

Abstract
Objective
The receptor for advanced glycation end products (RAGE) binds multiple ligands, including S100 proteins, high mobility group box chromosomal protein 1 (HMGB-1), and AGEs, all of which are present in articular cartilage. Stimulation of RAGE signaling can lead to MAP kinase activation and increased NF-κB activity. The objective of the present study was to determine if chondrocytes express functional RAGE.
Methods
The presence of chondrocyte RAGE was analyzed by immunohistochemistry using normal and osteoarthritic (OA) cartilage from young and old monkeys and humans, immunoblotting of chondrocyte lysates and human cartilage extracts, and reverse transcription–polymerase chain reaction (RT-PCR) analysis of RNA from chondrocytes treated with interleukin-1 (IL-1) and fibronectin fragments. RAGE signaling was evaluated by stimulating chondrocytes with S100B and HMGB-1 and analyzing for activation of the ERK MAP kinase and NF-κB. The ability of S100B and HMGB-1 to stimulate matrix metalloproteinase 13 (MMP-13) production was also assessed. A pull-down assay using biotin-labeled S100B was used to demonstrate binding to RAGE.
Results
RAGE was detected in sections of monkey knee cartilage and human knee and ankle cartilage. Increased immunostaining for RAGE was noted in cartilage from older adult monkeys and humans and was further increased in OA tissue. RAGE was also detected by immunoblotting and by RT-PCR, where IL-1β and fibronectin fragments were found to stimulate RAGE expression. Stimulation of chondrocytes with S100B or HMGB-1 increased phosphorylation of the ERK MAP kinase and the p65 subunit of NF-κB and increased the production of MMP-13. This signaling was inhibited in cells pretreated with soluble RAGE, and S100B was shown to bind to chondrocyte RAGE.
Conclusion
Articular chondrocytes express functional RAGE. The increase in RAGE noted in OA cartilage and the ability of RAGE ligands to stimulate chondrocyte MAP kinase and NF-κB activity and to stimulate MMP-13 production suggests that chondrocyte RAGE signaling could play a role in OA.
It is well recognized that age is a primary risk factor for the development of osteoarthritis (OA), but the mechanisms by which aging contributes to an increased susceptibility to OA are poorly understood. A number of aging-related changes in joint tissue structure and function have been described that could be involved (for review, see ref. 1). These changes include alterations in extracellular matrix components, as well as a reduced capacity of the resident cells to repair matrix damage. Aging is associated with the accumulation of advanced glycation end products (AGEs) in a number of different tissues (2–4). Although AGE formation occurs at an accelerated rate in many tissues of patients with diabetes because of nonenzymatic glycation, low-turnover tissues such as the articular cartilage may be particularly susceptible to the accumulation of AGEs independently of plasma glucose levels.
Cells can express receptors that are capable of binding AGEs and can modulate cell function. The best characterized AGE receptor is called the receptor for advanced glycation end products (RAGE). RAGE is a member of the immunoglobulin superfamily and has been shown to be expressed by diverse cell types, including macrophages, endothelial cells, smooth muscle cells, myocytes, and neuronal cells (5,6). RAGE signaling has also been found to be stimulated by members of the S100 family of proteins, including S100B (7,8), which has been shown to be present in articular cartilage (9). An additional RAGE ligand is high mobility group box chromosomal protein 1 (HMGB-1; amphoterin) (10), which has recently been found to be expressed in OA cartilage (11).
RAGE signaling activated by AGE binding, S100 proteins, or HMGB-1 induces oxidative stress and activates MAP kinase signaling, leading downstream to increased NF-κB activity (12,13). Oxidative damage (14) and activation of MAP kinases (15,16) have been reported in OA cartilage, and signaling that results in increased NF-κB could contribute to cartilage degradation through up-regulation of matrix metalloproteinase (MMP) expression (17). These findings suggest that activation of RAGE signaling could contribute to the susceptibility of older adults to OA.
To our knowledge, no study demonstrating RAGE expression by chondrocytes has previously been published. The objective of the present study was to determine if RAGE is expressed in cartilage and to correlate levels of RAGE with age and the presence of OA. Two known RAGE ligands, S100B and HMGB-1, were used to stimulate chondrocytes in order to demonstrate that chondrocyte RAGE was functional. We report that chondrocytes express RAGE, which appears to be increased in OA cartilage, and we demonstrate that chondrocyte RAGE signaling stimulates MAP kinase and NF-κB activity and can increase MMP-13 production.
MATERIALS AND METHODS
Reagents
Polyclonal (AB5484) and monoclonal (MAB5328) antibodies to RAGE were purchased from Chemicon (Temecula, CA). The immunogen for the polyclonal antibody was a synthetic peptide corresponding to amino acids 42–59 in the N-terminal domain of RAGE, whereas the monoclonal antibody was raised against a recombinant protein containing the extracellular domains of RAGE. A third anti-RAGE antibody (C-20; Santa Cruz Biotechnology, Santa Cruz, CA) was used, and the immunogen was a synthetic peptide corresponding to a region in the carboxy-terminus of RAGE.
ERK and NF-κB antibodies were purchased from Cell Signaling Technology (Beverly, MA) and the MMP-13 antibody was from Chemicon. Interleukin-1β (IL-1β) and recombinant soluble RAGE (sRAGE; RAGE extracellular domain/Fc chimera) were obtained from R&D Systems (Minneapolis, MN). Recombinant soluble tumor necrosis factor receptor (sTNFR; TNFR extracellular domain/Fc chimera; etanercept) was obtained from Amgen (Thousand Oaks, CA). The 110-kd fibronectin fragment was from Upstate Biotechnology (Lake Placid, NY). S100B was from Calbiochem (San Diego, CA), and HMGB-1 was from Sigma (St. Louis, MO). EZ-link NHS-LC-Biotin and streptavidin-linked agarose were from Pierce (Rockford, IL). Cell culture media and media supplements were obtained from Mediatech (Herndon, VA).
Immunohistochemistry
Paraformaldehyde-fixed paraffin-embedded sections of cartilage from monkeys and humans were deparaffinized, rehydrated, and blocked for endogenous peroxidase activity using 3% H2O2 at room temperature for 15 minutes. Protein Block (Serum-Free Protein Block; Dako, Carpinteria, CA) was applied for 10 minutes at room temperature prior to incubation with the primary antibody. Polyclonal (AB5484) or monoclonal (MAB5328) anti-RAGE antibodies were diluted 1:100 and incubated at room temperature for 1 hour. Antibody binding was detected using either biotinylated anti-goat link at 1:1,000 dilution for the polyclonal primary antibody or biotinylated anti-mouse link for the monoclonal primary (Jackson ImmunoResearch, West Grove, PA) and streptavidin–horseradish peroxidase label (Dako) for 20 minutes at room temperature. Vector Nova Red (Vector Laboratories, Burlingame, CA) and streptavidin–horseradish peroxidase were used for visualization, and sections were counterstained with Mayer’s hematoxylin (Sigma).
For the negative control sections for the polyclonal antibody, Negative Control SuperSensitive Goat Serum for SuperSensitive Antibodies (BioGenex, San Ramon, CA) was substituted for the primary antibody. For the negative control sections for the monoclonal antibody, Negative Control SuperSensitive Mouse Serum for SuperSensitive Antibodies was substituted for the primary antibody.
Monkey knee tissue was obtained at necropsy from 20 animals ranging from young adult to old adult (8–27 years; mean 16 years). Midcoronal sections of the distal femur and proximal tibia were examined. To quantify the degree of OA-like change, toluidine blue–stained sections were scored (0–7 scale, where 0 = normal and 7 = severe damage, as previously described [18]) by a single investigator (CSC) who was blinded to the immunostaining results. The sections used for the study had scores ranging from 0 to 7 (mean 2.2). RAGE immunostaining was performed on adjacent sections and scored by a different investigator (LSB) who was blinded to the age and OA score. The total area of articular cartilage and the area of articular cartilage containing chondrocytes with positive cellular immunostaining for RAGE were measured in each section, using an Osteomeasure Histomorphometry system (Osteometrics, Atlanta, GA). From these measurements, the percentage of articular cartilage area containing positive immunostaining was determined.
Human cartilage from 6 normal tissue donors (ages 21–85 years) and from 3 OA patients (ages 57, 64, and 76 years; cartilage obtained at the time of joint replacement) was also subjected to immunostaining, and the results were compared with those obtained for the monkey tissues.
Chondrocyte isolation and culture
Human ankle and knee articular cartilage was obtained from tissue donors within 48 hours of death through the Gift of Hope Organ and Tissue Donor Network, in accordance with institutional protocol and review board approval. Each donor specimen was graded for degenerative changes based on the 5-point Collins scale, as modified by Muehleman et al (19). OA cartilage was obtained after knee replacement surgery (discarded tissue) for OA. Cartilage was dissected from the joints and digested in a sequential manner with Pronase and then overnight with collagenase as previously described (20). Viability of isolated cells was determined using trypan blue dye, and cells were counted using a hemocytometer. Monolayer cultures were established by plating cells in 6-well plates at a density of 2 × 106 cells/ml in Dulbecco’s modified Eagle’s medium/Ham’s F-12 medium supplemented with 10% fetal bovine serum. Plates were maintained for ~5–7 days, with feedings every 2 days until they reached 100% confluency, prior to experimental use. Cells were not passaged.
RAGE immunoblotting
Whole cell lysates were prepared from freshly isolated chondrocytes and from cultured chondrocytes and were used for immunoblotting for RAGE. After washing monolayers with phosphate buffered saline (PBS), cells were lysed and scraped into a buffer containing 20 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton, X-100, 2.5 mM sodium pyrophosphate, 1 mM glycerol phosphate, 1 mM Na3VO4, 1 μg/ml of leupeptin, and 1 mM phenylmethylsulfonyl fluoride. Lysates were centrifuged to remove insoluble material, and the soluble protein concentration was determined with bicinchoninic acid (Pierce). In addition to isolated chondrocytes, cartilage slices removed from donor joints were extracted directly with cell lysis buffer at 4°C for 4 hours. Prior to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), some cell lysate samples were incubated with 25 units of glycopeptidase F (Calbiochem) at 37°C for 18 hours. Glycopeptidase F digestion was stopped by boiling the samples with SDS-PAGE sample buffer. Samples containing equal amounts of total protein were separated by SDS-PAGE and transferred to nitrocellulose for immunoblotting using the enhanced chemiluminescence system (Amersham, Piscataway, NJ). The intensity of bands on scanned blots from normal and OA samples was determined using Kodak 1D Image analysis software (Eastman Kodak, Rochester, NY).
RAGE transfection
Primary chondrocytes were transfected with a plasmid expressing full-length RAGE complementary DNA (cDNA) (21) using the Nucleofection method (Human Chondrocyte Nucleofector kit; Amaxa, Gaithersburg, MD). Cells in confluent monolayer were harvested by incubation with collagenase P and Pronase solution at a concentration of 1 mg/ml each for 5–10 minutes at 37°C and then washing prior to resuspending the cells in Human Chondrocyte Nucleofector Solution at a final concentration of 2 × 106 cells/100 μl. The cell–Nucleofector solution complex (100 μl) and the plasmid construct DNA (2 μg) were then transferred into a cuvette and gently mixed, followed by Nucleofection using the Nucleofector program setting H20. Next, 500 μl of prewarmed culture medium containing 20% serum was added to the cells, mixed, and the cell suspension was transferred into 6-well plates that had been precoated with poly-l-lysine (1:3 dilution in cold PBS; Sigma). The cells were then placed in an incubator and, after 24 hours, replenished with fresh culture medium containing 10% serum, followed by a further incubation for 24 hours prior to use in experiments.
Analysis of RAGE RNA
Expression of RAGE RNA was analyzed by reverse transcription–polymerase chain reaction (RT-PCR) and quantified using real-time PCR. RNA was isolated from confluent monolayer cultures using the RNeasy MiniKit (Qiagen, Valencia, CA). In some experiments, cells were changed overnight to serum-free medium and were then left untreated (control) or were stimulated for 16–18 hours with IL-1β (2 ng/ml) or fibronectin fragments (500 nM) prior to RNA isolation. For RT-PCR, 0.5–1 μg of total RNA was converted to cDNA and amplified using the ThermoScript RT-PCR system (Invitrogen, Carlsbad, CA).
The sequences of the primers used were based on the report of RAGE isoforms by Yonekura et al (22) and were as follows: RAGE full-length forward 5′-GCC-AGG-ACC-CTG-GAA-GGA-AGC-A-3′ and reverse 5′-CTG-ATG-GAT-GGG-ATC-TGT-CTG-TG-3′; RAGE N-truncated forward 5′-TGA-GGG-GAT-TTT-CCG-GTG-C-3′ and reverse 5′-CTG-ATG-GAT-GGG-ATC-TGT-CTG-TG-3′; and RAGE C-truncated forward 5′-GCC-AGG-ACC-CTG-GAA-GGA-AGC-A-3′ and reverse 5′-CTT-GTT-GAC-CAT-CCC-CCC-AG-3′. For real-time PCR, the RAGE primers were as follows: forward 5′-CAA-TAA-GGT-GGG-GAC-ATG-TG-3′ and reverse 5′-ATT-AGC-TCC-GAC-TGC-AGT-GTG-AAG-A-3′. Primers for GAPDH were used as a control, as previously described (23).
Conditions for real-time PCR and to obtain sufficient RAGE DNA for use as a standard were established using RNA isolated from the immortalized chondrocyte cell line C28I2 (provided by Dr. Mary Goldring, Harvard Bone and Joint Institute, Boston, MA). RT-PCR was run to amplify RAGE by using the primer set as above. The DNA product was run on a 1.5% agarose gel, and the resulting band was purified using Phase Lock Gel (Eppendorf, Westbury, NY). The RAGE cDNA product was sequenced for confirmation. The concentration of purified DNA was measured with a spectrophotometer, and serial dilutions were used to create a standard curve for real-time PCR using the Smart Cycler (Cepheid, Sunnyvale, CA). Samples of RNA (1 μg each) from primary cells treated as above were reverse transcribed, and then equal amounts of cDNA (1 μl) were used for real-time PCR along with the RAGE DNA standards.
Stimulation of chondrocyte RAGE signaling
Confluent primary monolayers were made serum-free overnight and then treated with S100B at 100 μg/ml or with HMGB-1 at 5 μg/ml. Some cells were preincubated for 45 minutes with 100 μg/ml of recombinant sRAGE to specifically inhibit RAGE activation or with 100 μg/ml of recombinant sTNFR as a control. Cells were stimulated for 30 minutes, media were removed, and cell lysates were prepared as described above. Samples with equal amounts of total protein were immunoblotted with antibodies recognizing phosphorylated ERK and the p65 subunit of NF-κB and, as a loading control, with non–phospho-specific antibodies. In other experiments, chondrocytes were stimulated overnight, and the conditioned media were collected and analyzed for MMP-13 by immunoblotting, as previously described (24).
RAGE–S100B pull-down assay
To directly demonstrate S100B binding to RAGE, a pull-down assay was used whereby cultured chondrocytes were incubated with biotin-labeled S100B and then bound proteins were pulled-down with streptavidin beads. S100B was biotinylated using EZ-link NHS-LC-Biotin (Pierce). Unreacted biotin was removed by dialyzing against PBS overnight. Biotinylated protein was then stored at 4°C in 0.1% sodium azide until ready for use. Cells in monolayer culture, pretreated with and without sRAGE for 45 minutes to block S100B–RAGE binding, were incubated with biotinylated S100B at 4°C for 2 hours. After incubation, cells were washed with PBS to remove unbound biotinylated S100B and then lysed using lysis buffer. The cell extract was then incubated with 50 μl of streptavidin-linked agarose beads (1:1 suspension) for 1 hour at 4°C. The beads were then washed thoroughly with PBS to remove any loosely bound proteins. Bound proteins were then eluted with 2× SDS sample buffer and subjected to SDS-PAGE followed by immunoblotting with anti-RAGE mouse monoclonal antibody.
Statistical analysis
Analysis of the relationship between RAGE immunostaining, the OA score, and age had to account for the fact that 52 sections come from 20 monkeys, each providing 1–4 sections from different joint sites. The distribution of the data was not suitable for a mixed-model analysis. Therefore, the nonparametric Spearman’s rank correlation was used to describe the relationship between the 3 variables. Since age varied between, but not within, monkeys, associations between age and the percentage of RAGE immunostaining and the OA score, respectively, were determined for within-monkey averages. For the percentage of RAGE immunostaining and the OA score, the within-monkey averages were first subtracted and then subjected to Spearman’s rank correlation analysis with appropriate reduction of degrees of freedom, in addition to the exclusion of animals providing only 1 section. The correlation between these 2 variables adjusted for age was based on the standard formula for partial correlations applied to Spearman’s instead of Pearson’s correlations. Real-time PCR results for RAGE expression and comparison of RAGE bands in OA and normal chondrocytes were analyzed by analysis of variance.
RESULTS
Expression of RAGE in normal articular cartilage with aging and in OA articular cartilage
The presence of RAGE in articular cartilage was first examined by immunohistochemistry. Initial immunostaining was performed using cartilage sections obtained from cynomolgus monkeys which, like humans, develop a naturally occurring OA, which most commonly affects the medial tibia and becomes more prevalent with increasing age (24). RAGE, detected using a polyclonal antibody to an epitope in the N-terminal region, was noted primarily in cartilage sections obtained from older adult animals or animals with histologic evidence of OA. Positive immunostaining was more prevalent in the tibia compared with the femur. Most of the staining was found in cells in the superficial zone of the cartilage, but staining extended into cells in deeper zones when OA changes were present (Figures 1A–C). Negative controls did not exhibit any immunostaining (data not shown). In addition, the pattern and intensity of immunostaining seen with the polyclonal antibody was found to be the same when a monoclonal antibody to the extracellular portion of RAGE was tested in 8 of the sections that had a range of ages and OA scores.
Figure 1.
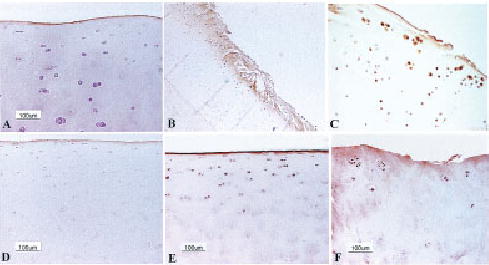
Immunohistochemical staining for receptor for advanced glycation end products (RAGE) in articular cartilage. A, Normal articular cartilage from a young adult monkey (9.8 years) shows only surface staining for RAGE. B, Positive immunostaining for RAGE in chondrocytes and matrix in monkey cartilage with surface fibrillation and cleft formation (original magnification × 15). C, Higher-magnification view demonstrates strong staining for RAGE in chondrocytes from monkey cartilage with early osteoarthritis (OA)–like changes (original magnification × 200). D, Normal human ankle cartilage from a 21-year-old subject shows faint immunostaining for RAGE in superficial chondrocytes. E, Positive immunostaining for RAGE in superficial zone chondrocytes and on the cartilage surface in normal human ankle cartilage from an 80-year-old subject. F, Positive cell and matrix staining in a human knee OA cartilage sample.
Immunostaining with the polyclonal antibody of 52 sections from a total of 20 animals was measured, and showed cellular staining of 0–100% of the articular cartilage area (mean ± SEM 14 ± 3%). Age showed a positive correlation with the percentage of RAGE immunostaining (r = 0.48, P = 0.031) and with the OA score (r = 0.50, P = 0.025). OA scores and percentages of RAGE immunostaining were significantly correlated (r = 0.69, P < 0.001) and remained so even when age was taken into account (r = 0.59, P < 0.001).
Immunostaining of normal (n = 6) and OA (n = 3) human knee cartilage also revealed the presence of RAGE (Figures 1D–F). In normal cartilage, RAGE was seen mainly in cells of the superficial zone. Cartilage from young adult humans (21 and 23 years old) had staining in a few superficial zone cells, while increased numbers of cells were positive in sections from older adults (80 and 85 years old). Sections of normal-appearing cartilage from older adults also showed staining in some deeper zone cells. Some sections also exhibited staining of the cartilage surface (Figure 1E). It is not clear if this is an artifact or if it is indicative of RAGE binding to the surface of cartilage. The latter could occur, since secreted RAGE has been found in serum (25) and, so, may be present in synovial fluid as well, although this has not been examined. Immunostaining was stronger and more extensive in cartilage from OA joints, with more cells staining in the deeper zones and with some matrix staining also noted (Figure 1F).
Isolated chondrocytes were examined for the presence of RAGE by immunoblotting of cell lysates obtained from human donors over the age of 50 years. Cell lysates were prepared from freshly isolated cells and from cells maintained in high-density monolayer culture until they were confluent (~5–7 days). Previous studies have shown that there are multiple forms of RAGE, which have different molecular weights due to both alternative splicing and posttranslational modifications, including glycosylation (5,21,22,26–28).
We performed immunoblotting on samples obtained from >25 human joints. Representative results are shown in Figure 2. In chondrocyte lysates, several protein bands of molecular weights ranging from just below 75 kd to ~37 kd were noted by immunoblotting with either the monoclonal or polyclonal anti-RAGE antibodies. RAGE was noted in freshly isolated as well as cultured chondrocytes obtained from either normal knee or ankle cartilage (Figure 2A). RAGE immunostaining demonstrated a similar pattern in knee and ankle cells, but the intensity of specific bands changed with culture. A band was noted just below 75 kd, which was usually more prominent after monolayer culture. The band at 55 kd was not consistently affected by culture, whereas a band at 50 kd was less intense after cell culture. A fourth band at 48 kd was more intense after cell culture, and the band at ~37 kd, the weakest band noted, did not change consistently with culture. Similar bands were seen in chondrocytes cultured in alginate (data not shown).
Figure 2.
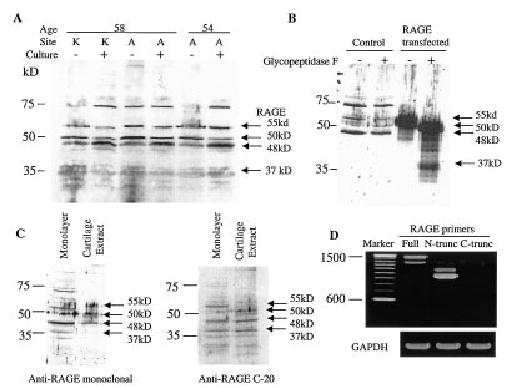
Detection of chondrocyte expression of receptor for advanced glycation end products (RAGE) by immunoblotting and reverse transcription–polymerase chain reaction (RT-PCR). A, Lysates were prepared from cells (obtained from 2 human donors) freshly isolated (culture −) from human knee (K) or ankle (A) articular cartilage or from cells isolated from the same joints but cultured in monolayer for 5–7 days (culture +). Equal amounts of total protein (10 μg) were run in each lane and immunoblotted with an anti-RAGE monoclonal antibody (MAB5328). Arrows to the right of the blot indicate potential RAGE isoforms. It is not known if the band just below 75 kd is related to RAGE. Molecular weight markers are shown at the left. B, Cell lysate samples were prepared from cells cultured in monolayer for 5 days (control) and from cells from the same donor that were transfected with a construct that expresses RAGE. One-half of the lysate sample was treated with glycopeptidase F prior to sodium dodecyl sulfate–polyacrylamide gel electrophoresis and immunoblotted for RAGE with the monoclonal antibody. C, Immunoblotting was performed with the monoclonal antibody using a cell lysate from monolayer-cultured cells and from a direct cartilage extract. The same blot was stripped and reprobed with a polyclonal antibody (C-20) to the RAGE C-terminus. D, RT-PCR was performed using RNA isolated from primary monolayer cultures, which was amplified using 3 different RAGE primer sets and GAPDH primers as a control. The RAGE primers were designed to amplify regions coding for full-length (full), N-truncated (N-trunc), and C-truncated (C-trunc) RAGE.
Further experiments were performed to identify the different forms of RAGE produced by human chondrocytes. Full-length RAGE that is N-glycosylated runs at ~55 kd, whereas full-length RAGE without glycosylation runs at 48–50 kd (21,22,28). Our results suggested that chondrocytes produce the full-length, fully glycosylated 55-kd RAGE as well as 48–50-kd forms (Figure 2). Lysates from primary chondrocytes and from chondrocytes transfected with a full-length RAGE expression construct were treated with glycopeptidase F to remove N-linked sugars and then immunoblotted for RAGE. In this experiment, the 55-kd band was found to shift to ~50 kd after glycopeptidase treatment (Figure 2B). It is not known if RAGE might contain O-linked sugars that could account for a difference in the 48-kd and 50-kd bands. The 48-kd band did not move, which suggests that it represents full-length RAGE without any glycosylation, and this would be consistent with the calculated molecular weight of RAGE based on amino acid content (394 amino acids not including a 22–amino acid signal sequence [5]). Although a high molecular weight form of RAGE between 66 kd and 75 kd has been reported and was seen on some of our blots, its identity is uncertain (5,21). We were able to detect only the 75-kd form on isolated chondrocytes and did not see it when whole cartilage slices were extracted with the cell lysis buffer (Figure 2C).
N-truncated (~37–42-kd) and C-truncated (~35-kd) RAGE isoforms, which are produced by alternative splicing, have also been described, and since the C-truncated form lacks the transmembrane and cytoplasmic domains, it is secreted (22,27). To determine if any of the bands on the blots represented C-truncated (secretory) RAGE, we probed blots of cell lysates and cartilage extracts with an antibody to the carboxy-terminus of RAGE, which would not be present in the C-truncated form. These blots demonstrated the presence of the 55-, 50-, 48-, and 37-kd bands, but the higher molecular weight band was not seen (Figure 2C). This suggested that perhaps the higher molecular weight band was a dimer of C-truncated RAGE retained in the cells, but RT-PCR using primers specific for this isoform of RAGE did not reveal any cDNA product, whereas RNA for both the full-length and N-truncated forms of RAGE was present (Figure 2D). We could not definitively detect the N-truncated protein form of RAGE on our blots, since antibodies specific for this isoform are not available and the antibody to the carboxy-terminus did not detect additional bands that were not detected by the N-terminal polyclonal or monoclonal antibodies.
When an additional immunoblot was performed using the N-terminal polyclonal antibody with cells isolated and cultured from 4 different OA knee joints and 3 normal ankle joints as controls, the intensity of the 55-kd RAGE band was significantly stronger (P = 0.003) in the OA samples compared with the normal samples (Figure 3).
Figure 3.
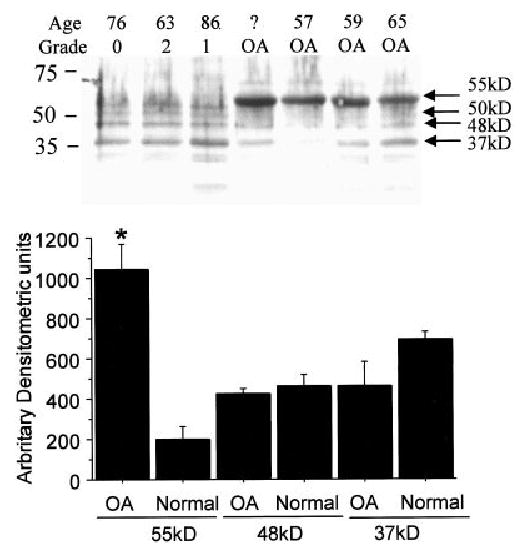
Receptor for advanced glycation end products (RAGE) in chondrocytes from normal and osteoarthritic (OA) human cartilage. Chondrocytes were isolated from OA knee and normal ankle cartilage, and lysates were prepared after 5–7 days in culture. Top, Samples with equal amounts of total protein (13 μg per lane) were immunoblotted with anti-RAGE polyclonal antibody (AB5484). Grade represents the modified Collins score for degenerative changes (see Materials and Methods). Age was not obtained for 1 of the OA samples, as indicated by a question mark. Arrows to the right of the blot indicate potential RAGE isoforms. Molecular weight markers are shown at the left. Bottom, Intensity of the 55-kd, 48-kd, and 37-kd bands in the OA (n = 4) and normal (n = 3) samples was measured by densitometry. Values are the mean and SEM. * = P = 0.003 versus normal samples with 55-kd bands.
Stimulation of RAGE expression by fibronectin fragments and IL-1β
The RAGE promoter contains 2 functional NF-κB binding sites (29); therefore, stimuli that increase NF-κB activity could potentially increase RAGE expression. This was tested in chondrocytes by treating cultured cells with the 110-kd fibronectin fragment and the cytokine IL-1β. Both these factors are found in OA cartilage (14,30) and have been shown to stimulate signaling pathways that result in increased NF-κB activity (31,32). Consistent with this, both fibronectin fragments and IL-1β stimulated RAGE expression (mean ± SEM 172 ± 18% of control and 185 ± 24% of control, respectively), measured using real-time PCR (Figure 4).
Figure 4.
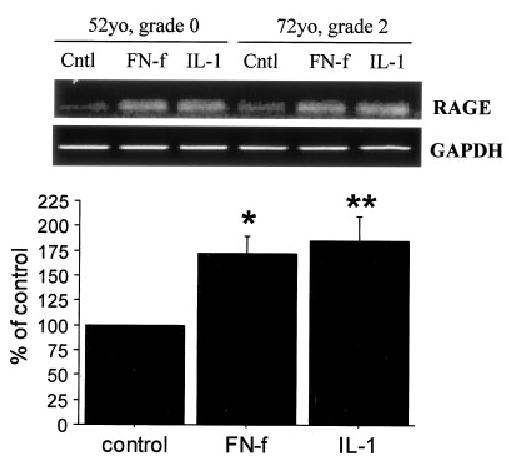
Chondrocyte expression of receptor for advanced glycation end products (RAGE) in response to fibronectin fragments (FN-f) and interleukin-1β (IL-1β). Top, RNA was isolated from monolayers of cultured human chondrocytes that had been left untreated (Cntl) or had been treated overnight with 500 nM fibronectin fragments or 2 ng/ml of IL-1β in serum-free medium and used for real-time reverse transcription–polymerase chain reaction (RT-PCR). RT-PCR for GAPDH was used for comparison. The PCR products were separated on an agarose gel and stained with ethidium bromide. Grade represents the modified Collins score for degenerative changes (see Materials and Methods). Bottom, Results of real-time RT-PCR expressed as the mean and SEM percentage of untreated control (n = 4). * = P = 0.017; ** = P = 0.008 versus control.
Activation of ERK MAP kinase and NF-κB and increase in MMP-13 production in cultured chondrocytes by RAGE signaling
In order to provide further evidence that chondrocytes express functional RAGE, cells were first treated with S100B, which is a known RAGE ligand (7,8) and which has previously been described in chondrocytes (9). Treatment with S100B resulted in phosphorylation of the ERK-1/2 MAP kinase and the p65 subunit of NF-κB (Figure 5), both of which are well-characterized mediators of RAGE signaling (12,13). Consistent with RAGE stimulation by S100B, the phosphorylation of ERK and NF-κB was inhibited by sRAGE but not by sTNFR. The latter was used as a negative control since, like sRAGE, it consists of the extracellular ligand binding region of the receptor fused to the Fc region of human IgG. Similar results were obtained when the cells were treated with HMGB-1 in place of S100B (data not shown).
Figure 5.
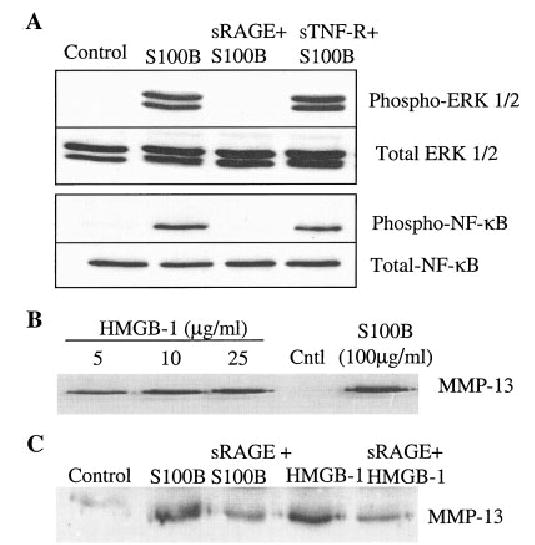
Stimulation of chondrocyte signaling for receptor for advanced glycation end products (RAGE) and production of matrix metalloproteinase 13 (MMP-13). A, Human chondrocytes in serum-free confluent monolayer culture were pretreated for 45 minutes with 100 μg/ml of recombinant soluble RAGE (sRAGE) or with 100 μg/ml of recombinant soluble tumor necrosis factor receptor (sTNFR) as a control. Cultures were then stimulated for 30 minutes with 100 μg/ml of S100B or were left untreated (control). Cell lysates were prepared and used for immunoblotting with phospho-specific antibodies to ERK-1/2 (phospho–ERK-1/2) or the phosphorylated p65 subunit of NF-κB (phospho–NF-κB). Blots were stripped and reprobed with non–phospho-specific (total) antibodies. B, Chondrocytes in serum-free monolayer culture were treated overnight with the indicated concentrations of high mobility group box chromosomal protein 1 (HMGB-1) or S100B. Conditioned media were collected and immunoblotted for MMP-13. C, Chondrocytes were pretreated with or without sRAGE as in A and then stimulated overnight with 100 μg/ml of S100B or 5 μg/ml of HMGB-1 and analyzed for MMP-13 production by immunoblotting of the conditioned media.
We next determined if RAGE-mediated signaling could stimulate the production of MMP-13 (collagenase 3), a key enzyme in OA cartilage due to its ability to cleave type II collagen (33,34). We found that both RAGE ligands, S100B and HMGB-1, could stimulate MMP-13 production and that the stimulation was inhibited by sRAGE (Figures 5B and C).
Binding of S100B to chondrocyte RAGE
In order to confirm that S100B was binding to chondrocyte RAGE, a pull-down assay was performed. Chondrocytes were incubated with biotin-labeled S100B, with and without sRAGE to compete for RAGE binding. The cells were treated with cell lysis buffer, and the proteins bound to biotin–S100B were then isolated by incubating the samples with streptavidin beads. The proteins bound to streptavidin were immunoblotted for RAGE, and 3 bands were noted at just below 75 kd, 55 kd, and at 48 kd, the most intense band (Figure 6A). No bands were seen in samples from cells incubated with biotin–S100B in the presence of sRAGE. Additional experiments were performed with chondrocytes transfected with the full-length RAGE construct, which revealed a 55-kd band after pull-down and RAGE immunoblotting (Figure 6B).
Figure 6.
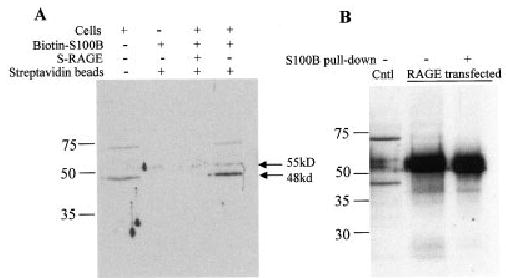
Binding of S100B to chondrocyte receptor for advanced glycation end products (RAGE). A, Human chondrocytes in monolayer culture were incubated for 2 hours at 4°C with biotin-labeled S100B, with or without soluble RAGE (sRAGE). Monolayers were washed to remove unbound protein, and cell lysates were prepared and then incubated with streptavidin–agarose beads to bind biotin–S100B and associated proteins (S100B pull-down). A sample of biotin–S100B that had not been incubated with cells was also incubated with the beads as an additional control. Proteins were eluted from the streptavidin beads in sodium dodecyl sulfate–polyacrylamide gel electrophoresis buffer and then were immunoblotted for RAGE with the monoclonal antibody. B, Primary chondrocytes transfected with a RAGE expression vector were also tested in the S100B pull-down assay and immunoblotted along with lysate samples from nontransfected cells (Cntl) and from transfected cells not subjected to the S100B pull-down assay. Arrows to the right of the blots indicate potential RAGE isoforms. Molecular weight markers are shown at the left.
DISCUSSION
To our knowledge, this is the first demonstration of RAGE expression by articular chondrocytes. Activation of RAGE is thought to play a role in mediating tissue injury in a diverse number of chronic degenerative and inflammatory conditions, including diabetic atherosclerosis and impaired wound healing, amyloidosis, arterial restenosis, and synovial inflammation in rheumatoid arthritis (RA) (35–37). The present findings suggest that RAGE is also a potential mediator of cartilage damage in OA. RAGE was detected by immunostaining in cartilage sections from both humans and monkeys, and the presence of chondrocyte RAGE was further confirmed by immunoblotting and RT-PCR. Examining RAGE in situ, there appeared to be an association between positive immunostaining for RAGE and both age and the presence of histologic changes of OA in monkeys and humans. Little to no RAGE was observed in normal cartilage from young adults, more extensive RAGE staining was noted in cartilage from older adults, and the strongest staining was noted in cartilage with OA changes.
RAGE has previously been shown to be expressed by synovial macrophages from humans with RA, and treatment of these cells with the RAGE ligand HMGB-1 was found to stimulate TNFα production (38). Inhibition of RAGE stimulation with sRAGE suppressed the development of arthritis in a collagen-induced model of inflammatory arthritis in mice (37). We found that stimulation of human chondrocytes with 2 known RAGE ligands, S100B and HMGB-1, increased MMP-13 production, suggesting that RAGE signaling in cartilage may also contribute to cartilage matrix degradation in arthritis. Stimulation of RAGE signaling resulted in ERK and NF-κB activation, which can further contribute to a proinflammatory state in cartilage. Inhibition by sRAGE provided evidence that the stimulation was due to RAGE binding and not an alternative receptor for S100B or HMGB-1. In addition, in a pull-down assay, we showed specific binding of S100B to chondrocyte RAGE.
RAGE expression has been shown to correlate with the presence of RAGE ligands (35). Our findings would be consistent with this, since AGEs are present at much higher levels in cartilage from older adults (39,40), and other RAGE ligands, including S100A4 (41) and HMGB-1 (11), appear to be increased in OA cartilage. The RAGE promoter has been found to contain NF-κB binding sites (29), and expression of RAGE is increased by RAGE ligands and inflammatory mediators including TNFα (35,42). We found that fibronectin fragments and IL-1β, which have both been found in cartilage and synovial fluid from patients with OA and RA (30,43), can stimulate chondrocyte RAGE expression.
The consistent detection of RAGE RNA and protein in cultured human chondrocytes suggests that the process of isolation and cell culture may stimulate some degree of chondrocyte RAGE expression. Chondrocytes were found to express several different forms of RAGE, which resulted from both posttranslational modifications, including N-linked glycosylation, as well as alternative splicing, which produced RNA coding for an N-truncated form of RAGE. The original report of RAGE isolated from bovine lung and endothelial cells noted proteins of ~50 kd and 30–40 kd, which we found in human chondrocytes, and a band close to the higher molecular weight band that we saw just below 75 kd but which has not been further identified (5). Subsequent studies (21,22,26–28) using various types of cells and tissues have also noted different forms of RAGE that are similar to the bands noted on our immunoblots.
Transfection of chondrocytes with a full-length RAGE cDNA construct revealed that the 55-kd RAGE band is likely full-length RAGE that has been N-glycosylated as previously shown (22), while the 50-kd and 48-kd bands are most likely forms of RAGE that have not been fully glycosylated. The pull-down assay revealed that S100B could bind to at least the 55-kd and 48-kd forms. When cells isolated from normal and OA joints were compared directly, the 55-kd RAGE band was more intense in OA. Whether glycosylation plays a role in modulating RAGE function is not known and should be the subject of future studies in order to determine the possible effects of increased production of 55-kd RAGE in OA. We detected RNA using RT-PCR with primers specific for the N-truncated isoform of RAGE, but we could not be certain if the protein was present since we lacked an antibody that could specifically detect this form of RAGE. We did not detect RNA or protein bands indicative of the C-truncated isoform.
In summary, we have shown that RAGE is present in articular cartilage and is increased with aging and the presence of OA. Factors that stimulate increased NF-κB activity, including IL-1β and fibronectin fragments, stimulate RAGE expression, likely by increasing RAGE promoter activity. Because chondrocyte RAGE signaling can be stimulated by HMGB-1 and S100 proteins, and because RAGE signaling activates inflammatory pathways through activation of NF-κB and increases MMP-13 production, RAGE may play an important role in the pathogenesis of cartilage damage in OA as well as RA. Additional work is needed to further define the role of RAGE in cartilage matrix turnover, including the function of the various RAGE isoforms, and to determine if RAGE inhibition could reduce the progression of cartilage damage in arthritis.
Acknowledgments
Thanks to the Gift of Hope Organ and Tissue Donor Network and donor families and Arkady Margulis for donor tissues, and to Gabriella Cs-Szabo and the Rush Department of Orthopaedic Surgery for OA tissue. We also thank Dorothee Aeppli for assistance with statistical analysis and Anne Undersander, Maggie Folk, and Carol Pacione for technical assistance.
Footnotes
Supported by the NIH (grants AG-16697, P50-AR-39239, NS-42855, and RR-14099).
References
- 1.Loeser RF, Shakoor N. Aging or osteoarthritis: which is the problem? Rheum Dis Clin North Am. 2003;29:653–73. doi: 10.1016/s0889-857x(03)00062-0. [DOI] [PubMed] [Google Scholar]
- 2.Sell DR, Monnier VM. Structure elucidation of a senescence cross-link from human extracellular matrix: implication of pentoses in the aging process. J Biol Chem. 1989;264:21597–602. [PubMed] [Google Scholar]
- 3.Makita Z, Vlassara H, Cerami A, Bucala R. Immunochemical detection of advanced glycosylation end products in vivo. J Biol Chem. 1992;267:5133–8. [PubMed] [Google Scholar]
- 4.Verzijl N, Bank RA, TeKoppele JM, DeGroot J. Ageing and osteoarthritis: a different perspective. Curr Opin Rheumatol. 2003;15:616–22. doi: 10.1097/00002281-200309000-00016. [DOI] [PubMed] [Google Scholar]
- 5.Neeper M, Schmidt AM, Brett J, Yan SD, Wang F, Pan YC, et al. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem. 1992;267:14998–5004. [PubMed] [Google Scholar]
- 6.Brett J, Schmidt AM, Yan SD, Zou YS, Weidman E, Pinsky D, et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol. 1993;143:1699–712. [PMC free article] [PubMed] [Google Scholar]
- 7.Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, et al. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- 8.Valencia JV, Mone M, Zhang J, Weetall M, Buxton FP, Hughes TE. Divergent pathways of gene expression are activated by the RAGE ligands S100b and AGE-BSA. Diabetes. 2004;53:743–51. doi: 10.2337/diabetes.53.3.743. [DOI] [PubMed] [Google Scholar]
- 9.Wolff DA, Stevenson S, Goldberg VM. S-100 protein immunostaining identifies cells expressing a chondrocytic phenotype during articular cartilage repair. J Orthop Res. 1992;10:49–57. doi: 10.1002/jor.1100100106. [DOI] [PubMed] [Google Scholar]
- 10.Hori O, Brett J, Slattery T, Cao R, Zhang J, Chen JX, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin: mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J Biol Chem. 1995;270:25752–61. doi: 10.1074/jbc.270.43.25752. [DOI] [PubMed] [Google Scholar]
- 11.Attur M, Dave MN, Akamatsu M, Nakagawa N, Miki J, Yang H, et al. Differential expression of high mobility group protein in human normal and arthritic cartilage; functional genomic analysis [abstract] Trans Orthop Res Soc. 2003;28:18. [Google Scholar]
- 12.Yan SD, Schmidt AM, Anderson GM, Zhang J, Brett J, Zou YS, et al. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J Biol Chem. 1994;269:9889–97. [PubMed] [Google Scholar]
- 13.Lander HM, Tauras JM, Ogiste JS, Hori O, Moss RA, Schmidt AM. Activation of the receptor for advanced glycation end products triggers a p21(ras)-dependent mitogen-activated protein kinase pathway regulated by oxidant stress. J Biol Chem. 1997;272:17810–4. doi: 10.1074/jbc.272.28.17810. [DOI] [PubMed] [Google Scholar]
- 14.Loeser RF, Carlson CS, del Carlo M, Cole A. Detection of nitrotyrosine in aging and osteoarthritic cartilage: correlation of oxidative damage with the presence of interleukin-1β and with chondrocyte resistance to insulin-like growth factor 1. Arthritis Rheum. 2002;46:2349–57. doi: 10.1002/art.10496. [DOI] [PubMed] [Google Scholar]
- 15.Clancy R, Rediske J, Koehne C, Stoyanovsky D, Amin A, Attur M, et al. Activation of stress-activated protein kinase in osteoarthritic cartilage: evidence for nitric oxide dependence. Osteoarthritis Cartilage. 2001;9:294–9. doi: 10.1053/joca.2000.0388. [DOI] [PubMed] [Google Scholar]
- 16.Pelletier JP, Fernandes JC, Brunet J, Moldovan F, Schrier D, Flory C, et al. In vivo selective inhibition of mitogen-activated protein kinase kinase 1/2 in rabbit experimental osteoarthritis is associated with a reduction in the development of structural changes. Arthritis Rheum. 2003;48:1582–93. doi: 10.1002/art.11014. [DOI] [PubMed] [Google Scholar]
- 17.Mengshol JA, Vincenti MP, Coon CI, Barchowsky A, Brinckerhoff CE. Interleukin-1 induction of collagenase 3 (matrix metalloproteinase 13) gene expression in chondrocytes requires p38, c-Jun N-terminal kinase, and nuclear factor κB: differential regulation of collagenase 1 and collagenase 3. Arthritis Rheum. 2000;43:801–11. doi: 10.1002/1529-0131(200004)43:4<801::AID-ANR10>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 18.Loeser RF, Shanker G, Carlson CS, Gardin JF, Shelton BJ, Sonntag WE. Reduction in the chondrocyte response to insulin-like growth factor 1 in aging and osteoarthritis: studies in a non-human primate model of naturally occurring disease. Arthritis Rheum. 2000;43:2110–20. doi: 10.1002/1529-0131(200009)43:9<2110::AID-ANR23>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 19.Muehleman C, Bareither D, Huch K, Cole AA, Kuettner KE. Prevalence of degenerative morphological changes in the joints of the lower extremity. Osteoarthritis Cartilage. 1997;5:23–37. doi: 10.1016/s1063-4584(97)80029-5. [DOI] [PubMed] [Google Scholar]
- 20.Loeser RF, Todd MD, Seely BL. Prolonged treatment of human osteoarthritic chondrocytes with insulin-like growth factor-I stimulates proteoglycan synthesis but not proteoglycan matrix accumulation in alginate cultures. J Rheumatol. 2003;30:1565–70. [PubMed] [Google Scholar]
- 21.Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, et al. RAGE and amyloid-β peptide neurotoxicity in Alzheimer’s disease. Nature. 1996;382:685–91. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- 22.Yonekura H, Yamamoto Y, Sakurai S, Petrova RG, Abedin MJ, Li H, et al. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem J. 2003;370:1097–109. doi: 10.1042/BJ20021371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forsyth CB, Pulai J, Loeser RF. Fibronectin fragments and blocking antibodies to α2β1 and α5β1 integrins stimulate mitogen-activated protein kinase signaling and increase collagenase 3 (matrix metalloproteinase 13) production by human articular chondrocytes. Arthritis Rheum. 2002;46:2368–76. doi: 10.1002/art.10502. [DOI] [PubMed] [Google Scholar]
- 24.Carlson CS, Loeser RF, Purser CB, Gardin JF, Jerome CP. Osteoarthritis in cynomolgus macaques. III. Effects of age, gender, and subchondral bone thickness on the severity of disease. J Bone Miner Res. 1996;11:1209–17. doi: 10.1002/jbmr.5650110904. [DOI] [PubMed] [Google Scholar]
- 25.Park L, Raman KG, Lee KJ, Lu Y, Ferran LJ, Jr, Chow WS, et al. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat Med. 1998;4:1025–31. doi: 10.1038/2012. [DOI] [PubMed] [Google Scholar]
- 26.Schmidt AM, Yan SD, Brett J, Mora R, Nowygrod R, Stern D. Regulation of human mononuclear phagocyte migration by cell surface-binding proteins for advanced glycation end products. J Clin Invest. 1993;91:2155–68. doi: 10.1172/JCI116442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malherbe P, Richards JG, Gaillard H, Thompson A, Diener C, Schuler A, et al. cDNA cloning of a novel secreted isoform of the human receptor for advanced glycation end products and characterization of cells co-expressing cell-surface scavenger receptors and Swedish mutant amyloid precursor protein. Brain Res Mol Brain Res. 1999;71:159–70. doi: 10.1016/s0169-328x(99)00174-6. [DOI] [PubMed] [Google Scholar]
- 28.Yan SS, Wu ZY, Zhang HP, Furtado G, Chen X, Yan SF, et al. Suppression of experimental autoimmune encephalomyelitis by selective blockade of encephalitogenic T-cell infiltration of the central nervous system. Nat Med. 2003;9:287–93. doi: 10.1038/nm831. [DOI] [PubMed] [Google Scholar]
- 29.Li J, Schmidt AM. Characterization and functional analysis of the promoter of RAGE, the receptor for advanced glycation end products. J Biol Chem. 1997;272:16498–506. doi: 10.1074/jbc.272.26.16498. [DOI] [PubMed] [Google Scholar]
- 30.Homandberg GA, Wen C, Hui F. Cartilage damaging activities of fibronectin fragments derived from cartilage and synovial fluid. Osteoarthritis Cartilage. 1998;6:231–44. doi: 10.1053/joca.1998.0116. [DOI] [PubMed] [Google Scholar]
- 31.Grall F, Gu X, Tan L, Cho JY, Inan MS, Pettit AR, et al. Responses to the proinflammatory cytokines interleukin-1 and tumor necrosis factor α in cells derived from rheumatoid synovium and other joint tissues involve nuclear factor κB–mediated induction of the Ets transcription factor ESE-1. Arthritis Rheum. 2003;48:1249–60. doi: 10.1002/art.10942. [DOI] [PubMed] [Google Scholar]
- 32.Im HJ, Pacione C, Chubinskaya S, van Wijnen AJ, Sun Y, Loeser RF. Inhibitory effects of insulin-like growth factor-1 and osteogenic protein-1 on fibronectin fragment- and interleukin-1β-stimulated matrix metalloproteinase-13 expression in human chondrocytes. J Biol Chem. 2003;278:25386–94. doi: 10.1074/jbc.M302048200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mitchell PG, Magna HA, Reeves LM, Lopresti-Morrow LL, Yocum SA, Rosner PJ, et al. Cloning, expression, and type II collagenolytic activity of matrix metalloproteinase-13 from human osteoarthritic cartilage. J Clin Invest. 1996;97:761–8. doi: 10.1172/JCI118475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reboul P, Pelletier JP, Tardif G, Cloutier JM, Martel-Pelletier J. The new collagenase, collagenase-3, is expressed and synthesized by human chondrocytes but not by synoviocytes: a role in osteoarthritis. J Clin Invest. 1996;97:2011–9. doi: 10.1172/JCI118636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmidt AM, Yan SD, Yan SF, Stern DM. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest. 2001;108:949–55. doi: 10.1172/JCI14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sakaguchi T, Yan SF, Yan SD, Belov D, Rong LL, Sousa M, et al. Central role of RAGE-dependent neointimal expansion in arterial restenosis. J Clin Invest. 2003;111:959–72. doi: 10.1172/JCI17115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hofmann MA, Drury S, Hudson BI, Gleason MR, Qu W, Lu Y, et al. RAGE and arthritis: the G82S polymorphism amplifies the inflammatory response. Genes Immun. 2002;3:123–35. doi: 10.1038/sj.gene.6363861. [DOI] [PubMed] [Google Scholar]
- 38.Taniguchi N, Kawahara K, Yone K, Hashiguchi T, Yamakuchi M, Goto M, et al. High mobility group box chromosomal protein 1 plays a role in the pathogenesis of rheumatoid arthritis as a novel cytokine. Arthritis Rheum. 2003;48:971–81. doi: 10.1002/art.10859. [DOI] [PubMed] [Google Scholar]
- 39.DeGroot J, Verzijl N, Bank RA, Lafeber FP, Bijlsma JW, TeKoppele JM. Age-related decrease in proteoglycan synthesis of human articular chondrocytes: the role of nonenzymatic glycation. Arthritis Rheum. 1999;42:1003–9. doi: 10.1002/1529-0131(199905)42:5<1003::AID-ANR20>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 40.Verzijl N, DeGroot J, Thorpe SR, Bank RA, Shaw JN, Lyons TJ, et al. Effect of collagen turnover on the accumulation of advanced glycation endproducts. J Biol Chem. 2000;275:39027–31. doi: 10.1074/jbc.M006700200. [DOI] [PubMed] [Google Scholar]
- 41.Aigner T, Zien A, Gehrsitz A, Gebhard PM, McKenna L. Anabolic and catabolic gene expression pattern analysis in normal versus osteoarthritic cartilage using complementary DNA–array technology. Arthritis Rheum. 2001;44:2777–89. doi: 10.1002/1529-0131(200112)44:12<2777::aid-art465>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 42.Tanaka N, Yonekura H, Yamagishi S, Fujimori H, Yamamoto Y, Yamamoto H. The receptor for advanced glycation end products is induced by the glycation products themselves and tumor necrosis factor-α through nuclear factor-κB, and by 17β-estradiol through Sp-1 in human vascular endothelial cells. J Biol Chem. 2000;275:25781–90. doi: 10.1074/jbc.M001235200. [DOI] [PubMed] [Google Scholar]
- 43.Xie DL, Meyers R, Homandberg GA. Fibronectin fragments in osteoarthritic synovial fluid. J Rheumatol. 1992;19:1448–52. [PubMed] [Google Scholar]