were considered as true identifications based on the following filtering criteria: ΔCn (Δcorrelation) ≥ 0.08 and Xcorr (cross-correlation) ≥2.0 for 1+, ≥2.2 for 2+, and ≥3.3 for 3+ peptides. The SEQUEST summary files were submitted to INTERACT, and most of the comparative analysis was done via the web interface of INTERACT (13).
RESULTS
We first compared the two ion traps using identical data acquisition parameters. With the scan rate of LCQ Deca (~1 scan/s), the top 2 acquisition protocol usually gives the maximum number of identifications, and using the top 5 protocol resulted in a much lower number of protein identifications. Hence, on LCQ Deca, we chose to analyze the sample only by the top 2 protocol. However, with the ~3× faster scan rate on the 2-D ion trap, we also attempted top 5 and top 9 protocols. As shown in Fig. 1, even with the top 2 protocol, the 2-D ion trap performs ~3-fold better in identifying peptides and thereby proteins with stringent scores. Replicate analyses of the same sample using the top 5 and top 9 protocols resulted in a further increase in peptide and protein identification (Fig. 1). There is approximately a 4-fold increase in the number of proteins and a 5-fold increase in the number of peptides identified with the top 5 scheme (Fig. 1, a and b). A similar trend was observed when proteins with multiple peptide hits were selected (Fig. 1b). Overall we observed a 4–6-fold increase in the number of peptides and proteins identified on the 2-D ion trap when six different complex protein mixtures were separately analyzed.
Fig. 1. Rigorous comparison of the 2-D and the 3-D ion trap in protein identification.
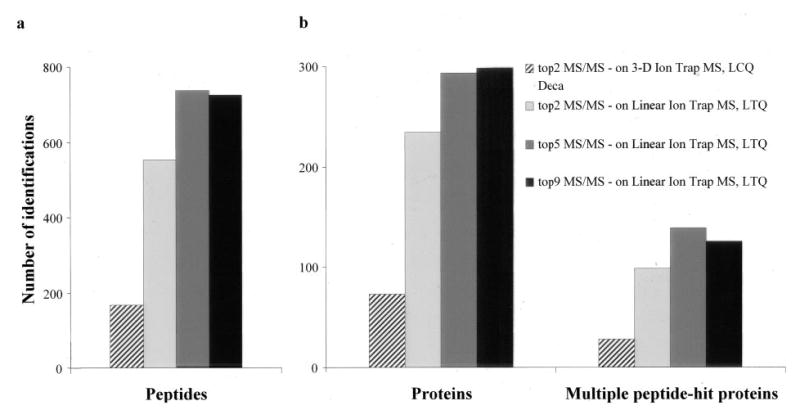
The results from three different acquisition protocols are shown. The top 2 acquisition protocol, which performs MS/MS of the two most intense ions, highlights the performance differences between the two platforms. The approximately 3-times-faster scan rate of the LTQ permits MS/MS of five and even nine most intense ions from the full scan acquisition in a data-dependent manner. The increase in the number of identified peptides scales linearly with the increase in the identification capacity of the ion trap except for the top 9 acquisition protocol on the linear ion trap mass spectrometer (a). The protein sample analyzed was derived from the 45–51-kDa gel region of the Jurkat T cell extracts.
Proteins identified by a single peptide usually require manual inspection of CID spectra. Confidence in protein identification increases with multiple unique peptides identified. It has been shown before that the false-positive rate decreases dramatically even for proteins identified with two unique peptides (14). Hence we compared the sequence coverage of proteins identified both on the 2-D ion trap and the 3-D ion trap from the Jurkat T cellular extract corresponding to the 45–51-kDa region. We found that most of the proteins with single peptide identification on the 3-D ion trap were identified with better coverage on the 2-D ion trap (Table I). For most proteins, the top 5 acquisition protocol on the 2-D ion trap provided further increase in sequence coverage (Table I). We observed a similar trend in the peptide sequence coverage with five other complex mixtures (data not shown). Thus, on a comparative basis, the 2-D ion trap provides better sequence coverage and thereby increases the confidence in protein identification.
Table I. Sequence coverage (given by the number of unique peptides identified) of proteins that were identified both on LTQ and LCQ Deca.
Tryptic peptides from the Jurkat T cell extracts corresponding to the 45–51-kDa region were analyzed.
| Number of peptides identified
|
|||
|---|---|---|---|
| Proteina | top 2 on LCQ Deca | top 2 on LTQ | top 5 on LTQ |
| GPN:AF343348_1 | 1 | –b | 1 |
| GP:AK024002_1 | 1 | 1 | – |
| GPN:AK035340_1 | 1 | 1 | – |
| GPN:AY223417_1 | 1 | 2 | – |
| GPN:BC018664_1 | 1 | 2 | 1 |
| GPN:BC036816_1 | 1 | 1 | – |
| GPN:Z70680_10 | 1 | – | 1 |
| PIR1:KIHUG | 1 | 1 | 1 |
| PIR2:T34545 | 1 | – | 1 |
| SW:ABL2_HUMAN | 1 | 1 | – |
| SW:ALBU_HUMAN | 1 | – | 2 |
| SW:CAP1_HUMAN | 1 | – | 1 |
| SW:CGL1_HUMAN | 1 | 3 | 3 |
| SW:CPS1_HUMAN | 1 | 1 | – |
| SW:DCT2_HUMAN | 1 | 5 | 7 |
| SW:DD48_HUMAN | 1 | 6 | 7 |
| SW:DJA1_HUMAN | 1 | 5 | 8 |
| SW:EFTU_HUMAN | 1 | 1 | 9 |
| SW:FEN1_HUMAN | 1 | 2 | 3 |
| SW:FUMH_HUMAN | 1 | 8 | 11 |
| SW:GLYM_HUMAN | 1 | 5 | 5 |
| SW:GRF1_HUMAN | 1 | – | 1 |
| SW:HE47_HUMAN | 1 | 6 | 8 |
| SW:HS72_HUMAN | 1 | 2 | 3 |
| SW:HS9B_HUMAN | 1 | 8 | 6 |
| SW:IF36_HUMAN | 1 | 7 | 8 |
| SW:K22E_HUMANc | 1 | 1 | 2 |
| SW:METK_HUMAN | 1 | 3 | 4 |
| SW:NPL1_HUMAN | 1 | 4 | 2 |
| SW:OST4_HUMAN | 1 | 4 | 4 |
| SW:PSD6_HUMAN | 1 | 3 | 5 |
| SW:PSDB_HUMAN | 1 | 9 | 9 |
| SW:PSDC_HUMAN | 1 | 7 | 11 |
| SW:PTD4_HUMAN | 1 | 3 | 6 |
| SW:PUR6_HUMAN | 1 | 7 | 13 |
| SW:ROF_HUMAN | 1 | 3 | 5 |
| SW:SEP6_HUMAN | 1 | 4 | 4 |
| SW:SEP7_HUMAN | 1 | 7 | 7 |
| SW:SNXE_HUMAN | 1 | – | 1 |
| SW:ST13_HUMAN | 1 | 8 | 5 |
| SW:TBA2_HUMAN | 1 | 3 | 1 |
| SW:TBB2_HUMAN | 1 | 5 | 5 |
| SW:TBB5_HUMAN | 1 | 1 | 1 |
| SW:TBBX_HUMAN | 1 | – | 1 |
| SW:THIL_HUMAN | 1 | 1 | 1 |
| SW:TPP1_HUMAN | 1 | 1 | 1 |
| SW:TRAL_HUMAN | 1 | – | 2 |
| SW:UCR2_HUMAN | 1 | 9 | 8 |
| SW:YB1_HUMAN | 1 | 4 | 2 |
| GPN:BC021139_1 | 4 | 6 | 14 |
| PIR2:T12456 | 4 | 5 | 10 |
| SW:6PGD_HUMAN | 4 | 8 | 11 |
| SW:ACTA_HUMAN | 7 | 5 | 8 |
| SW:ACTB_HUMAN | 4 | 5 | 7 |
| SW:ATPB_HUMAN | 8 | 15 | 15 |
| SW:CO1A_HUMAN | 9 | 5 | 8 |
| SW:CRTC_HUMAN | 3 | 11 | 9 |
| SW:DBPA_HUMAN | 3 | 3 | 3 |
| SW:EF11_HUMAN | 7 | 10 | 12 |
| SW:EF1G_HUMAN | 8 | 14 | 15 |
| SW:ENOA_HUMAN | 14 | 14 | 16 |
| SW:GDIA_HUMAN | 3 | 6 | 4 |
| SW:GDIB_HUMAN | 3 | 8 | 8 |
| SW:IDHP_HUMAN | 3 | 8 | 12 |
| SW:IF35_HUMAN | 4 | 4 | 9 |
| SW:IF41_HUMAN | 7 | 11 | 14 |
| SW:KPY1_HUMAN | 4 | 11 | 14 |
| SW:LA_HUMAN | 5 | 6 | 8 |
| SW:P2G4_HUMAN | 6 | 8 | 7 |
| SW:PGK1_HUMAN | 13 | 17 | 17 |
| SW:PLSL_HUMAN | 4 | 15 | 15 |
| SW:RBB4_HUMAN | 3 | 2 | 6 |
| SW:RL4_HUMAN | 5 | 5 | 8 |
| SW:SAHH_HUMAN | 3 | 7 | 11 |
| SW:TBA1_HUMAN | 10 | 12 | 12 |
| SW:TBB1_HUMAN | 15 | 17 | 17 |
| SW:TRYP_PIGc | 3 | 4 | 5 |
| GPN:BC056886_1 | 2 | 2 | 3 |
| SW:ERF1_HUMAN | 2 | 10 | 7 |
| SW:HS9A_HUMAN | 2 | 8 | 15 |
| SW:PDA6_HUMAN | 2 | 9 | 11 |
Acession numbers in NCBI, SWISS-Prot, or PIR database.
The protein was not identified in this mode of acquisition.
Autocatalysis products from trypsin and peptides from contaminants like keratin were included as they were part of the identical samples analyzed.
We next compared the identification of regulatory and presumed low abundant proteins on the 2-D and the 3-D ion traps. Working with the faster scan rate and top 5 acquisition protocol of the 2-D ion trap, we identified transcription factors; kinases; signaling adapter proteins; proteins/enzymes involved in chromatin modulation, vesicular transport, protein translocation, and post-translational modification; and membrane proteins that were not identified on the 3-D ion trap (Table II). The 2-D ion trap also identified more of the abundant proteins such as the cytoskeletal and structural proteins, metabolic enzymes, heat shock proteins, proteasomal components, ribonuclear proteins, and many housekeeping proteins involved in fundamental functions of transcription and translation (Table I). Therefore, the 2-D ion trap is more efficient in the identification of potentially less abundant and regulatory proteins. We have substantiated this finding by extrapolating the absolute protein abundance of yeast homologs to human proteins (Supplemental Fig. 1 and Tables 1–3).
Table II. List of regulatory proteins identified by multiple peptides.
Tryptic peptides from the Jurkat T cell extracts corresponding to the 45–51-kDa region were analyzed both on LTQ (top 5 protocol) and LCQ Deca (top 2 protocol).
| Protein IDa | Data base annotation and functional information |
|---|---|
| GP:U94174_1 | Mammary tumor-associated protein INT6 gene |
| GPN:BC000404_1 | Thyroid hormone receptor interactor 13 |
| GPN:BC009597_1 | Basic leucine zipper and W2 domains 2 |
| GPN:BC004302_1 | COP9 constitutive photomorphogenic homolog subunit 4 (Arabidopsis) |
| GPN:BC021139_1 | Nuclear distribution gene C homolog (Aspergillus nidulans)b |
| GPN:BC053350_1 | Solute carrier family 9 (sodium/hydrogen exchanger) |
| GPN:BC056886_1 | LUC7-like 2 (S. cerevisiae) (is localized to spliceosome complex in yeast)b |
| PIR2:A54857 | Transcription factor NF-AT 45-kDa chain |
| PIR2:JC4525 | Nucleic acid-binding protein E5.1 |
| PIR2:S68209 | SDS22 protein homolog (a type 1 serine/threonine phosphatase) |
| SW:2ABA_HUMAN | Serine/threonine protein phosphatase 2A, 55-kDa regulatory subunit B |
| SW:A1M1_HUMAN | Adaptor-related protein complex 1, clathrin assembly protein |
| SW:ATXX_HUMAN | Ataxin-10 (involved in neurodegenerative diseases) |
| SW:CGA2_HUMAN | Cyclin A2 |
| SW:CGL1_HUMAN | Cytosolic nonspecific dipeptidaseb |
| SW:CO1A_HUMAN | Coronin-like protein p57 (promotes barbed end assembly of actin filaments) |
| SW:CSK_HUMAN | Tyrosine-protein kinase CSK (c-Src kinase) |
| SW:CSN2_HUMAN | COP9 signalosome complex subunit 2 |
| SW:DCT2_HUMAN | Dynactin complex 50-kDa subunit |
| SW:DD17_HUMAN | Probable RNA-dependent helicase p72 |
| SW:DEK_HUMAN | Dek protein (induces alterations in superhelical density of DNA in chromatin) |
| SW:ENPL_HUMAN | Endoplasmin precursor (gp96 homolog) |
| SW:FKB4_HUMAN | FK506-binding protein 4 (peptidyl-prolyl cis-trans isomerase) |
| SW:GD1A_HUMAN | Rab GDP dissociation inhibitor αb |
| SW:GD1B_HUMAN | Rab GDP dissociation inhibitor βb |
| SW:GLYC_HUMAN | Serine hydroxymethyltransferase |
| SW:GLYM_HUMAN | Serine hydroxymethyltransferase, mitochondrial precursorb |
| SW:HAT1_HUMAN | Histone acetyltransferase type B catalytic subunit |
| SW:HE47_HUMAN | Probable ATP-dependent RNA helicase p47b |
| SW:IEFS_HUMAN | Stress-induced phosphoprotein 1 |
| SW:ILK1_HUMAN | Integrin-linked protein kinase 1 |
| SW:IMA2_HUMAN | Importin α-2 subunit |
| SW:KAP0_HUMAN | cAMP-dependent protein kinase type I-α regulatory chain |
| SW:LPAX_HUMAN | Leupaxin (most homologous to paxillin, which is found in focal adhesions) |
| SW:MPK1_HUMAN | Dual specificity mitogen-activated protein kinase kinase 1 |
| SW:OST4_HUMAN | Dolichyl-diphosphooligosaccharide-protein glycosyltransferase |
| SW:P2G4_HUMAN | Proliferation-associated protein 2G4b |
| SW:PDA6_HUMAN | Protein-disulfide isomerase A6 precursorb |
| SW:PFTA_HUMAN | Protein farnesyltransferase α subunit |
| SW:PTD4_HUMAN | Putative GTP-binding proteinb |
| SW:RBB7_HUMAN | Histone acetyltransferase type B subunit 2 (retinoblastoma-binding protein 7) |
| SW:SEP6_HUMAN | Septin 6 (involved in cytokinesis)b |
| SW:ST24_HUMAN | Serine/threonine protein kinase 24 (mammalian STE20-like protein kinase 3) |
| SW:VAT1_HUMAN | Synaptic vesicle membrane protein VAT-1 homolog |
| SW:WD12_HUMAN | WD repeat protein 12 |
| SW:YB1_HUMAN | CCAAT-binding transcription factor I subunit Ab |
| SWN:SMY3_HUMAN | Zinc finger MYND domain-containing protein 1 |
Acession numbers in NCBI, Swiss-Prot, or PIR database.
These proteins were also identified on the 3-D ion trap (LCQ Deca) at least by a single peptide.
Higher Xcorr scores reflect better matching of acquired CID spectra to peptide sequences in the data base (7). Furthermore higher Xcorr score filtering criteria for true peptide identification decreases the false-positive rate (14). Hence we compared the Xcorr scores of peptides that were identified both on the 2-D ion trap and the 3-D ion trap mass spectrometer. To be unbiased toward specific instruments, we first considered peptides identified both from the 2-D ion trap and the 3-D ion trap using significantly relaxed SEQUEST filtering criteria (ΔCn ≥0.08 and Xcorr ≥1.0 for 1+, ≥1.2 for 2+, and ≥2.3 for 3+ peptides). We next selected the common peptide pairs with at least one high scoring match (ΔCn ≥0.08 and Xcorr ≥1.5 for 1+, ≥2.0 for 2+, and ≥3.3 for 3+ peptides). We subsequently compared the Xcorr score of singly, doubly, and triply charged common peptides identified from the 3-D and the 2-D ion traps. Approximately 80% of the singly charged peptides showed a decrease in Xcorr score on the 2-D ion trap mass spectrometer (Fig. 2a). In contrast, more than 70% of the doubly and triply charged peptides showed improvement in Xcorr scores on the 2-D ion trap (Fig. 2, c and e). There is an average increase of ~0.6 in Xcorr scores for doubly and triply charged peptides on the 2-D ion trap. This trend is consistent all across the large data set comprising over 100,000 MS/MS spectra.
Fig. 2. Rigorous comparison of the 2-D and the 3-D ion traps in peptide identification scores.
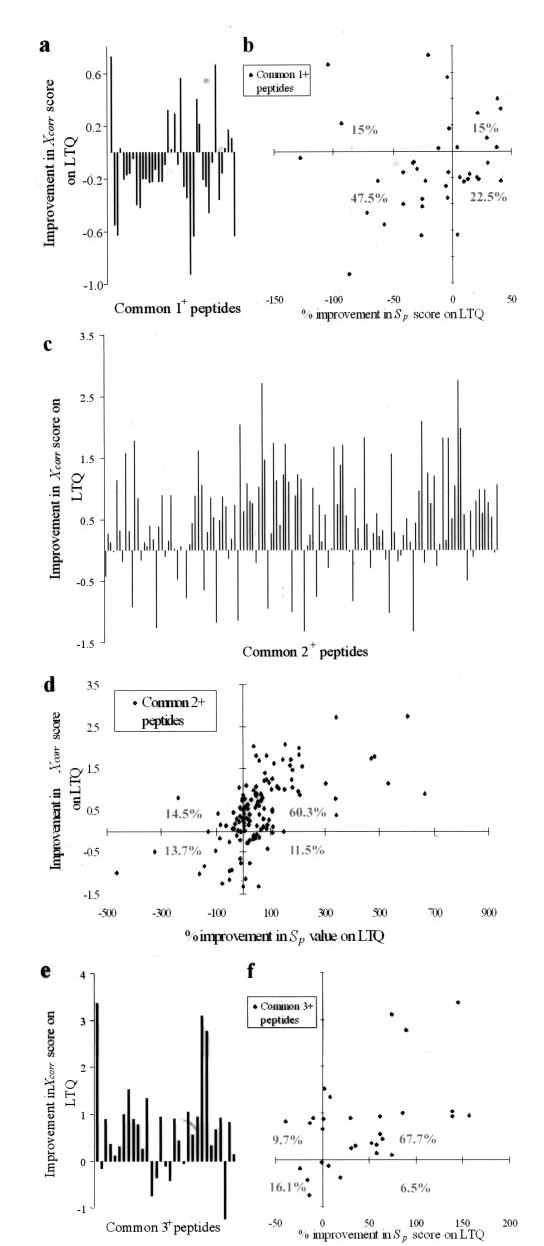
The detailed comparison of improvements in Xcorr and Sp scores from common peptides with singly, doubly, and triply charged states are shown. The improvement in Xcorr is represented as vertical bars for each of the common peptides. Negative values, indicating the deterioration in Xcorr, are most striking for the singly charged peptides, while significant improvement is seen for most of the doubly and triply charged peptides (a, c, and e). Similarly combined analysis of the Sp score (x axis), which reflects the information content of the CID spectra, with the Xcorr (y axis) showed improvement for the doubly and triply charged peptides as indicated (b, d, and f, rhomboid symbol). The percentages indicate the distribution of peptides in each of the four quadrants. Improvement in Xcorr score on the linear ion trap is given as ((Sp LTQ − Sp LCQ Deca) × 100)/(minimum of Sp LTQ and Sp LCQ Deca). The lowest minimum of Sp scores from the two instruments was used in each case to avoid bias in representation of the data. The protein sample analyzed was derived from the 45–51- and 51–64-kDa gel regions of the Jurkat T cell extracts.
Remarkable improvement in Xcorr scores for the doubly charged peptides is likely due to better quality of the CID spectra of these peptides on the 2-D ion trap. We used the Sp score, a preliminary score used by SEQUEST to prune the list of peptides to do rigorous cross-correlation analysis, as a quantitative measure of the quality or the information content of the CID spectra. The Sp score is based on the number of fragment ions that matches the predicted b and y ions of the peptide in consideration, their relative abundances, and the continuity in the ion series (7). There is a positive correlation (correlation coefficient of 0.59) between “improvement in Xcorr” and “percent improvement in Sp score” on the 2-D ion trap, implying that it gives more informative CID spectra of doubly charged peptides than LCQ Deca (Fig. 2d). Similar trends were obtained for triply charged peptides (Fig. 2f). Surprisingly the 2-D ion trap provides less informative CID spectra for singly charged peptides (Fig. 2b).
We next examined the CID spectra of several doubly charged peptides in detail by manual inspection. Among the common peptides identified by both the 2-D and the 3-D ion traps, we found that additional b ions and proximal ion series from either the N or C terminus were detected on the 2-D ion trap mass spectrometer (Fig. 3). It is known that these ions are not produced efficiently during the CID of doubly charged tryptic peptides (15). However, the 2-D ion trap mass spectrometer detected some of these less abundant fragment ions, providing rich fragmentation information (Fig. 3). Most of the tryptic peptides (>90%) ionize as doubly or triply charged peptides under typical solvent condition. Since the 2-D ion trap mass spectrometer provides more informative CID spectra for multiply charged peptides, the confidence in peptide identification is significantly increased for these peptides.
Fig. 3. Improved quality of fragment ion series on the 2-D ion trap.
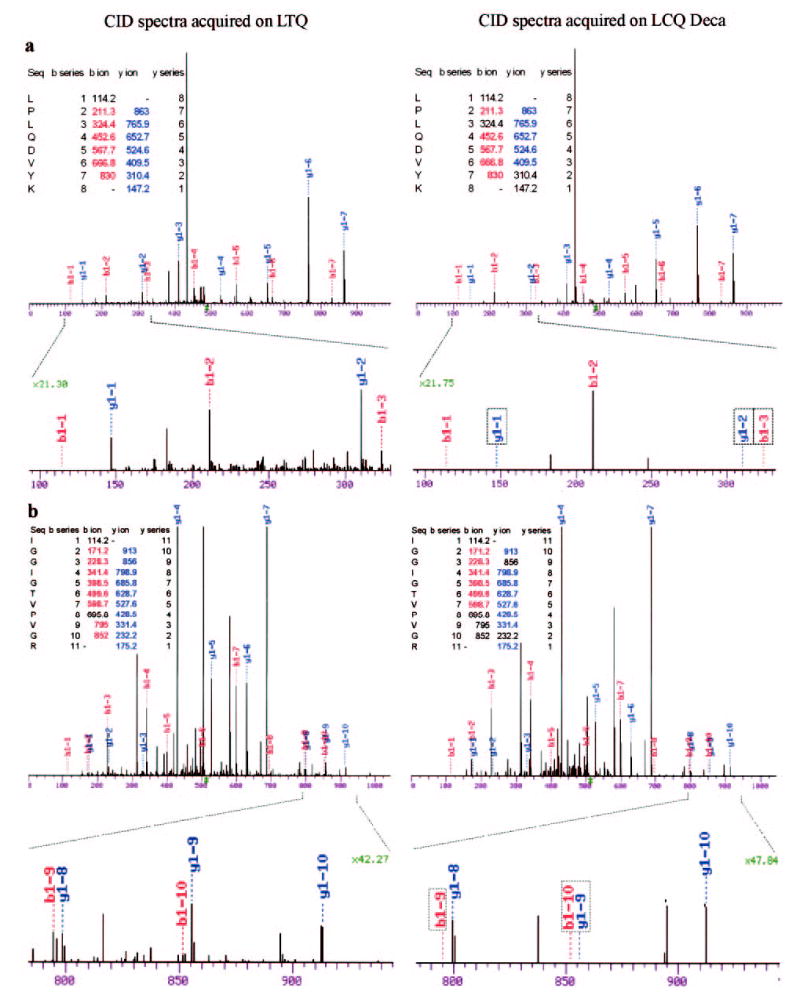
Significant improvement in the quality of the CID spectra is seen on the LTQ, and two representative spectra are illustrated (a and b). The b and y ions detected on the 2-D ion trap but not on the 3-D ion trap are indicated in the enlarged region of fragmentation spectra (boxed). The b and y ions that were not detected are indicated in the ion series tables (uncolored). The magnification of the y axis is also indicated.
Based on the number of high scoring peptides and proteins identified on the 2-D ion trap mass spectrometer, we anticipated that proteins that were identified by the 2-D trap would encompass all of the proteins identified by the 3-D ion trap. However, the 2-D ion trap identified only 65% of the peptides and 79% of the proteins identified by the 3-D ion trap (Fig. 4a). Interestingly, when we compared the multiple peptide-hit proteins, the 2-D ion trap identified all but two of the proteins identified by the 3-D ion trap. We made similar observations with data acquired on the 2-D ion trap with top 5 and top 9 acquisition protocols; 55% of the high scoring peptides, 65% of the proteins, and 81% of proteins identified by multiple unique peptides were common to the two data sets (Fig. 4b). Similar observations have been reported with replicate analyses of complex tryptic peptide mixtures (16, 17). We attribute the poor overlap in peptides to overwhelming complexity of the sample and data-dependent and dynamic exclusion features of acquisition protocols.
Fig. 4. Assessment of overlap in the identified proteome using the 2-D and the 3-D ion traps.
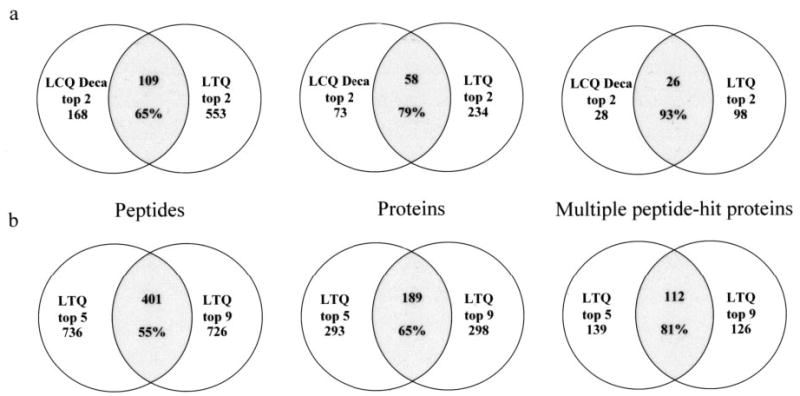
a, the proteome overlap identified by the LTQ and the LCQ using the top 2 protocol is shown. The peptide overlap (left), the protein overlap (middle), and the overlap in multiple peptide-hit proteins (right) are shown. The total number of identified peptides, proteins, and multiple peptide-hit proteins for the LCQ Deca or the LTQ are indicated. The intersected (shaded) region represents the overlap, and the number of common peptides, proteins, and multiple peptide-hit proteins are indicated. The percentage refers to the proteome overlap of LTQ over the LCQ Deca. Although the LTQ did not completely identify the peptides and proteins identified on the LCQ, it identified almost all the multiple peptide-hit proteins. b, the proteome overlap identified by the top 5 and top 9 protocols on the LTQ is shown. Trends in the identified peptides, proteins, and the multiple peptide-hit proteins are as observed in a. The percentage refers to the proteome overlap of the top 9 over the top 5 protocol on the LTQ.
DISCUSSION
Our analyses based on >100,000 MS/MS spectra clearly favor 2-D ion trap instruments such as LTQ for proteomic analyses. Overall we found a 4–6-fold increase in the number of peptides/proteins identified on the 2-D ion trap mass spectrometer (Fig. 1). As observed with some of our samples, this enhancement is more than what can be expected from an ~3× faster scan rate of the 2-D ion trap. We believe that additional enhancement in the number of peptides/proteins identified is attributable to improvement in SEQUEST Xcorr scores, which is the direct result of improvement in the quality and information content of CID spectra of doubly and triply charged peptides on the 2-D ion trap (Fig. 2). The 2-D ion trap mass spectrometer allowed the detection of some of the less abundant fragment ions, providing more fragmentation information (Fig. 3). This enhancement in sensitivity is likely due to improved trapping efficiency and ion capacity of LTQ (10). On a comparative basis, the 2-D ion trap also provided better sequence coverage (Table I). Thus, the 2-D ion trap mass spectrometer contributes both to confidence in peptide identification and the size of the proteome defined. Moreover, by identifying more regulatory proteins, the 2-D ion trap also provides more information critical for biological discovery (Table II, Supplemental Fig. 1, and Supplemental Tables 1–3).
We have also explored the dynamic range of these two platforms and found that the 2-D ion trap has superior internal dynamic range in MS/MS spectra and improved peptide detection. For the internal dynamic range of MS/MS spectra, we compared the lowest and the highest fragment ions of a standard peptide, angiotensin I. The 3-D ion trap (LCQ Deca) showed an internal dynamic range of ~1:40, while the value for the 2-D ion trap (LTQ) was ~1:1000. Similar results have been reported for the 2-D ion trap (22). For several standard peptides, we observed a detection limit of ~300 amol on the 3-D ion trap with the selected ion monitoring and selected reaction monitoring scan modes. The 2-D ion trap detection limit, however, is ~500 zmol. This data is consistent with recent report demonstrating 550 zmol detection of angiotensin I on the LTQ mass spectrometer (22).
In this study, we did not utilize all of the available ion traps such as the newer Agilent 3-D ion trap (MSD Trap XCT Plus) or the Finnigan LCQ Deca XP+. The Finnigan LCQ Deca XP+ scans significantly slower than LTQ and is only about 25% faster than the LCQ Deca. However, the scan rate of the Agilent MSD trap is comparable to that of the Finnigan LTQ. Accordingly the theoretical identification capacity of this mass spectrometer should be comparable to that of LTQ 2-D ion trap. Therefore, we cannot make a sweeping generalization that the 2-D ion traps are better than the 3-D ion traps.
Peptide identification capacity is limited by the scan rate of the mass spectrometers as well as by the chromatography time window where all the peptides elute. If most of the peptides are eluting within a 35-min window and if the 2-D mass spectrometer performs ~2 MS/MS scans/s (top 2 protocol), the identification capacity is roughly 4200 peptides (35 min × 60 s × 2). The identification capacities of the top 5 and top 9 methods are slightly increased due to the increasing MS/MS scan rates of 2.75 and 3 scans/s, respectively. Similarly a 3-D ion trap such as the LCQ Deca with the top 2 protocol will allow the identification capacity of 1260 peptides (35 min × 60 s × 0.6 MS/MS scan/s). The increase in the number of peptides identified scales linearly with the increase in the theoretical identification capacity of the 2-D ion trap except for the top 9 acquisition protocol (Fig. 1). The list of the most intense parent ions for sequencing attempts is updated more frequently in the top 5 protocol (once every ~2 s) than in the top 9 protocol (once every ~3 s). Working in a data-dependent fashion, the top 5 scheme has a better chance of picking the most intense ions at any instance of time. With higher order “top n” acquisition protocols, the co-eluting peaks and their respective intensities may change significantly from the survey scan. Therefore, a number of MS/MS attempts may not yield high quality CID spectra when the signal to noise ratio of the peptide peaks become much lower than the survey scan. Moreover the increase in theoretical identification capacity of the tandem mass spectrometer begins to saturate. Due to these reasons, beyond a certain top n protocol, an increase in the theoretical peptide identification capacity of the ion trap does not translate to an increase in the number of peptides identified.
Further improvements in the scan rate of tandem mass spectrometry approaches are needed to handle the complexity of cellular proteomes. The human protein sequence data base has ~5000 proteins between 45 and 51 kDa, which give rise to ~60,000 tryptic peptides within the m/z range detectable by ion traps. Repositories of serial analysis of gene expression (SAGE) data and cDNA libraries of various tissue and cell types suggest that, on average, ~25% of the genes in the human genome are expressed in cells. Even if we make a conservative estimate that only half of these expressed proteins are adequately abundant, our samples had ~7500 analyzable tryptic peptides above femtomole amounts. This is more than the theoretical identification capacity of the 2-D ion trap-based proteomic platform. With the consideration of post-translational modifications, complexity reaches combinatorial proportions. Thus, complexity of the proteome is going to remain a challenging issue. Alternatively peptide identification based on accurate and high resolution mass measurement along with certain constraints may hold the promise (18).
We were quite surprised by the observation of incomplete overlap between the proteomes defined by different acquisition protocols on the 3-D and 2-D ion trap mass spectrometers (Fig. 4). Although we used stringent Xcorr filtering criteria, the presence of false-positive peptides may be partially contributing to the poor overlap of peptides. However, the presence of potential false-positives is not the main reason for the incomplete overlap. We attribute the poor overlap to a combination of the following factors: 1) overwhelming complexity of the samples, 2) data-dependent and dynamic exclusion features of acquisition protocols, and 3) variation in chromatographic elution of peptides. These add random character to the process of ion selection for sequencing attempts. Thus, during replicate analyses different sets of ions are selected for and then excluded from MS/MS attempts. The randomness in ion selection is further pronounced due to complexity of samples. As described earlier, our samples were very complex, and as a result, a considerable number of multiple peptide-hit proteins, which are likely to be quite abundant, were not identified (Fig. 4b). Similar observations and explanations are found in the literature wherein replicate analyses were carried out with complex tryptic peptide mixtures (16, 17). If the samples are less complex than the identification capacity achievable by the proteomic platforms, we anticipate a more complete overlap in replicate analyses. From our results, it is clear that even the 2-D ion trap mass spectrometer does not identify all or most of the analyzable peptides and also fails to identify all the peptides identified by the 3-D ion trap mass spectrometer. Estimating the complexity and then using suitable fractionation and enrichment techniques are more likely to provide biological leads and insights (19).
Different SEQUEST scoring criteria have been used before along with the inclusion of partially tryptic peptides as true identifications (8, 14, 20, 21). The following filtering criteria were reported to give less than 1% false-positives for fully tryptic peptides based on reverse data base testing in the case of yeast proteome: ΔCn ≥0.08 and Xcorr ≥2.0 for 1+, ≥1.5 for 2+, and ≥3.3 for 3+ peptides (14). However, the above criterion for doubly charged peptides yielded a lot of false-positives in our case when we validated the identification using the protein molecular weight information and manual inspection of the spectra. The likely reason for the high occurrence of false-positives with the above Xcorr score criterion may be due to the complexity of the human proteome, which is at least 5 times larger than the yeast proteome. Hence we chose a more stringent filtering criterion of Xcorr ≥ 2.2 for doubly charged peptides.
2-D ion trap designs have been incorporated into hybrid mass analyzers. In one such hybrid mass spectrometer, the 2-D ion trap LTQ acts as the front end of a Fourier transform ion cyclotron resonance mass analyzer with features to operate both mass analyzers simultaneously (22). Thus, high mass accuracy of Fourier transform ion cyclotron resonance mass analyzers can be combined with other advantages provided by 2-D ion traps to increase both the size of the proteome identified and confidence in peptide identification (23). In another hybrid instrument, the third quadrupole of a triple quadrupole instrument was designed to function either as a conventional quadrupole mass filter or as a liner ion trap enabling novel scan features to characterize post-translational modifications (24). Also, in two recent studies, the 2-D ion trap was used to perform novel MS3 (second MS/MS scan of the most intense fragment ion from the first MS/MS scan) scan functions for phosphoproteomic applications and also to further increase the confidence in peptide identification (25, 26). It has also been used to explore a novel tandem mass spectrometry approach termed electron transfer dissociation of peptides (27). Apart from routine identification and profiling studies, we have begun to use LTQ for quantification studies due to its improved dynamic range and sensitivity (22). Thus, 2-D ion traps promise to be of valuable utility in proteomic studies for complex biological samples where more efficient protein identification, protein quantification, and determination of post-translational modification are required.
Acknowledgments
We thank Debbie Lundgren for theoretical estimates of the complexity of our samples and Steven Gygi and the members of his laboratory for technical input on nanospray automation and sample handling with microautosamplers. We also thank Scott Gerber and Edger Naegele for technical information regarding the Thermo LCQ Deca XP+ and Agilent MSD trap XCT Plus, respectively.
Footnotes
This work was supported by National Institutes of Health Grants RO1 HL67569, PO1 HL70694, and RR13186 (to D. H.).
The on-line version of this article (available at http://www.mcponline.org) contains supplemental material.
The abbreviations used are: MS/MS, tandem mass spectrometry; CID, collision-induced dissociation; ΔCn, Δcorrelation score provided by SEQUEST; Xcorr, cross-correlation score provided by SEQUEST; 3-D, three-dimensional; 2-D, two-dimensional.
References
- 1.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 2.Yates JR., III Mass spectral analysis in proteomics. Annu Rev Biophys Biomol Struct. 2004;33:297–316. doi: 10.1146/annurev.biophys.33.111502.082538. [DOI] [PubMed] [Google Scholar]
- 3.Dongre AR, Eng JK, Yates JR., III Emerging tandem-mass spectrometry techniques for the rapid identification of proteins. Trends Biotechnol. 1997;15:418–425. doi: 10.1016/S0167-7799(97)01110-4. [DOI] [PubMed] [Google Scholar]
- 4.Jonscher KR, Yates JR., III The quadrupole ion trap mass spectrometer—a small solution to a big challenge. Anal Biochem. 1997;244:1–15. doi: 10.1006/abio.1996.9877. [DOI] [PubMed] [Google Scholar]
- 5.Aebersold R, Goodlett DR. Mass spectrometry in proteomics. Chem Rev. 2001;101:269–295. doi: 10.1021/cr990076h. [DOI] [PubMed] [Google Scholar]
- 6.Sadygov RG, Eng J, Durr E, Saraf A, McDonald H, MacCoss MJ, Yates JR., III Code developments to improve the efficiency of automated MS/MS spectra interpretation. J Proteome Res. 2002;1:211–215. doi: 10.1021/pr015514r. [DOI] [PubMed] [Google Scholar]
- 7.Eng JK, McCormack AL, Yates JR., III An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 8.Washburn MP, Wolters D, Yates JR., III Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 9.Peng J, Gygi SP. Proteomics: the move to mixtures. J Mass Spectrom. 2001;36:1083–1091. doi: 10.1002/jms.229. [DOI] [PubMed] [Google Scholar]
- 10.Schwartz JC, Senko MW, Syka JE. A two-dimensional quadrupole ion trap mass spectrometer. J Am Soc Mass Spectrom. 2002;13:659–669. doi: 10.1016/S1044-0305(02)00384-7. [DOI] [PubMed] [Google Scholar]
- 11.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 12.Gatlin CL, Kleemann GR, Hays LG, Link AJ, Yates JR., III Protein identification at the low femtomole level from silver-stained gels using a new fritless electrospray interface for liquid chromatography-microspray and nanospray mass spectrometry. Anal Biochem. 1998;263:93–101. doi: 10.1006/abio.1998.2809. [DOI] [PubMed] [Google Scholar]
- 13.Han DK, Eng J, Zhou H, Aebersold R. Quantitative profiling of differentiation-induced microsomal proteins using isotope-coded affinity tags and mass spectrometry. Nat Biotechnol. 2001;19:946–951. doi: 10.1038/nbt1001-946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peng J, Elias JE, Thoreen CC, Licklider LJ, Gygi SP. Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC MS/MS) for large-scale protein analysis: the yeast proteome. J Proteome Res. 2003;2:43–50. doi: 10.1021/pr025556v. [DOI] [PubMed] [Google Scholar]
- 15.Elias JE, Gibbons FD, King OD, Roth FP, Gygi SP. Intensity-based protein identification by machine learning from a library of tandem mass spectra. Nat Biotechnol. 2004;22:214–219. doi: 10.1038/nbt930. [DOI] [PubMed] [Google Scholar]
- 16.Spahr CS, Davis MT, McGinley MD, Robinson JH, Bures EJ, Beierle J, Mort J, Courchesne PL, Chen K, Wahl RC, Yu W, Luethy R, Patterson SD. Towards defining the urinary proteome using liquid chromatography-tandem mass spectrometry. I Profiling an unfractionated tryptic digest. Proteomics. 2001;1:93–107. doi: 10.1002/1615-9861(200101)1:1<93::AID-PROT93>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 17.Yi EC, Marelli M, Lee H, Purvine SO, Aebersold R, Aitchison JD, Goodlett DR. Approaching complete peroxisome characterization by gas-phase fractionation. Electrophoresis. 2002;23:3205–3216. doi: 10.1002/1522-2683(200209)23:18<3205::AID-ELPS3205>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 18.Goodlett DR, Bruce JE, Anderson GA, Rist B, Pasa-Tolic L, Fiehn O, Smith RD, Aebersold R. Protein identification with a single accurate mass of a cysteine-containing peptide and constrained database searching. Anal Chem. 2000;72:1112–1118. doi: 10.1021/ac9913210. [DOI] [PubMed] [Google Scholar]
- 19.Patterson SD. How much of the proteome do we see with discovery-based proteomics methods and how much do we need to see? Curr Proteomics. 2004;1:3–12. [Google Scholar]
- 20.Florens L, Washburn MP, Raine JD, Anthony RM, Grainger M, Haynes JD, Moch JK, Muster N, Sacci JB, Tabb DL, Witney AA, Wolters D, Wu Y, Gardner MJ, Holder AA, Sinden RE, Yates JR, Carucci DJ. A proteomic view of the Plasmodium falciparum life cycle. Nature. 2002;419:520–526. doi: 10.1038/nature01107. [DOI] [PubMed] [Google Scholar]
- 21.Link AJ, Eng J, Schieltz DM, Carmack E, Mize GJ, Morris DR, Garvik BM, Yates JR., III Direct analysis of protein complexes using mass spectrometry. Nat Biotechnol. 1999;17:676–682. doi: 10.1038/10890. [DOI] [PubMed] [Google Scholar]
- 22.Syka JE, Marto JA, Bai DL, Horning S, Senko MW, Schwartz JC, Ueberheide B, Garcia B, Busby S, Muratore T, Shabanowitz J, Hunt DF. Novel linear quadrupole ion trap/FT mass spectrometer: performance characterization and use in the comparative analysis of histone H3 post-translational modifications. J Proteome Res. 2004;3:621–626. doi: 10.1021/pr0499794. [DOI] [PubMed] [Google Scholar]
- 23.Olsen JV, Ong SE, Mann M. Trypsin cleaves exclusively C-terminal to arginine and lysine residues. Mol Cell Proteomics. 2004;3:608–614. doi: 10.1074/mcp.T400003-MCP200. [DOI] [PubMed] [Google Scholar]
- 24.Le Blanc JC, Hager JW, Ilisiu AM, Hunter C, Zhong F, Chu I. Unique scanning capabilities of a new hybrid linear ion trap mass spectrometer (Q TRAP) used for high sensitivity proteomics applications. Proteomics. 2003;3:859–869. doi: 10.1002/pmic.200300415. [DOI] [PubMed] [Google Scholar]
- 25.Beausoleil SA, Jedrychowski M, Schwartz D, Elias JE, Villen J, Li J, Cohn MA, Cantley LC, Gygi SP. Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc Natl Acad Sci U S A. 2004;101:12130–12135. doi: 10.1073/pnas.0404720101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olsen JV, Mann M. Improved peptide identification in proteomics by two consecutive stages of mass spectrometric fragmentation. Proc Natl Acad Sci U S A. 2004;101:13417–13422. doi: 10.1073/pnas.0405549101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Syka JE, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci U S A. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]