

Abstract
Botulinum toxin is the etiologic agent responsible for the disease botulism, which is characterized by peripheral neuromuscular blockade. Botulism is ordinarily encountered as a form of oral poisoning. The toxin is absorbed from the lumen of the gut to reach the general circulation and is then distributed to peripheral cholinergic nerve endings. However, there is a widespread presumption that botulinum toxin can also act as an inhalation poison, which would require that it be absorbed from the airway. Experiments have been done to show that both pure toxin and progenitor toxin (a complex with auxiliary proteins) are inhalation poisons. Interestingly, the data indicate that auxiliary proteins are not necessary to protect the toxin or to facilitate its absorption. When studied on rat primary alveolar epithelial cells or on immortalized human pulmonary adenocarcinoma (Calu-3) cells, botulinum toxin displayed both specific binding and transcytosis. The rate of transport was greater in the apical-to-basolateral direction than in the basolateral-to-apical direction. Transcytosis was energy dependent, and it was blocked by serotype-specific antibody. The results demonstrated that the holotoxin was not essential for the process of binding and transcytosis. Both in vivo and in vitro experiments showed that the heavy-chain component of the toxin was transported across epithelial monolayers, which indicates that the structural determinants governing binding and transcytosis are found in this fragment. The heavy chain was not toxic, and therefore it was tested for utility as an inhalation vaccine against the parent molecule. This fragment was shown to evoke complete protection against toxin doses of at least 104 times the 50% lethal dose.
Botulinum neurotoxin (BoNT), which is produced by the organisms Clostridium botulinum, Clostridium beratii, and Clostridium butyricum, is synthesized as a single-chain polypeptide with a molecular mass of ca. 150 kDa (29). This relatively inactive protoxin undergoes posttranslation modification, during which a trypsin-like enzyme cleaves (nicks) the single-chain molecule to generate a dichain molecule in which a heavy-chain polypeptide (ca. 100 kDa) is linked by a disulfide bond to a light-chain polypeptide (ca. 50 kDa). The dichain molecule is the agent responsible for the disease botulism.
BoNT is synthesized in seven immunologically distinct forms designated A, B, C1, D, E, F, and G (25, 26). Under natural conditions, each of these serotypes is released from clostridia in association with two classes of proteins: a family of hemagglutinins (HA) and a single nontoxin, nonhemagglutinin (NTNH) (24). The complex that is composed of a toxin and noncovalently associated proteins is known as progenitor toxin. Each of the serotypes responsible for human illness (mainly A, B, and E) forms a unique progenitor complex with associated proteins, but these associated proteins play no role in the signs and symptoms characteristic of clinical botulism (10, 15, 22).
The target organ for BoNT is the cholinergic nerve ending (4). Substantial progress has been made in defining the cellular and subcellular actions of the toxin, beginning with the discovery that the toxin progresses through a sequence of well-defined steps. These include binding to the plasma membrane, penetration of the plasma membrane by receptor-mediated endocytosis, penetration of the endosome membrane by pH-induced translocation, and intracellular expression of enzymatic activity (30). BoNT is a zinc-dependent endoprotease that cleaves several polypeptides that are essential for exocytosis (12, 14). Toxin-induced cleavage of these substrates leads to blockade of transmitter release from peripheral cholinergic neurons, and this in turn accounts for all major signs and symptoms of botulism, including flaccid paralysis and respiratory arrest.
For BoNT to cause poisoning, it must first enter the body and gain access to nerve endings. Most cases of botulism are oral in nature, and therefore the toxin must escape the lumen of the gut and reach the general circulation. A number of reports indicate that toxin movement across gut membranes likely occurs in the small intestine (2, 31). Recent work on isolated and polarized human gut epithelial cells has provided additional information about this phenomenon (20). BoNT binds to serotype-specific receptors on the apical (mucosal) sides of cells, undergoes receptor-mediated endocytosis and transcytosis, and is ultimately released on the basolateral (serosal) sides of cells. The intact 150-kDa holotoxin is not essential for the process of binding and transcytosis; the 50-kDa carboxy-terminal half of the heavy chain is sufficient to mediate this process. Furthermore, the entire message for ligand binding resides within the toxin molecule. Associated proteins, such as HA and NTNH, are not essential for the toxin to bind or penetrate gut epithelial cells (21).
In contrast to the volume of work on the gastrointestinal system, little work has been done on BoNT and the respiratory system. This is due to the fact that, in the past, clinical botulism has been recognized as a form of oral poisoning rather than inhalation poisoning (but see reference 13). Therefore, in this report, an attempt has been made to evaluate the respiratory system as a portal for toxin entry into the body. More precisely, this report (i) characterizes the process of inhalation poisoning by the toxin, (ii) identifies a likely mechanism for transcytosis of the toxin from the airway lumen to the general circulation, and (iii) describes a mechanism for using the inhalation route to induce immunity against the toxin. In each case, new data on toxin interaction with airway epithelial cells are compared with existing data on toxin interaction with gut epithelial cells.
MATERIALS AND METHODS
Materials.
Human pulmonary adenocarcinoma (Calu-3) cells and Madin-Darby canine kidney (MDCK) cells were obtained from the American Type Culture Collection (Manassas, Va.). Tissue culture media and sera were purchased from Life Technologies, Inc. (Baltimore, Md.), and the American Type Culture Collection. [3H]inulin and 125I-Bolton-Hunter reagent were purchased from NEN Life Science Products (Boston, Mass.). Sephadex G-25 gel filtration columns and Western blotting detection reagent were obtained from Amersham Pharmacia Biotech (Piscataway, N.J.). DNase I, rat tail collagen type I, and rat immonuglobulin G (IgG) were purchased from Sigma Chemical Co. (St. Louis, Mo.). Elastase was purchased from Worthington Biochemical Corporation (Worthington, N.J.). Other reagents were purchased from Sigma Chemical Co., and additional laboratory supplies were obtained from Fisher Scientific (Malvern, Pa.).
Animals.
Female Swiss-Webster mice (25 to 30 g) and male Sprague-Dawley rats (125 to 150 g), which were purchased from Ace Animals (Boyertown, Pa.), were housed in an accredited animal colony (American Association for Accreditation of Laboratory Animal Care) and allowed unrestricted access to food and water. All procedures involving animals were reviewed and approved by the Thomas Jefferson University Institutional Animal Care and Use Committee.
Toxin.
BoNT type A (BoNT/A) and its heavy-chain component (HC) were purified to homogeneity by procedures decribed in the literature (8, 27). Progenitor BoNT/A was purchased from WAKO Fine Chemicals (Dallas, Tex.). Protein concentrations were routinely determined by the Bradford assay (3). For progenitor BoNT/A and pure BoNT/A, concentrations were determined spectrophotometrically using the relationships 1.66 A278 = 1 mg/ml and 1.63 A278 = 1 mg/ml, respectively, where A278 is the absorbance at 278 nm (8).
Iodination of protein.
BoNT/A and the HC were iodinated using 125I-Bolton-Hunter reagent essentially according to the manufacturer's instructions, as described previously (20). The reaction time was reduced to diminish loss of toxicity of the resulting product. The toxin was labeled to an average specific activity of 500 Ci/mmol with a residual toxicity of >90%. Sample protein concentrations and associated counts were used to calculate the specific activity.
Cell culture.
Calu-3 cells were grown in Eagle's minimum essential medium supplemented with 10% newborn calf serum, 100 U of sodium penicillin G/ml, and 100 μg of streptomycin/ml. MDCK cells were grown in Dulbecco's modified Eagle's medium supplemented with 5% newborn calf serum, 100 U of sodium penicillin G/ml, and 100 μg of streptomycin/ml. The cultures were maintained at 37°C in 5% CO2. Calu-3 cells were fed every 3 days and passaged (1:2) when they were 80 to 90% confluent, approximately every 7 days. MDCK cells were fed every 3 days and passaged (1:5) when they were 95% confluent, approximately every 3 days.
Isolation and culture of alveolar epithelial cells.
Primary cultures of rat alveolar epithelial cells, which establish polarized monolayers with tight junctions (≥1,500 Ω/cm2), were prepared as described in detail elsewhere (16, 17). Briefly, the lungs of pathogen-free male Sprague-Dawley rats (125 to 150 g) were perfused via the pulmonary artery, lavaged with Ca2+- and Mg2+-free Ringer's solution, and exteriorized. The isolated lungs were instilled intratracheally with porcine pancreatic elastase (2.5 U/ml) and gently agitated for 30 min at 37°C. The elastase-treated lungs were minced, and the resulting crude cell mixtures were filtered sequentially through 150-, 37-, and 15-μm-pore-size filters (Cross Wire Cloth Manufacturing Co. Bellmawr, N.J.). The filtered cell mixture was purified by adhering blood-borne cells, which contain Fc receptors, to immobilized IgG (9). Nonadhering cells (>90% type II pneumocytes) were pooled and pelleted at 100 × g for 10 min, and the pellet was resuspended in culture medium consisting of Eagle's modified essential medium supplemented with 100 U of sodium penicillin G/ml, 100 μg of streptomycin/ml, and 10% total calf serum. The cells were further purified by panning them on tissue culture petri dishes in an incubator (5% CO2 in air) at 37°C for 45 min. Nonadhering cells were pooled and seeded onto tissue culture-treated polycarbonate filter cups (Transwell; Corning-Costar, Cambridge, Mass.) at a density of 1.5 × 106 cells/cm2. The media were changed on the third day after plating and every other day thereafter. The cultures were maintained in a humidified 5% CO2 incubator at 37°C. Cell viability (>90%) was measured by trypan blue dye exclusion.
Transcytosis assay.
For assay of transcytosis, cells were grown in Transwell porous-bottom dishes on polycarbonate membranes with a 0.4-μm pore size. Prior to being seeded with cells, the Transwell dishes were coated with 10 μg of rat tail collagen type I/cm2 as described previously (20). The coated wells were allowed to dry at room temperature overnight. After drying, the wells were sterilized under UV light for 1 h, followed by preincubation with cell culture medium (30 min) (see above). The preincubation medium was removed immediately prior to the addition of cells and fresh medium.
Calu-3 cells and MDCK cells were plated at confluent density (∼1.5 × 105 cells) into Transwell dishes with 0.5 ml of medium in the upper chamber and 1.0 ml in the lower chamber. In these experiments, medium in the upper chamber bathed the apical surfaces of the cells, and medium in the lower chamber bathed the basolateral surfaces of the cells. The culture medium was changed every 2 days. The cultures were allowed to differentiate for a minimum of 10 days before assay of transcytosis.
The transcytosis assay was initiated by adding 10−8 M 125I-labeled BoNT/A or HC to either the upper or the lower chamber. Transport of radiolabeled molecules was monitored by collecting all of the medium from the appropriate chamber. An aliquot (0.5 ml) from each sample was filtered through a Sephadex G-25 column, and 0.5-ml fractions were collected. The amount of radioactivity in the fractions was determined in a γ counter. Labeled molecules eluted at void volume, and the radioactivity contained in the void volume fractions was summed to determine the total amount of protein present.
The energy dependence of the transcytosis process was confirmed by examining the phenomenon at low temperature. Calu-3 monolayers were preincubated on ice for 1 h and then washed three times with iced phosphate-buffered saline. The monolayers were placed in a Transwell apparatus, and iced medium was placed in both chambers. The Transwell plates were transferred to an aerobic jar in which the temperature was maintained at 4°C and CO2 tension was maintained with a CampyPak (Becton Dickinson, Cockeysville, Md.).
Monolayer permeability and integrity.
The transepithelial electrical resistance (TEER) and inulin transport were measured to evaluate the structural integrity of Calu-3, MDCK, and alveolar epithelial cells for transcytosis experiments. An EVOM Epithelial Voltohmmeter (World Precision Instruments, Sarasota, Fla.) was used to record electrical resistance across the cell monolayers before and after each experiment. A polycarbonate membrane immersed in medium was used as a control, and resistance was calculated in ohms per square centimeter.
To control for membrane integrity and to estimate diffusion between cells, the rate of passage of [3H]inulin from the upper to the lower reservoir was determined essentially as described previously (19). According to this procedure, the media in both reservoirs contained unlabeled 1 mM inulin. The assay was initiated by adding 1 mM [3H]inulin to the upper reservoir. The rates of inulin flux from the upper to the lower chambers were measured over a period of 18 h.
Structure of transcytosed material.
Polyacrylamide gel electrophoresis was combined with autoradiography to confirm that transcytosed toxin maintained its native structure. 125I-BoNT/A was added to the apical sides of Calu-3 monolayers, and medium on the basolateral sides was collected 18 h later. The media from 12 wells were combined and then concentrated using a Centriplus centrifugal filter device (Millipore, Bedford, Mass.). The concentrate was filtered as described above on a Sephadex G-25 column, and the labeled toxin that eluted at void volume was concentrated again using a Centricon microconcentrator (Millipore). Samples were separated on 8% polyacrylamide gels under reducing and nonreducing conditions. The gels were fixed for 30 min in destaining solution, dried on blotting paper, and exposed to Biomax film (Kodak, Rochester, N.Y.).
Administration of BoNT/A and HC.
Proteins were administered to animals in three different protocols: (i) pharmacokinetics of absorption, (ii) induction of immunity, and (iii) toxicity assay. For each protocol, BoNT/A or the HC was administered by the intranasal (i.n.) or intraperitoneal (i.p.) route. For i.n. administration, mice were lightly anesthetized with isoflurane (Iso-thesia; Abbott Laboratories, North Chicago, Ill.). Protein was administered by a single application of a 20-μl solution to the nares. The heads of the animals were maintained in an upright position to minimize drainage into the posterior pharynx. For i.p. administration, animals received a single injection of 0.1 ml. For both i.n. and i.p. administration, proteins were given in a phosphate-buffered saline solution (pH 7.0).
During pharmacokinetic experiments, mice (n = 5 per group) received 36.4 μg of 125I-BoNT/A or 24.4 μg of 125I-HC/kg of body weight by the i.n. route. Individual groups were sacrificed with CO2 at 1, 2, 3, 4, or 6 h, and blood was collected by cardiac puncture. Plasma was separated from the blood by centrifugation at 3,000 × g for 10 min and then stored at −20°C until it was assayed. Plasma samples (100 μl) were subsequently mixed with phosphate-buffered saline (400 μl) and filtered through a Sephadex G-25 column. Fractions (0.5 ml) were collected, and the amount of radioactivity in the fractions was measured in a γ counter. Labeled BoNT/A and HC eluted at void volume, and the radioactivity contained in the void volume fractions was summed to determine the total amount of protein present.
For immunization experiments, mice (n = 5 per group) received 0.1, 1.0, or 10 μg of the purified HC by the i.n. route, as described above. The protein was administered on days 0, 7, 14, 21, and 28. The animals were challenged i.p. with 3 × 104 50% lethal doses (LD50) of BoNT/A on day 35 and then observed for an additional 5 days.
Toxicity experiments were done to compare the relative potencies of progenitor BoNT/A and pure BoNT/A by the i.n. route. The techniques for protein administration were the same as those described above. The doses of protein used in toxicity assays were 0.1, 1, or 10 μg of BoNT/A or 0.267, 2.67, or 26.7 μg of progenitor toxin A per mouse. The animals (n = 15 per group per dose) were observed for 15 days.
Preparation of anti-BoNT/A antiserum.
Mice were injected i.p. with 5 μg of HC mixed with Freund's complete adjuvant (50:50). The animals subsequently received 5 μg of HC on days 14 and 28 and 5 μg of BoNT/A on days 42 and 56, each with Freund's incomplete adjuvant (50:50).
ELISAs.
Enzyme-linked immunosorbent assays (ELISAs) were performed as described by Siegel (28) with only minor modifications. Highly purified (>95%) BoNT/A was diluted to 5 μg/ml in phosphate-buffered saline, pH 7.4, and then added to microtiter plates (100 μl/well) that were incubated at 4°C overnight in a sealed container. One percent bovine serum albumin in Tris-buffered saline with 0.1% Tween 20 was used to block nonspecific binding. Serum samples were initially diluted 1:30 and then serially diluted fourfold for a total of seven dilutions (1:30 to 1:122,880). The diluted sera were added in duplicate to toxin-coated wells (100 μl/well). The secondary antibody was alkaline phosphate-conjugated goat anti-human IgA or IgG diluted 1:1,000. The primary and secondary antibodies were incubated for 60 min at 37°C. p-Nitrophenyl phosphate (100 μl/well) was added as a substrate. The plates were incubated at room temperature for 30 min, and absorbance was measured with a microplate reader at 405 nm. ELISA titers were defined as the reciprocal of the highest serum dilution giving an absorbance of 0.2 (U) above background.
Statistical analyses.
All data are presented as means ± standard deviations. The equality of variances was tested using the t test.
RESULTS
BoNT/A is an inhalation poison.
Most cases of naturally occurring botulism are oral in nature, and they can be characterized as either primary intoxication or primary infection followed by secondary intoxication. In primary intoxication, patients ingest food that is tainted with preformed toxin. In primary infection, patients ingest clostridia that colonize the gut and produce the toxin in situ.
Experiments were done to determine whether the respiratory system could serve as a portal of entry for primary intoxication. For this purpose, mice were challenged with three doses of pure BoNT or progenitor BoNT. Challenge doses were administered i.n. and i.p.
As shown in Fig. 1, pure BoNT is an inhalation poison. The potency of the material is substantial, though not as great as when the portal of entry is bypassed and the toxin is administered directly into the peritoneal cavity. A closer examination of the data reveals another interesting point. Figure 1 plots the geometric means of survival times versus the log10 of the administered dose. The resulting relationship is apparently linear over the range of doses tested. This is similar to the outcome that has been reported for oral, i.p., and intravenous administration of toxin.
FIG. 1.

Relationship between concentration of BoNT/A and mean survival time of mice. I.p. and i.n. administrations were studied, and each point represents the average value (plus standard deviation) obtained with 15 mice.
Experiments were also done to compare the potency of pure BoNT with that of progenitor BoNT when administered i.n. Interestingly, the presence of HA and NTNH in the progenitor complex did not enhance toxicity. These data make it clear that it is the pure neurotoxin that is an inhalation poison.
Transcytosis of pure BoNT/A.
Human gut epithelial cells grown in monolayer cultures have the ability to bind and transcytose BoNT/A (20). This phenomenon operates in both the apical-to-basolateral (A→B) and basolateral-to-apical (B→A) directions, although the former is more efficient. Experiments were done to determine whether human pulmonary adenocarcinoma (Calu-3) cells, when allowed to differentiate and polarize, would behave in a similar manner. Experiments were also done with MDCK cells, which have the ability to differentiate, polarize, and transcytose certain substances but which have not been implicated in BoNT/A action.
125I-BoNT/A (10−8 M) was added either to the upper (viz., apical) or lower (viz., basolateral) sides of Calu-3 monolayers in a Transwell apparatus, and the rate of appearance of iodinated toxin on the contralateral side was monitored over a period of 18 h. Interestingly, transcytosis of BoNT/A was detected in both directions across the Calu-3 monolayers, and the rate of transcytosis was apparently constant over the duration of the experiment (Fig. 2A). The actual rate constants, which are given in Table 1, indicate that the unidirectional flux of 125I-labeled BoNT/A in the A→B direction was approximately twofold greater than that in the B→A direction (P < 0.01).
FIG. 2.

Time course of BoNT/A transport in A→B and B→A directions across Calu-3 cells (A) and rat alveolar epithelial cells (B). Monolayers were exposed for various times at 37°C to 125I-BoNT/A (10−8 M), which was added to the donor medium. At the indicated times, receptor media were removed and measured for radioactivity as described in Materials and Methods. Each data point represents an average (plus standard deviation) of three separate experiments with three replicates each.
TABLE 1.
Transcytosis rates of BoNT/A and HC across Calu-3 and rat alveolar epithelial cells
| Molecule | Cells | Direction | Transcytosis rate (fmol/h/cm2) |
|---|---|---|---|
| BoNT/A | Calu-3 | A→B | 0.422 ± 0.076 |
| B→A | 0.206 ± 0.037 | ||
| Rat alveolar epithelial | A→B | 0.376 ± 0.014 | |
| B→A | 0.159 ± 0.027 | ||
| MDCK | A→B | 0.060 ± 0.014 | |
| B→A | 0.045 ± 0.006 | ||
| HC | Calu-3 | A→B | 0.198 ± 0.007 |
| B→A | 0.112 ± 0.033 | ||
| Rat alveolar epithelial | A→B | 0.140 ± 0.050 | |
| B→A | 0.132 ± 0.026 |
BoNT/A was also transported across MDCK cells, but the rate of transcytosis was substantially less than that in Calu-3 cells (Table 1). This reinforces an earlier observation that transcytosis of BoNT/A across MDCK cells was much lower than that across gut epithelial (T-84) cells (20).
Additional experiments were done to determine whether a primary cell line of rat alveolar origin would bind and transcytose the toxin. Cells were isolated and plated as described in Materials and Methods, and they were used in transcytosis experiments as described above for cell lines. Figure 2B shows the results of experiments in which 125I-BoNT/A (10−8 M) was added to either the apical or basolateral sides of monolayers. The rate constants calculated from the data (Table 1) indicate that (i) transcytosis in the A→B direction was greater than that in the B→A direction (P < 0.001) and (ii) regardless of direction, transcytosis in the Calu-3 cell line was slightly greater than that in the primary cell culture. These rates are highly likely to reflect transcytosis rather than paracellular movement, because monolayers had patent tight junctions (see below).
Structural integrity of transcytosed material.
Active BoNT is a dichain molecule linked by a disulfide bond. When this bond is reduced, the holotoxin cannot poison intact cells. Therefore, an effort was made to confirm that transcytosed material retained its structural integrity.
125I-BoNT/A (10−8 M) was added to the upper wells, and the entire contents of the lower wells were collected after 18 h. This material was concentrated and then submitted to polyacrylamide gel electrophoresis and autoradiography. The results demonstrated that transcytosed toxin was almost entirely in the active dichain form (Fig. 3). When this material was reduced, it yielded an HC and a light-chain component that were indistinguishable from those of native toxin.
FIG. 3.
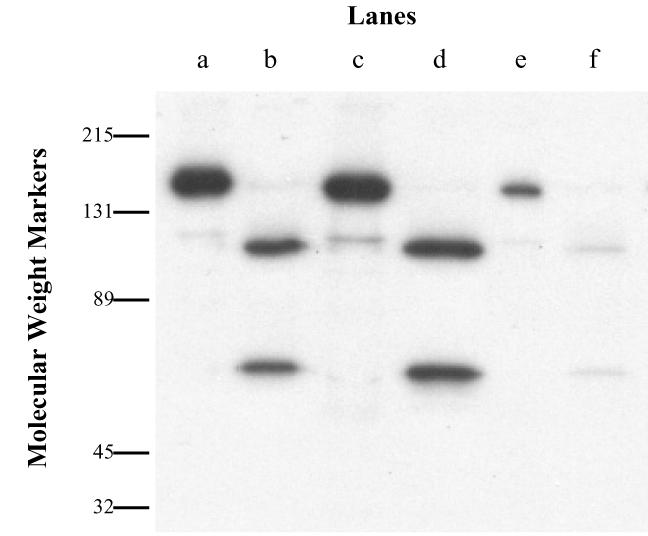
Autoradiogram of transcytosed 125I-BoNT/A across Calu-3 cells. 125I-BoNT/A that had passed through Calu-3 cells was separated on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (10%). Lanes a, c, and e, nonreducing conditions; lanes b, d, and f, reducing conditions; lanes a and b, labeled starting material; lanes c and d, material incubated with medium at 37°C for 18 h; lanes e and f, transcytosed material.
Transcytosis of the HC of BoNT/A.
Earlier work on gut epithelial cells and on cholinergic nerve endings showed that the BoNT/A holotoxin is not necessary for binding and internalization. The HC, and perhaps even the carboxy-terminal half of the HC, may be sufficient (18). However, there is some disagreement about the significance of binding and internalization of the HC. Questions have been raised about whether, at nerve endings, this process is relevant to the course of events in poisoning with holotoxin (7). On the other hand, binding and transcytosis of the HC in gut epithelial cells appears to be similar to that of holotoxin (A. Maksymowych, unpublished observation).
Experiments were done to determine whether the isolated HC of BoNT/A could bind and cross either Calu-3 cells or primary alveolar cells. Transcytosis of the HC, which was measured in both the A→B and B→A directions, was examined in the same manner as transcytosis of BoNT/A. The data for Calu-3 cells, which are shown in Fig. 4A and Table 1, show that (i) 125I-HC was transported in both directions, although the A→B rate was greater than the B→A rate (P < 0.05), and (ii) the rate of transport of the HC was lower than that of BoNT/A (P < 0.01). The primary cultures of alveolar epithelial cells also transported [125I]-HC (Fig. 4B), but there was not a statistically significant difference between the A→B rate and the B→A rate (Table 1). Nevertheless, similarly to Calu-3 cells, the rate of transport of holotoxin in primary cells was slightly greater than that of the HC (P < 0.001).
FIG. 4.

Time course of HC transport in A→B and B→A directions across Calu-3 cells (A) and rat alveolar epithelial cells (B). Monolayers were exposed for various times at 37°C to 125I-HC (10−8 M), which was added to the donor medium. At the indicated times, receptor media were removed and measured for radioactivity as described in Materials and Methods. Each data point represents an average (plus standard deviation) of three separate experiments with three replicates each.
Integrity of polarized monolayers and movement of toxin across monolayers.
There are two broad categories of mechanisms by which a large protein such as BoNT can cross a monolayer: (i) active transcellular movement, such as transcytosis, and (ii) inactive paracellular movement, such as simple diffusion. Earlier work on BoNT/A movement across polarized monolayers of gut epithelial cells demonstrated the involvement of an active transport process. The work presented above suggests that BoNT/A can cross polarized monolayers of alveolar cells by a similar mechanism. Four additional types of experiments were done to demonstrate that this assumption is correct: (i) measurement of transcellular resistance, (ii) monitoring of paracellular movement of [3H]inulin, (iii) determining the effect of low temperature on BoNT/A movement, and (iv) determining the effect of serotype-specific antibodies on BoNT/A movement.
The principal barrier to paracellular movement is the formation of tight junctions. When tight junctions are formed, there is a pronounced increase in TEER. Daily measurements of TEER in monolayers of Calu-3 cells and MDCK cells revealed that a high level of resistance (>1,000 Ω/cm2) was attained by day 10, and this high level of resistance was normally maintained for ∼14 days. Monolayers of primary alveolar cells reached a TEER of ∼1,500 Ω/cm2 by day 4, and this was maintained for ∼2 days. For all experiments described in this report, monolayers were used only when they displayed a TEER of >1,000 Ω/cm2.
The structural integrity of the monolayer was further confirmed by monitoring the movement of [3H]inulin from the apical to the basolateral sides of cells in a Transwell apparatus. The experiments were done identically to those reported earlier for BoNT, except that a higher concentration of inulin was employed (1 mM). The average values obtained in three experiments indicated that the A→B rate in Calu-3 cells fell within the range of 0.01 to 0.07 nmol/h/cm2. The A→B rates for MDCK and primary alveolar cells were even lower. These values are a clear indication that paracellular diffusion of inulin was hindered, and thus, that the structural integrity of cells and tight junctions was intact (see Discussion).
The next set of experiments was designed to test the premise that movement of toxin across the polarized monolayer is an energy-dependent process. This was done by comparing the rates of transcytosis at physiological temperature (37°C) and at a low temperature (4°C). As shown in Fig. 5, reducing the temperature had a profound effect, decreasing transcytosis by ca. 81% (P < 0.001).
FIG. 5.

Effects of temperature and preincubation with BoNT/A-specific antiserum on A→B BoNT/A flux across Calu-3 cells. Each data point represents an average (plus standard deviation) of three separate experiments with three replicates each.
Finally, work was done to show that the movement of toxin across the monolayer was dependent on structural determinants within the toxin molecule, which would serve as another indicator of an active process as opposed to a passive one. For this purpose, 125I-BoNT/A was preincubated with either nonimmune serum or immune serum (1 h; 37°C). The incubation mixture was then added to upper wells, and the amount of transcytosis was measured over an 18-h period. The data showed that there was a slight reduction in transcytosis with nonimmune serum (ca. 20%) (Fig. 5). On the other hand, preincubation with immune serum reduced transcytosis by ca. 76% (P < 0.001).
Systemic absorption of BoNT/A and HC after i.n. administration.
In vivo studies with mice (Fig. 1) demonstrated that BoNT/A is an inhalation poison. Transcytosis experiments (Fig. 2 and 4 and Table 1) demonstrated that both BoNT/A and its HC are actively transported from the apical to the basolateral side of polarized alveolar epithelial cells. Therefore, additional experiments were done to quantify both the time course and the amount of BoNT/A and HC appearance in blood following i.n. administration.
Animals (n = 5 per group per data point [Fig. 6]) were given either 125I-BoNT/A or 125I-HC by inhalation, and at various times after protein administration, animals were sacrificed and blood was collected. The animals that received BoNT/A were monitored for 3 h, at which time toxin-induced flaccid paralysis was apparent. The animals that received the HC were monitored for 6 h, and as expected, they did not develop flaccid paralysis or any other adverse effects.
FIG. 6.

Concentrations of BoNT/A and HC in plasma after i.n. administration of 125I-BoNT/A (36.4 μg/kg) or 125I-HC (24.4 μg/kg). The values represent the means (± standard deviations) for five mice.
As shown in Fig. 6, the time point for maximum protein concentration in the blood was ∼2 h. However, the peak value for BoNT/A was higher than that for the HC. For both proteins, there was rapid clearance after attainment of peak values.
Evaluation of HC as an inhalation vaccine against BoNT/A.
There is an extensive literature demonstrating that the HCs of BoNT and tetanus toxin are the most immunogenic portions of the holotoxins, and furthermore, that these components can be used as parenteral vaccines against the parent molecules (1, 5, 23). The data in Fig. 6 indicate that the HC of BoNT/A can be administered by the i.n. route to reach the general circulation, and it does not evoke toxicity. Therefore, this component was tested as a potential inhalation vaccine.
Mice (n = 5 per group) received various doses of the HC (0.1, 1.0, and 10 μg/mouse) at 0, 1, 2, 3, and 4 weeks. Each dose was administered by the i.n. route. At 5 weeks, all animals were challenged with 3 × 104 LD50 of BoNT/A by the i.p. route. Animals that had not been immunized with the HC died within 1 h of challenge, and the same result was obtained with animals that received 0.1 μg/mouse. However, 20% of the animals that received 1.0 μg survived, and 100% of the animals that received 10 μg survived (Fig. 7). In view of the substantial challenge dose that was administered, the protection at the high dose of the HC was impressive.
FIG. 7.

Survival rate and anti-BoNT/A IgG responses after i.n. administration of HC. Mice were immunized i.n. with three different doses of the HC on days 0, 7, 14, 21, and 28. Challenges with 3 × 104 LD50 of BoNT/A were given i.p. on day 35. Serum samples were also taken on day 35, and serum antibody titers were determined by a BoNT/A-specific IgG ELISA. The values represent the means (plus standard deviations) for five mice.
Animals that had been immunized with the HC were also used to measure evoked levels of IgG. Similar to the challenge experiments, the IgG experiments yielded a dose-response curve. The ELISA titer at the 1-μg dose was 0.93 log10 units, and the titer at 10 μg was 4.00 log10 units. Thus, the combined data indicate that a sufficient titer of IgG, and resistance to challenge with 3 × 104 LD50, occurs at an inhalation dose between 1 and 10 μg.
DISCUSSION
There are a variety of reasons for studying the interaction between BoNT and the respiratory system, two of which are especially compelling. First, the possibility that the toxin can be absorbed through the respiratory system means that special attention must be focused on this phenomenon in the context of bioterrorism. Oral poisoning due to BoNT has long been recognized as a potential threat, but the potential threat due to inhalation poisoning has not been widely appreciated. The data presented in this report show that both oral and inhalation poisoning should be sources of concern. On a completely different note, the possibility that the toxin—or a fragment derived from it—can penetrate epithelial membranes raises the possibility that the toxin can be reengineered to create an inhalation vaccine. More precisely, a derivative could be administered at doses that trigger a substantial IgG response without eliciting any adverse effects. This would represent a completely unique therapeutic application of BoNT. Though strikingly different in their implications for human health and well being, both the potential threat as an agent of bioterrorism and the potential utility as an inhalation vaccine serve as strong motivation to develop a better understanding of BoNT interaction with the respiratory system.
BoNT/A is an inhalation poison.
The overwhelming majority of cases of naturally occurring botulism can be traced to an oral etiology (i.e., primary intoxication or primary infection with secondary intoxication). Furthermore, virtually all known cases of the disease are due to progenitor toxin rather than pure toxin (but see below). The ability of progenitor toxin to produce naturally occurring poisoning is linked to two of its characteristics. First, the intertwined structure of the progenitor toxin is highly resistant to the harsh conditions of low pH and proteolytic enzymes in the gut (2, 6, 31). Second, the toxin itself is capable of binding to receptors on epithelial cells that mediate internalization and transcytosis (20). Thus, the complex of the toxin and associated proteins has both the ability to survive degradation in the gut and the ability to escape the lumen and reach the general circulation.
It is important to note that the role of associated proteins is relative rather than absolute. For example, laboratory animal experiments have shown that progenitor toxin is a very potent oral poison, but higher doses of pure toxin can also act as an oral poison (21). As another example, at least one episode of human poisoning has been attributed to a strain of clostridia that does not possess the gene for HA (Eric Johnson, personal communication). Thus, that protein could not have contributed to poisoning.
The fact that BoNT/A by itself can penetrate gut epithelial cells raises the obvious question of whether it might penetrate other epithelial cells, and especially those that serve as a boundary between the outside world and the general circulation. Also, the fact that associated proteins play the principal role of protecting the toxin from low pH and proteolytic enzymes raises the question of whether they would be essential in portals of entry that do not have such harsh conditions. For obvious reasons, airway epithelial cells and the inhalation route are logical choices as cells and systems in which to answer these questions about BoNT.
The literature on BoNT as an inhalation poison is very limited (13). Indeed, there has never been a publication that fully describes the phenomenon of primary intoxication. Nevertheless, there has been a general acceptance of two premises: (i) the toxin can act by the inhalation route, and (ii) most research dealing with this phenomenon is unpublished work related to bioterrorism.
In the present study, an attempt has been made to begin the process of characterizing the interaction between BoNT/A and the respiratory system. This has involved experiments on homogeneous epithelial cell lines of human alveolar origin and a primary epithelial cell culture of rat alveolar origin and inhalation studies of mice. The work done here on Calu-3 cells and primary cell cultures shows that pure BoNT/A can cross polarized monolayers that have tight junctions. Work done in vivo shows that both progenitor toxin and pure toxin can cross epithelial membranes and cause toxicity. Somewhat less neurotoxin crossed cells when it was in complex with associated proteins. Similar findings have been obtained when the abilities of progenitor toxin and pure toxin to cross intestinal cells have been compared (A. Maksymowych, personal communication).
It is highly likely that toxin crossed airway cells by transcytosis rather than paracellular movement. Several lines of evidence support this conclusion, including the following: (i) TEER measurements demonstrated a high level of resistance, which is consistent with patent tight junctions; (ii) paracellular movement of inulin, a molecule that is ∼2 orders of magnitude smaller than BoNT/A, was hindered, and this too is consistent with structurally intact tight junctions; (iii) low temperature greatly diminished the movement of BoNT/A across epithelial cells, which is suggestive of active transport rather than simple diffusion; and (iv) serotype-specific antibody virtually abolished toxin movement across cell layers, making simple diffusion unlikely.
The cumulative data strongly support the premise that BoNT/A is an inhalation poison and that poisoning is due to the active process of binding and transcytosis across airway epithelial cells. In contrast to oral poisoning, inhalation poisoning does not benefit from the presence of auxiliary proteins.
It is unlikely that the ability of the toxin to enter the body through the respiratory system will ever have major significance in the context of naturally occurring poisoning. With the exception of specialized settings, such as laboratories that handle the toxin, the prospects for exposure by inhalation are slight. This concept is amply verified by the clinical literature on the incidence of botulism. However, there is the unfortunate prospect that poisoning could occur in an unnatural manner. The data reported here are consistent with the premise that BoNT could be exploited as an inhalation agent in biological warfare or bioterrorism (11).
The HC of BoNT/A mediates absorption.
Research on gut epithelial cells has shown that the HC is both necessary and sufficient to mediate binding and transcytosis. Work on cholinergic nerve endings, which are the target organ for toxin action, has shown that the HC is necessary for binding, but the question of whether it is sufficient remains unresolved. It has been reported that the HC binds to nerve endings and is internalized, but its intraneuronal fate is different from that of the holotoxin (7).
The work reported here demonstrates that the HC of BoNT/A can penetrate airway epithelial cells both in vivo and in vitro. The HC appears to be slightly less efficient than the holotoxin, but the magnitude of the disparity is not great. The slight difference in efficiency could be due to one of two broad causes: (i) when the HC is separated from the rest of the toxin molecule, its structure is altered in a way that diminishes affinity for receptors on epithelial cells, and (ii) there is some portion of the light chain that acts in concert with the HC to produce maximal binding affinity. Work to further characterize the mechanisms that underlie BoNT/A binding to epithelial cells is under way.
BoNT/A can be modified to create an inhalation vaccine.
The finding that a truncation fragment of BoNT/A can be absorbed by the inhalation route has an immediate and obvious clinical implication. There is a substantial body of research indicating that the HCs of clostridial neurotoxins are immunogenic and that parenteral administration of these compounds can evoke immunity against the parent toxins. Indeed, clinical trials are under way in which the carboxy-terminal half of the HC of BoNT/A is being tested as an injection (subcutaneous) vaccine against native toxin (5). The fact that the HC is an efficient immunogen, combined with the fact that it can be absorbed through the airway, raises the prospect that the HC can serve as an inhalation vaccine against botulism.
In an initial series of experiments, the abilities of three doses of the HC to evoke immunity against a substantial challenge dose of native toxin (3 × 104 LD50; i.p.) were tested. At the highest dose tested (10 μg/animal/administration), there was complete immunity. In relation to these experiments, it should be noted that inhalation administration of the HC leads to systemic appearance of the protein, and the lethal dose of BoNT/A was administered systemically. This suggests that immunity was IgG mediated but does not necessarily exclude a mucosal mechanism. To address the latter possibility, immunity experiments have been initiated in animals that lack Peyer's patches and therefore are deficient in the ability to initiate a mucosal immune response.
Regardless of whether immunity ultimately proves to be systemic, mucosal, or a combination of the two, the data show that the HC can serve as an inhalation vaccine. The resistance that this vaccine evokes could confer protection against botulism that is either naturally occurring or the product of malice.
Acknowledgments
This work was supported in part by National Institutes of Health grants NS22153 and GM57342.
Editor: J. T. Barbieri
REFERENCES arsid6160434
- 1.Atassi, M. Z., and M. Oshima. 1999. Structure, activity, and immune (T and B Cell) recognition of botulinum neurotoxins. Crit. Rev. Immunol. 19:219-260. [PubMed] [Google Scholar]
- 2.Bonventre, P. F. 1979. Absorption of botulinal toxin from the gastrointestinal tract. Rev. Infect. Dis. 1:663-667. [DOI] [PubMed] [Google Scholar]
- 3.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 4.Burgen, A. S. V., F. Dickens, and L. J. Zatman. 1949. The action of botulinum toxin on the neuromuscular junction. J. Physiol. (London) 109:10-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Byrne, M., and L. A. Smith. 2000. Development of vaccines for prevention of botulism. Biochimie 82:955-966. [DOI] [PubMed] [Google Scholar]
- 6.Chen, F., G. M. Kuziemko, and R. C. Stevens. 1998. Biophysical characterization of the stability of the 150-kilodalton botulinum toxin, the nontoxic component, and the 900-kilodalton botulinum toxin complex species. Infect. Immun. 66:2420-2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daniels-Holgate, P. U., and J. O. Dolly. 1996. Productive and nonproductive binding of botulinum neurotoxin A to motor nerve endings are distinguished by its heavy chain. J. Neurosci. Res. 44:263-271. [DOI] [PubMed] [Google Scholar]
- 8.DasGupta, B. R., and V. Sathyamoorthy. 1984. Purification and amino acid composition of type A botulinum neurotoxin. Toxicon 22:415-424. [DOI] [PubMed] [Google Scholar]
- 9.Dobbs, L. G., M. C. Williams, and R. Gonzalez. 1986. An improved method for isolating type II cells in high yield and purity. Am. Rev. Respir. Dis. 134:141-145. [DOI] [PubMed] [Google Scholar]
- 10.Ferrari, N. D., and M. E. Weisse. 1995. Botulism. Adv. Pediatr. Infect. Dis. 10:81-91. [PubMed] [Google Scholar]
- 11.Franz, D. R., P. B. Jahrling, A. M. Friedlander, D. J. McClain, D. L. Hoover, W. R. Bryne, J. A. Pavlin, G. W. Christopher, and E. M. Eitzen. 1997. Clinical recognition and management of patients exposed to biological warfare agents. JAMA 278:399-411. [DOI] [PubMed] [Google Scholar]
- 12.Herreros, J., G. Lalli, C. Montecucco, and G. Schiavo. 2000. Tetanus toxin fragment C binds to a protein present in neuronal cell lines and motoneurons. J. Neurochem. 74:1941-1950. [DOI] [PubMed] [Google Scholar]
- 13.Holzer, V. E. 1962. Botulismus durch inhalation. Med. Klin. 57:1735-1738. [PubMed] [Google Scholar]
- 14.Humeau, Y., F. Doussau, N. J. Grant, and B. Poulain. 2000. How botulinum and tetanus neurotoxins block neurotransmitter release. Biochimie 82:427-446. [DOI] [PubMed] [Google Scholar]
- 15.Johnson, E. A., and M. C. Goodnough. 1998. Botulism, p. 723-741. In L. Coliter, A. Ballows, and M. Sussman (ed.), Topley and Wilson's microbiology and microbial infections: bacterial infections. Arnold, London, United Kingdom.
- 16.Kim, K. J., J. M. Cheek, and E. D. Crandall. 1991. Contribution of active Na+ and Cl− fluxes to net ion transport by alveolar epithelium. Respir. Physiol. 85:245-256. [DOI] [PubMed] [Google Scholar]
- 17.Kim, K. J., D. J. Suh, R. L. Lubman, S. I. Danto, Z. Borok, and E. D. Crandall. 1992. Ion fluxes across alveolar epithelial cell monolayers. J. Tissue Culture Methods 14:187-194. [Google Scholar]
- 18.Lalli, G., J. Herreros, S. L. Osborne, C. Montecucco, O. Rossetto, and G. Schiavo. 1999. Functional characterization of tetanus and botulinum neurotoxin binding domains. J. Cell Sci. 112:2715-2724. [DOI] [PubMed] [Google Scholar]
- 19.Madara, J. L., and K. Dharmsathaphorn. 1985. Occluding junction structure-function relationships in a cultured epithelial monolayer. J. Cell Biol. 101:2124-2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maksymowych, A. B., and L. L. Simpson. 1998. Binding and transcytosis of botulinum neurotoxin by polarized human colon carcinoma cells. J. Biol. Chem. 273:21950-21957. [DOI] [PubMed] [Google Scholar]
- 21.Maksymowych, A. B., M. Reinhard, C. J. Malizio, M. C. Goodnough, E. A. Johnson, and L. L. Simpson. 1999. Pure botulinum neurotoxin is absorbed from the stomach and small intestine and produces peripheral neuromuscular blockade. Infect. Immun. 67:4708-4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maselli, R. A. 1998. Pathogenesis of human botulism. Ann. N. Y. Acad. Sci. 841:122-139. [DOI] [PubMed] [Google Scholar]
- 23.Middlebrook, J. L., and J. E. Brown. 1995. Immunodiagnosis and immunotherapy of tetanus and botulinum neurotoxins. Curr. Top. Microbiol. Immunol. 195:89-122. [DOI] [PubMed] [Google Scholar]
- 24.Oguma, K., Y. Fujinaga, and K. Inoue. 1995. Structure and function of Clostridium botulinum toxins. Microbiol. Immunol. 39:161-168. [DOI] [PubMed] [Google Scholar]
- 25.Popoff, M. R., and J.-C. Marvaud. 1999. Structural and genomic features of clostridial neurotoxins, p. 174-201. In J. E. Alouf and J. Freer (ed.), Bacterial toxins: a comprehensive sourcebook. Academic Press, London, United Kingdom.
- 26.Sakaguchi, G. 1983. Clostridium botulinum toxins. Pharmacol. Ther. 19:165-194. [DOI] [PubMed] [Google Scholar]
- 27.Sathyamoorthy, V., and B. R. DasGupta. 1985. Separation, purification, partial characterization and comparison of the heavy and light chains of botulinum neurotoxin types A, B, and E. J. Biol. Chem. 260:10461-10466. [PubMed] [Google Scholar]
- 28.Siegel, L. S. 1988. Human immune response to botulinum pentavalent (ABCDE) toxoid determined by a neutralization test and by an enzyme-linked immunosorbent assay. J. Clin. Microbiol. 26:2351-2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simpson, L. L. 1989. Botulinum neurotoxin and tetanus toxin. Academic Press, San Diego, Calif.
- 30.Simpson, L. L. 1981. The origin, structure, and pharmacological activity of botulinum toxin. Pharmacol. Rev. 33:155-188. [PubMed] [Google Scholar]
- 31.Sugii, S., I. Ohishi, and G. Sakaguchi. 1977. Intestinal absorption of botulinum toxins of different molecular sizes in rats. Infect. Immun. 17:491-496. [DOI] [PMC free article] [PubMed] [Google Scholar]