

Abstract
Packaging of the adenovirus (Ad) genome into a capsid is absolutely dependent upon the presence of a cis-acting region located at the left end of the genome referred to as the packaging domain. The functionally significant sequences within this domain consist of at least seven similar repeats, referred to as the A repeats, which have the consensus sequence 5′ TTTG-N8-CG 3′. In vitro and in vivo binding studies have demonstrated that the adenovirus protein IVa2 binds to the CG motif of the packaging sequences. In conjunction with IVa2, another virus-specific protein binds to the TTTG motifs in vitro. The efficient formation of these protein-DNA complexes in vitro was precisely correlated with efficient packaging activity in vivo. We demonstrate that the binding activity to the TTTG packaging sequence motif is the product of the L4 22-kDa open reading frame. Previously, no function had been ascribed to this protein. Truncation of the L4 22-kDa protein in the context of the viral genome did not reduce viral gene expression or viral DNA replication but eliminated the production of infectious virus. We suggest that the L4 22-kDa protein, in conjunction with IVa2, plays a critical role in the recognition of the packaging domain of the Ad genome that leads to viral DNA encapsidation. The L4 22-kDa protein is also involved in recognition of transcription elements of the Ad major late promoter.
The packaging of the adenovirus (Ad) 36,000-bp, double-stranded DNA genome into a capsid is absolutely dependent upon a cis-acting region referred to as the packaging domain. This domain is located at the left end of the genome between nucleotides (nt) 200 and 400 (reviewed in reference 25). Neither the inverted terminal repeats nor the terminal proteins covalently linked to the 5′ ends of the genome are required in cis for packaging (11, 24). To achieve optimal packaging activity, however, the packaging domain must be near an end of the genome (16). The molecular role of the packaging domain is unknown, but a likely possibility is that it targets the genome to an immature procapsid via proteins that bind to packaging sequences (reviewed in reference 23). Identification of all of the DNA binding proteins relevant to packaging would assist in our understanding of the molecular details of Ad assembly and viral DNA encapsidation.
Results of mutational analyses of the packaging domain demonstrated that the critical sequences for packaging consist of a series of related elements referred to as A repeats (see Fig. 1A) (12, 16, 30). Although the seven A repeats are not identical in their DNA sequence, there is a shared consensus sequence of nucleotides TTTG, eight variable nucleotides, and a CG pair (5′ TTTG-N8-CG 3′). The identification of the core nucleotide sequences involved in packaging permitted the production of simplified packaging domains consisting of multiple copies of one A repeat or several A repeats (30, 31). The sequences of the spacer nucleotides and/or flanking nucleotides must be important for packaging function, since the different A repeats had variable efficiencies for packaging activity when used as multimeric copies in place of the authentic packaging domain sequences. Synthetic packaging sequences that functioned at wild-type or nearly wild-type levels in the context of the Ad genome were used to detect DNA binding proteins (5, 31). Several cellular DNA binding proteins that bind Ad packaging repeats were identified, but they were not found to be relevant to the packaging process (5, 26, 31). Because the packaging domain overlaps with the transcriptional enhancer region for the early genes of Ad, the packaging domain binds proteins not only important for packaging but also proteins important for Ad transcription (1-3). Therefore, it is important to demonstrate that any protein that binds to packaging sequences in vitro is relevant for packaging activity in vivo.
FIG. 1.

Ad5 packaging domain and binding proteins. (A) Schematic representation of the left end of the Ad5 genome. The genome beyond the inverted terminal repeat (ITR) is indicated by the horizontal line, and nucleotide coordinates relative to the left terminus represent the boundaries of the packaging domain. Packaging A repeats 1 through 7 are indicated by arrows above the genome. The inverted terminal repeat is represented by the rectangle, and the E1A gene is indicated. The consensus sequence for the A repeats is shown under the schematic. (B) The nucleotide sequence overlapping Ad5 A repeats 1 (A1) and 2 (A2) (underlined) is presented; consensus motifs TTTG and CG are shown in bold type. Complexes 1, 2, and 3 that form on A repeats 1 and 2 in gel mobility shift assays with Ad5-infected cell nuclear extract are shown above the sequence. Ovals represent the IVa2 and virus-induced proteins. (C) Depiction of the coding sequences for the L4 33-kDa and 22-kDa proteins. Numbers correspond to Ad5 nucleotides. The intron for the L4 33-kDa protein-coding region is indicated by the gap and angled lines. An amber termination codon was introduced in the unique coding sequences of the L4 22-kDa protein (indicated by an arrow and TAG) and is referred to as v22k− in the text.
Utilizing packaging repeats A1 and A2 as a probe, Zhang and Imperiale showed that the Ad intermediate gene product IVa2 bound to the critical CG nucleotides of the A repeats in vitro (34). The results of chromatin immunoprecipitation confirmed that IVa2 bound the packaging domain in vivo (26, 28). The IVa2 protein is involved in Ad packaging, and it is required for the assembly of virus particles late during infection (35, 36). The IVa2 protein interacts with the L1 52/55-kDa protein (14), another viral gene product involved in the packaging process (13, 15). Interestingly, the core sequences of the packaging domain are also found in the DE elements (34), transcriptional elements located downstream of the Ad major late promoter (MLP) (33). The IVa2 protein was shown to bind these sequences and transactivate the MLP (20, 32). While mutation of the IVa2 binding sites in the MLP alone modestly reduced late gene expression, the combination of a termination codon at position six in the IVa2 coding region with a mutation of the USF transcription factor binding site in the MLP resulted in a nonviable mutant virus (27). These results likely are explained by functional redundancy of transcription elements in the MLP (32). There is no evidence, however, that binding of IVa2 at the DE elements is involved in viral DNA packaging. Taken together, these results suggest that IVa2 is a multifunctional protein in the Ad life cycle, playing roles in transcription at the MLP and in the packaging of viral DNA to produce virions.
In addition to IVa2 binding at the CG motif of the core packaging sequence, an additional, virus-specific protein of unknown origin was found to bind to the TTTG motif of the packaging A repeats (34). The binding of this second, virus-specific protein, in addition to IVa2, comprised a complex crucial to packaging activity in vivo (26). We surmised that possible candidates for a protein(s) that binds to the TTTG motif of the packaging repeats were possibly the Ad L4 33-kDa protein or the putative L4 22-kDa protein. The L4 33-kDa protein is a product of a spliced late region 4 transcript (22). The L4 22-kDa protein could be translated from the unspliced version of this transcript. The L4 33-kDa and 22-kDa proteins would share a common N terminus (105 amino acids) but have different C termini (124 and 91 amino acids, respectively). Several reports indicated a role for the L4 33-kDa protein in Ad assembly (7, 8, 19). Stop codons were introduced at several locations in the open reading frame of the L4 33-kDa protein in human and bovine Ads. With two of the three viral mutants analyzed, the L4 22-kDa protein-coding sequences were also affected (7, 19). These mutations resulted in viruses that were reduced in virus production or the mutations were lethal. Importantly, all three of the mutations appeared to have an effect late in the virus life cycle consistent with a packaging defect. Mutant viruses replicated and expressed late viral gene products, but virus assembly and/or packaging of the genomes was reduced or absent (7, 8, 19).
In this article, we report that the L4 22-kDa protein binds to the TTTG motif of Ad packaging sequences. Previously, no function had been ascribed to the L4 22-kDa protein. Truncation of the L4 22-kDa protein in the context of the viral genome did not reduce viral gene expression or viral DNA replication but eliminated the production of infectious virus. We suggest that the L4 22-kDa protein, in conjunction with IVa2, plays a critical role in the recognition of the packaging domain of the Ad genome and leads to viral DNA encapsidation. The L4 22-kDa protein is also involved in recognition of the DE elements of the Ad major late promoter.
MATERIALS AND METHODS
Cells, viruses, and infections.
The E1-expressing cell line N52.E6 (29) and a derivative expressing Cre recombinase were a gift from G. Schiedner and S. Kochanek, Center for Molecular Medicine (ZMMK), University of Ulm, Ulm, Germany. HeLa cells were obtained from the ATCC. N52 cells were maintained in Dulbecco's modified Eagle medium supplemented with 10% Fetalclone III (HyClone), penicillin, and streptomycin. HeLa cells were maintained in Dulbecco's modified Eagle medium supplemented with 10% calf serum (HyClone), penicillin, and streptomycin. An N52.E6-Cre subclone that constitutively expresses Ad type 2 (Ad2) IVa2 was generated by transfection of cells with the pCDNA3-IVa2 plasmid (described below). Stable cell subclones were obtained by selection for neomycin resistance (200 μg/ml Geneticin). Clones were screened for IVa2 expression by Western blot analysis using a rabbit polyclonal antibody to IVa2 (26). The IVa2 cell line 6 showed optimal expression and was maintained in Dulbecco's modified Eagle medium supplemented with 10% Fetalclone III, penicillin, streptomycin, and 200 μg/ml Geneticin.
Cells were infected with Ad5 (wt300) at a multiplicity of infection of 10 PFU per cell, as previously described (5). Plasmid pTG3602 contains the complete Ad5 genome (4) and was a gift from Transgene, S.I. (Strasbourg, France). pTG3602-L4-22k− contains a termination codon (TAG) after amino acid 113 of the L4 22-kDa protein (Ad5 nt 26534 to 26536). The TAG codon was introduced in the context of a new AvrII restriction site (CCTAGG [the TAG codon underlined], Ad5 nt 26532 to 26537) using PCR. The mutated segment was recombined into pTG3602 using the methods of Evans and Hearing (6). PacI digestion of pTG3602 or pTG3602-L4-22k− released the Ad5 genomes. Transfection of PacI-digested plasmids into N52.E6 cre cells was done by using Fugene 6 transfection reagent (Roche) following the recommended protocol or by calcium phosphate precipitation. Cells were harvested 48 h after transfection and used to (i) prepare cellular extracts to measure infectious virus yield, (ii) prepare whole-cell extracts for protein quantification (see below), and (iii) prepare viral DNA by the method of Hirt (17).
Protein expression and antibody production.
Expression plasmids were generated using pCDNA3 (Invitrogen Life Technologies). pCDNA3-IVa2, a plasmid expressing the Ad2 IVa2 gene, was made in the following way. A DNA fragment containing the Ad sequences encompassing the second exon for Ad2 IVa2 (Ad2 nt 4081 to 5417) was obtained by PCR using a 5′ oligonucleotide containing the sequences for the first four amino acids of IVa2 from the first exon and additional sequences from the 5′ end of the second exon and a 3′ oligonucleotide containing sequences within the 3′ end of the second exon. The PCR product was cloned into the polylinker region of pCDNA3 and was confirmed to encode IVa2 by DNA sequencing. Expression plasmids for the Ad5 L4 33-kDa and 22-kDa proteins were generated by PCR cloning of the coding regions from Ad5 nt 26195 to 26510 joined to nt 26713 to 27086 for the L4 33-kDa protein and from Ad5 nt 26195 to 26785 for the L4 22-kDa protein into the pCDNA3 polylinker region.
The IVa2 and L4 22-kDa protein-coding regions were cloned into baculovirus transfer vectors and subsequently into infectious baculoviruses using the Bac-to-Bac system (GIBCO/Invitrogen Life Technologies). The IVa2 protein was expressed in its native form, and the L4 22-kDa protein was expressed with a C-terminal six-histidine tag. Virus stocks were generated by using the procedures recommended by the manufacturer. Sf9 cells were infected at a multiplicity of infection of 5, and nuclear extracts were prepared 72 h after infection as described below. The coding sequences unique to the L4 22-kDa protein were cloned into vector pGEX-KG to express a glutathione S-transferase-L4-22-kDa fusion protein in Escherichia coli. The fusion protein was purified by preparative sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and used to develop rabbit polyclonal antiserum against the L4 22-kDa protein (Lampire Biologicals, Reamstown, PA).
Preparation of nuclear extracts and gel mobility shift assays.
Nuclear extracts from Ad-infected or plasmid-transfected cells were prepared 24 to 48 h postinfection or posttransfection as previously described (26) and stored at −80°C. Proteins in nuclear extracts were quantified using the Bio-Rad protein reagent (Bio-Rad). The gel mobility shift assays were performed by the method of Ostapchuk et al. (26), with the modification that nuclear extract was incubated with probe for 30 min at room temperature. Packaging sequence probes used in the assays contained A repeats 1 and 2, corresponding to Ad5 nt 236 to 282. PM2, PM3, PM12, and PM13 probes are the same as the wild-type probe except for the point mutations illustrated below (see Fig. 5A) and previously described (26). A probe containing the DE1 and DE2 sequences from the MLP was used that corresponds to +84 to +114 relative to the transcription initiation site. The probes have additional nucleotides, TCGAC at the 5′ ends and G at the 3′ ends that are not part of the Ad5 sequences. These sequences were added for radiolabeling and cloning purposes. Oligonucleotide probes were labeled using Klenow DNA polymerase and [α-32P]dCTP. Specific activities of probes were determined by trichloroacetic acid precipitation, and ∼50,000 cpm/∼5 fmol of probe were added to each binding reaction mixture.
FIG. 5.

Point mutations in the TTTG packaging sequence motif affect binding of the L4 22-kDa protein. (A) The Ad5 sequences in the A repeat 1 and 2 probe are shown. The TTTG and CG packaging sequence motifs are shown in bold type and underlined. Point mutations introduced into A repeats 1 and 2 are shown (PM2, PM3, PM12, and PM13). (B) A gel mobility shift assay was done with probes containing wild-type (WT) and mutant A repeats 1 and 2 (indicated above the gel) using nuclear extract from cells that constitutively express the IVa2 protein and transfected with an L4 33-kDa protein expression vector. The positions of complexes 1, 2, and 3 are indicated by arrows.
Western blot analyses.
Either nuclear extracts or whole-cell lysates were used as sources of protein. In the case of whole-cell lysates, approximately 3 × 106 to 5 × 106 cells were washed twice with phosphate-buffered saline and then incubated with 400 μl of radioimmunoprecipitation assay buffer (150 mM sodium chloride, 1.0% NP-40, 0.5% deoxycholate, 0.1% sodium dodecyl sulfate, 50 mM Tris, pH 8.0) for 20 min at room temperature. The lysates were clarified by centrifugation at 13,000 × g for 10 min at 4°C. Supernatants were recovered, and protein was quantified using the BCA protein assay kit (Pierce). Nuclear extracts were prepared as described above. Proteins were separated by SDS-PAGE and transferred overnight at 4°C to Hybond P (Amersham). Membranes were blocked with a solution of 3% bovine serum albumin in Tris-buffered saline for 1 h at room temperature. Primary antibodies were diluted in the same buffer. Filters were exposed to rabbit polyclonal antibodies directed against IVa2 (26) or hexon and penton proteins (Carl Anderson, Brookhaven National Laboratory) for 1 h at room temperature or to a rabbit polyclonal antibody directed against the L4 33-kDa protein (10) or a monoclonal antibody against viral DNA binding protein (DBP) (Arnold Levine, Princeton University) for 18 h at 4°C. Filters were washed three times with Tris-buffered saline containing 5% nonfat powdered milk and 0.5% Tween 40 and then exposed to goat anti-rabbit immunoglobulin G antibody conjugated to alkaline phosphatase (Zymed) for 30 min at room temperature. The filters were washed again and exposed to the fluorescent alkaline phosphatase substrate AttoPhos (Promega) for 1 min. Images were obtained by scanning with a Storm 860 instrument (Molecular Dynamics) using the blue fluorescence setting.
Southern blot analysis and quantification of infectious virus yield.
DNA in the Hirt supernatant fraction was digested with AvrII plus DpnI, and DNA fragments were separated on a 0.8% agarose gel. The relative DNA levels in different samples were quantitated by Southern blot analysis using a fluorescent probe of the Ad5 genome labeled using the AlkPhos direct labeling system (Amersham). Southern blots were quantified using a phosphorimager (Molecular Dynamics Storm 860) and quantified using ImageQuant software version 1.2 (Molecular Dynamics). A titration of Ad5 DNA was included in each blot to ensure that the fluorescent signals obtained were in the linear range of the assay. Infectious virus yield was quantified by a fluorescent focus assay. HeLa cells were infected using different dilutions of cellular lysates prepared after transfection of cells with plasmid pTG3602 or pTG3602-L4-22k−. Eighteen hours later, cells were processed for immunofluorescence using an antibody directed against DBP as previously described (6). Infectious virus yields were calculated by determining the number of DBP-positive cells at a given dilution of the cellular lysate used for infection.
RESULTS
It was shown previously that Ad IVa2 protein, in conjunction with another virally induced protein, bound A1 and A2 repeats to form three DNA-protein complexes that we termed 1, 2, and 3 (Fig. 1B) (26). All three complexes contained the IVa2 protein. Complex 1 was due to the binding of IVa2 at the CG motif in the A1 repeat. Complex 2 was the result of binding, in addition to IVa2, a virus-induced protein at the TTTG motif in the A2 repeat. Complex 3 was likely due to the binding of an additional IVa2 protein at the CG sequence in the A2 repeat on the basis of the homology of the A1 and A2 CG motifs. The binding of IVa2 and the virally induced protein to packaging repeats in vitro was directly correlated with efficient packaging in vivo (26). The results of recent analyses of mutant human and bovine Ad viruses with stop codons in the reading frame for the putative L4 33-kDa protein and/or the L4 22-kDa protein suggested that these proteins might be involved in the packaging process (7, 8, 19). Therefore, we asked whether the formation of complexes 1, 2, and 3 on packaging repeats involved either the L4 33-kDa or 22-kDa protein.
We utilized an Ad5 mutant (v33k.1) carrying a termination codon in the regions common to both the L4 33-kDa and 22-kDa proteins (Fig. 1C) (7). Nuclear extracts were prepared at late times after infection of cells with either wild-type Ad5 or L4 mutant v33K.1 (7). The formation of complexes specific to Ad5 packaging sequences was assessed using a mobility shift assay and a probe containing A repeats 1 and 2 (Fig. 2A). Complexes 1, 2, and 3 formed with Ad5-infected cell extract (lane 2) as previously described (26). Only complex 1 formed with extracts from cells infected with the L4 mutant virus (lane 3), indicating that the L4 33-kDa and/or 22-kDa protein may represent the virus-induced activity involved in these complexes. Cytomegalovirus promoter expression vectors that contained the coding region for either the L4 33-kDa protein or L4 22-kDa protein were generated. The protein-coding region for the L4 33-kDa protein initiates at nt 26195 and contains a splice between nt 26510 and 26713, resulting in a shift in the reading frame for the remainder of the protein that terminates at nt 27086 (illustrated in Fig. 1C). The reading frame for the L4 22-kDa protein initiates at the same AUG codon and reads through the 33-kDa splice site to nt 26785 (Fig. 1C). Therefore, both proteins share an N-terminal domain of 105 amino acids. Western blot analyses were performed using an antibody raised against the L4 33-kDa protein to compare protein expression patterns from cells transfected with L4 protein expression vectors to those obtained with nuclear extracts prepared from Ad5-infected cells (Fig. 2B). The L4 33-kDa protein from Ad-infected cells runs anomalously, migrating on SDS-polyacrylamide gels at a molecular mass of 39 kDa (9, 10) compared to its predicted molecular mass of 25,162 Da. An immunoreactive band that migrated at approximately 39 kDa in the nuclear extracts from cells transfected with the L4 33-kDa expression plasmid (lane 3) that was not seen in the extracts of untransfected cells (lane 5) was observed. This band migrated slightly faster than a protein detected using Ad5-infected nuclear extracts (lane 1). We also observed a protein migrating with a molecular mass of ∼30 kDa that was specific to extracts from cells transfected with the L4 22-kDa expression plasmid (lane 4). A band migrating at approximately the same mobility was observed in Ad5-infected cell extract (lane 1) that was not seen in the mock-infected protein extract (lane 2). In repeated experiments, we found that the mobilities of these two proteins varied depending on the conditions used for SDS-PAGE (the percentage of acrylamide in the gel and the rate of electrophoresis).
FIG. 2.

L4 protein expression and DNA binding. (A) A gel mobility shift assay was performed using a probe containing Ad5 A repeats 1 and 2 with nuclear extract from cells infected with wild-type (WT) Ad5 (lane 2) or the L4 viral mutant v33k.1 (lane 3). Mock-infected cell extract was used for the binding reaction mixture in lane 1. The positions of complexes 1, 2, and 3 are indicated by the arrows to the right of the gel. (B) Western blot analysis of proteins in nuclear extracts prepared from Ad5-infected cells (lane 1), mock-infected cells (lane 2), IVa2-expressing cells transfected with an L4 33-kDa protein expression vector (lane 3), IVa2-expressing cells transfected with an L4 22-kDa protein expression vector (lane 4), and IVa2-expressing cells transfected with empty vector (−) (lane 5). The membrane was probed with an anti-L4 33-kDa protein antibody. The positions of the L4 33-kDa and 22-kDa proteins are indicated to the left of the gel. (C) Western blot analysis of proteins in nuclear extracts prepared from cells infected with Ad5 (lane 1) or L4 mutant virus v33k.1 (lane 2) and probed with an anti-L4 33-kDa protein antibody. The positions of the L4 33-kDa and 22-kDa proteins are indicated to the left of the gel. (D) Western blot analysis of cellular extracts prepared from untransfected cells (lane 1) or cells transfected with the wild-type (WT) Ad5 infectious clone (lane 3) or the Ad5 infectious clone containing a termination codon in the L4 22-kDa protein-coding sequences (lane 2; v22k−) and probed with an anti-L4 33K antibody. The positions of the L4 33-kDa and 22-kDa proteins are indicated to the left of the gel.
Confirmation that these products in fact represent proteins expressed from the L4 33/22-kDa reading frames came from the analysis of extracts prepared from cells infected with the L4 33-kDa viral mutant v33K.1 (Fig. 2C). Neither the 39-kDa nor 30-kDa protein was expressed by the mutant virus, in comparison to wild-type Ad5 (lanes 1 and 2). We engineered site-specific mutations in the unique part of the L4 22-kDa protein to introduce a termination codon (TAG) in the L4 22-kDa protein-coding region (Fig. 1C) in the background of an Ad5 infectious clone. The termination would give rise to an L4 22-kDa protein truncated after amino acid 113 in the 196-amino-acid L4 22-kDa protein-coding region. Cells were transfected with the wild-type or L4 22k− clones, and cellular extracts were prepared and analyzed for expression of the L4 33-kDa and 22-kDa proteins (Fig. 2D). A protein doublet was detected following transfection of the wild-type clone (lane 3), but only the upper protein was detected with the L4 22k− mutant (lane 2). These results show that the 22-kDa protein-coding region of L4 does indeed encode a protein that is expressed in Ad5-infected cells.
Gel mobility shift assays were done using a probe containing A repeats 1 and 2 with nuclear extracts prepared from cells that constitutively express the IVa2 protein, with or without transfection of either the L4 33-kDa or L4 22-kDa expression vector (Fig. 3A). Complexes 1, 2, and 3 formed with nuclear extracts prepared from Ad5-infected cells (lane 2). Only complex 1 was observed with nuclear extracts prepared from cells that constitutively express IVa2 (lane 3). No additional binding other than complex 1 was observed using nuclear extracts from cells expressing the IVa2 and L4 33-kDa proteins (lane 5). However, all three complexes were reproduced using nuclear extracts prepared from cells expressing IVa2 and the L4 22-kDa proteins (lane 4). These results suggest that the binding activity to the critical TTTG motif of the Ad packaging repeats is the L4 22-kDa protein. In agreement with this idea, the same three complexes were observed when nuclear extracts prepared from cells cotransfected with expression vectors for the IVa2 and L4 22-kDa proteins were used as the source of protein (Fig. 3B, lane 3). Only complex 1 was seen with nuclear extracts prepared from cells transfected with the IVa2 expression vector alone (lane 4), and no specific binding was seen with nuclear extracts prepared from cells transfected with only the L4 22-kDa expression vector (lane 2). Two approaches were taken to verify that the L4 22-kDa protein is contained in complexes 2 and 3. First, we developed a rabbit polyclonal antibody directed against the unique coding region of the L4 22-kDa protein. Addition of this antiserum to a binding reaction mixture containing Ad5-infected cell nuclear extract resulted in a reduction in the mobility of complexes 2 and 3 (Fig. 3C, lane 3, supershift), whereas the addition of preimmune serum had no effect (Fig. 3C, lane 2). We individually expressed the IVa2 and L4 22-kDa protein in insect cells using baculovirus vectors and analyzed their binding to A repeats 1 and 2 (Fig. 3D). The IVa2 protein was able to form complex 1, and the addition of the L4 22-kDa protein resulted in the formation of complexes 2 and 3 (lanes 1 and 2). Baculovirus-expressed L4 22-kDa protein carries a six-histidine tag. The addition of anti-His monoclonal antibody supershifted complexes 2 and 3 but had no effect on complex 1 (lane 3). The addition of an isotype-matched, unrelated monoclonal antibody did not alter the mobility of any of these complexes (lane 4). We conclude that the L4 22-kDa protein binds to the TTTG motif of Ad packaging sequences.
FIG. 3.

The L4 22-kDa protein binds to Ad5 packaging sequences in vitro. Gel mobility shift assays were done with a probe containing A repeats 1 and 2 using different sources of Ad5 IVa2 and L4 22-kDa proteins. (A) Lanes 1 and 2, nuclear extracts from mock-infected or Ad5-infected cells, respectively; lanes 3 to 5, cells that constitutively express the IVa2 protein were transfected with empty vector (−) (lane 3), an L4 22-kDa protein expression vector (22k) (lane 4), or an L4 33-kDa protein expression vector (33k) (lane 5). (B) Cells were transfected with empty vector (lane 1), an L4 22-kDa protein expression vector (lane 2), expression vectors for the IVa2 and L4 22-kDa proteins (lane 3), or a IVa2 protein expression vector (lane 4). (C) Nuclear extract from cells infected with wild-type Ad5 was used for the binding reactions. No antibody was added (lane 1) or anti-L4- 22-kDa protein antibody (α-22k Ab) (lane 3) or preimmune (preimm.) serum (lane 2) was added to the binding reaction mixture prior to the addition of probe DNA. (D) Ad5 IVa2 and L4 22-kDa proteins were expressed in insect cells using baculovirus vectors, and nuclear extracts were prepared. Lanes 1 and 2, addition of IVa2 or IVa2 and L4 22-kDa proteins to the binding reaction mixtures, respectively; lane 3, IVa2 and L4 22-kDa proteins added to the binding reaction mixture in addition to an anti-His monoclonal antibody (αhis Ab); lane 4, IVa2 and L4 22-kDa proteins added to the binding reaction mixture in addition to an isotype-matched unrelated monoclonal antibody (anti-E2F4) (ns Ab). The positions of complexes 1, 2, and 3 are indicated by arrows.
We examined the growth properties of the L4 22-kDa mutant (v22k−) in the context of the Ad5 infectious clone. A termination codon in the L4 22-kDa protein-coding region was designed to introduce an AvrII restriction enzyme site as a marker for the mutation. Cells were transfected with wild-type and L4 22-kDa mutant plasmids linearized with PacI, and viral DNA was isolated 48 h after transfection. The DNAs were digested with AvrII and DpnI. AvrII was used to detect the additional restriction enzyme site in the mutant genome, and DpnI was used to digest input plasmid DNA newly replicated viral DNAs will not be methylated and thus will be DpnI resistant. Plasmid and viral DNAs were analyzed by Southern blotting (Fig. 4A). Equivalent amounts of input plasmid DNAs were evident with the wild-type and mutant clones, and both clones replicated efficiently (lanes 3 and 4). The diagnostic AvrII cleavage patterns were evident with both clones in comparison to plasmid DNAs digested by AvrII plus PacI (lanes 1 and 2). We next examined viral early and late gene expression by Western blotting (Fig. 4B). The cells transfected with the wild-type and L4 22-kDa mutant plasmids expressed equivalent levels of the early gene product DBP and the late gene products L1 52/55-kDa, hexon, and penton. Finally, we examined infectious virus yield using a fluorescent focus assay with cell lysates from transfected cells. Transfection with the wild-type Ad5 infectious clone gave rise to 5 × 104 focus-forming units/ml, whereas no infectious virus was produced with the L4 22-kDa mutant. This phenotype is consistent with a role for the L4 22-kDa protein in virus assembly and/or viral DNA packaging.
FIG. 4.

Analysis of an L4 22-kDa viral mutant. Cells were transfected with either a wild-type (WT) Ad5 infectious clone or an infectious clone containing an termination codon in the unique coding region of the L4 22-kDa protein. (A) Viral DNAs were isolated and analyzed by Southern blotting using an Ad5 genomic probe. Lanes 1 and 2 contain plasmid DNAs digested with PacI plus AvrII (lane 1, wild-type virus; lane 2, v22k−). Four virus-specific bands were observed with the wild-type clone (top part of gel) along with the plasmid backbone (lower part of gel). Five virus-specific bands were observed with the L4 22-kDa mutant due to the introduction of an AvrII cleavage site at the site of the mutations. Lanes 3 and 4 contain AvrII- and DpnI-digested viral DNAs (lane 3, wild-type virus; lane 4, v22k−). The bands near the top of the gel correspond to newly replicated, DpnI-resistant viral DNAs (replic. vDNA). The bands near the bottom of the gel correspond to the DpnI-sensitive input plasmid DNAs, as indicated to the right of the gels. (B) Western blot analysis of DBP, hexon, and penton protein expression with extracts prepared from untransfected (uninfected [uninf.]) cells (lane 1), cells transfected with the wild-type (WT) Ad5 infectious clone (lane 2), and cells transfected with the infectious clone containing an termination codon in the unique coding region of the L4 22-kDa protein (v22k−).
The formation of complex 2 previously had been shown to be sensitive to various point mutations within the TTTG sequences of A repeats 1 and 2 (illustrated in Fig. 5A). We tested whether the binding of the L4 22-kDa protein to the TTTG motif was affected by these mutations in the same way as were the natural Ad5-infected cell complexes. Mobility shift assays were done using nuclear extracts prepared from cells that constitutively express IVa2 and that were transfected with an expression vector for the L4 22-kDa protein. These results are shown in Fig. 5B. PM12 did not disrupt the formation of complex 2 or 3 when nuclear extracts expressing only Ad proteins IVa2 and L4 22-kDa protein were used (lane 3). Mutations PM2, PM3, and PM13 markedly reduced or eliminated the formation of complexes 2 and 3 (lanes 2, 4, and 5, respectively). These results correlate precisely with those obtained when the mutant packaging repeat probes were used with nuclear extracts prepared from Ad5-infected cells (26). Thus, the binding of the L4 22-kDa protein to packaging repeats in vitro (Fig. 5B) correlates precisely with packaging activity in vivo (26). As anticipated, the binding of IVa2 to the CG motif was unaffected by the point mutations, and complex 1 was observed in all the lanes.
The Ad5 IVa2 protein interacts with the DE elements downstream of the major late promoter transcription initiation site, and IVa2 serves as a transcriptional activator of the MLP (21, 32). IVa2 is involved in the formation of two complexes at the MLP termed DEF-A and DEF-B (20, 32). DEF-B corresponds to a dimer of IVa2; DEF-A appears to represent IVa2 in association with an unknown, virus-induced activity (20, 32). We asked whether the L4 22-kDa protein is a binding partner with IVa2 on the DE sequences of the MLP. Gel mobility shift assays were done using a DE probe and nuclear extracts prepared from cells that express IVa2 and that were transfected with either the L4 33-kDa or L4 22-kDa expression vector (Fig. 6). The results of these analyses demonstrated that coexpression of the IVa2 and L4 22-kDa proteins resulted in the formation of two complexes on the DE probe (lane 4) that comigrated with complexes observed using Ad5-infected cell extract (lane 2). Weak binding of the IVa2 protein alone was observed (lane 3), and expression of the L4 33-kDa protein did not change this binding pattern (lane 5). We conclude that a virus-induced activity that is found with IVa2 on the MLP DE sequences corresponds to the L4 22-kDa protein.
FIG. 6.
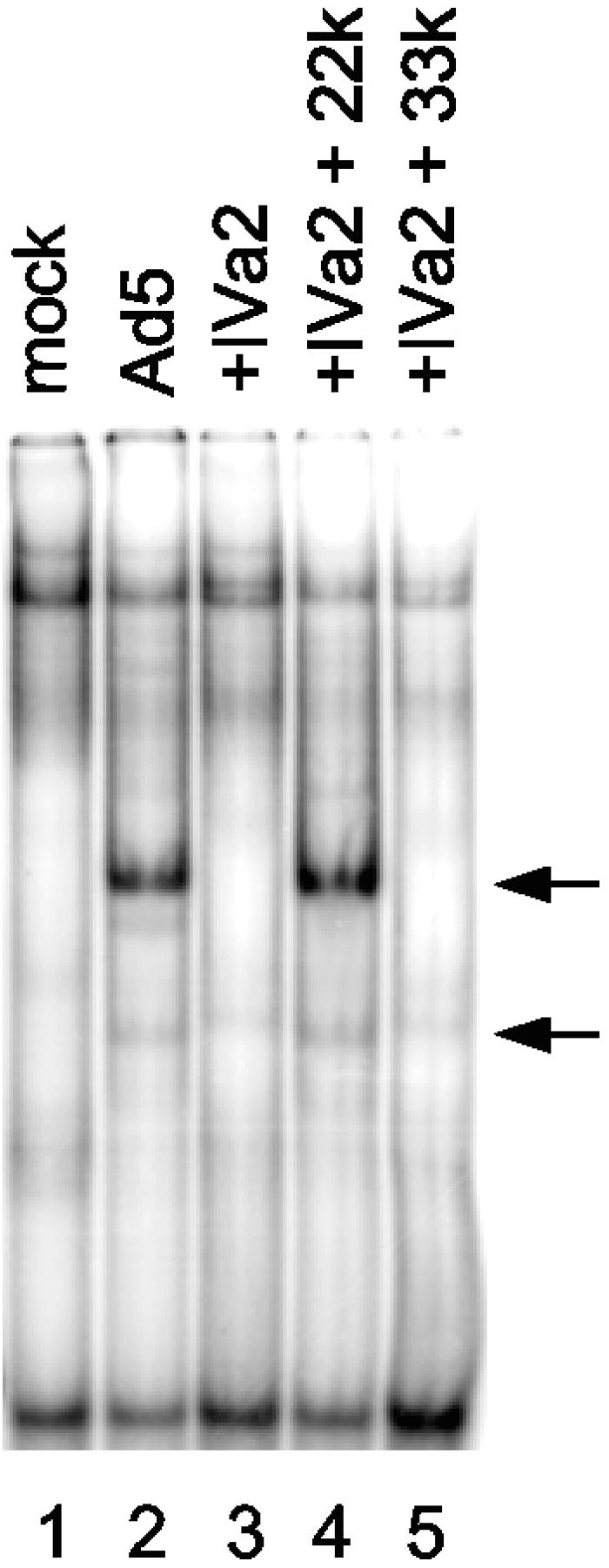
The L4 22-kDa protein binds to the DE element in the major late promoter. A gel mobility shift assay was performed with a DE probe and nuclear extract prepared from mock-infected or Ad5-infected cells (lanes 1 and 2, respectively) or cells transfected with expression vectors for the IVa2 protein (lane 3), IVa2 and L4 22-kDa proteins (22k) (lane 4), or the IVa2 and L4 33-kDa proteins (33k) (lane 5). The positions of Ad5 infection-specific complexes are indicated by arrows.
DISCUSSION
We have demonstrated that the product of an unspliced L4 transcript, encoding the L4 22-kDa protein, is synthesized in Ad-infected cells. An amber mutation introduced into the coding region of the L4 22-kDa protein resulted in a block to infectious virus production. The reduction in virus growth was not at the level of viral DNA replication (Fig. 4A), nor does it appear to be at the level of viral gene expression as seen for DBP, L1 52/55-kDa, hexon, and penton proteins (Fig. 4B). These results suggest that the block occurs after late gene expression consistent with a defect in virus assembly and/or viral DNA packaging. The introduction of an opal mutation in coding sequences shared by the L4 33-kDa and 22-kDa proteins eliminated the detection by Western blotting of two protein species that were recognized by an anti-33-kDa protein antiserum (Fig. 2C). The upper species migrated in a similar, but not identical, manner with the L4 33-kDa protein expressed in a transfection assay. The slight difference in the mobilities of these two products may reflect differences in posttranslation modification, perhaps phosphorylation (see below). The faster-migrating species comigrated with the L4 22-kDa protein expressed in a transfection assay. Confirmation that the faster species corresponds to the L4 22-kDa protein was obtained following transfection of an Ad5 infectious clone containing mutations in coding sequences unique to the L4 22-kDa protein that would truncate this gene product (Fig. 2D). The faster-migrating species was evident following transfection with the wild-type Ad5 infectious clone, but not with the L4 22-kDa mutant. In contrast, the slower-migrating species (the L4 33-kDa protein) was evident following transfection with both wild-type and mutant clones.
Consistent with the idea that the L4 22-kDa protein has a role in viral DNA packaging are the results of gel mobility shift assays. The results showed that the formation of complexes 1, 2, and 3 on Ad5 packaging repeats 1 and 2 (depicted in Fig. 1B; described in reference 25) was recapitulated by IVa2 and L4 22-kDa proteins expressed using transfection assays (Fig. 3A and B) or with baculovirus vectors (Fig. 3D). Complex 1 was formed by the expression of IVa2 alone and binding of IVa2 to the CG motif in the A1 repeat. The L4 22-kDa protein, in addition to IVa2, is responsible for the formation of complex 2 and requires the TTTG motif of the A2 repeat. Complex 3 is likely due to the binding of an additional IVa2 protein to the CG sequence in the A2 repeat on the basis of the homology of the A1 and A2 CG motifs. We did not observe binding of the L4 22-kDa protein to the TTTG motif in the absence of IVa2 in vitro. One might imagine that a series of complexes consisting of the IVa2 and L4 22-kDa proteins could be reiterated on the authentic packaging domain consisting of the seven tandem A repeat elements (Fig. 1A). Previous results showed that an additional protein, the L1 52/55-kDa protein, also associated with the packaging domain in vivo (26, 28), and viruses with mutations in this gene are defective in the packaging process (13, 15). The binding of these proteins to the packaging domain of the Ad genome may result in a complex initiating the formation of a portal entry site in an immature virus particle.
It had been shown that the efficient formation of complexes 2 and 3 in vitro correlated precisely with efficient Ad packaging in vivo (26). Mutations introduced into the critical TTTG motif of the A repeat that reduced Ad packaging in vivo also eliminated the formation of complexes 2 and 3 in vitro. We conclude that the binding of the IVa2 and L4 22-kDa proteins to wild-type and mutant packaging repeat probes was identical to that of Ad5-infected cell extracts in comparison to extracts containing the two proteins expressed in the absence of other Ad products. The binding of both the IVa2 and L4 22-kDa proteins to packaging sequences is required for Ad packaging, since mutations of the critical CG nucleotides that disrupt binding of IVa2 and certain mutations in the TTTG motif that disrupt L4 22-kDa protein binding were lethal in the context of the virus (26).
The L1 52/55-kDa protein is able to interact with Ad packaging sequences in vivo in the absence of the IVa2 protein (28), but it remains to be determined whether the L4 22-kDa protein may play a role in L1 52/55-kDa protein recruitment, since the latter protein does not appear to bind DNA on its own. The L4 33-kDa protein, which shares an N-terminal domain with the L4 22-kDa protein, did not detectably bind packaging repeats. Therefore, it seems likely that the unique C-terminal domain of the L4 22-kDa protein is required for DNA binding. Sequence comparisons of L4 22-kDa proteins from human adenoviruses with adenoviruses from species as diverse as dogs, mice, fowl, and amphibians show that there is a series of conserved amino acids within the C termini of these proteins. The observed conservation of amino acids suggests that the L4 22-kDa product is synthesized by Ads that infect species other than humans and that these C-terminal amino acids may be important for DNA binding, since A-repeat-like sequences are identifiable in the left ends of evolutionarily divergent Ads.
The L4 22-kDa protein, like the L4 33-kDa protein, runs anomalously on SDS-polyacrylamide gels. The L4 22-kDa protein runs at approximately 30 kDa, and the L4 33-kDa protein runs at approximately 39 kDa. It was suggested that the anomalous migration of the L4 33-kDa protein was due to the high percentage of glutamic acid and proline residues (22). Seventy-five percent of glutamic acid residues and 89% of proline residues are found in the N-terminal portion of the L4 22-kDa protein—the region shared by the 22-kDa and 33-kDa proteins, which likely explains why the L4 22-kDa protein runs anomalously as well. Analyses of the tryptic peptides of the L4 33-kDa protein suggested that a peptide encompassing amino acids 7 to 70 was phosphorylated (22). This region contains seven serine residues and is identical in both the L4 33-kDa and 22-kDa proteins, raising the possibility that the L4 22-kDa protein may be phosphorylated as well. Gambke and Deppert (10) showed that the L4 33-kDa protein could be specifically immunoprecipitated from 32P-labeled Ad-infected cell extracts. However, they did not observe the L4 22-kDa protein in these assays. They used the same antibody that we used for Western blotting to detect the L4 22-kDa protein (Fig. 2). It may be that the L4 22-kDa protein is not immunoprecipitated by this antibody or that the L4 22-kDa protein is not phosphorylated, or at least not to the same extent as the L4 33-kDa protein. Whether these proteins are differentially modified awaits future analyses. Finally, Jansen-Durr and colleagues determined that the size of a virus-induced protein that binds to the DE elements in the Ad MLP was ∼40 kDa (18). Our data (Fig. 6) suggest that this protein of unknown origin may be the L4 22-kDa protein. The apparent discrepancy between the molecular mass estimates of this product in our studies and those of the Kedinger group may reflect the anomalous behavior of the L4 22-kDa protein depending on the conditions used for SDS-PAGE.
The amber mutation that we introduced into the L4 22-kDa protein resulted in a lethal phenotype that appears to be due to a defect that occurs after late gene expression. This result is consistent with a role for the L4 22-kDa protein in virus assembly and/or viral DNA packaging. We did not detect the 113-amino-acid truncated protein that would be expressed with the L4 22-kDa mutant (Fig. 2D). It is possible that the truncated protein is not stable in cells. Other viral mutants have been described in the human and bovine L4 33-kDa/22-kDa protein-coding regions (7, 8, 19). The phenotypes observed with these mutants are similar to that found with our L4 22-kDa mutant and support the notion that these proteins play important roles in virus assembly and/or packaging. Stop codons were introduced into the human Ad and bovine Ad3 L4 22-kDa/33-kDa protein-coding regions. The mutations were introduced early in the open reading frame, after amino acid 20 in human Ad and after amino acid 6 in bovine Ad (7, 19). These mutations would affect both the L4 22-kDa and 33-kDa proteins. These mutations were not lethal, but rather the mutant viruses exhibited reductions in the yield of virus. One might anticipate that preventing the synthesis of a protein involved in packaging would be lethal. Interestingly, results from the bovine system showed that a truncated form of the products of the L4 22-kDa/33-kDa reading frame was made with an L4 mutant (19). Kulshreshtha et al. (19) generated an antibody to the C-terminal 197 amino acids of the L4 22-kDa reading frame. Therefore, this polyclonal antibody could recognize both L4 22-kDa and 33-kDa proteins by virtue of antibodies with epitopes in the shared N terminus. Three bands migrating at 42 kDa, 38 kDa, and 33 kDa were observed in bovine Ad-infected cell extracts utilizing this antibody. Western blot analysis of extracts from the bovine L4 33-kDa/22-kDa mutant, BAV-33S1, showed a single immunoreactive protein migrating slightly faster than the 33-kDa band. This smaller protein may have initiated at methionine 87 in the open reading frame of the L4 33-kDa/22-kDa proteins, leading to the production of a truncated protein that was able to support virus growth at a reduced level. It seems likely that the human Ad L4 33-kDa/22-kDa mutant virus, v33K.1 (7), likewise could produce a truncated form of the L4 33-kDa/22-kDa proteins by initiation at one of the downstream methionines at residue 35, 36, or 77. Hence, the mutation generates a virus that is defective rather than lethal. Consistent with this idea are our observations from mobility shift assays with nuclear extracts made from cells infected with the human Ad L4 33-kDa/22-kDa mutant virus v33K.1. Complex 1 formed due to binding of the IVa2 protein. The formation of complexes 2 and 3 was severely impaired with this mutant virus; however, two new complexes that migrated faster than complexes 2 and 3 were weakly evident on longer exposures of the gels (data not shown), consistent with the initiation of translation at an internal methionine residue(s). Also of note is the lethality of a truncation mutation introduced into the unique C-terminal segment of the L4 33-kDa protein with a mutant phenotype that is consistent with a virus assembly defect (8). Together, these published results and those reported in this article argue that both the L4 22-kDa and 33-kDa proteins play important roles in the Ad assembly and/or packaging process.
Acknowledgments
We thank several colleagues for the generous gifts of antibodies including Wolfgang Deppert for antibody to the L4 33-kDa protein, Carl Anderson for antibodies to hexon and penton, and Arnold Levine for antibody against DBP. We thank Hamish Young for the viral mutant v33K.1. We acknowledge the excellent technical assistance of Chris Gordon for baculovirus generation and protein expression. We thank members of our laboratory for informed discussions.
This work was supported by NIH grant AI041636.
REFERENCES
- 1.Bolwig, G. M., J. T. Bruder, and P. Hearing. 1992. Different binding site requirements for binding and activation for the bipartite enhancer factor EF-1A. Nucleic Acids Res. 20:6555-6564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bruder, J. T., and P. Hearing. 1991. Cooperative binding of EF-1A to the E1A enhancer region mediates synergistic effects on E1A transcription during adenovirus infection. J. Virol. 65:5084-5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bruder, J. T., and P. Hearing. 1989. Nuclear factor EF-1A binds to the adenovirus E1A core enhancer element and to other transcriptional control regions. Mol. Cell. Biol. 9:5143-5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chartier, C., E. Degryse, M. Gantzer, A. Dieterle, A. Pavirani, and M. Mehtali. 1996. Efficient generation of recombinant adenovirus vectors by homologous recombination in Escherichia coli. J. Virol. 70:4805-4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Erturk, E., P. Ostapchuk, S. I. Wells, J. Yang, K. Gregg, A. Nepveu, J. P. Dudley, and P. Hearing. 2003. Binding of CCAAT displacement protein CDP to adenovirus packaging sequences. J. Virol. 77:6255-6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Evans, J. D., and P. Hearing. 2003. Distinct roles of the adenovirus E4 ORF3 protein in viral DNA replication and inhibition of genome concatenation. J. Virol. 77:5295-5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fessler, S. P., and C. S. Young. 1999. The role of the L4 33K gene in adenovirus infection. Virology 263:507-516. [DOI] [PubMed] [Google Scholar]
- 8.Finnen, R. L., J. F. Biddle, and J. Flint. 2001. Truncation of the human adenovirus type 5 L4 33-kDa protein: evidence for an essential role of the carboxy-terminus in the viral infectious cycle. Virology 289:388-399. [DOI] [PubMed] [Google Scholar]
- 9.Gambke, C., and W. Deppert. 1981. Late nonstructural 100,000- and 33,000-dalton proteins of adenovirus type 2. I. Subcellular localization during the course of infection. J. Virol. 40:585-593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gambke, C., and W. Deppert. 1981. Late nonstructural 100,000- and 33,000-dalton proteins of adenovirus type 2. II. Immunological and protein chemical analysis. J. Virol. 40:594-598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goncalves, M. A., I. van der Velde, J. M. Janssen, B. T. Maassen, E. H. Heemskerk, D. J. Opstelten, S. Knaan-Shanzer, D. Valerio, and A. A. de Vries. 2002. Efficient generation and amplification of high-capacity adeno-associated virus/adenovirus hybrid vectors. J. Virol. 76:10734-10744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grable, M., and P. Hearing. 1990. Adenovirus type 5 packaging domain is composed of a repeated element that is functionally redundant. J. Virol. 64:2047-2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gustin, K. E., and M. J. Imperiale. 1998. Encapsidation of viral DNA requires the adenovirus L1 52/55-kilodalton protein. J. Virol. 72:7860-7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gustin, K. E., P. Lutz, and M. J. Imperiale. 1996. Interaction of the adenovirus L1 52/55-kilodalton protein with the IVa2 gene product during infection. J. Virol. 70:6463-6467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hasson, T. B., P. D. Soloway, D. A. Ornelles, W. Doerfler, and T. Shenk. 1989. Adenovirus L1 52- and 55-kilodalton proteins are required for assembly of virions. J. Virol. 63:3612-3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hearing, P., R. J. Samulski, W. L. Wishart, and T. Shenk. 1987. Identification of a repeated sequence element required for efficient encapsidation of the adenovirus type 5 chromosome. J. Virol. 61:2555-2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirt, B. 1967. Selective extraction of polyomavirus DNA from infected mouse cell cultures. J. Mol. Biol. 26:365-369. [DOI] [PubMed] [Google Scholar]
- 18.Jansen-Durr, P., G. Mondesert, and C. Kedinger. 1989. Replication-dependent activation of the adenovirus major late promoter is mediated by the increased binding of a transcription factor to sequences in the first exon. J. Virol. 63:5124-5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kulshreshtha, V., L. A. Babiuk, and S. K. Tikoo. 2004. Role of bovine adenovirus-3 33K protein in viral replication. Virology 323:59-69. [DOI] [PubMed] [Google Scholar]
- 20.Lutz, P., and C. Kedinger. 1996. Properties of the adenovirus IVa2 gene product, an effector of late-phase-dependent activation of the major late promoter. J. Virol. 70:1396-1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mondesert, G., C. Tribouley, and C. Kedinger. 1992. Identification of a novel downstream binding protein implicated in late-phase-specific activation of the adenovirus major late promoter. Nucleic Acids Res. 20:3881-3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oosterom-Dragon, E. A., and C. W. Anderson. 1983. Polypeptide structure and encoding location of the adenovirus serotype 2 late, nonstructural 33K protein. J. Virol. 45:251-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ostapchuk, P., and P. Hearing. 2005. Control of adenovirus packaging. J. Cell. Biochem. 96:25-35. [DOI] [PubMed] [Google Scholar]
- 24.Ostapchuk, P., and P. Hearing. 2003. Minimal cis-acting elements required for adenovirus genome packaging. J. Virol. 77:5127-5135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ostapchuk, P., and P. Hearing. 2003. Regulation of adenovirus packaging. Curr. Top. Microbiol. Immunol. 272:165-185. [DOI] [PubMed] [Google Scholar]
- 26.Ostapchuk, P., J. Yang, E. Auffarth, and P. Hearing. 2005. Functional interaction of the adenovirus IVa2 protein with adenovirus type 5 packaging sequences. J. Virol. 79:2831-2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pardo-Mateos, A., and C. S. Y. Young. 2004. Adenovirus IVa2 protein plays an important role in transcription from the major late promoter in vivo. Virology 327:50-59. [DOI] [PubMed] [Google Scholar]
- 28.Perez-Romero, P., R. E. Tyler, J. R. Abend, M. Dus, and M. J. Imperiale. 2005. Analysis of the interaction of the adenovirus L1 52/55-kilodalton and IVa2 proteins with the packaging sequence in vivo and in vitro. J. Virol. 79:2366-2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schiedner, G., S. Hertel, and S. Kochanek. 2000. Efficient transformation of primary human amniocytes by E1 functions of Ad5: generation of new cell lines for adenoviral vector production. Hum. Gene Ther. 11:2105-2116. [DOI] [PubMed] [Google Scholar]
- 30.Schmid, S. I., and P. Hearing. 1997. Bipartite structure and functional independence of adenovirus type 5 packaging elements. J. Virol. 71:3375-3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmid, S. I., and P. Hearing. 1998. Cellular components interact with adenovirus type 5 minimal DNA packaging domains. J. Virol. 72:6339-6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tribouley, C., P. Lutz, A. Staub, and C. Kedinger. 1994. The product of the adenovirus intermediate gene IVa2 is a transcriptional activator of the major late promoter. J. Virol. 68:4450-4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Young, C. S. Y. 2003. The structure and function of the adenovirus major late promoter. Curr. Top. Microbiol. Immunol. 272:213-250. [DOI] [PubMed] [Google Scholar]
- 34.Zhang, W., and M. J. Imperiale. 2000. Interaction of the adenovirus IVa2 protein with viral packaging sequences. J. Virol. 74:2687-2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang, W., and M. J. Imperiale. 2003. Requirement of the adenovirus IVa2 protein for virus assembly. J. Virol. 77:3586-3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang, W., J. A. Low, J. B. Christensen, and M. J. Imperiale. 2001. Role for the adenovirus IVa2 protein in packaging of viral DNA. J. Virol. 75:10446-10454. [DOI] [PMC free article] [PubMed] [Google Scholar]