

Abstract
A role for the RNA interference (RNAi) pathway in the establishment of heterochromatin is now well accepted for various organisms. Less is known about its relevance and precise role in mammalian cells. We previously showed that tandem insertion of a 1,000-copy inducible transgene into the genome of baby hamster kidney (BHK) cells initiated the formation of an extremely condensed chromatin locus. Here, we characterized the inactive transgenic locus as heterochromatin, since it was associated with heterochromatin protein 1 (HP1), histone H3 trimethylated at lysine 9, and cytosine methylation in CpG dinucleotides. Northern blot analysis did not detect any transgene-derived small RNAs. RNAi-mediated Dicer knockdown did not disrupt the heterochromatic transgenic locus or up-regulate transgene expression. Moreover, neither Dicer knockdown nor overexpression of transgene-directed small interfering RNAs altered the bidirectional transition of the transgenic locus between the heterochromatic and euchromatic states. Interestingly, tethering of HP1 to the transgenic locus effectively induced transgene silencing and chromatin condensation in a Dicer-independent manner, suggesting a role for HP1 in maintaining the heterochromatic locus. Our results suggest that the RNAi pathway is not required for the assembly and maintenance of noncentromeric heterochromatin initiated by tandem transgene repeats in mammalian cells.
In eukaryotic cells, DNA is packaged with chromosomal proteins into chromatin and higher-order chromosome structures. Chromosomes are composed of two types of domains, euchromatin and heterochromatin. While euchromatin is generally considered to be the transcriptionally active portion of the genome, heterochromatin was originally defined through cytological studies as genomic regions that remain visibly condensed and deeply stained throughout the cell cycle (42). From the biochemical aspect, DNA methylation, histone H3 methylated at lysine 9 (H3K9me), and heterochromatin protein 1 (HP1) have been identified as important markers for heterochromatic domains (42). Heterochromatin mediates various functions in the nucleus, including centromere function, gene silencing, and nuclear organization (14). In large blocks of heterochromatin surrounding functional chromosome structures, such as centromeres and telomeres, heterochromatin is also thought to stabilize repetitive DNA sequences and transposable elements (13, 29).
Recent studies of diverse organisms have provided new insights into the mechanism of heterochromatin organization. The requirement for an RNA interference (RNAi) pathway for heterochromatin assembly was originally discovered, and is now well established, in the fission yeast Schizosaccharomyces pombe, in which the RNAi machinery is required for the assembly of silent condensed heterochromatin at centromeres and the mating-type region (16, 51). RNAi-mediated heterochromatin formation has subsequently been identified in other organisms, including Arabidopsis thaliana (52) and Drosophila melanogaster (39). Studies of vertebrates further revealed a similar reliance on RNAi for centromeric heterochromatin formation in a chicken-human hybrid DT40 cell line (12). The first evidence linking the RNAi pathway to heterochromatin formation in mammals was provided by a recent report showing that Dicer-deficient mouse embryonic stem (ES) cells are defective in centromeric silencing (24). Thus, the role of RNAi in heterochromatin formation appears to be conserved among diverse species and probably reflects genome defense mechanisms against invasive sequences, transposons, and related repeats (32).
Studies of fission yeast have proposed a collective model suggesting a crucial role for components of the RNAi pathway in heterochromatin assembly at centromeric regions (14, 49). Similar to centromeres, in which the repetitive DNA content seems to be the major target for heterochromatin assembly, transgene repeat-induced heterochromatin formation has been observed in diverse organisms (7, 10, 18, 21, 28). However, the mechanisms that define the transgene array as the site of heterochromatin remain unknown.
Using the lac operator/repressor system (43), we have developed a system to visualize a transgenic locus and its protein product directly in living cells (47). This system consists of a stable cell line, clone 2, in which a 1,000-copy inducible reporter plasmid, p3216PCbeta (GenBank/EMBL/DDBJ accession number, AB236435), was tandemly inserted into a single site in the genome of baby hamster kidney (BHK) cells. The reporter plasmid is 18.52 kb in length and composed of 256 copies (9.7 kb) of direct lac operator repeats, followed by 96 tandemly arranged copies (4 kb) of the tetracycline-responsive element (TRE; also known as the tet operator) controlling a minimal cytomegalovirus (CMVm) promoter regulating the expression of a cyan fluorescent protein (CFP) with a peroxisome-targeting signal (Ser-Lys-Leu tripeptide; SKL) as a reporter. The transgenic locus remains an extremely condensed structure in interphase cells, as visualized by binding of a fluorescently labeled lac repressor (LacR) to the lac operator array. Upon induction by doxycycline (Dox), Tet-On, which is a fusion protein of a reverse Tet repressor (rTetR) and the VP16 acidic activation domain (AAD), is recruited to the TRE repeats and subsequently induces transgene activation and large-scale chromatin decondensation (47).
The highly condensed transgenic locus in clone 2 cells resembles centromeric heterochromatin from the aspect of its repetitive DNA content. The integrated reporter plasmid contains a large array (13.5 kb) of tandemly repeated lac operator and TRE sequences, and the 1,000-copy transgene array extends over a chromosome region close to 20 Mb in length, which is even larger than the satellite repeat array (0.5 to 5 Mb) found in pericentromeric heterochromatin in mammals (29). While previous studies have mainly focused on the unfolding of the large-scale chromatin structure (3, 21, 37, 47, 48), particularly interesting questions that remain to be addressed are how the tandem transgene repeats form such an extremely condensed chromatin structure and how the condensed locus is maintained throughout the cell cycle. Given the prevalence of RNAi-dependent centromeric heterochromatin assembly and the structural analogy of the tandem transgene repeats to centromeric repeats, it is worth investigating whether RNAi also plays a role in mammalian heterochromatin induced by transgene repeats.
In the present study, we characterized the condensed transgenic locus in clone 2 cells as heterochromatin, since it was associated with H3K9me3, HP1, and CpG methylation. Northern blot analysis revealed that the inactive transgene repeats did not produce any small RNAs that might be functionally involved in the heterochromatinization. We further demonstrated that neither RNAi-mediated Dicer knockdown nor overexpression of transgene-directed small interfering RNAs (siRNAs) altered the transgene expression and large-scale chromatin structure at the transgenic locus. In contrast, tethering of HP1 to the transgenic locus effectively antagonized VP16-induced transgene expression, and heterochromatin decondensation in Dicer-depleted cells, similar to its effects in control cells. Taken together, these data suggest that the assembly and maintenance of the heterochromatin locus at the tandem transgene array do not require the RNAi pathway.
MATERIALS AND METHODS
Plasmid construction.
All the oligonucleotide sequences used in this study are shown in Table S1 in the supplemental material. Human HP1α, HP1β, HP1γ, and SUV39H1 cDNA fragments were PCR amplified with Pfu DNA polymerase from Human HeLa (S3) QUICK-Clone cDNAs (Clontech) using primers bearing BsiWI and BamHI sites. The amplified DNA fragments were cloned into the EcoRV site of pBluescriptII KS(−) (Stratagene), sequenced, and then digested with BsiWI and BamHI for subcloning.
The phTet-On and phTet-Off plasmids used for transgene activation were humanized codon versions of pTet-On and pTet-Off (Clontech), respectively. The humanized version of the rTetR portion of the phTet-On plasmid was synthesized by sequential PCRs using overlapping oligonucleotides covering the entire rTetR coding region. The humanized version of the normal TetR sequence was generated by the same procedure. The humanized sequences were designed to adapt codon usage for use in mammalian cells and to eliminate potential cryptic splice donor and acceptor sites. The humanized TetR and rTetR sequences and clones are available from W. Reith upon request (Walter.Reith@medecine.unige.ch).
For expression and tethering to the transgenic locus, fusion proteins with rTetR were constructed based on the phTet-On plasmid. One BamHI site between the neomycin resistance gene and the ampicillin resistance gene was removed by digestion, blunt ended, and self-ligated. To insert a FLAG epitope into phTet-On, a BsiWI site was recreated in phTet-On by site-directed mutagenesis at a position corresponding to that in the original plasmid. The resulting plasmid was digested with BsiWI and partly filled in with Klenow DNA polymerase (Takara) in the presence of dGTP and dTTP. A pair of annealed oligonucleotides encoding a FLAG tag was inserted at this site to give phTet-On-FLAG-VP16. phTet-On-FLAG-fused HP1s were created by inserting BsiWI/BamHI-digested HP1 fragments into BsiWI/BamHI-digested phTet-On-FLAG-VP16.
To insert a nuclear localization signal (NLS) into phTet-On-FLAG-VP16, a BsiWI (blunt ended with Klenow)/BamHI fragment containing VP16 AAD was excised from phTet-On-FLAG-VP16 and inserted into PstI (blunt ended with Klenow)/BamHI-digested pECFP-NLS-C1/C3, which was constructed by inserting a pair of annealed oligonucleotides encoding an NLS of the simian virus 40 (SV40) large T antigen into SacI/EcoRI-digested pECFP-C1. The XhoI (blunt ended with Klenow)/BamHI-digested fragment containing NLS-VP16 AAD was then ligated back into BsiWI (blunt ended with Klenow)/BamHI-digested phTet-On-FLAG-VP16, resulting in phTet-On-FLAG-NLS-VP16. Self-ligation of the BsiWI/BamHI (both blunt ended with Klenow)-digested phTet-On-FLAG-NLS-VP16 fragment resulted in a control plasmid, phTet-On-FLAG-NLS-ΔVP16, which encoded a fusion protein without VP16 AAD.
To construct plasmids expressing enhanced cyan fluorescent protein (ECFP)-fused HP1α and SUV39H1 under the control of the SV2 promoter, a BsiWI (blunt ended with Klenow)/BamHI-digested fragment was inserted into EcoRI (blunt ended with Klenow)/BamHI-digested pSV2-ECFP-C1, which was identical to pECFP-C1 (Clontech), except that the original CMV promoter was replaced with the SV2 promoter. The HP1α V22M point mutation was generated by PCR amplification using a mutational primer pair and pBluescriptII KS(−) containing the HP1α cDNA as a template. Following self-ligation of the amplified fragment, the mutated HP1α fragment was released and subcloned into pSV2-ECFP-C1 as described above.
To generate a plasmid expressing a triple-fusion protein consisting of enhanced yellow fluorescent protein (EYFP)-LacR-VP16 AAD, a BsiWI site was introduced by PCR-based mutagenesis just upstream of the stop codon of pEYFP-lacR, which is a CMV promoter-containing plasmid corresponding to pSV2-EYFP-lacR (also known as pSV2-EYFP-lac repressor) (47). The resulting plasmid was digested with BsiWI and BamHI, and the BsiWI/BamHI-digested VP16 AAD fragment from phTet-On-FLAG-VP16 was inserted to give pEYFP-lacR-VP16.
pSilencer plasmids expressing transgene-directed siRNAs were generated by ligating pairs of annealed oligonucleotides with the BamHI/HindIII-linearized pSilencer 3.1-H1 puro siRNA expression vector (Ambion), according to the manufacturer's instructions. The pSilencer 3.1-H1 puro negative control plasmid (pSilencer-negative; Ambion) encoding an siRNA with limited homology to any known sequences in the human, mouse, and rat genomes was used as a negative control.
Antibodies.
The rabbit polyclonal antibodies against human HP1β and HP1γ used for immunofluorescence staining and chromatin immunoprecipitation (ChIP) assays were produced in rabbits immunized with a synthetic peptide, CNEDDDKKDDKN (including amino acids corresponding to 176 to 185 of human HP1β) (unpublished data) and WHSCPEDEAQ (residues 174 to 183 of human HP1γ) (23), respectively. The rabbit polyclonal antibody against H3K9me3 used in the ChIP assays was obtained from Abcom (ab8898). The secondary antibody used in the immunofluorescence staining was an Alexa Fluor 594-conjugated goat anti-rabbit immunoglobulin G (IgG) (H+L) antibody (A11037; Molecular Probes). The primary antibodies used for Western blot analyses were mouse monoclonal antibody against human HP1α (05-689; Upstate), rabbit polyclonal antibody against Dicer (9) (a gift from K. Saigo, University of Tokyo, Tokyo, Japan), rabbit polyclonal antibody against human erythrocyte catalase (01-05-030000; Athens Research and Technology), and anti-FLAG M2 monoclonal antibody (F3165; Sigma). The secondary antibodies used in Western blot analyses were horseradish peroxidase-conjugated donkey anti-rabbit IgG (NA934; Amersham Biosciences) and sheep anti-mouse IgG (NA931; Amersham Biosciences) antibodies.
Cell culture and plasmid transfection.
Clone 2 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum and 150 U/ml hygromycin B (Wako) at 37°C with 5% CO2. BHK cells were cultured in a similar manner, but without hygromycin B. The cells were plated 24 h prior to transfections and harvested for biochemical analyses and fluorescence microscopy at the time points indicated in specific experiments. Plasmid transfections were performed with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions, using 4 μg of plasmids and 10 μl of reagent per 2 ml of medium in a 35-mm dish.
Immunofluorescence staining and fluorescence microscopy.
Clone 2 cells were cultured on coverslips coated with poly-l-lysine (Sigma). At 24 h after plasmid transfection, the cells were washed with phosphate-buffered saline (PBS) and permeabilized in cold hypotonic buffer (10 mM HEPES-KOH, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM dithiothreitol) containing 0.1% Triton X-100 for 10 min on ice. Next, the cells were fixed for 1 min by adding a half-volume of 4% paraformaldehyde (PFA) in PBS, followed by routine fixation in 4% PFA for 1 h. The fixed cells were treated with 0.1% Triton X-100 in PBS for 15 min and blocked with 1% bovine serum albumin in PBS for 1 h. The cells were then incubated in PBS containing 0.1% Tween 20 and antibodies against HP1β (9.8 μg/ml) or HP1γ (52 μg/ml) for 40 min at room temperature with shaking, followed by incubation at 4°C overnight. Next, the cells were refixed in 4% PFA for 1 h and incubated with a secondary antibody (1:500 dilution) for 8 h. Several washes with PBS were performed between the different steps described above. The immunostained cells were mounted on slides with 90% glycerol in PBS and viewed using a Zeiss Axiovert 200 M inverted fluorescence microscope equipped with an ApoTome.
For fluorescence microscopic observation of cells without immunofluorescence staining, cells were cultured on uncoated coverslips, routinely fixed in 4% PFA for 1 h, mounted on slides, and viewed using an Olympus BX-60 or Zeiss Axiovert 200 M inverted fluorescence microscope. Images were taken without the ApoTome. The acquired images were processed using Adobe Photoshop.
ChIP assay.
ChIP assays were performed using a ChIP assay kit (Upstate) according to the manufacturer's instructions, with some modifications. Briefly, clone 2 cells grown in 35-mm dishes were either mock transfected with 4 μg of pBluescriptII KS(−) or transcriptionally activated by transfection with phTet-Off. At 24 h posttransfection, formaldehyde (37%) was added to the medium to a final concentration of 1% (vol/vol), and the cells were incubated for 10 min at room temperature with gentle shaking. After removal of the medium, the cells were rinsed once with ice-cold PBS containing 20 mM glycine and then twice with ice-cold PBS. Next, the cells were scraped into 1 ml of ice-cold PBS containing protease inhibitors (Roche Applied Science), centrifuged into a pellet, resuspended in 200 μl of sodium dodecyl sulfate (SDS) lysis buffer containing protease inhibitors, and incubated for 10 min on ice. The cell lysates were sonicated with a Bioruptor (Cosmo Bio) for 30 s at the maximum setting 20 times with 1-min intervals, yielding DNA fragments 300 to 500 bp in length. After removal of the cell debris by centrifugation, the supernatants were diluted with 1.8 ml of ChIP dilution buffer containing protease inhibitors and precleared with 80 μl of salmon sperm DNA/protein A agarose slurry for 1 h at 4°C with agitation. Immunoprecipitation was carried out overnight at 4°C with rotation, using antibodies specific for HP1β (5 μg), HP1γ (5 μg), or H3K9me3 (2 μg). The immune complexes were recovered using 60 μl of the salmon sperm DNA/protein A agarose slurry at 4°C for 1 h with rotation and sequentially washed once with low-salt immune complex wash buffer, once with high-salt immune complex wash buffer, once with LiCl immune complex wash buffer, and twice with Tris-EDTA (TE) buffer. The immune complexes were eluted from the agarose beads with 500 μl of freshly prepared elution buffer (1% SDS, 0.1 M NaHCO3), and the cross-links were reversed by incubation with 0.2 M NaCl and 10 μg of RNase A (Wako) at 65°C for 6 h. Following the addition of 10 μl of 0.5 M EDTA, 20 μl of 1 M Tris-HCl (pH 6.5), and 1 μl of 20 μg/μl proteinase K (Takara), the samples were incubated for 1 h at 45°C and then extracted with phenol-chloroform. The DNA obtained was either recovered by ethanol precipitation using glycogen as a carrier (for H3K9me3) (see Fig. 3) or purified using a Qiaquick PCR Purification kit (QIAGEN) and finally eluted in 50 μl of TE buffer. A 4-μl aliquot of the recovered DNA was subjected to quantitative real-time PCR using SYBR Green Supermix (Bio-Rad) in an iCycler (Bio-Rad) according to the manufacturer's instructions. The PCR cycling conditions were 3 min at 95°C, followed by 40 cycles of 45 s at 94°C, 30 s at 55°C, and 30 s at 72°C. For amplification of the β-actin promoter, the annealing temperature was set to 64°C.
FIG. 3.

The CMVm promoter and transcribed CFP regions of the transgene are associated with HP1β, HP1γ, and H3K9me3. Cells mock transfected with pBluescriptII KS(−) (silent, indicated as OFF) or transcriptionally activated by hTet-Off (indicated as ON) were subjected to ChIP assays using antibodies against HP1β, HP1γ, and H3K9me3. The IP DNA and input DNA were analyzed by quantitative real-time PCR. The relative enrichment was determined by normalizing the immunoprecipitation percentage of transgene sequences (CMVm, black bars; CFP, gray bars), defined as the ratio of IP DNA to input DNA, against that of the active β-actin promoter region (white bars). The mean values and standard deviations from three independent experiments are shown.
CpG methylation analysis by bisulfite genomic sequencing.
Bisulfite sequencing of genomic DNA was performed as described previously (15) with some modifications. Briefly, clone 2 cells grown in 60-mm dishes were either mock transfected with 8 μg of pBluescriptII KS(−) or activated by transfection with phTet-Off. At 24 h posttransfection, genomic DNA was isolated using a GenElute Mammalian Genomic DNA Miniprep kit (Sigma). Next, 12 μg of the genomic DNA was fragmented by digestion with EcoRI and PstI. The reaction mixture was extracted with phenol-chloroform, precipitated with ethanol, and then dissolved in 12 μl of TE buffer. A 3-μl aliquot of the fragmented genomic DNA was denatured by incubation in 0.3 M NaOH in a final volume of 20 μl at 37°C for 15 min. Next, the reaction solution was mixed with 107 μl of 4.04 M sodium bisulfite (Sigma), 7 μl of 10 mM hydroquinone (Sigma), and 6 μl of 6 N NaOH and incubated at 95°C for 1 h in the dark. The bisulfite-treated DNA was subsequently desalted using a Wizard DNA Clean-Up System (Promega) and dissolved in 50 μl of TE buffer. The obtained DNA samples were incubated in 0.3 M NaOH at room temperature for 5 min, neutralized with sodium acetate, precipitated with ethanol in the presence of 20 μg of glycogen as a carrier, and finally dissolved in 20 μl of TE buffer. A 1-μl aliquot of the bisulfite-treated DNA was PCR amplified in a 25-μl reaction mixture containing reaction buffer, 200 μM each deoxynucleoside triphosphate (dNTP), 0.2 U/μl Taq DNA polymerase (Takara), and 0.4 μM phosphorylated forward and reverse primers. The primers were designed to contain several non-CpG cytosines but exclude CpG dinucleotides, thereby allowing amplification of the DNA only when the cytosines within the primer binding sites were bisulfite converted to uracil. The PCR cycling conditions were 5 min at 94°C; 40 cycles of 30 s at 95°C, 30 s at 52°C, and 1 min at 72°C; and a final extension for 7 min at 72°C. The PCR products were phenol-chloroform extracted, ethanol precipitated, and then blunt ended by Pfu DNA polymerase treatment in the presence of 200 μM each dNTP for 10 min at 70°C. The 240-bp PCR fragment was subcloned into the EcoRV site of pBluescriptII KS(−) and sequenced.
Northern blot analysis of small RNAs.
Small RNAs were isolated from BHK and clone 2 cells using a mirVana miRNA Isolation kit (Ambion) and subjected to low-stringency Northern blot analysis according to the manufacturer's instructions with some modifications. Briefly, a 7.5-μg portion of the small-RNA fraction was mixed with an equal volume (15 μl) of gel loading buffer II (Ambion), heat denatured, and separated in a 17.5% denaturing polyacrylamide gel containing 7 M urea. The separated RNAs were transferred onto a Hybond N+ nylon membrane (Amersham Biosciences) by electroblotting using a semidry blotting apparatus (ATTO) and then cross-linked by exposure to UV light. Prehybridization was carried out for 2 h at 37°C in a hybridization solution (6× SSC [1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate], 5× Denhardt's solution, 0.5% SDS, 0.1 mg/ml denatured and sheared salmon sperm DNA). Hybridization was performed at 37°C in the same solution containing DNA probes, which had been labeled with [α-32P]dCTP (Amersham Biosciences) using a BcaBEST Labeling kit (Takara). After overnight hybridization, the membranes were washed twice in 6× SSC containing 0.2% SDS at 37°C for a total of 30 min. Finally, the blots were exposed to an imaging plate overnight and analyzed using a BAS-2500 BioImaging Analyzer (Fuji Film). Digoxigenin-labeled 22-nucleotide (nt) and 25-nt oligonucleotides (gifts from T. Natsuaki, Utsunomiya University, Utsunomiya, Japan) were used as molecular-size markers. The sequences of the DNA probes corresponded to nucleotides in different regions of the integrated plasmid p3216PCbeta (AB236435) as follows: lac operator (35 to 181), TRE (9391 to 9652), CMVm promoter (13471 to 13616), CFP (13630 to 14360), and nontranscribed vector (15434 to 15689).
Cloning of a hamster Dicer partial cDNA.
Total RNA was isolated from BHK cells using an RNeasy mini kit (QIAGEN) according to the manufacturer's instructions. The first-strand cDNA was synthesized from 1.5 μg of total RNA in a 20-μl reverse transcription reaction mixture containing reaction buffer, 1 mM each dNTP, 2 U/μl ReverTra Ace reverse transcriptase (Toyobo), and 0.25 μM mouse Dicer reverse primer. The mixture was incubated at 42°C for 60 min and then inactivated at 99°C for 5 min. A 5-μl aliquot of the heat-inactivated reverse transcription product was PCR amplified using Pfu DNA polymerase with mouse Dicer forward and reverse primers. The PCR cycling conditions were 1 min at 94°C and 35 cycles of 30 s at 94°C, 30 s at 57°C, and 1.5 min at 72°C, with a final extension for 2 min at 72°C. The 437-bp amplified fragment was subcloned into the EcoRV site of pBluescriptII KS(−) and sequenced.
RNAi-mediated Dicer knockdown.
Hamster Dicer-specific siRNA transfection was carried out with Lipofectamine 2000 according to the manufacturer's instructions, using 5 μg of reagent per 35-mm dish. To achieve complete Dicer knockdown, clone 2 cells were pretreated with three consecutive siRNA (20 pmol) transfections at 48-h intervals, followed by a fourth siRNA (5 pmol) transfection, together with 4 μg of plasmids as indicated for specific experiments. The siRNA transfection efficiency was monitored by cotransfection with 1 μg of pEYFP-SKL (17), followed by calculation of the percentage of cells showing peroxisomal EYFP signals. The synthetic Dicer siRNA duplex (sense, 5′-GUACUCAAACCUAGAAGUAdTdT-3′, and antisense, 5′-UACUUCUAGGUUUGAGUACdTdT-3′) was purchased from Proligo. A siRNA duplex (sense, 5′-GAUCUGCUCGUACAAGAAUdTdT-3′, and antisense, 5′-AUUCUUGUACGAGCAGAUCdTdT-3′) targeted to the angiotensin-converting enzyme (ACE) gene of the silkworm Bombyx mori was used as a control (a gift from H. Kawasaki, Utsunomiya University, Utsunomiya, Japan).
Quantitative real-time RT-PCR.
Total RNA was isolated from clone 2 cells using an RNeasy Mini kit (QIAGEN) with on-column DNase I treatment. Quantitative real-time reverse transcription (RT)-PCR analysis was performed in an iCycler (Bio-Rad) using a one-step QuantiTect SYBR Green RT-PCR kit (QIAGEN) according to the manufacturer's instructions. The reverse transcription was carried out at 50°C for 30 min, followed by an initial PCR activation step at 95°C for 15 min. The subsequent PCR conditions were 30 cycles of 15 s at 94°C, 30 s at 55°C, and 30 s at 72°C. The number of cycles required to produce a detectable product above the background level was measured and used to calculate the difference in the starting mRNA level for each sample. The relative mRNA levels of Dicer and CFP were normalized to that of β-actin.
Western blot analysis.
Total cell lysates were separated by SDS-polyacrylamide gel electrophoresis (PAGE) and transferred onto ClearBlot membrane-P (ATTO) by electroblotting using a semidry blotting apparatus (ATTO) according to the manufacturer's instructions. After blocking with 5% ECL blocking reagent (Amersham Biosciences), the membrane was probed with appropriate primary antibodies specific for Dicer (1:200 dilution), FLAG tag (1:500 dilution), or catalase (1:200 dilution) and subsequently with horseradish peroxidase-conjugated secondary antibodies (donkey anti-rabbit IgG, 1:2,500 dilution; sheep anti-mouse IgG, 1:7,500 dilution). ECL or ECL Plus reagent (Amersham Biosciences) was used for immunological detection of the labeled proteins. Chemiluminescence was detected using an LAS1000UV mini-imager (Fuji Film) and quantified using Image Gauge software (Fuji Film).
Nucleotide sequence accession number.
The GenBank/EMBL/DDBJ accession number of the 437-bp amplified hamster Dicer partial cDNA is AB248867.
RESULTS
Endogenous HP1β and HP1γ are enriched at the condensed transgenic locus.
HP1 is a major component of heterochromatin. To characterize the transgenic locus in clone 2 cells, we first performed immunofluorescence staining to examine the enrichment of endogenous HP1 at the condensed transgenic locus. Cells transiently expressing EYFP-LacR were immunofluorescently labeled with anti-human HP1β or HP1γ antibody. The distributions of endogenous HP1β and HP1γ were analyzed by fluorescence microscopy in both silent and transcriptionally activated cells. As shown in Fig. 1, in silent cells, endogenous HP1β (Fig. 1B) and HP1γ (Fig. 1D) were enriched at the condensed locus (Fig. 1A and C) marked by EYFP-LacR. In contrast, upon “double activation” by tethering EYFP-LacR-VP16 and hTet-Off to the transgenic locus, HP1β (Fig. 1F) and HP1γ (Fig. 1H) were hardly detectable at the highly decondensed locus (Fig. 1E and G) marked by EYFP-LacR-VP16. Upon single activation by tethering hTet-Off alone, HP1β and HP1γ were present at lower, but detectable, levels in the decondensed locus (data not shown), suggesting incomplete decondensation of the lac operator array, which shares about 50% of the reporter plasmid in length. Thus, endogenous HP1β and HP1γ are enriched at the inactive condensed locus.
FIG. 1.

Endogenous HP1β and HP1γ are enriched at the condensed transgenic locus but depleted upon transcriptional activation. At 24 h posttransfection with EYFP-LacR (A to D) (silent) or a combination of EYFP-LacR-VP16 and hTet-Off (E to H) (double activation), cells were immunofluorescently labeled with antibodies against HP1β (B and F) or HP1γ (D and H). The transgenic locus is highly condensed in silent cells (A and C; marked by EYFP-LacR), but significantly decondensed upon double activation (E and G; marked by EYFP-LacR-VP16). The images shown are optical sections taken with an ApoTome. Scale bar, 5 μm.
ECFP-HP1α and ECFP-SUV39H1 localize to the condensed transgenic locus.
Since we did not have an appropriate antibody for detecting enrichment of endogenous HP1α at the condensed locus, we sought to examine the intracellular localization of ECFP-HP1α in clone 2 cells. As shown in Fig. 2, concentrated localization of ECFP-HP1α was observed at the condensed locus in silent cells (Fig. 2A and B) but not at the decondensed locus upon double activation (Fig. 2C and D). Moreover, ECFP-HP1α carrying the point mutation V22M, which is known to virtually eliminate binding of HP1 to H3K9me in vitro (36) and localization of ECFP-HP1 to heterochromatin regions in vivo (5), did not localize to the condensed locus in silent cells (Fig. 2E and F). Similar results were obtained using ECFP-fused HP1β, HP1γ, and their point mutants (HP1β V23M and HP1γ V22M, respectively) (data not shown). In addition, ECFP-fused SUV39H1, a histone methyltransferase responsible for H3K9me3 in human pericentric heterochromatin (41), localized to the condensed locus in silent cells (Fig. 2G and H) but not to the decondensed locus upon single activation (Fig. 2I and J), suggesting that H3K9me3 is enriched at the inactive locus.
FIG. 2.

ECFP-HP1α and ECFP-SUV39H1 localize to the condensed transgenic locus. Cells were transiently transfected with combinations of plasmids encoding proteins as follows: EYFP-LacR and ECFP-HP1α (A and B) (silent); EYFP-LacR-VP16, ECFP-HP1α, and hTet-Off (C and D) (double activation); EYFP-LacR and ECFP-HP1α V22M (E and F) (silent); EYFP-LacR and ECFP-SUV39H1 (G and H) (silent); and EYFP-LacR, ECFP-SUV39H1, and hTet-Off (I and J) (single activation). The left panels show the transgenic loci, and the right panels show the distribution patterns of the ECFP fusion proteins. The cytoplasmic punctate fluorescence (D and J) represents peroxisomal CFP signals in the activated cells. The triangles indicate the sites of the transgenic loci. Scale bar, 5 μm.
The promoter and transcribed regions of the silent transgene array are associated with HP1β, HP1γ, and H3K9me3.
Next, we performed ChIP assays to further examine the association of the transgene array with HP1 and H3K9me3. Clone 2 cells mock transfected with pBluescriptII KS(−) (silent) or transfected with phTet-Off were used for ChIP assays with antibodies specific for HP1β, HP1γ, and H3K9me3. The immunoprecipitated (IP) DNA and input DNA were analyzed by quantitative real-time PCR using primers specific for the CMVm promoter region and the transcribed CFP region of the transgene, as well as the promoter region of the active β-actin gene. The CMVm primers were designed to eliminate amplification of the CMV promoter in phTet-Off. The relative enrichment was determined by normalizing the immunoprecipitation percentage of transgene sequence, defined as the ratio of IP DNA to input DNA, against that of the β-actin promoter region.
Figure 3 shows that, in silent cells, significant enrichment of HP1β (12.8-fold) and H3K9me3 (9.8-fold) and relatively moderate enrichment of HP1γ (6.4-fold) was present in the inactive CMVm promoter region relative to the active β-actin promoter region. Upon single activation, the enrichment of HP1β was significantly reduced to 5.2-fold, while the enrichment of H3K9me3 and HP1γ was moderately reduced to 6.9- and 4.9-fold, respectively. Very similar results were obtained using primers specific for the CFP region. The lack of clear reduction in the enrichment of H3K9me3 and HP1γ upon activation may be due to the low transfection efficiency of phTet-Off or variation in the extent of unfolding of the transgene array among transfected cells or transgene copies in individual cells. The relatively moderate enrichment of HP1γ at the inactive transgene array may also reflect its tendency to localize in euchromatic regions (19). Thus, the CMVm promoter and transcribed CFP regions in the silent transgene array are associated with endogenous HP1β, HP1γ, and H3K9me3.
CpG methylation in the promoter and 5′ untranslated region of the transgene.
CpG methylation is an important epigenetic marker of constitutive heterochromatin in mammals. We next performed bisulfite genomic sequencing to analyze the CpG methylation status of the transgene array in both silent and single-activated cells. Under each condition, a total of 20 individual clones were sequenced to reveal the methylation pattern of the 20 CpG sites in the CMVm promoter and 5′ untranslated region of the transgene (Fig. 4A). As shown in Fig. 4B, 16 of the 20 CpG sites were more heavily methylated in silent cells than in activated cells. Notably, a more significant reduction in CpG methylation upon activation was observed at sites 12 to 18 in the 5′ untranslated region than in other regions. These results indicate that the stable transgene array is methylated in silent cells and undergoes demethylation following transcriptional activation.
FIG. 4.

Bisulfite genomic sequencing analysis of CpG methylation in the transgene array. (A) Distribution of 20 CpG dinucleotides in the CMVm promoter and 5′ untranslated region of the transgene. (B) Percentage of methylated clones for each CpG. Genomic DNA isolated from cells mock transfected with pBluescriptII KS(−) (silent, black bars) or activated by hTet-Off (gray bars) was used for bisulfite sequencing. A total of 20 individual clones were sequenced to reveal the methylation pattern of the 20 CpG sites over the amplified region.
Taken together, the condensed transgenic locus in clone 2 cells is heterochromatin in nature, since it shows all the important hallmarks of constitutive heterochromatin in mammals. Upon transcriptional activation, the heterochromatic locus becomes decondensed, accompanied by depletion of these heterochromatic markers, indicating a structural transition, at least partially, to the euchromatic state.
The inactive transgene repeats do not generate small RNAs.
An important feature of RNAi-mediated heterochromatin assembly is the generation of siRNAs homologous to their target loci. Previous studies have identified numerous siRNAs corresponding to repetitive DNA sequences, which are often associated with heterochromatin (1, 14, 30, 40). In the case of transgene silencing, siRNAs derived from transgene arrays have been detected in Drosophila cells (38). Having demonstrated that the tandem transgene repeats are assembled into a heterochromatin locus in clone 2 cells, we next investigated whether these tandem repeats are potential sources for the generation of small RNAs that might be functionally involved in the assembly and maintenance of the heterochromatic locus. To this end, we performed low-stringency Northern blot analyses using DNA probes with homologous sequences to different regions of the transgene to examine the existence of transgene-derived small RNAs. As shown in Fig. 5, probe specific for the lac operator sequence did not detect any small RNAs of around 22 to 25 nt in clone 2 cells (lane 2). The other longer or shorter bands detected in clone 2 cells were similar to those in parental BHK cells lacking the transgene (lane 1), indicating that they were unrelated RNAs, probably identified due to the low-stringency hybridization. In contrast, exogenously expressed siRNAs were readily detected in clone 2 cells transfected with pSilencer-lac operator (lane 3) but not in those transfected with pSilencer-negative (lane 4), even though only 1/15 of the amount of small RNAs was loaded in lanes 3 and 4. Similar results were obtained using probes specific for the sequences of TRE (lanes 5 to 8), CMVm promoter (lanes 9 to 12), and CFP (lanes 13 to 16). These results indicate that the inactive transgene repeats do not generate detectable amounts of small RNAs.
FIG. 5.

Northern blot analyses of transgene-derived small RNAs. Low-stringency Northern blot hybridization using radiolabeled DNA probes homologous to the lac operator (lanes 1 to 4), TRE (lanes 5 to 8), CMVm promoter (lanes 9 to 12), and CFP (lanes 13 to 16) sequences in the transgene was performed to detect transgene-specific small RNAs. Small RNA fractions isolated from parental BHK cells (7.5 μg/lane for lanes 1, 5, 9, and 13), clone 2 cells (7.5 μg/lane for lanes 2, 6, 10, and 14), or clone 2 cells transfected with pSilencer plasmids (0.5 μg/lane for lanes 3, 4, 7, 8, 11, 12, 15, and 16) were loaded for probing. The closed triangles (lanes 3, 7, 11, and 15) represent transiently expressed siRNAs. The electrophoretic mobilities of the digoxigenin-labeled 22-nt and 25-nt DNA oligonucleotides are indicated. N, pSilencer-negative.
RNAi-mediated Dicer knockdown.
A key step in the RNAi pathway is the cleavage of double-stranded RNA (dsRNA) precursors by Dicer to produce siRNAs. RNAi-mediated Dicer knockdown in mammalian cells leads to a defect in the generation of small RNAs from their dsRNA precursors (4, 20). Although our Northern blot analysis did not detect any transgene-derived small RNAs, we could not exclude the possibility that even undetectable amounts of small RNAs derived from the transgene repeats are sufficient for heterochromatinization. To address this point, we carried out RNAi-mediated Dicer knockdown to further deplete these undetectable (if present) small RNAs. After four consecutive Dicer siRNA transfections, the Dicer was specifically reduced by 73% and 79% at the mRNA (Fig. 6A) and protein (Fig. 6B) levels, respectively. In a separate experiment, a 65% depletion of Dicer caused a 61% reduction in the expression level of siRNAs processed by Dicer from short-hairpin RNA (shRNA) precursors transcribed from transiently transfected pSilencer-lac operator (Fig. 6C). It indicated that Dicer-depleted cells were physiologically defective in processing dsRNA precursors into siRNAs. Separate controls showed that one siRNA treatment with a transfection efficiency of about 50% resulted in an approximately 50% reduction in the Dicer mRNA level. These results suggested that Dicer was substantially depleted in the fraction of cells that were consecutively transfected with Dicer siRNA.
FIG. 6.

RNAi-mediated Dicer knockdown. (A) Quantitative real-time RT-PCR analysis of the Dicer mRNA levels. The relative Dicer mRNA levels in siRNA-transfected cells were determined by normalizing the ratios of Dicer mRNA to β-actin mRNA against that of a nontransfected sample. An siRNA targeting the silkworm ACE gene was used as a negative control. The mean values and standard deviations from three independent experiments using the average value of triplicate RT-PCRs are shown. (B) Western blot analysis of the Dicer protein levels. Total cell lysates were resolved by 8% SDS-PAGE and detected with an anti-Dicer antibody. The levels of catalase are shown as a loading control. The indicated number (0.21) in the Dicer siRNA-transfected cells was determined by normalizing the ratio of Dicer to catalase against that of the control siRNA-transfected cells. (C) Dicer knockdown caused a defect in siRNA generation from shRNA precursors. Top, Western blot analysis of Dicer knockdown. Bottom, Northern blot analysis of the siRNA expression levels. Following transfection of pSilencer-lac operator into control and Dicer-depleted cells, small RNAs were detected by Northern blot analysis using radiolabeled probes homologous to the lac operator sequence. The levels of 5S rRNA stained with ethidium bromide are shown as a loading control. The indicated number (0.39) in the Dicer-depleted cells was determined by normalizing the ratio of siRNA to 5S rRNA against that of control cells.
Dicer knockdown does not cause up-regulation of transgene expression.
In a number of organisms in which RNAi has been implicated in the assembly of centromeric heterochromatin, Dicer knockout results in aberrant accumulation of centromeric transcripts (12, 24, 35, 51). If transgene-derived small RNAs are indeed responsible for the transgene-induced heterochromatinization in clone 2 cells, depletion of these small RNAs by Dicer knockdown would be predicted to relieve the heterochromatic nature of the transgenic locus and thus cause up-regulation of transgene expression. To address this point, at the fourth siRNA transfection, clone 2 cells were either mock transfected with pBluescriptII KS(−) (silent) or transfected with phTet-Off and incubated for 24 h in the presence (weak activation) or absence (strong activation) of Dox (0.1 μg/ml). The effect of Dicer knockdown on transgene expression was evaluated by real-time RT-PCR analysis of the CFP mRNA levels. Figure 7A shows that, after mock transfection, the CFP mRNA level in Dicer-depleted cells was lower than that in control cells. In addition, the CFP mRNA levels in Dicer-depleted and control cells were close to the background levels (Fig. 7A) determined by direct PCR without the reverse transcription step. These results indicated that the CFP mRNA was hardly detectable in silent cells and that Dicer knockdown did not cause leakage of the transgene expression. Moreover, in cells transfected with control siRNA, weak activation (Fig. 7B) resulted in a CFP mRNA level that was much higher than the background level observed in silent cells (Fig. 7A) but about 480-fold lower than that in strongly activated cells (Fig. 7C), indicating that the presence of Dox prevented hTet-Off from binding to the TRE sequences and strongly impaired transgene activation. Consistently, regardless of whether the transgene was weakly or highly activated, Dicer knockdown did not increase the CFP mRNA levels compared to control siRNA-transfected cells (Fig. 7B and C), indicating that Dicer knockdown did not up-regulate transgene expression.
FIG. 7.

Dicer knockdown does not cause up-regulation of transgene expression. Control or Dicer-depleted cells were either mock transfected with pBluescriptII KS(−) (A) (silent) or transfected with phTet-Off and incubated for 24 h in the presence (B) (weak activation) or absence (C) (strong activation) of Dox. The relative CFP mRNA levels were determined as the ratios of CFP mRNA to β-actin mRNA. The mean values and standard deviations from three independent experiments using the average value of triplicate RT-PCRs are shown. The black bars represent the data from RT-PCRs, while the gray bars in panel A represent the data from direct PCRs without the reverse transcription step.
Dicer knockdown does not cause deconstruction of the heterochromatic locus.
It has been shown that loss of Dicer leads to deconstruction of centromeric heterochromatin in Dicer-deficient fission yeast (51), chicken-human hybrid DT40 cells (12), and mouse ES cells (24). To evaluate the effect of Dicer knockdown on the heterochromatic nature of the transgenic locus, we first used fluorescence microscopy to examine the cytological morphology of the transgenic locus, as well as its association with HP1 and SUV39H1 in Dicer-depleted cells. The results showed that the transgenic locus retained a condensed structure in cells transfected with Dicer siRNA, as shown in the representative cells (Fig. 8A, C, E and G). Moreover, enrichment of ECFP-HP1α (Fig. 8B) and ECFP-SUV39H1 (Fig. 8D), as well as endogenous HP1β (Fig. 8F) and HP1γ (Fig. 8H), was observed at the condensed locus in cells transfected with Dicer siRNA (Fig. 8A-H), similar to the case for the control siRNA-transfected cells (data not shown). These results suggested that the HP1 binding sites, i.e., H3K9me3, were still present in the transgene array after Dicer knockdown. To further confirm this finding, we performed ChIP assays to examine whether H3K9me3 was altered in the transgene array following Dicer knockdown. As shown in Fig. 8I, Dicer knockdown did not cause any reduction in the enrichment of H3K9me3 in the CMVm promoter region and CFP region. It should be noted that, although we did not perform immunofluorescence staining to confirm the Dicer depletion in the individual cells shown in Fig. 8A to H, the expression of EYFP-LacR or coexpression of EYFP-LacR and ECFP fusion proteins in the cells chosen for observation indicated that cotransfection of plasmids with Dicer siRNA had indeed occurred, at least at the fourth siRNA transfection. In addition, the almost 80% reduction in the Dicer protein level revealed by Western blot analysis suggested that Dicer was substantially depleted in the majority of the cells. Collectively, these data suggest that Dicer knockdown does not cause deconstruction of the heterochromatic locus.
FIG. 8.
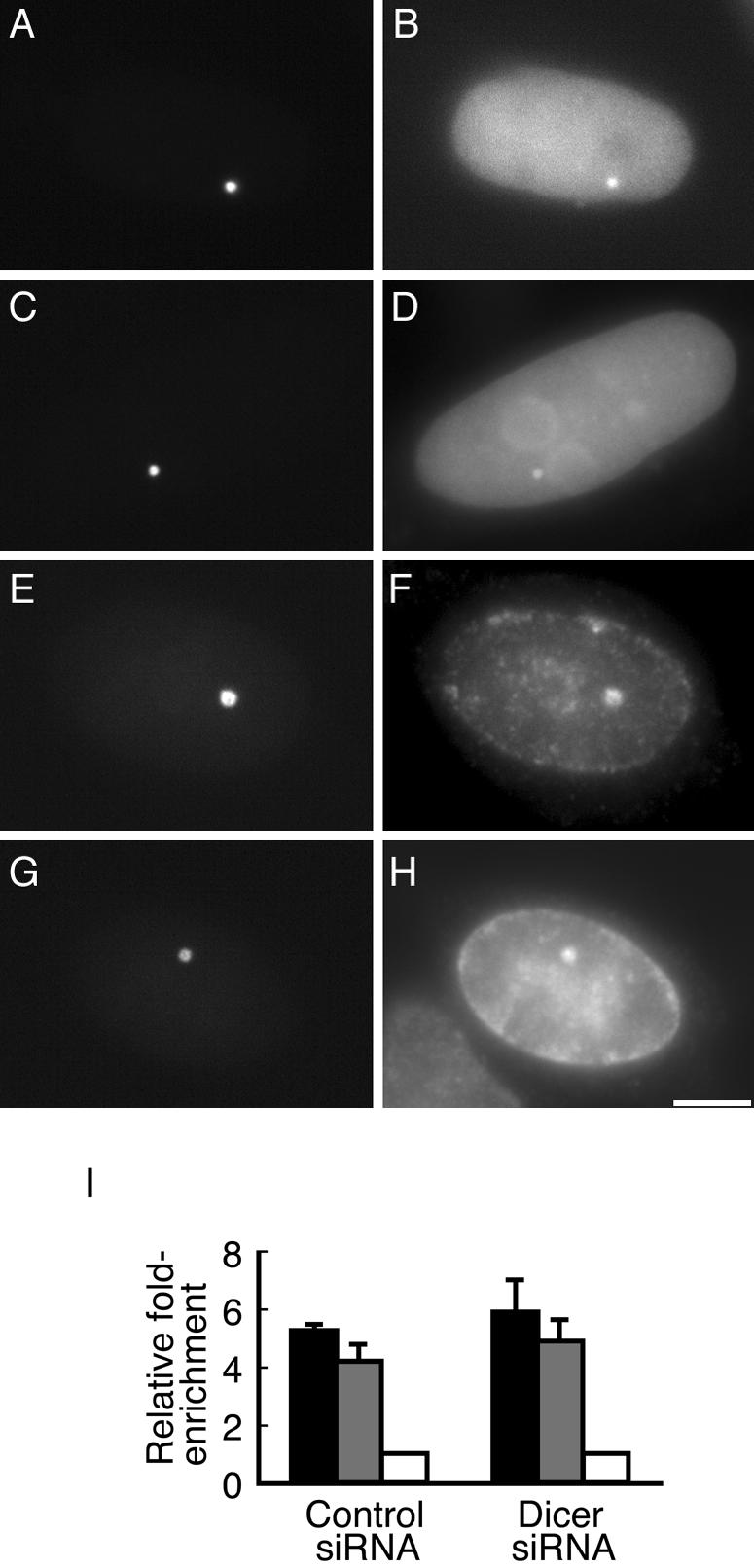
Dicer knockdown does not cause deconstruction of the heterochromatic transgenic locus. ECFP-HP1α (B) and ECFP-SUV39H1 (D) localized to the condensed locus (A and C) in Dicer siRNA-transfected cells. Enrichment of endogenous HP1β (F) and HP1γ (H) at the condensed locus (E and F) in Dicer siRNA-transfected cells was detected by immunofluorescence staining. The transgenic locus is marked by EYFP-LacR (A, C, E, and G). Scale bar, 5 μm. (I) ChIP analysis of H3K9me3 in the CMVm promoter region (black bars) and transcribed CFP region (gray bars), in control or Dicer siRNA-transfected cells. The mean values and standard deviations from three independent experiments are shown.
Dicer knockdown does not alter the bidirectional transition of the transgenic locus between the heterochromatic and euchromatic states.
As mentioned earlier, the transgenic locus changed from a closed (heterochromatic) to an open (euchromatic) structure upon activation by hTet-Off; we further observed that removal of hTet-Off from activated transgene arrays by adding Dox caused quick reversion of the decondensed locus to its former heterochromatic state. This feature is an advantage that allows the effect of Dicer knockdown on the bidirectional transition of the transgenic locus between the heterochromatic and euchromatic states to be evaluated.
To this end, clone 2 cells were cotransfected with EYFP-LacR and hTet-Off at the fourth siRNA transfection and then incubated for 24 h in the absence of Dox. Subsequently, the cells were either fixed or allowed to grow for an additional 2.5 h in the presence of Dox (0.1 μg/ml). A total of 200 EYFP-positive cells were randomly chosen from the Dicer-depleted and control cells for analysis of their transgenic locus morphologies (open or closed). The results showed that only 9% of the Dicer-depleted cells exhibited a closed locus at 24 h postactivation, similar to the percentage observed in control cells (10%), indicating that binding of hTet-Off to the transgene array efficiently induced large-scale chromatin decondensation and that Dicer knockdown did not facilitate chromatin decondensation. Moreover, the percentage of cells showing a closed locus markedly increased to 61.5% in Dicer-depleted cells at 2.5 h after Dox exposure, similar to the percentage observed in control cells (63.5%), indicating that a similar large proportion (close to 60%) of the decondensed loci underwent recondensation during transcription shutdown, regardless of the Dicer expression level. Thus, Dicer knockdown does not facilitate transition of the heterochromatic locus to the euchromatic state or inhibit reversion of the euchromatic locus to the heterochromatic state.
Exogenous expression of transgene-directed siRNAs does not antagonize chromatin decondensation or facilitate chromatin recondensation at the transgenic locus.
A recent paper showed that siRNA targeted to the promoter region of an integrated transgene in mammalian cells induces transcriptional gene silencing (33). If a very low concentration of transgene-derived small RNAs is sufficient to maintain the heterochromatic locus, overexpression of transgene-directed siRNAs could induce a silent chromatin structure and thus antagonize transition of the heterochromatic locus to the euchromatic state or facilitate reversion of the euchromatic locus to the heterochromatic state. To address this point, pSilencer plasmids carrying siRNAs targeted to different regions of the transgene were cotransfected into clone 2 cells, along with EYFP-LacR and hTet-Off, and the effects of transgene-directed siRNAs on transgene expression, as well as chromatin decondensation and recondensation, were evaluated as described above.
As shown in Fig. 9, compared to control cells cotransfected with pSilencer-negative, the CFP mRNA level was reduced by 20% and 26% in cells expressing siRNAs targeted to the sequences of the lac operator and TRE, respectively. However, siRNAs targeted to regions of the CMVm promoter, nontranscribed vector sequence, or a combination of regions of the lac operator, TRE, and vector sequence did not reduce the CFP mRNA level. As expected, a CFP-targeted siRNA significantly reduced the CFP mRNA level by 87% due to its posttranscriptional gene-silencing activity. The presence of siRNAs targeted to regions of the lac operator, TRE, CMVm, and CFP were confirmed by Northern blot analyses using radiolabeled probes (Fig. 5, lanes 3, 7, 11, and 15, respectively). The presence of vector sequence-directed shRNA was confirmed by Northern blot analysis using digoxigenin-labeled probe (data not shown).
FIG. 9.

Effects of overexpressed transgene-directed siRNAs on transgene expression. Cells were activated by transfection with phTet-Off, together with the indicated pSilencer plasmids. The relative CFP mRNA levels in cells expressing transgene-directed siRNAs were determined by normalizing the ratios of CFP mRNA to β-actin mRNA against that of control cells cotransfected with pSilencer-negative. The mean values and standard deviations from three independent experiments using the average value of triplicate RT-PCRs are shown.
Table 1 shows that, in 100 EYFP-positive cells that were randomly chosen, only 12 to 19% of the cells expressing transgene-directed siRNAs exhibited a closed locus at 24 h postactivation, similar to the percentage observed in control cells (16%). Moreover, after 2.5 h of Dox exposure, 60 to 70% of the cells expressing transgene-directed siRNAs showed a closed locus, similar to the percentage observed in control cells (65%). Thus, overexpression of transgene-directed siRNAs does not antagonize heterochromatin decondensation during transcriptional activation or facilitate chromatin recondensation after removal of the activator.
TABLE 1.
Overexpression of transgene-directed siRNAs does not antagonize chromatin decondensation (Dox−) or facilitate chromatin recondensation (Dox−→Dox+)
| pSilencer | Closed locusa
|
|
|---|---|---|
| Dox−, 24 h | Dox−, 24 h →Dox+, 2.5 h | |
| lac operator | 14 | 68 |
| TRE | 13 | 68 |
| CMV | 12 | 60 |
| CFP | 19 | 64 |
| Vector | 14 | 63 |
| lac operator + TRE + vector | 18 | 70 |
| Negative | 16 | 65 |
Data shown are the number of cells with a closed locus (homogeneous and compact) among a total of 100 EYFP-positive cells.
HP1 tethering induces transgene silencing and chromatin condensation.
Having demonstrated that HP1s are enriched at the heterochromatic transgenic locus, we further examined whether they are capable of regulating the transgene expression and large-scale chromatin structure and whether the HP1 function depends on the Dicer-related RNAi pathway. To this end, clone 2 cells were cotransfected with phTet-On-FLAG-HP1 and phTet-On-FLAG-NLS-VP16 at the fourth siRNA transfection, followed by incubation for 18 h in the presence of Dox (1.0 μg/ml). The effects of HP1 tethering on VP16-induced transgene expression and heterochromatin decondensation were examined as described above. As shown in Fig. 10A, in control siRNA-transfected cells, tethering of HP1α, HP1β, or HP1γ significantly reduced the CFP mRNA levels by 95 to 99% compared to cells cotransfected with phTet-On-FLAG-NLS-VP16 and pBluescriptII KS(−). As a control, tethering of hTet-On-FLAG-NLS-ΔVP lacking VP16 AAD caused a smaller reduction (13%) in the CFP mRNA level. Western blot analysis revealed that hTet-On-FLAG-NLS-VP16 was expressed at similar levels among the five samples (Fig. 10B, compare bands indicated by arrows in lanes 1 to 5) and that hTet-On-FLAG-NLS-ΔVP was expressed at a level similar to or higher than that of hTet-On-FLAG-HP1 (Fig. 10B, compare band indicated by open triangle in lane 2 with bands indicated by closed triangles in lanes 3 to 5). These results indicated that HP1 tethering effectively antagonized the transgene activation induced by the simultaneously tethered VP16. Furthermore, HP1 tethering effectively repressed transgene expression in Dicer-depleted cells, similar to its effect in control siRNA-transfected cells (Fig. 10A), indicating that HP1-mediated transcriptional repression is independent of the Dicer-related RNAi pathway.
FIG. 10.

HP1 tethering represses VP16-induced transgene activation in a Dicer-independent manner. (A) Real-time RT-PCR analysis of CFP mRNA levels. Cells consecutively transfected with control (black bars) or Dicer (gray bars) siRNA were cotransfected with plasmids as indicated. The relative CFP mRNA levels were determined by normalizing the ratios of CFP mRNA to β-actin mRNA against those of cells cotransfected with control siRNA, phTet-On-FLAG-NLS-VP16, and pBluescriptII KS(−). The remaining levels of Dicer mRNA and protein in Dicer-depleted cells relative to that of control siRNA-transfected cells are shown in parentheses (left, mRNA; right, protein). The mean values and standard deviations from three independent experiments are shown. (B) Western blot analysis of FLAG-tagged fusion proteins in control siRNA-transfected cells. Total cell lysates were resolved by 10% SDS-PAGE and detected with an anti-FLAG antibody. pBlue, pBluescriptII KS(−); VP16, hTet-On-FLAG-NLS-VP16 (bands indicated by arrows in lanes 1 to 5); ΔVP, hTet-On-FLAG-NLS-ΔVP (band indicated by open triangle in lane 2); HP1, hTet-On-FLAG-HP1 (bands indicated by closed triangles in lanes 3 to 5).
Table 2 shows the effect of HP1 tethering on VP16-induced heterochromatin decondensation. In control siRNA-transfected cells, cotransfection of phTet-On-FLAG-NLS-VP16 and pBluescriptII KS(−) resulted in 90% of the EYFP-positive cells (n = 50) showing a decondensed locus with peroxisomal CFP signals, whereas cells showing a closed locus without peroxisomal CFP signals were very rare. Similarly, in cells cotransfected with phTet-On-FLAG-NLS-VP16 and phTet-On-FLAG-NLS-ΔVP, 72% of the cells showed a decondensed locus with peroxisomal CFP signals and very rare cells showed a closed locus without peroxisomal CFP signals. In contrast, in cells cotransfected with phTet-On-FLAG-NLS-VP16 and phTet-On-FLAG-HP1, very rare cells had a decondensed locus with peroxisomal CFP signals, whereas a large proportion (62 to 68%) of the cells appeared to have a closed locus without peroxisomal CFP signals, indicating that HP1 tethering effectively repressed chromatin decondensation induced by VP16. Very similar results were obtained in Dicer-depleted cells (Table 2). Thus, HP1 tethering effectively induced chromatin compaction independently of the Dicer-related RNAi pathway. Taken together, these results suggest that HP1 may play a key role in maintaining the transgene-induced heterochromatic locus.
TABLE 2.
HP1 tethering antagonizes VP16-induced chromatin decondensation in a Dicer-independent manner
| Plasmid | No. of cells showing chromatin morphology and peroxisomal CFP signals (n = 50)
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Control siRNA-transfected cells
|
Dicer siRNA-transfected cells
|
|||||||
| Opena
|
Closedb
|
Open
|
Closed
|
|||||
| Per+c | Per− | Per+ | Per− | Per+ | Per− | Per+ | Per− | |
| VP16+pBlued | 45 | 0 | 3 | 2 | 44 | 3 | 2 | 1 |
| VP16+ΔVP16 | 36 | 3 | 9 | 2 | 34 | 3 | 6 | 7 |
| VP16+HP1α | 4 | 12 | 3 | 31 | 4 | 9 | 1 | 36 |
| VP16+HP1β | 1 | 15 | 1 | 33 | 2 | 6 | 3 | 39 |
| VP16+HP1γ | 3 | 11 | 2 | 34 | 6 | 8 | 0 | 36 |
Open, heterogeneous and extended locus.
Closed, homogeneous and compact locus.
Per, peroxisomal CFP signals.
pBlue, pBluescriptII KS(−).
DISCUSSION
In the present study, we used a BHK-derived cell line containing a tandem 1,000-copy inducible transgene to test whether the RNAi pathway is responsible for the transgene-induced heterochromatinization in mammals. Through a combination of fluorescence microscopy of individual cells and cell population analyses, such as ChIP assays and bisulfite genomic sequencing, we demonstrated that the condensed locus is heterochromatic and that VP16-induced large-scale chromatin decondensation is accompanied by loss of the heterochromatic markers of the locus. These results are consistent with an earlier study of a human cell line containing a similar transgenic locus (21).
We suggest that the RNAi pathway is not responsible for the assembly and maintenance of the transgene repeat-induced heterochromatic locus in clone 2 cells. Several lines of evidence support this conclusion. First, Northern blot analysis did not detect any transgene-specific small RNAs, indicating that the tandem inducible transgene repeats do not generate small RNAs that might be functionally involved in the heterochromatinization. This result seems reasonable, since the only potential promoter for generating RNA in the reporter plasmid is the CMVm promoter, which is completely inactive without transcriptional activation. Consistently, RT-PCR analysis showed that the CFP mRNA level in silent cells is close to the background level, indicating that no double-stranded transcripts are available for Dicer to generate small RNAs. Moreover, no mammalian-derived sequences, except for a short rat cDNA encoding SKL, are present in the reporter plasmid, so there is little chance that endogenous small RNAs show accidental similarity to the transgene.
Second, Dicer knockdown does not cause any leakage of the transgene transcription or disruption of the heterochromatic locus in silent cells. Given that Dicer-depleted cells are defective in processing siRNAs from their exogenously expressed shRNA precursors and that the cells underwent more than 10 divisions during the time course (168 h) of the consecutive siRNA transfections, we predict that the abundance of undetectable transgene-derived small RNAs, if any, should have been largely reduced in the fraction of Dicer-depleted cells. In addition, Dicer knockdown does not up-regulate transgene expression or facilitate heterochromatin decondensation during transcriptional activation. At this point, one may wonder whether, if Dicer really does act in heterochromatinization, it may not be sufficient to overcome VP16-induced heterochromatin decondensation and transgene expression in activated cells. We attempted to address this by showing that, in a special situation (weak activation) in which VP16 is incapable of decondensing the transgenic locus (data not shown) and the transgene activation is largely impaired, Dicer knockdown does not cause any elevation of the CFP mRNA level. This suggests that, even in the presence of weakly bound VP16, Dicer knockdown does not facilitate the accessibility of VP16 to the transgenic locus, which would be expected to increase transgene transcription. Moreover, the quick condensation of the unfolded locus after removal of VP16 in Dicer-depleted cells, similar to that observed in wild-type cells, further suggests that the Dicer-related RNAi pathway is not responsible for reassembly of the condensed locus.
Third, overexpression of transgene-directed siRNAs does not antagonize chromatin decondensation or facilitate chromatin recondensation, suggesting that these siRNAs do not have the potential to induce a silent chromatin state. Based on these results with the aforementioned Dicer knockdown experiments, we can rule out the possibility that undetectable amounts of transgene-derived small RNAs, if any, account for the heterochromatin assembly and maintenance at the transgenic locus. On the other hand, a preliminary experiment to identify the factors responsible for maintenance of the heterochromatic locus showed that HP1 tethered to the transgenic locus simultaneously induces transgene silencing and chromatin condensation in Dicer-depleted cells, similar to its effects in control cells. This result is consistent with, and advances, the results of previous related studies (8, 50). Since the heterochromatic locus is composed of 1,000 copies of a reporter plasmid, it is possible that, in some cases, some reporter plasmid copies may accidentally show euchromatic properties in silent cells. It remains unclear whether the tethered HP1 induces heterochromatin formation by further tightening the transgene array or by recruiting HP1-interacting factors that antagonize the VP16 effects. Experiments are being carried out to identify the molecular determinants for HP1-mediated heterochromatin maintenance and transcriptional gene silencing.
In clone 2 cells, a strong promoter, enhancer, and endogenous replication origin are absent from the tandemly arranged transgene repeats. If free methyltransferase in the nucleus is enzymatically active and randomly methylates histone H3, such a large block of repetitive DNA sequences tends to be assembled into a condensed chromosome domain in a sequence-independent manner. Once established, the heterochromatic domain can be maintained as a consequence of modifications of the DNA and surrounding histones and subsequent binding of nonhistone chromosomal proteins, such as HP1. Since the majority of mammalian genomes is comprised of intergenic DNA and should be kept in the silent state, the idea that the default pathway is heterochromatinization is reasonable. This hypothesis may explain the previous observations that a large tandem array of the lac operator without a strong promoter (21, 28, 47), but not a tandem array with the mouse mammary tumor virus promoter (34), forms a highly condensed chromatin structure after integration into the genome.
It is noteworthy that, although a requirement for RNAi in centromeric heterochromatin formation has been demonstrated in diverse organisms, RNAi-independent pathways for heterochromatin formation also occur in organisms in which RNAi takes place (6, 11, 22, 25). In addition, to date, the evidence that siRNAs can induce a silent chromatin status along a homologous DNA region remains inconclusive (26, 33, 45, 46). Moreover, there are contradictory reports that the pericentric heterochromatin in Dicer-null mouse ES cells is partially disrupted (24) or remains completely intact (35). Besides, although RNA transcripts corresponding to both strands of the centromeric major satellite repeats have been detected in mouse cells (24, 27, 31, 35, 44), centromeric small RNAs have not been identified in mammals (2). These results suggest that RNAi may not play a ubiquitous role in the maintenance of heterochromatin in mammals and that there are probably subtle differences in the pathways that establish heterochromatin in different genomic regions. With regard to the transgene array investigated in the present study, our observations suggest that the assembly and maintenance of the heterochromatic transgenic locus are independent of the RNAi pathway, although we still do not know whether this mechanism acts on endogenous genes. The RNAi-mediated pathways in the nuclei of mammalian cells remain attractive topics for further investigations.
Supplementary Material
Acknowledgments
We thank K. Saigo (University of Tokyo, Tokyo, Japan) for the anti-Dicer antibody, T. Natsuaki (Utsunomiya University, Utsunomiya, Japan) for helpful suggestions and some reagents for the Northern blot analyses, and H. Kawasaki (Utsunomiya University, Utsunomiya, Japan) for the control siRNA. We also thank members of our laboratory for helpful discussions in support of this work.
Footnotes
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Aravin, A. A., M. Lagos-Quintana, A. Yalcin, M. Zavolan, D. Marks, B. Snyder, T. Gaasterland, J. Meyer, and T. Tuschl. 2003. The small RNA profile during Drosophila melanogaster development. Dev. Cell 5:337-350. [DOI] [PubMed] [Google Scholar]
- 2.Bernstein, E., and C. D. Allis. 2005. RNA meets chromatin. Genes Dev. 19:1635-1655. [DOI] [PubMed] [Google Scholar]
- 3.Carpenter, A. E., S. Memedula, M. J. Plutz, and A. S. Belmont. 2005. Common effects of acidic activators on large-scale chromatin structure and transcription. Mol. Cell. Biol. 25:958-968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chendrimada, T. P., R. I. Gregory, E. Kumaraswamy, J. Norman, N. Cooch, K. Nishikura, and R. Shiekhattar. 2005. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 436:740-744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheutin, T., A. J. McNairn, T. Jenuwein, D. M. Gilbert, P. B. Singh, and T. Misteli. 2003. Maintenance of stable heterochromatin domains by dynamic HP1 binding. Science 299:721-725. [DOI] [PubMed] [Google Scholar]
- 6.Chicas, A., C. Cogoni, and G. Macino. 2004. RNAi-dependent and RNAi-independent mechanisms contribute to the silencing of RIPed sequences in Neurospora crassa. Nucleic Acids Res. 32:4237-4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chicas, A., E. C. Forrest, S. Sepich, C. Cogoni, and G. Macino. 2005. Small interfering RNAs that trigger posttranscriptional gene silencing are not required for the histone H3 Lys9 methylation necessary for transgenic tandem repeat stabilization in Neurospora crassa. Mol. Cell. Biol. 25:3793-3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Danzer, J. R., and L. L. Wallrath. 2004. Mechanisms of HP1-mediated gene silencing in Drosophila. Development 131:3571-3580. [DOI] [PubMed] [Google Scholar]
- 9.Doi, N., S. Zenno, R. Ueda, H. Ohki-Hamazaki, K. Ui-Tei, and K. Saigo. 2003. Short-interfering-RNA-mediated gene silencing in mammalian cells requires Dicer and eIF2C translation initiation factors. Curr. Biol. 13:41-46. [DOI] [PubMed] [Google Scholar]
- 10.Dorer, D. R., and S. Henikoff. 1994. Expansions of transgene repeats cause heterochromatin formation and gene silencing in Drosophila. Cell 77:993-1002. [DOI] [PubMed] [Google Scholar]
- 11.Freitag, M., D. W. Lee, G. O. Kothe, R. J. Pratt, R. Aramayo, and E. U. Selker. 2004. DNA methylation is independent of RNA interference in Neurospora. Science 304:1939. [DOI] [PubMed] [Google Scholar]
- 12.Fukagawa, T., M. Nogami, M. Yoshikawa, M. Ikeno, T. Okazaki, Y. Takami, T. Nakayama, and M. Oshimura. 2004. Dicer is essential for formation of the heterochromatin structure in vertebrate cells. Nat. Cell Biol. 6:784-791. [DOI] [PubMed] [Google Scholar]
- 13.Grewal, S. I., and D. Moazed. 2003. Heterochromatin and epigenetic control of gene expression. Science 301:798-802. [DOI] [PubMed] [Google Scholar]
- 14.Grewal, S. I., and J. C. Rice. 2004. Regulation of heterochromatin by histone methylation and small RNAs. Curr. Opin. Cell Biol. 16:230-238. [DOI] [PubMed] [Google Scholar]
- 15.Grunau, C., S. J. Clark, and A. Rosenthal. 2001. Bisulfite genomic sequencing: systematic investigation of critical experimental parameters. Nucleic Acids Res. 29:E65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hall, I. M., G. D. Shankaranarayana, K. Noma, N. Ayoub, A. Cohen, and S. I. Grewal. 2002. Establishment and maintenance of a heterochromatin domain. Science 297:2232-2237. [DOI] [PubMed] [Google Scholar]
- 17.Hashiguchi, N., T. Kojidani, T. Imanaka, T. Haraguchi, Y. Hiraoka, E. Baumgart, S. Yokota, T. Tsukamoto, and T. Osumi. 2002. Peroxisomes are formed from complex membrane structures in PEX6-deficient CHO cells upon genetic complementation. Mol. Biol. Cell 13:711-722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Henikoff, S. 1998. Conspiracy of silence among repeated transgenes. Bioessays 20:532-535. [DOI] [PubMed] [Google Scholar]
- 19.Horsley, D., A. Hutchings, G. W. Butcher, and P. B. Singh. 1996. M32, a murine homologue of Drosophila heterochromatin protein 1 (HP1), localises to euchromatin within interphase nuclei and is largely excluded from constitutive heterochromatin. Cytogenet. Cell Genet. 73:308-311. [DOI] [PubMed] [Google Scholar]
- 20.Hutvagner, G., J. McLachlan, A. E. Pasquinelli, E. Balint, T. Tuschl, and P. D. Zamore. 2001. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 293:834-838. [DOI] [PubMed] [Google Scholar]
- 21.Janicki, S. M., T. Tsukamoto, S. E. Salghetti, W. P. Tansey, R. Sachidanandam, K. V. Prasanth, T. Ried, Y. Shav-Tal, E. Bertrand, R. H. Singer, and D. L. Spector. 2004. From silencing to gene expression: real-time analysis in single cells. Cell 116:683-698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jia, S., K. Noma, and S. I. Grewal. 2004. RNAi-independent heterochromatin nucleation by the stress-activated ATF/CREB family proteins. Science 304:1971-1976. [DOI] [PubMed] [Google Scholar]
- 23.Kametaka, A., M. Takagi, T. Hayakawa, T. Haraguchi, Y. Hiraoka, and Y. Yoneda. 2002. Interaction of the chromatin compaction-inducing domain (LR domain) of Ki-67 antigen with HP1 proteins. Genes Cells. 7:1231-1242. [DOI] [PubMed] [Google Scholar]
- 24.Kanellopoulou, C., S. A. Muljo, A. L. Kung, S. Ganesan, R. Drapkin, T. Jenuwein, D. M. Livingston, and K. Rajewsky. 2005. Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev. 19:489-501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kanoh, J., M. Sadaie, T. Urano, and F. Ishikawa. 2005. Telomere binding protein Taz1 establishes Swi6 heterochromatin independently of RNAi at telomeres. Curr. Biol. 15:1808-1819. [DOI] [PubMed] [Google Scholar]
- 26.Kawasaki, H., and K. Taira. 2004. Induction of DNA methylation and gene silencing by short interfering RNAs in human cells. Nature 431:211-217. [DOI] [PubMed] [Google Scholar]
- 27.Lehnertz, B., Y. Ueda, A. A. Derijck, U. Braunschweig, L. Perez-Burgos, S. Kubicek, T. Chen, E. Li, T. Jenuwein, and A. H. Peters. 2003. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr. Biol. 13:1192-1200. [DOI] [PubMed] [Google Scholar]
- 28.Li, G., G. Sudlow, and A. S. Belmont. 1998. Interphase cell cycle dynamics of a late-replicating, heterochromatic homogeneously staining region: precise choreography of condensation/decondensation and nuclear positioning. J. Cell Biol. 140:975-989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lippman, Z., and R. Martienssen. 2004. The role of RNA interference in heterochromatic silencing. Nature 431:364-370. [DOI] [PubMed] [Google Scholar]
- 30.Llave, C., K. D. Kasschau, M. A. Rector, and J. C. Carrington. 2002. Endogenous and silencing-associated small RNAs in plants. Plant Cell 14:1605-1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martens, J. H., R. J. O'Sullivan, U. Braunschweig, S. Opravil, M. Radolf, P. Steinlein, and T. Jenuwein. 2005. The profile of repeat-associated histone lysine methylation states in the mouse epigenome. EMBO J. 24:800-812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matzke, M. A., and J. A. Birchler. 2005. RNAi-mediated pathways in the nucleus. Nat. Rev. Genet. 6:24-35. [DOI] [PubMed] [Google Scholar]
- 33.Morris, K. V., S. W. Chan, S. E. Jacobsen, and D. J. Looney. 2004. Small interfering RNA-induced transcriptional gene silencing in human cells. Science 305:1289-1292. [DOI] [PubMed] [Google Scholar]
- 34.Muller, W. G., D. Walker, G. L. Hager, and J. G. McNally. 2001. Large-scale chromatin decondensation and recondensation regulated by transcription from a natural promoter. J. Cell Biol. 154:33-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murchison, E. P., J. F. Partridge, O. H. Tam, S. Cheloufi, and G. J. Hannon. 2005. Characterization of Dicer-deficient murine embryonic stem cells. Proc. Natl. Acad. Sci. USA 102:12135-12140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nielsen, A. L., M. Oulad-Abdelghani, J. A. Ortiz, E. Remboutsika, P. Chambon, and R. Losson. 2001. Heterochromatin formation in mammalian cells: interaction between histones and HP1 proteins. Mol. Cell 7:729-739. [DOI] [PubMed] [Google Scholar]
- 37.Nye, A. C., R. R. Rajendran, D. L. Stenoien, M. A. Mancini, B. S. Katzenellenbogen, and A. S. Belmont. 2002. Alteration of large-scale chromatin structure by estrogen receptor. Mol. Cell. Biol. 22:3437-3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pal-Bhadra, M., U. Bhadra, and J. A. Birchler. 2002. RNAi related mechanisms affect both transcriptional and posttranscriptional transgene silencing in Drosophila. Mol. Cell 9:315-327. [DOI] [PubMed] [Google Scholar]
- 39.Pal-Bhadra, M., B. A. Leibovitch, S. G. Gandhi, M. Rao, U. Bhadra, J. A. Birchler, and S. C. Elgin. 2004. Heterochromatic silencing and HP1 localization in Drosophila are dependent on the RNAi machinery. Science 303:669-672. [DOI] [PubMed] [Google Scholar]
- 40.Reinhart, B. J., and D. P. Bartel. 2002. Small RNAs correspond to centromere heterochromatic repeats. Science 297:1831. [DOI] [PubMed] [Google Scholar]
- 41.Rice, J. C., S. D. Briggs, B. Ueberheide, C. M. Barber, J. Shabanowitz, D. F. Hunt, Y. Shinkai, and C. D. Allis. 2003. Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol. Cell 12:1591-1598. [DOI] [PubMed] [Google Scholar]
- 42.Richards, E. J., and S. C. Elgin. 2002. Epigenetic codes for heterochromatin formation and silencing: rounding up the usual suspects. Cell 108:489-500. [DOI] [PubMed] [Google Scholar]
- 43.Robinett, C. C., A. Straight, G. Li, C. Willhelm, G. Sudlow, A. Murray, and A. S. Belmont. 1996. In vivo localization of DNA sequences and visualization of large-scale chromatin organization using lac operator/repressor recognition. J. Cell Biol. 135:1685-1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rudert, F., S. Bronner, J. M. Garnier, and P. Dolle. 1995. Transcripts from opposite strands of gamma satellite DNA are differentially expressed during mouse development. Mamm. Genome. 6:76-83. [DOI] [PubMed] [Google Scholar]
- 45.Schramke, V., and R. Allshire. 2003. Hairpin RNAs and retrotransposon LTRs affect RNAi and chromatin-based gene silencing. Science 301:1069-1074. [DOI] [PubMed] [Google Scholar]
- 46.Sigova, A., N. Rhind, and P. D. Zamore. 2004. A single Argonaute protein mediates both transcriptional and posttranscriptional silencing in Schizosaccharomyces pombe. Genes Dev. 18:2359-2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsukamoto, T., N. Hashiguchi, S. M. Janicki, T. Tumbar, A. S. Belmont, and D. L. Spector. 2000. Visualization of gene activity in living cells. Nat. Cell Biol. 2:871-878. [DOI] [PubMed] [Google Scholar]
- 48.Tumbar, T., G. Sudlow, and A. S. Belmont. 1999. Large-scale chromatin unfolding and remodeling induced by VP16 acidic activation domain. J. Cell Biol. 145:1341-1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Verdel, A., S. Jia, S. Gerber, T. Sugiyama, S. Gygi, S. I. Grewal, and D. Moazed. 2004. RNAi-mediated targeting of heterochromatin by the RITS complex. Science 303:672-676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Verschure, P. J., I. van der Kraan, W. de Leeuw, J. van der Vlag, A. E. Carpenter, A. S. Belmont, and R. van Driel. 2005. In vivo HP1 targeting causes large-scale chromatin condensation and enhanced histone lysine methylation. Mol. Cell. Biol. 25:4552-4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Volpe, T. A., C. Kidner, I. M. Hall, G. Teng, S. I. Grewal, and R. A. Martienssen. 2002. Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science 297:1833-1837. [DOI] [PubMed] [Google Scholar]
- 52.Zilberman, D., X. Cao, and S. E. Jacobsen. 2003. ARGONAUTE4 control of locus-specific siRNA accumulation and DNA and histone methylation. Science 299:716-719. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.