

Abstract
Asp kinase catalyzes the first step of the Asp-derived essential amino acid pathway in plants and microorganisms. Depending on the source organism, this enzyme contains up to four regulatory ACT domains and exhibits several isoforms under the control of a great variety of allosteric effectors. We report here the dimeric structure of a Lys and S-adenosylmethionine–sensitive Asp kinase isoform from Arabidopsis thaliana in complex with its two inhibitors. This work reveals the structure of an Asp kinase and an enzyme containing two ACT domains cocrystallized with its effectors. Only one ACT domain (ACT1) is implicated in effector binding. A loop involved in the binding of Lys and S-adenosylmethionine provides an explanation for the synergistic inhibition by these effectors. The presence of S-adenosylmethionine in the regulatory domain indicates that ACT domains are also able to bind nucleotides. The organization of ACT domains in the present structure is different from that observed in Thr deaminase and in the regulatory subunit of acetohydroxyacid synthase III.
INTRODUCTION
In plants and microorganisms, the Asp-derived amino acid pathway leads to the synthesis of the essential (regarding animals) amino acids Thr, Lys, Met, and Ile. Enzymes of this pathway have a high biotechnological importance. First, they constitute targets for development of new agrochemical and pharmaceutical compounds controlling weeds, fungi, and bacteria. Also, deregulation of this pathway in plants has been proven to increase the soluble content of essential amino acids thus improving the nutritional quality of animal diet (Zhu and Galili, 2003). In plants, this pathway is highly controlled at the level of seven allosteric enzymes (Asp kinase, EC 2.7.2.4; homoserine dehydrogenase, EC 1.1.1.3; dihydrodipicolinate synthase, EC 4.2.1.52; Thr synthase, EC 4.2.3.1; Thr deaminase, EC 4.3.1.19; acetohydroxyacid synthase, EC 2.2.1.6; and isopropylmalate synthase, EC 2.3.3.13). Four of these enzymes (Asp kinase [AK], Asp kinase-homoserine dehydrogenase [AK-HSDH], Thr deaminase [TD], and acetohydroxyacid synthase [AHAS]) have regulatory domains that belong to the ACT domain family (Pfam 01842).
ACT domains are small regulatory domains found in a large number of proteins involved in amino acid or purine metabolism (Aravind and Koonin, 1999; Chipman and Shaanan, 2001). ACT domains have a characteristic βαββαβ topology initially identified on AK (A), chorismate mutase (C), and prephenate dehydrogenase (T, for TyrA) by PSI-BLAST search using the regulatory domain of AHAS as template (Aravind and Koonin, 1999; Chipman and Shaanan, 2001). Only four proteins containing this domain have been crystallized to date: phosphoglycerate dehydrogenase (PGDH) (Schuller et al., 1995; Thompson et al., 2005), TD (Gallagher et al., 1998), and the regulatory subunit of AHAS III (Kaplun et al., 2006) from Escherichia coli and Phe hydroxylase (PheOH) (Kobe et al., 1999) from Rattus norvegicus. Structure determinations showed that the ACT domains of these four proteins have a similar fold. However, PGDH and PheOH contain only one ACT domain, whereas TD and AHAS III contain two ACT domains per monomer. Among these proteins, only PGDH has been crystallized with its effector Ser (Schuller et al., 1995).
A fascinating aspect of the ACT domains is the extreme variety of ligands that they can bind. Considering this point, AK, on which we will now focus, is a case study. In plants and bacteria, the first step of the Asp-derived amino acid pathway involves monofunctional AK and bifunctional AK-HSDH. Plant AK-HSDH displays seven different allosteric effectors (Curien et al., 2005). By contrast, AK-HSDH from E. coli is either not regulated (Umbarger, 1978) or inhibited only by Thr (Patte et al., 1966; Bearer and Neet, 1978). Similar complexity is observed for monofunctional AK. Depending on the source organism, monofunctional AKs are inhibited by Thr (Saccharomyces cerevisiae; Marina et al., 2004), Lys (Bacillus subtilis AK2; Moir and Paulus, 1977); Arabidopsis thaliana AK2 and AK3; G. Curien and R. Dumas, unpublished data), or mesodiaminopimelate (B. subtilis AK1; Rosner and Paulus, 1971). Other monofunctional AKs are inhibited in synergy by Lys and Leu (E. coli; Mazat and Patte, 1976), Lys and Thr (B. subtilis AK3; Graves and Switzer, 1990), or Lys and S-adenosylmethionine (SAM) (Arabidopsis AK1; Rognes et al., 1980; (G. Curien and R. Dumas, unpublished data). Monofunctional and bifunctional plant AKs contain two ACT domains per monomer. By contrast, the number of ACT domains per monomer of AK is extremely variable in microorganisms, ranging from zero to four (according to the Pfam Database, PF01842). In summary, the existence of different allosteric effectors and the variable copy number of ACT domains found in AK illustrate well the diversity of control linked to the presence of ACT domains. Determination of the structure of an AK is a prerequisite to understanding the structural basis of this versatility.
In this study, we crystallized Arabidopsis AK1 in complex with SAM and Lys. This work describes the structure of an AK and the structure of an enzyme containing two ACT domains cocrystallized with its effectors. In addition, this structure provides new insights into the mechanism of allosteric inhibition and gives an explanation for the synergistic inhibition of the enzyme by Lys and SAM.
RESULTS
Overall Model
The structure of Arabidopsis AK1 presented here contains one dimer (944 amino acid residues), four Lys residues, two SAMs, and 178 water molecules per asymmetric unit (Figures 1A and 1B). It was refined to an R factor of 20.2% at 2.85-Å resolution (Table 1). Dimer subunits are related by a noncrystallographic twofold axis. The model is of good quality, with all the residues falling in the most favorable (89.8%), additionally allowed (10%), and generously allowed (0.2%) regions of the Ramachandran plot, as assessed by PROCHECK (Laskowski et al., 1993). The N-terminal (residues 1 to 24) and the C-terminal regions (495 to 496) of the structure are disordered, as no electron density was observed for these regions. However, the lack of density for the first 24 residues has no consequence for the structural analysis of the enzymatic mechanism. Indeed, in the course of the work on AK1, a shorter form of the protein was produced starting at the Met immediately preceding the conserved KFGG site. The protein was inhibited by Lys and SAM as observed for the longer form (data not shown). Altogether, these observations suggest that the first 24 residues have no functional significance.
Figure 1.

Structure of AK1.
(A) Ribbon diagram of the dimer of AK1 in complex with Lys and SAM. One monomer is colored in orange and the other in blue. SAM molecules are shown in violet. Lys molecules found in the active site and in the regulatory binding site are indicated in green and red, respectively. This diagram, as well as the following diagrams of structures, was produced with the program PyMol (http://pymol.sourceforge.net/).
(B) A 90° rotated view, with respect to the horizontal axis, of the dimer showing that the regulatory domains make a core of 16 strands containing two perpendicular sheets of eight strands surrounded on both sides by four helices exposed to the solvent and flanked by the two catalytic domains.
Table 1.
Data Collection, Phasing, and Refinement Statistics
| Native | Se (λ1) | Se (λ2) | Se (λ3) | |
|---|---|---|---|---|
| Diffraction Data | ||||
| Unit cell (Å) | a = 117.3, b = 117.3, c = 255.3 | a = 117.75, b = 117.75, c = 255.82 | ||
| Wavelength (Å) | 0.9797 | 0.97924 (peak) | 0.97939 (edge) | 0.97563 (remote) |
| Resolution (Å) | 2.85 | 3.1 | 3.1 | 3.1 |
| Space group | I41 | I4122 | I4122 | I4122 |
| No. of reflections | 117,971 | 52,416 | 52,519 | 52,339 |
| No. of unique reflections | 38,537 | 27,580 | 27,649 | 27,543 |
| Completeness | 96.2 (97.9) | 88.8 (91.0) | 88.6 (90.9) | 88.6 (90.9) |
| Multiplicity | 3.1 (3.0) | 1.9 | 1.9 | 1.9 |
| R-sym (%)a | 6.9 (43) | 13.5 (79.8) | 13.9 (79.8) | 13.1 (76.2) |
| I/σ(I) | 15.1 (2.2) | 7.1 (1.6) | 7.1 (1.6) | 7.6 (1.7) |
| Phasing | ||||
| Refined f′ (e-) | – | −8.56 | −10.3 | −5.34 |
| Refined f″ (e-) | – | 5.06 | 2.5 | 1.13 |
| Mean figure of merit | – | 0.36 | ||
| No. of Se sites | – | 10 | ||
| Structure Refinement | ||||
| Rfactor (%)b | 20.2 | – | – | – |
| Rfree (%) | 24.4 | – | – | – |
| No. of protein atoms | 7312 | – | – | – |
| No. of solvent atoms | 176 | – | – | – |
| No. of ligand atoms | 90 | – | – | – |
| RMS deviation of bond lengths (Å) | 0.007 | – | – | – |
| RMS deviation of bond angles (°) | 1.12 | – | – | – |
| RMS deviation of dihedral angles (°) | 9.35 | – | – | – |
| Average B factor (Å2) | 62.3 | – | – | – |
The numbers in parentheses indicate the value in the outer resolution shell. Rfree was calculated using 5% of reflections that were kept apart from the refinement during the whole process. RMS, root mean square.
R-sym =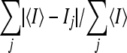 .
.
R-factor = 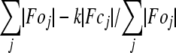 .
.
Domain Organization
Each monomer is composed of an N-terminal domain (25 to 324) connected via a hinge (325 to 332) to a regulatory C-terminal domain (333 to 494) (Figures 2A and 2B).
Figure 2.

Structure of the Subunit.
(A) Stereo view of the ribbon diagram of one subunit. The N-terminal catalytic domain belongs to the amino acid kinase family. It is composed of two lobes: an N-terminal lobe involved in amino acid binding (violet) and a C-terminal lobe involved in ATP binding (orange). A Lys molecule bound in the active site is shown (green sticks). The C-terminal regulatory domain contains two domains belonging to the ACT domain family: ACT1 (in yellow) and ACT2 (in cyan). ACT1 is involved in the binding of Lys (red sticks) and SAM (violet sticks).
(B) Scheme for the secondary structure of AK1 made with the program DSSP (Hooft et al., 1996). The N-terminal lobe, the C-terminal lobe, ACT1, and ACT2 are colored in violet, orange, yellow, and cyan, respectively.
The N-terminal domain consists of an eight-stranded β-sheet with a topology β3-β5-β2-β1-β8-β10-β11-β9, the two last strands β9 and β11 being antiparallel to the rest. The sheet is sandwiched between three other strands (β4-β6-β7) and four α-helices (α6 to α9) on one side and a three α-helix bundle (α3, α4, and α5) and two other α-helices (α1 and α2) on the other side (Figures 2A and 2B).
The N-terminal domain belongs to the amino acid kinase family (Pfam 00696). Apart from the two additional helices α2 and α3, the AK1 N-terminal domain shows strong structural similarity to the catalytic domain of three members of this family: acetylglutamate kinase (NAGK; PDB code 1gs5; 163/259 Cα atoms matched, RMS distance 1.8 Å) (Ramon-Maiques et al., 2002), urydilate kinase (UMPK; PDB code 2bmu; 150/225 Cα atoms matched, RMS distance 1.7 Å), and carbamate kinase-like carbamoyl phosphate synthetase (CBMK; PDB code 1e19; 128/313 Cα atoms matched, RMS distance 1.7 Å) (Ramon-Maiques et al., 2000) structures.
The regulatory domain consists of two subdomains, ACT1 and ACT2, belonging to the ACT domain family (Pfam 01842). Both ACT domains have a similar fold. Each subdomain folds as a four-stranded antiparallel sheet with two α-helices parallel to the sheet and located on one side of the sheet (Figures 2A and 2B). ACT1 (340 to 419) has a topology βαββαβ (β13-α10-β14-β15-α11-β16) and a fold similar to that determined for the ACT domain from PGDH (PDB code 1ygy; 123/152 Cα atoms matched, RMS distance 1.3 Å) (Schuller et al., 1995). By contrast, ACT2 is made of two nonsequential regions (333 to 339 and 420 to 494) and has an unusual ββαββα topology (β12-β17-α12-β18-β19-α13) with its first strand located before ACT1 (Figures 2A and 2B). As can be seen in Figures 1B and 2A, ACT1 and ACT2 are perpendicular to each other and are connected by two short hairpins.
Dimer Organization
Dimerization of AK involves interactions between the ACT domains (Figures 1A and 1B). The surface area of each subunit buried in the dimer interface is 2830 Å2, which amounts to ∼16% of each subunit surface area. No contact occurs between the two catalytic domains within the dimer. ACT1 of one monomer interacts with ACT1 of the other monomer, making a long sheet of eight strands with four α-helices on one side of the sheet (Figure 1B). Similarly, ACT2 of one monomer interacts with ACT2 of the other monomer (Figure 1B). Interactions between ACT1 and ACT2 occur at the level of their two long sheets, one being perpendicular to the other (Figure 1B). In summary, the regulatory domain of the dimer is composed of a core of 16 strands containing two perpendicular sheets surrounded on both sides by four helices exposed to the solvent and flanked by the two catalytic domains (Figure 1B).
A dimer of dimers can also be generated by crystallographic symmetry. However, the number of interactions between the two dimers is low, with only eight H-bonds, and the surface of each subunit buried in the tetramer interface is only 525 Å2, which amounts to ∼2.4% of each subunit surface area. These results are in agreement with native gel electrophoresis experiments showing that AK1 behaves predominantly (95%) as a dimeric enzyme in equilibrium with a tetramer (data not shown).
Regulatory Domain
Two Lys molecules (real space R factor: 16.5%; see Methods) and two SAM molecules (real space R factor: 24%) were found in the regulatory domain of the dimer (Figure 3A). Surprisingly, these four effectors are located only in the ACT1 domains (Figure 3A). Both Lys molecules are well defined in the sigma-weighted difference electron density map 2mFo-DFc contoured at 1 sigma (Figure 3A). In addition, they exhibit a mean B-factor (50 Å2) equivalent to those of the surrounding residues of ACT1, indicating that they are tightly bound inside their binding site. Both Lys binding sites are equivalent and involve mainly the main chain atoms of both ACT1 domains (Figures 3B and 3C). Indeed, the α-NH3+ group of one Lys molecule interacts with loop 1 (348 to 356; real space R factor: 29%) and helix α10 of monomer B, whereas its α-COO− group interacts with loop 2 (369 to 372; real space R factor: 20%) of monomer A and loop 1 of monomer B (Figure 3C). For ease of understanding, loops have been numbered from 1 to 5 starting at the N-terminal part of each ACT domain.
Figure 3.

Lys and SAM Binding Sites of ACT1.
(A) Electron density maps around the Lys and SAM molecules bound to ACT1 of monomers A and B.
(B) Lys and SAM binding sites. Two equivalent Lys binding sites are located at the interface of two ACT1 domains. ACT1 of monomer A (ACT1A) and ACT1 of monomer B (ACT1B) are colored in orange and blue, respectively. SAM binding sites are located between loops 2 and 4 of each ACT1.
(C) Stereo view of the interactions of Lys and SAM molecules with amino acid residues belonging to ACT1A (residues depicted in orange sticks) and ACT1B (residues depicted in blue sticks). Bond lengths <3.4 Å are indicated. Lys and SAM molecules are depicted as red and violet sticks, respectively.
The side chain of this Lys molecule points toward strands β14 of both monomers (Figure 3C). As mentioned above, the second Lys binding site is equivalent and involves the corresponding residues of the other monomer. Although interactions mainly involve main chain atoms, sequence comparison with the Lys-sensitive E. coli enzyme shows that >50% of the residues belonging to the Lys binding site are conserved between the plant and the bacterial enzyme (see Supplemental Figure 1 online). Furthermore, mutants of the E. coli Lys-sensitive AK affected in their response to Lys were obtained by screening mutagenized E. coli strains for their resistance to Lys (Kikuchi et al., 1999). Three mutant forms of the enzyme (Gly323Asp, Thr344Met, and Ser345Leu) were obtained, which displayed higher K0.5 values for Lys inhibition compared with the wild-type enzyme. Targeted amino acids were located in the conserved region and correspond to Gly-356, Thr-377, and Ser-378 in Arabidopsis AK1 (Figure 3C; see Supplemental Figure 1 online), confirming the importance of these amino acids in the formation of the Lys binding site.
Unlike Lys, each SAM molecule interacts only with residues belonging to the ACT1 domain of only one monomer (Figures 3B and 3C). The two SAM binding sites are equivalent. As shown in Figures 3B and 3C, the adenosine and the ribose moieties of the SAM molecule are sandwiched between loop 2 and loop 4 (387 to 397; real space R factor: 30%), involving mainly H-bonds and a stacking of adenosine with Trp-392. The conformation of the two SAM molecules is similar. The mean B-factor of the adenine moiety (58.7 Å2) is equal to those of the surrounding residues, indicating a tight binding. By contrast, the mean B-factor of the ribose and the Met moiety is higher (91.5 Å2) in agreement with the fact that the Met moiety of SAM from both monomers points toward the solvent (Figures 3B and 3C). SAM 1 interacting with monomer A is well defined, whereas the Met moiety of SAM 2 is disordered between the Cδ and the sulfur atom (Figure 3A). There is no direct interaction between SAM and Lys molecules. However, the different effectors interact indirectly, as loop 2 is involved in both the Lys and the SAM binding sites (Figure 3C).
Catalytic Domain
As described for the members of the amino acid kinase family, the catalytic domain of AK1 is organized in two lobes: an N-terminal lobe (25 to 227) containing a part of the long sheet with helices α1 to α6 and a C-terminal lobe containing the rest of the domain (232 to 322) (Figures 2A and 2B).
Interestingly, during the model refinement of AK1, electron density was found in the N-terminal lobe around the Asp binding site (Figure 4A). Density observed in the active site permitted the modeling of a Lys molecule (Figure 4B) (real space R factor: 21%; see Methods). The α-NH3+ group of this Lys molecule interacts with Glu-148 (bond length of 2.5 Å), whereas its ζ-NH3+ group points toward Ser-233 (2.7 Å). The α-COO− interacts weakly with Thr-70 (3.2 Å). The mean B-factor of the Lys molecule (61 Å2) is similar to those of the surrounding residues of the active site (60 Å2). However, by contrast with the Lys molecule located in the regulatory domain, the agreement with surrounding B-factors was obtained by reducing the occupancy of the Lys molecule of the active site to 60 to 70%. The previously described density was attributed to a Lys, rather than a piece of polyethylene glycol (PEG). To further discriminate between Lys and PEG, the effect of PEG 3350 on enzyme activity was tested in the presence of a saturating concentration of SAM (200 μM). We did not observe any inhibitory effect of PEG at the concentration used in the crystallization conditions (20% [v/v]). Furthermore, at this concentration, PEG 3350 does not displace the K0.5 value for Lys inhibition toward higher values (K0.5 value for Lys inhibition of 3.4 ± 0.2 and 4.5 ± 0.2 μM with and without PEG, respectively; 200 μM SAM), indicating the absence of competition between PEG and Lys. These additional experiments indicate therefore that PEG does not interact with the substrate and the regulatory binding sites.
Figure 4.

Substrate Binding Site.
(A) Electron density maps around the Lys molecule bound to the active site of monomers A and B.
(B) Interactions of Lys molecule (green sticks) inside the active site. Bond lengths <3.4 Å are indicated.
DISCUSSION
ACT Domains of AK1 Exhibit a Novel Organization
The structures of only four proteins containing ACT domains have been determined previously. The first one, PGDH, is a tetramer containing only one ACT domain per monomer (Schuller et al., 1995; Thompson et al., 2005). The structure of PGDH in complex with its effector (Ser) shows that two Ser molecules bind at the interface of two ACT domains. The second one, TD, is a dimer of dimers containing two ACT-like domains per monomer. “ACT-like” denotes that the fold of the regulatory domains of TD is similar to the fold of the ACT domain of PGDH. However, the regulatory domain of TD was not detected using the PSI-BLAST search of Aravind and Koonin (1999). Mutants of the effector binding sites (Wessel et al., 2000), designed using the three-dimensional structure of the E. coli enzyme crystallized without its effectors (Ile and Val) (Gallagher et al., 1998), indicated that the Ile and the Val binding sites are located at the interface of the two different ACT domains of each monomer of TD. The third one, AHAS, is a dimer containing two ACT domains per monomer (Kaplun et al., 2006). The last one, PheOH, is a dimer containing one ACT domain per monomer (Kobe et al., 1999). By contrast with PGDH, TD, and AHAS III, dimerization of PheOH does not involve interaction between ACT domains.
Arabidopsis AK1 has two different ACT domains per monomer, as described for TD and AHAS III. The present structure shows that the structural organization of ACT1 and ACT2 domains in AK1 could not have been predicted from previous studies on TD and AHAS III. Indeed, ACT1 of one monomer interacts with ACT1 of the second monomer in AK1 (Figure 3B) (similarly, ACT2 of one monomer interacts with ACT2 of the second monomer), whereas ACT1 interacts with ACT2 of the same monomer in TD (Gallagher et al., 1998). Consequently, the interaction of the ACT1 domains generates in the case of AK1 two equivalent binding sites for Lys (Figure 3B), as determined for the Ser binding site of PGDH (Schuller et al., 1995). By contrast, interaction of two different ACT domains generates two nonequivalent binding sites, one for Ile and one for Val in the case of TD (Wessel et al., 2000). Comparison with the structure of AHAS III indicates a similar interaction between ACT1 of each monomer. However, by contrast with AK1, the ACT2 domains are positioned back to back with their two α-helices on both sides of the two sheets (Kaplun et al., 2006).
The structure of AK1 also shows additional unexpected features. Indeed, ACT1 is also able to bind a nucleotide (Figure 3B). Furthermore, the two SAM binding sites are not located at the interface between two ACT domains but are sandwiched between specific loops of ACT1 (Figure 3B). Finally, ACT2 does not interact with an effector as suggested for TD (Wessel et al., 2000) and AK-HSDH (Paris et al., 2003).
Comparison of ACT1 with ACT2 Reveals That ACT2 Is Unsuitable for Effector Binding
Figure 5A shows that ACT1 and ACT2 have a similar fold (140/162 Cα atoms matched, RMS distance 1.5 Å). The subtle differences between the ACT domains mainly relate to the length of loops. Loops 1 and 4 of ACT1 are longer than their counterparts in ACT2 (Figure 5A). By contrast, loop 3 (379 to 380, between β14 and β15; real space R factor: 17%) of ACT1 is smaller (Figure 5A). In addition, helix α12 of ACT2 contains three supplementary residues by comparison with the corresponding helix (α10) of ACT1. Comparison of the Lys binding site of ACT1 with the corresponding residues of ACT2 shows that the three supplementary residues of helix α12 decrease the length of loop 1 of ACT2, preventing the binding of Lys (Figure 5A). As shown in Figures 3B and 3C, each SAM binding site involves loops 2 and 4 of ACT1 of the same monomer. Comparison of the SAM binding site of ACT1 with the corresponding residues of ACT2 shows that loop 4 is much smaller in ACT2 and does not contain the residues involved in the interaction with SAM (Figure 5A). Altogether, these features exclude Lys and SAM binding in the ACT2 domain of the plant AK1.
Figure 5.

Superimposition of ACT Domains.
(A) Superimposition of ACT1 domains of monomer A (ACT1A; orange) and B (ACT1B; blue) with ACT2 domains of monomer A (light green) and B (light blue) of AK1. The figure shows that ACT2 domains are unable to bind Lys and SAM molecules.
(B) Superimposition of ACT1A (orange) and ACT1B (blue) of AK1 with ACT domains of PGDH (monomers A and B of PGDH are colored in green and brown, respectively). The figure shows that Lys and Ser binding sites are located at the same place.
(C) A 90° rotated view, with respect to the horizontal axis, showing a zoom on the Ser (black sticks) and Lys (red sticks) molecules depicted in (B).
(D) Superimposition of ACT1A (orange) and ACT1B (blue) of AK1 with ACT domains of one monomer of TD (ACT1 and ACT2 of one monomer of TD are colored in yellow and red, respectively). In the case of TD, two different ACT domains interact, generating two nonequivalent binding sites. Lys and SAM molecules are depicted as red and violet sticks, respectively.
Comparison of the Lys Binding Site with the Ser Binding Site of PGDH
The present structure allows a comparison of the Lys and Ser binding sites of AK1 and PGDH, respectively. Figure 5B shows that the fold of both ACT1 domains of the AK1 dimer is similar to the fold of the ACT domains of the PGDH dimer (123/152 Cα atoms matched, RMS distance 1.3 Å). However, apart from loop 2, the conformation and the length of the four other loops differ (Figure 5B). In particular, loops 1 (involved in the Lys binding site) and 4 (involved in the SAM binding site) are longer for AK1. Comparison of the Ser and Lys binding sites also shows that the interactions of Ser and Lys are different. Whereas the α-amino group of Lys interacts with the main chain of loops 1 and 2, the α-amino group of Ser interacts with side chain residues of loop 1 and strand β1. The interactions of Ser and Lys side chains with their binding sites are also different. Indeed, whereas the ζ-NH3+ of Lys interacts with two strands, the Oγ of Ser points toward loop 2. Despite these differences, Figures 5B and 5C show that the Ser binding sites in PGDH superimpose with the Lys binding sites in AK1.
Specificities of the SAM Binding Site
Figures 3B and 3C show that SAM is sandwiched between loops 2 and 4 of each ACT1. Interactions involve mainly hydrogen bonds and a stacking of the adenine moiety with a Trp residue of loop 4. Comparison of the SAM binding site of AK1 with the corresponding residues of PGDH (Figure 5B) and TD (Figure 5C) shows that loop 4 is much smaller in PGDH and TD (loop 4 of AK1 has eight and six supplementary residues compared with the equivalent loops of PGDH and TD, respectively) and does not contain the residues involved in the interaction with SAM in AK1. These differences may account for the ability of loop 4 of AK1 to accommodate SAM, whereas the equivalent loop of PGDH and TD cannot (Figures 5B and 5D). Interestingly, loop 2 of ACT1 in AK1 has a conformation and a length very similar to its counterpart in PGDH. This observation suggests therefore that the specificity of AK1 toward SAM is mainly due to loop 4. In agreement with this finding, sequence comparison with SAM-insensitive AK from microorganisms shows that residues involved in the SAM interaction, W392SR394, are not found in the bacterial and fungal sequences (see Supplemental Figure 1 online). By contrast, sequence comparison with the two SAM-insensitive Arabidopsis AK isoforms AK2 and AK3 shows only a few differences at the level of this loop (see Supplemental Figure 1 online). However, the Trp residue is not found in AK2, and the loop is longer in AK3 (see Supplemental Figure 1 online). As AK2 and AK3 were not inhibited by SAM even at high concentration (400 μM SAM; G. Curien and R. Dumas, unpublished data), this result suggests that Trp-392 and the presence of a large loop 4 are key determinants of the specific interaction of AK1 with SAM.
Finally, the SAM binding site of AK1 shows a topology different from those observed in plant Thr synthase, the other allosteric enzyme controlled by SAM in the Asp-derived amino acid pathway (Mas-Droux et al., 2006). Indeed, the crystallographic structure of Thr synthase in the presence of SAM and pyridoxal-5′-phosphate revealed four SAM molecules located in two different binding sites at the dimer interface. Its topology is also different compared with other SAM binding proteins of known structures, such as the Rossmann fold present in methyl transferases (Martin and McMillan, 2002), SAM radical–dependent enzymes (Layer et al., 2003; Berkovitch et al., 2004; Lepore et al., 2005), or Met repressor MetJ (Somers and Phillips, 1992). The structure of AK1 therefore contains a new SAM binding fold.
Mechanism of Synergistic Inhibition by Lys and SAM
As described above, three isoforms of monofunctional AK are involved in the first step of the pathway in plants (G. Curien and R. Dumas, unpublished data). These isoforms display similar kinetic properties with respect to Asp and ATP. However the allosteric controls of the three isoforms differ markedly. Indeed, AK2 and AK3 are inhibited by low concentrations of Lys (K0.5 value for Lys inhibition of 10 and 7 μM, respectively), whereas AK1 is only marginally inhibited at these concentrations (K0.5 value for Lys inhibition of 570 μM). SAM by itself is not inhibitory. Interestingly, only AK1 displayed an increase in the apparent affinity for Lys in the presence of SAM (in the presence of 20 μM SAM, the K0.5 value for Lys inhibition decreased to 80 μM). Reciprocally, increasing Lys concentration also leads to a huge decrease of the K0.5 value for SAM, demonstrating that Lys and SAM inhibit AK1 in a synergistic manner (G. Curien and R. Dumas, unpublished data). Note that the K0.5 value for inhibition of AK1 by Lys alone is approximately eightfold higher than the physiological concentration of Lys (70 μM; Giovanelli et al., 1989). Hence, the presence of SAM and Lys in the ACT domain is required for the inhibition by Lys to be efficient under physiological conditions (G. Curien and R. Dumas, unpublished data). The structure of AK1 in complex with Lys and SAM provides an explanation of the synergistic inhibition. Indeed, as loop 2 belongs to the binding sites of both effectors, SAM and Lys binding sites of AK1 are strongly interconnected (Figures 3B and 3C).
Conformation of the ATP Binding Site of AK1 Complexed with Its Effectors Is Appropriate for β- and γ-Phosphate Interactions but Unsuitable for Adenosine Binding
Superimposition of the structure of AK1 with the structures of NAGK, UMPK, and CBMK cocrystallized with 5′-(β,γ-imido)-triphosphate (AMP-PNP) (an ATP analogue) or ADP allowed the identification of the ATP binding site of AK1. Furthermore, previous studies, including sequence comparisons, site-directed mutagenesis, and a theoretical model of the catalytic domain of the E. coli AK3 (built on the basis of the structure of NAGK), suggested that residues corresponding in NAGK and AK3 to Lys-32, Gly-35, and Ser-64 of AK1 are involved in the interaction with the β- and γ-phosphate moiety of ATP (Gil-Ortiz et al., 2003; Marco-Marin et al., 2003). In agreement with these studies, superimposition of the catalytic domain of AK1 with the structures of NAGK cocrystallized with AMP-PNP shows that Lys-32, Gly-35, and Ser-64 of AK1 have a position suitable for interaction with the β- and γ-phosphate of ATP (Figure 6A). Furthermore, the structure of AK1 shows an interaction between Asp-234 and Lys-32 (Figure 6A) as determined for NAGK. In summary, all the residues interacting with β- and γ-phosphate of ATP in the structure of NAGK have a similar position in AK1.
Figure 6.

ATP Binding Site of AK1.
(A) Position of AMP-PNP upon superimposition of the ATP binding sites of NAGK and AK1. The figure shows that (1) Lys-32, Gly-35, and Ser-64 of AK1 have a position suitable for interaction with the β- and γ-phosphate of AMP-PNP, (2) Asp-234 interacts with Lys-32 as observed in the structure of NAGK, and (3) the position of loops A (254 to 273) and B (289 to 296) (in black) of AK1 are incompatible for interaction with the adenosine moiety of AMP-PNP. AMP-PNP is depicted in light blue, whereas AK1 is colored in orange.
(B) Superimposition of the C-terminal lobe of NAGK cocrystallized with AMP-PNP (light blue), CBMK cocrystallized with ADP (yellow), and AK1 (orange). The figure shows that the three structures superimpose very well, except loops A and B of AK1 (in black). Loop B of AK1 makes a steric clash with the nucleotide of NAGK and CBMK. Loop A of AK1 is too distant to interact with the adenosine moiety of AMP-PNP or ADP. (In particular, Asp-254/Asp-262 and Leu-259 cannot interact with the ribose and the adenine moiety of AMP-PNP of NAGK.)
(C) Superimposition of the structure of NAGK (light blue) with the whole monomer of AK1 (orange). Loop A, loop B, helix α8, helix α9, β12, β17, and β19 are indicated. The figure shows that the loops of NAGK corresponding to loops B and A of AK1 make a steric clash (shown in the dashed ovals) with helix α13 and strands β12, β17, and β19 of ACT2 of AK1.
Superimposition of the AK1 structure with the NAGK, UMPK, and CBMK structures cocrystallized with AMP-PNP or ADP also shows that most of the C-terminal lobe superimposed well, except loop A (254 to 273, located between strands β8 and β9), helix α8, and loop B (289 to 296, located between helices α8 and α9) of AK1 (Figure 6B). (For a better understanding, only superimposition of NAGK, CBMK, and AK1 is shown in Figure 6B; UMPK was omitted in the superimposition.) Loops A and B interact with the adenine and ribose moiety of the nucleotide in NAGK, UMPK, and CBMK. However, when the nucleotide molecule of NAGK, UMPK, or CBMK is positioned in the ATP binding site of AK1, loops A and B of AK1 are found at a position that would prevent the interaction with the adenosine moiety of AMP-PNP or ADP (Figures 6A and 6B). Indeed, Figures 6A and 6B show that loop A of AK1 would be too far from the adenosine moiety of AMP-PNP (or ADP) to interact with it. Furthermore, loop B of AK1 would make a steric clash with the adenosine moiety of AMP-PNP (or ADP).
In addition to these observations, loops A and B of AK1 cannot adopt the position observed in the structure of NAGK, UMPK, or CBMK. Indeed, superimposition of the structures of UMPK, NAGK, and CBMK with the whole structure of AK1 shows that the corresponding loops involved in adenosine binding in UMPK, NAGK, and CBMK make a steric clash with β12, α13, and β19 of ACT2 in AK1 (only superimposition of NAGK and AK1 is shown in Figure 6C).
Comparison of the present structure with the structures of NAGK, UMPK, or CBMK suggests therefore that the AK1-Lys-SAM complex would be unable to interact efficiently with the adenosine moiety of ATP. This hypothesis is in agreement with steady state kinetic measurements that indicate that in the presence of Lys and SAM, AK1 displays a lower affinity for ATP compared with the free enzyme (G. Curien and R. Dumas, unpublished data).
Possible Role of the ACT Domains in the Conformation of the ATP Binding Site
Interactions between the catalytic domain and the regulatory domain of AK1 mainly involve loop A, helix α8, and loop B of the catalytic domain. Loop A interacts with β12, α13, and β19 of ACT2 from the same monomer. Helix α8 interacts with β12 and β19 of ACT2 and with loop 4 of ACT1 (involved in the SAM binding site) from the same monomer. Helix α8 also interacts with the C terminus of β14 (involved in the SAM binding site) of ACT1 from the other monomer. Finally, loop B interacts with loop 1 (involved in the Lys binding site) of ACT1 from the other monomer.
The extensive network of interaction between ACT1/ACT2 and loop A/helix α8/loop B of the active site in AK1-Lys-SAM complex might explain why the conformation of the ATP binding site in this complex is different from that observed in UMPK, NAGK, and CBMK. Upon binding of Lys and SAM on ACT1, both ACTs might undergo a conformational change leading to a conformation of the ATP binding site (i.e., loop A/helix α8/loop B) unsuitable for the binding of the adenosine moiety of ATP. Crystallization of AK1 with its substrates and without its effectors is under way and should help support this hypothesis.
Asp Binding Site
The crystallographic structure of NAGK with its substrate and the mutagenesis studies performed on E. coli AK3 suggested that the AK3 residues corresponding to Glu-148 and Arg-230 of AK1 interact with the α-NH3+ and the α-COO− part of Asp, respectively (Marco-Marin et al., 2003). Density corresponding to a Lys molecule was found in the Asp binding site of AK1 (crystals were grown with Lys in the absence of Asp; see Methods) (Figure 4). As shown in Figure 7, a molecule of Asp can be modeled manually inside the active site using the coordinates of the α-COO− and the α-NH3+ group of the Lys molecule and by positioning the γ-COO− group toward the γ-phosphate of ATP. Figure 7 shows that the α-NH3+ of the Asp molecule modeled in the active site interacts with Glu-148 (Figure 4) as suggested previously (Marco-Marin et al., 2003). However, Figure 7 shows that Arg-230 cannot interact with the α-COO− part of the Asp molecule but interacts with ACT1 at the level of the binding sites of both effectors. Indeed, the side chain of Arg-230 interacts with the SAM binding loop (loop 4, via the main chain oxygen of Leu-391) of the same monomer and with the Lys binding loop (loop 2, via the main chain oxygen of Gly-353) of the other monomer (Figure 7). If binding of Asp by Arg-230 is required for the reaction, then interaction of Arg-230 with ACT1 would decrease the affinity for Asp. In agreement with kinetic data showing that Lys and SAM inhibit AK1 by decreasing the affinity for Asp (and ATP) (G. Curien and R. Dumas, unpublished data), the present structure suggests therefore that the AK1-Lys-SAM complex would also be unable to bind Asp efficiently.
Figure 7.

Interaction of the Substrate Binding Site with ACT1.
Asp (red sticks) was modeled manually using the coordinates of Lys (green sticks) observed in the active site. The α-NH3+ of Asp is coordinated with the conserved Glu-148, as demonstrated for the E. coli AK3 (Marco-Marin et al., 2003). The COO− side chain of Asp points toward the γ-phosphate moiety of AMP-PNP (light blue) (bond length between Oγ of Asp and O of the γ-phosphate moiety of AMP-PNP of 2.7 Å). The figure shows that the conserved Arg-230 is not involved in the interaction with Asp but interacts with both the Lys and the SAM binding sites of ACT1 via Leu-391 of the same monomer (monomer A) and Gly-353 (blue sticks) of the other monomer (monomer B). Glu-148, Arg-230, Ile-391, and the main chain of loop 227 to 233 of monomer A are shown with orange sticks.
METHODS
Protein Expression and Purification
Protein expression was performed in Escherichia coli BL21-DE3-RILX strain transformed with plasmid pET-23d-AK1 encoding the mature AK1 enzyme as described by G. Curien and R. Dumas (unpublished data). The purification procedure and the biochemical characterization of the three Arabidopsis thaliana AK isoforms will be described elsewhere (G. Curien and R. Dumas, unpublished data). The seleno-methionine enzyme (Se-AK1) was expressed similarly except that the growth was conducted in 1 liter of a 2× M9 minimal medium containing MgSO4 (2 mM), FeSO4 (25 mg/L), glucose (4 g/L), vitamins (thiamine, pyridoxine, riboflavin, and niacinamide at 1 mg/L), a mix of all amino acids except Met (40 mg/L), and seleno-methionine (40 mg/L). The purification procedure of the seleno-methionine enzyme was identical to the procedure used for the native enzyme, but DTT (5 mM) was added to all buffers. The native molecular weight of AK1 was estimated on native gel electrophoresis. AK1 migrated predominantly (95%) as a dimer of 120 kD in equilibrium with a tetramer of 240 kD.
Crystallization of AK1
Crystals of the native enzyme were grown at 20°C in 10-μL sitting drops containing 7 μL protein mix (30 μg AK1, 1 mM Lys, and 1 mM SAM) and 3 μL reservoir solution (0.2 M disodium tartrate dehydrate and 20% [w/v] PEG 3350). Crystals of the seleno-methionine enzyme were grown at 20°C in 12-μL hanging drops containing 8 μL protein mix (30 μg AK1, 1 mM Lys, 1 mM SAM, and 5 mM DTT) and 4 μL reservoir solution (0.2 M disodium tartrate dehydrate and 20% [w/v] PEG 3350). Cryoconditions consisted of 20% (v/v) ethylene glycol, 1 mM Lys, and 1 mM SAM (and 1 mM DTT for crystals grown from the seleno-methionine AK1) added to the reservoir solution. SAM was purified as previously described (Curien et al., 1998).
Structure Determination
Seleno-methionine derivative data (3.1 Å) were collected on beamline ID29 at the European Synchrotron Radiation Facility (ESRF), using an ADSC CCD detector. Data were processed with XDS (Kabsch, 1993). The crystals contain one monomer per asymmetric unit (64% of solvent) and belong to the space group I4122 (Table 1). Ten Se sites were located and used for phasing with SOLVE/RESOLVE (Terwilliger and Berendzen, 1999). Automated model building was performed by cycling with RESOLVE (Terwilliger, 2002) and REFMAC (Murshudov et al., 1997). The model was built manually in the experimental electron density maps displayed in O (Jones et al., 1991).
Model Improvement with a Native Data Set
A native data set (2.85 Å) was collected on beamline FIP-BM30A at the ESRF, using a MAR-CCD 165 detector. The first seleno-methionine derivative model was used for molecular replacement in the native data set using MOLREP (Vagin and Teplyakov, 1997). This model was adjusted in space group I4122 by rigid body using CNS (Brunger et al., 1998) followed by several cycles of refinement with CNS, including annealing and temperature factor refinement and manual rebuilding with O. Two molecules of Lys and one molecule of SAM were modeled per monomer. As Rfree did not go below 33%, we have tested lower symmetry space groups. Space group I41 was eventually chosen, giving one dimer per asymmetric unit with strong interactions between the two monomers. New refinement with restrained noncrystallographic symmetry (for the protein only) was performed with CNS, as well as addition of water molecules. Then REFMAC (Winn et al., 2001) was used for a final translation libration screw rotation refinement (Table 1). Real space R factors were calculated with respect to a 2Fo-Fc omit map using CNS (Brunger et al., 1998). The values provided above are averaged over the two monomers.
Structure Comparisons
Amino acid residues topologically equivalent were defined by the program LSQMAN (Kleywegt and Jones, 1999). After optimal superposition of these residues, the RMS deviation between the structurally aligned Cα atoms (cutoff: 3.5 Å) was determined using the program LSQMAN (Kleywegt and Jones, 1999).
Accession Number
The coordinates and the structure factors have been deposited to the Protein Data Bank (www.rcsb.org/pdb/) as entry 2cdq.
Supplemental Data
The following material is available in the online version of this article.
Supplemental Figure 1. Sequence Alignment of AK from Plants and Microorganisms.
Supplementary Material
Acknowledgments
We thank Valérie Biou (Laboratoire d'Enzymologie et Biochimie Structurales, Gif sur Yvette, France), Lilian Jacquamet, and Franck Borel (Institut de Biologie Structurale) for helpful discussions during this work. We also acknowledge staff of ID23 and FIP-BM30A beamlines at the ESRF for their help during data collection. We are grateful to Paul O'Maille (Salk Institute, San Diego, CA) for his proofreading.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (www.plantcell.org) is: Renaud Dumas (rdumas@cea.fr).
Online version contains Web-only data.
Article, publication date, and citation information can be found at www.plantcell.org/cgi/doi/10.1105/tpc.105.040451.
References
- Aravind, L., and Koonin, E.V. (1999). Gleaning non-trivial structural, functional and evolutionary information about proteins by iterative database searches. J. Mol. Biol. 287 1023–1040. [DOI] [PubMed] [Google Scholar]
- Bearer, C.F., and Neet, K.E. (1978). Threonine inhibition of the aspartokinase–homoserine dehydrogenase I of Escherichia coli. Threonine binding studies. Biochemistry 17 3512–3516. [DOI] [PubMed] [Google Scholar]
- Berkovitch, F., Nicolet, Y., Wan, J.T., Jarrett, J.T., and Drennan, C.L. (2004). Crystal structure of biotin synthase, an S-adenosylmethionine-dependent radical enzyme. Science 303 76–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger, A.T., et al. (1998). Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54 905–921. [DOI] [PubMed] [Google Scholar]
- Chipman, D.M., and Shaanan, B. (2001). The ACT domain family. Curr. Opin. Struct. Biol. 11 694–700. [DOI] [PubMed] [Google Scholar]
- Curien, G., Job, D., Douce, R., and Dumas, R. (1998). Allosteric activation of Arabidopsis threonine synthase by S-adenosylmethionine. Biochemistry 37 13212–13221. [DOI] [PubMed] [Google Scholar]
- Curien, G., Ravanel, S., Robert, M., and Dumas, R. (2005). Identification of six novel allosteric effectors of Arabidopsis thaliana aspartate kinase-homoserine dehydrogenase isoforms. Physiological context sets the specificity. J. Biol. Chem. 280 41178–41183. [DOI] [PubMed] [Google Scholar]
- Gallagher, D.T., Gilliland, G.L., Xiao, G., Zondlo, J., Fisher, K.E., Chinchilla, D., and Eisenstein, E. (1998). Structure and control of pyridoxal phosphate dependent allosteric threonine deaminase. Structure 6 465–475. [DOI] [PubMed] [Google Scholar]
- Gil-Ortiz, F., Ramon-Maiques, S., Fita, I., and Rubio, V. (2003). The course of phosphorus in the reaction of N-acetyl-L-glutamate kinase, determined from the structures of crystalline complexes, including a complex with an AlF(4)(-) transition state mimic. J. Mol. Biol. 331 231–244. [DOI] [PubMed] [Google Scholar]
- Giovanelli, J., Mudd, S.H., and Datko, A.H. (1989). Regulatory structure of the biosynthetic pathway for the aspartate family of amino acids in Lemna paucicostata Hegelm. 6746, with special reference to the role of aspartokinase. Plant Physiol. 90 1584–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves, L.M., and Switzer, R.L. (1990). Aspartokinase III, a new isozyme in Bacillus subtilis 168. J. Bacteriol. 172 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooft, R.W., Sander, C., Scharf, M., and Vriend, G. (1996). The PDBFINDER database: A summary of PDB, DSSP and HSSP information with added value. Comput. Appl. Biosci. 12 525–529. [DOI] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard. (1991). Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 (Pt 2), 110–119. [DOI] [PubMed] [Google Scholar]
- Kabsch, W. (1993). Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 26 795–800. [Google Scholar]
- Kaplun, A., Vyazmensky, M., Zherdev, Y., Belenky, I., Slutzker, A., Mendel, S., Barak, Z., Chipman, D.M., and Shaanan, B. (2006). Structure of the regulatory subunit of acetohydroxyacid synthase isozyme III from Escherichia coli. J. Mol. Biol. 357 951–963. [DOI] [PubMed] [Google Scholar]
- Kikuchi, Y., Kojima, H., and Tanaka, T. (1999). Mutational analysis of the feedback sites of lysine-sensitive aspartokinase of Escherichia coli. FEMS Microbiol. Lett. 173 211–215. [DOI] [PubMed] [Google Scholar]
- Kleywegt, G.J., and Jones, T.A. (1999). Software for handling macromolecular envelopes. Acta Crystallogr. D Biol. Crystallogr. 55 941–944. [DOI] [PubMed] [Google Scholar]
- Kobe, B., Jennings, I.G., House, C.M., Michell, B.J., Goodwill, K.E., Santarsiero, B.D., Stevens, R.C., Cotton, R.G., and Kemp, B.E. (1999). Structural basis of autoregulation of phenylalanine hydroxylase. Nat. Struct. Biol. 6 442–448. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., Moss, D.S., and Thornton, J.M. (1993). Main-chain bond lengths and bond angles in protein structures. J. Mol. Biol. 231 1049–1067. [DOI] [PubMed] [Google Scholar]
- Layer, G., Moser, J., Heinz, D.W., Jahn, D., and Schubert, W.D. (2003). Crystal structure of coproporphyrinogen III oxidase reveals cofactor geometry of radical SAM enzymes. EMBO J. 22 6214–6224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepore, B.W., Ruzicka, F.J., Frey, P.A., and Ringe, D. (2005). The x-ray crystal structure of lysine-2,3-aminomutase from Clostridium subterminale. Proc. Natl. Acad. Sci. USA 102 13819–13824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco-Marin, C., Ramon-Maiques, S., Tavarez, S., and Rubio, V. (2003). Site-directed mutagenesis of Escherichia coli acetylglutamate kinase and aspartokinase III probes the catalytic and substrate-binding mechanisms of these amino acid kinase family enzymes and allows three-dimensional modelling of aspartokinase. J. Mol. Biol. 334 459–476. [DOI] [PubMed] [Google Scholar]
- Marina, P., Martinez-Costa, O.H., Calderon, I.L., and Aragon, J.J. (2004). Characterization of the aspartate kinase from Saccharomyces cerevisiae and of its interaction with threonine. Biochem. Biophys. Res. Commun. 321 584–591. [DOI] [PubMed] [Google Scholar]
- Martin, J.L., and McMillan, F.M. (2002). SAM (dependent) I AM: The S-adenosylmethionine-dependent methyltransferase fold. Curr. Opin. Struct. Biol. 12 783–793. [DOI] [PubMed] [Google Scholar]
- Mas-Droux, C., Biou, V., and Dumas, R. (2006). Allosteric threonine synthase: Reorganization of the pyridoxal phosphate site upon asymmetric activation through S-adenosylmethionine binding to a novel site. J. Biol. Chem. 281 5188–5196. [DOI] [PubMed] [Google Scholar]
- Mazat, J.P., and Patte, J.C. (1976). Lysine-sensitive aspartokinase of Escherichia coli K12. Synergy and autosynergy in an allosteric V system. Biochemistry 15 4053–4058. [DOI] [PubMed] [Google Scholar]
- Moir, D., and Paulus, H. (1977). Properties and subunit structure of aspartokinase II from Bacillus subtilis VB217. J. Biol. Chem. 252 4648–4651. [PubMed] [Google Scholar]
- Murshudov, G.N., Vagin, A.A., and Dodson, E.J. (1997). Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53 240–255. [DOI] [PubMed] [Google Scholar]
- Paris, S., Viemon, C., Curien, G., and Dumas, R. (2003). Mechanism of control of Arabidopsis thaliana aspartate kinase-homoserine dehydrogenase by threonine. J. Biol. Chem. 278 5361–5366. [DOI] [PubMed] [Google Scholar]
- Patte, J.C., Truffa-Bachi, P., and Cohen, G.N. (1966). The threonine-sensitive homoserine deshydrogenase and aspartokinase of Escherichia coli. Evidence that the two activities are carried by a single protein. Biochim. Biophys. Acta 128 426–439. [Google Scholar]
- Ramon-Maiques, S., Marina, A., Gil-Ortiz, F., Fita, I., and Rubio, V. (2002). Structure of acetylglutamate kinase, a key enzyme for arginine biosynthesis and a prototype for the amino acid kinase enzyme family, during catalysis. Structure 10 329–342. [DOI] [PubMed] [Google Scholar]
- Ramon-Maiques, S., Marina, A., Uriarte, M., Fita, I., and Rubio, V. (2000). The 1.5 A resolution crystal structure of the carbamate kinase-like carbamoyl phosphate synthetase from the hyperthermophilic Archaeon pyrococcus furiosus, bound to ADP, confirms that this thermostable enzyme is a carbamate kinase, and provides insight into substrate binding and stability in carbamate kinases. J. Mol. Biol. 299 463–476. [DOI] [PubMed] [Google Scholar]
- Rognes, S.E., Lea, P.J., and Miflin, B.J. (1980). S-adenosylmethionine–A novel regulator of aspartate kinase. Nature 287 357–359. [DOI] [PubMed] [Google Scholar]
- Rosner, A., and Paulus, H. (1971). Regulation of aspartokinase in Bacillus subtilis. The separation and properties of two isofunctional enzymes. J. Biol. Chem. 246 2965–2971. [PubMed] [Google Scholar]
- Schuller, D.J., Grant, G.A., and Banaszak, L.J. (1995). The allosteric ligand site in the Vmax-type cooperative enzyme phosphoglycerate dehydrogenase. Nat. Struct. Biol. 2 69–76. [DOI] [PubMed] [Google Scholar]
- Somers, W.S., and Phillips, S.E. (1992). Crystal structure of the met repressor-operator complex at 2.8 A resolution reveals DNA recognition by beta-strands. Nature 359 387–393. [DOI] [PubMed] [Google Scholar]
- Terwilliger, T.C. (2002). Automated structure solution, density modification and model building. Acta Crystallogr. D Biol. Crystallogr. 58 1937–1940. [DOI] [PubMed] [Google Scholar]
- Terwilliger, T.C., and Berendzen, J. (1999). Automated MAD and MIR structure solution. Acta Crystallogr. D Biol. Crystallogr. 55 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J.R., Bell, J.K., Bratt, J., Grant, G.A., and Banaszak, L.J. (2005). Vmax regulation through domain and subunit changes. The active form of phosphoglycerate dehydrogenase. Biochemistry 44 5763–5773. [DOI] [PubMed] [Google Scholar]
- Umbarger, H.E. (1978). Amino acid biosynthesis and its regulation. Annu. Rev. Biochem. 47 532–606. [DOI] [PubMed] [Google Scholar]
- Vagin, A., and Teplyakov, A. (1997). MOLREP: An automated program for molecular replacement. J. Appl. Crystallogr. 30 1022–1025. [Google Scholar]
- Wessel, P.M., Graciet, E., Douce, R., and Dumas, R. (2000). Evidence for two distinct effector-binding sites in threonine deaminase by site-directed mutagenesis, kinetic, and binding experiments. Biochemistry 39 15136–15143. [DOI] [PubMed] [Google Scholar]
- Winn, M.D., Isupov, M.N., and Murshudov, G.N. (2001). Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 57 122–133. [DOI] [PubMed] [Google Scholar]
- Zhu, X., and Galili, G. (2003). Increased lysine synthesis coupled with a knockout of its catabolism synergistically boosts lysine content and also transregulates the metabolism of other amino acids in Arabidopsis seeds. Plant Cell 15 845–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}