

Abstract
The transforming growth factor β (TGF-β) superfamily, including the bone morphogenetic protein (BMP) and TGF-β/activin A subfamilies, is regulated by secreted proteins able to sequester or present ligands to receptors. KCP is a secreted, cysteine-rich (CR) protein with similarity to mouse Chordin and Xenopus laevis Kielin. KCP is an enhancer of BMP signaling in vertebrates and interacts with BMPs and the BMP type I receptor to promote receptor-ligand interactions. Mice homozygous for a KCP null allele are hypersensitive to developing renal interstitial fibrosis, a disease stimulated by TGF-β but inhibited by BMP7. In this report, the effects of KCP on TGF-β/activin A signaling are examined. In contrast to the enhancing effect on BMPs, KCP inhibits both activin A- and TGF-β1-mediated signaling through the Smad2/3 pathway. These inhibitory effects of KCP are mediated in a paracrine manner, suggesting that direct binding of KCP to TGF-β1 or activin A can block the interactions with prospective receptors. Consistent with this inhibitory effect, primary renal epithelial cells from KCP mutant cells are hypersensitive to TGF-β and exhibit increased apoptosis, dissociation of cadherin-based cell junctions, and expression of smooth muscle actin. Furthermore, KCP null animals show elevated levels of phosphorylated Smad2 after renal injury. The ability to enhance BMP signaling while suppressing TGF-β activation indicates a critical role for KCP in modulating the responses between these anti- and profibrotic cytokines in the initiation and progression of renal interstitial fibrosis.
The transforming growth factor β (TGF-β) superfamily encodes growth and differentiation factors that regulate a wide variety of cellular processes. The members of the TGF-β superfamily include two major branches, bone morphogenetic proteins (BMPs) and TGF-βs/activins (19). TGF-β signaling is initiated by the binding of the ligand to a specific pair of type I and type II transmembrane receptors, leading to the phosphorylation of the cytoplasmic serine/threonine kinase domains (19, 25). The type I receptors, also known as activin-linked kinases (ALKs), then transduce signals downstream by phosphorylation of receptor-activated Smad proteins. The Smad family can be divided into three distinct subfamilies: receptor-activated Smads (R-Smad), common-partner Smads (Co-Smad), and inhibitory Smads. Whereas Smad1, Smad5, and Smad8 are phosphorylated by BMP type I receptors (ALK-2, ALK-3, and ALK-6), Smad2 and Smad3 are activated by TGF-β and activin type I receptors (ALK-5 and ALK-4). The R-Smad and the Co-Smad (Smad4) form a heterooligomer which translocates into the nucleus and regulates expression of ligand-responsive genes. Inhibitory Smads include Smad6 and Smad7, which prevent the activation of R- and Co-Smad.
While members of the TGF-β superfamily use similar mechanisms to signal to the nucleus, the two major branches achieve specificity by using different elements. Outside the cell, each ligand of the TGF-β superfamily binds to specific pairs of the receptor serine/threonine kinases, type I and type II (12). There are two distinct modes of the ligand-receptor interaction in the TGF-β family signaling. One is represented by the members of the BMP subfamily and the other exemplified by TGF-βs and activins. BMP ligands, such as BMP-2 and BMP-4, exhibit a high affinity for the extracellular ligand binding domains of the type I BMP receptors and a low affinity for the type II receptors. The preassembled type I receptor-ligand complex has a higher binding affinity for the type II receptor (19). In contrast to the BMPs, TGF-βs and activins display a high affinity for the type II receptors. The ligand binds tightly to the type II receptor first, which allows the subsequent incorporation of the type I receptor, forming a large ligand-receptor complex involving a ligand dimer and four receptor molecules (19).
Within the TGF-β superfamily, regulation of signaling can occur at multiple levels. The active TGF-β family ligand is a disulfide-linked homo- or heterodimer that is processed from large inactive precursor proteins. A diverse family of secreted proteins can inhibit TGF-β superfamily signaling by sequestering ligands away from receptors. In the Golgi complex, processing of the TGF-β proprotein results in the formation of a latent complex (2, 6). The latency-associated protein LAP1, which is the cleaved amino terminus of the TGF-β proprotein, together with latent TGF-β binding protein 1 (LTBP) and the active TGF-β homodimer constitutes the large latent complex, which associates with the extracellular matrix and can be released and activated by proteolytic cleavage. Extracellular inhibitors of BMP signaling include vertebrate Chordin, which binds directly to BMPs through the cysteine-rich (CR) domains containing CXXCXC and CCXXC motifs (5, 10, 19). While Chordin blocks BMP/receptor interactions, the CR domain protein KCP enhances BMP/receptor interactions to increase the efficacy of signaling (11). Similarly, the CR domain protein connective tissue growth factor (CTGF) also enhances TGF-β-mediated signaling while suppressing the BMP-dependent pathway (1).
The KCP protein contains 18 cysteine-rich domains and is expressed in the developing kidney at both early and late stages (11). KCP expression corresponds to the formation of epithelial structures within the intermediate mesoderm and to the formation of the proximal tubules in the more developed metanephric kidney. Unlike Chordin or CTGF, KCP enhances BMP-mediated signaling in a paracrine manner by interacting with the type I receptor to facilitate the binding of BMP7 to BMP receptor 1A (11). Mice homozygous for a mutant KCP allele show no gross developmental abnormalities but exhibit enhanced susceptibility to developing renal interstitial fibrosis, a process known to be regulated both by BMPs and by TGF-β, in two different animal models (11).
In this report, we characterized the effects of the CR domain protein KCP on TGF-β- and activin-mediated signaling. By use of Smad2/3-dependent reporter assays, KCP is able to suppress activin- and TGF-β-dependent transcription and Smad phosphorylation. KCP purified from conditioned media can interact directly with either activin or TGF-β1. Furthermore, expression of KCP in MDCK cells enhances the formation of tubules in response to hepatocyte growth factor and inhibits the negative effects of activin A in vitro. These data indicate a suppressor function for KCP in the regulation of TGF-β superfamily signaling. The ability to enhance BMP-mediated signaling while suppressing TGF-β/activin-mediated signaling may have important consequences in the progression of renal disease and may underlie the profibrotic phenotype found in KCP mutants.
MATERIALS AND METHODS
Plasmid DNAs.
The full-length coding sequence of KCP was inserted into a pCMV plasmid (where CMV is cytomegalovirus) containing a myc epitope tag at the carboxyl-terminal end and the neomycin resistance gene. To increase the secretion efficiency for the nickel affinity pull-down experiments, the full-length KCP was also inserted into the pSecTag B plasmid (Invitrogen) in frame via EcoRI and XbaI sites. The 3TP-Lux reporter construct was kindly provided by A. Roberts (National Cancer Institute, Bethesda, Md.).
Luciferase reporter assays.
NIH 3T3 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum and 100 U/ml penicillin and streptomycin under conditions of humidified 5% CO2 and 95% air at 37°C. Cells were transfected using FuGene 6 as described in the manufacturer's protocol (Roche Molecular Biochemicals). Cells were harvested 48 h after transfection. NIH 3T3 parental and KCP stably transfected cells were cotransfected with 3TP-Lux plasmid and β-galactosidase plasmid, used to monitor transfection efficiency. Activin A or TGF-β1 (R&D Systems) was added into the media at indicated concentrations 24 h after transfection. Cells were harvested, luciferase activity was measured by using a luciferase assay system (Promega), and luciferase value was normalized to β-galactosidase activity. The luciferase value under the condition without any stimulation was expressed as 1; the other values that represented the responses to activin A or TGF-β1 were divided by the basic value and expressed as increasing amounts (n-fold). Each experiment was repeated three times, and the results are presented as averages ± 1 standard deviation from the mean.
Collection of the conditioned media of KCP stable cells.
The KCP stably transfected cells were grown to confluence in DMEM with 10% fetal bovine serum and Geneticin or zeocin. The monolayer was washed with phosphate-buffered saline (PBS), and the medium was changed to serum-free DMEM with 1× insulin-transferrin-selenium-X (Invitrogen) for another 48 h. The conditioned medium was harvested and centrifuged to remove the cell debris. The clarified medium was concentrated about 10-fold by using an Amicon Ultra-15 NMWL 100K centrifugal filter device with a membrane at the speed of 3,100 rpm for 20 min, as described in the manufacturer's protocol (Millipore), and stored at −70°C. Control medium was collected in parallel from NIH 3T3 parental cells.
Western blot analysis.
Western blot analysis was performed with equal amounts of protein obtained by lysis of cells in 1% NP lysis buffer (15 mM sodium chloride, 1% NP-40, 50 mM Tris [pH 8.0]) and a mixture of protease inhibitors (Roche Molecular Biochemicals). The lysates were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotted with antibodies as indicated. Horseradish peroxidase-conjugated secondary antibodies were used to detect antigen-antibody complexes by an ECL detection system (Amersham Biosciences). The antibodies used were anti-Myc (9E10, 1:2,000; BabCO), anti-phosphorylated Smad2 Ser 465/467 (1:1,000; Cell Signaling), anti-Smad2 (1:1,000; Santa Cruz), anti-activin A (1:500; R&D Systems), anti-TGF-β1 (1:500; R&D Systems), antitubulin (1:10,000; Sigma), and anti-activated caspase-3 (1:500; Cell Signaling Technology).
For quantitation of Western blots, either 60 μg (total Smad2) or 120 μg (phosphorylated Smad2 [P-Smad2]) of total kidney protein lysates was fractionated on 8% polyacrylamide gels and transferred to a polyvinylidene difluoride membrane. Enhanced chemiluminescence reagents were used to visualize the secondary antibody, with attention paid to ensure that the film was not saturated. Images were scanned from the film with an Agfa Arcus II scanner, and band density quantification was performed by using Multi-Analyst software (Bio-Rad Laboratories). Results were expressed as means ± standard errors. Statistical analysis was carried out by unpaired Student's t test. Differences with P values of <0.05 were considered significant.
Pull-down assays.
For biotinylation, cells were grown to confluence, washed in PBS, and incubated with 0.5 mg/ml of N-hydroxysulfosuccinimide-biotin (Pierce) at 4°C for 20 min. After being quenched with 50 mM ammonium chloride, cells were washed in PBS, and lysates were prepared in 1% NP-40 lysis buffer. Protein complexes were pulled down with neutravidin agarose, washed three times in PBS, and incubated with 100 ng of TGF-β1 or activin A (R&D Systems) in a total volume of 200 ml PBS at 4°C overnight. After three more washing steps, proteins were boiled in 2× SDS-PAGE buffer and analyzed on 12% gels.
For nickel affinity pull-downs, the concentrated conditioned media of parental and His-tagged KCP stably expressing cells were run through ProBond Ni-nitrilotriacetic acid agarose resin (QIAGEN) and washed under native conditions according to the manufacturer's protocol. The beads were washed with PBS, incubated with 0.5 ng/μl of TGF-β1 or 5 ng/μl of activin A in PBS containing 1.0 mM Mg2+ and 1.3 mM Ca2+ overnight, and then washed with PBS three times and eluted in 2× SDS-PAGE sample buffer. The Ni bead pull-down fraction was detected by Western blotting.
Isolation and analysis of primary kidney cells.
Proximal tubule cells were isolated by collagenase digestion, sieving, and use of Percoll gradients as originally described by Humes et al. (7). A defined serum-free medium was used as described by Sens et al. (18). Cells were expanded and frozen in aliquots. Samples were thawed in six-well plates and used for testing TGF-β responsiveness without any additional passages. For immunostaining, cells were fixed for 10 min in 4% paraformaldehyde, washed twice in PBS and 0.1% Tween 20, and stained with anti-E-cadherin (Cell Signaling Technologies) and Cy3-conjugated anti-smooth muscle actin (SMA) monoclonal antibody (Sigma).
Animal models of renal injury.
For the induction of acute tubular necrosis (ATN), age-matched mice were injected intraperitoneally with 250 mg folic acid/kg of body weight (8) and sacrificed at 7, 14, and 28 days postinjury. Mice were observed daily after injection, and serum was collected at the time of death. At least seven animals of each genotype were analyzed at each time point. KCP null animals were defined as described previously (11).
For the induction of renal fibrosis, the unilateral urethral obstruction (UUO) model was utilized. Mice were anesthetized by intraperitoneal injection of ketamine and xylazine. Through a midline abdominal incision, the right ureter was exposed and tied off at the midurethral level with fine suture materials (4-0 silk) to induce a complete obstruction. Mice were allowed to recover from anesthesia and were kept with an ad libitum supply of food and water until the indicated time of sacrifice (7, 13, and 28 days). At least four mice of wild-type and KCP null genotypes were analyzed at each time point. Both obstructed and contralateral kidneys were harvested for protein analysis.
RESULTS
As previous work had demonstrated KCP-mediated enhancement of BMP signaling (11), we examined whether KCP could also affect other ligands of the TGF-β superfamily. The Smad2/3-dependent transcription reporter 3TP-Lux (14) was used in both transiently and stably transfected KCP cell lines to test for TGF-β and activin responses (Fig. 1). A dose-response curve demonstrated a maximal fivefold response of 3TP-Lux to TGF-β1 in control cells (Fig. 1A) and sixfold activation with 100 ng/ml activin A (Fig. 1B). However, expression of transiently or stably transfected KCP reduced the level of TGF-β1-mediated activation to 1.5- or 2-fold, respectively (Fig. 1A). Similarly, activin-dependent activation was inhibited approximately two- or threefold by KCP in transiently or stably expressing cells (Fig. 1B). These results are in sharp contrast to the levels of activation observed with BMP7 and a Smad1 reporter, where KCP enhanced activity 5- to 10-fold (11).
FIG. 1.

KCP suppresses TGF-β1- and activin A-mediated reporter gene expression. The effect of TGF-β1 (A) or activin A (B) on activation of the 3TP-Lux reporter was examined under various conditions. The 3TP-Lux reporter was transiently transfected into control NIH 3T3 cells (white bars), cotransfected transiently with KCP into NIH 3T3 cells (black bars), or transiently transfected into KCP stably expressing NIH 3T3 cell lines (gray bars). TGF-β1 (A) or activin A (B) was added to the media at the indicated concentrations. All experiments were done in triplicate, with average values shown, adjusted for a β-galactosidase internal standard. Error bars are 1 standard deviation from the mean (★, P < 0.05). (C and D) Paracrine assay using conditioned media from NIH 3T3 parental (white bars) or KCP stably expressing cells (black bars) added to the 3TP-Lux transiently transfected NIH 3T3 cells prior to addition of TGF-β1 or activin A. The response to TGF-β1 (C) or activin A (D) is decreased via the conditioned media of KCP stable cells. All experiments were performed a minimum of three times, and results are shown as the average values, corrected for transfection efficiency, as measured by β-galactosidase control plasmid. The luciferase value under the condition without any stimulation was assigned a basal value of 1. Error bars are 1 standard deviation from the mean (★, P < 0.05).
To demonstrate that inhibition by KCP was mediated by a paracrine mechanism, KCP conditioned media were added to the cells transfected with 3TP-Lux. Conditioned media from KCP-expressing cells inhibited both TGF-β1-dependent (Fig. 1C) and activin A-dependent (Fig. 1D) activation by one-third compared to the conditioned media from parental, nonexpressing cells. These data suggest that the KCP-mediated inhibition of Smad2/3 signaling works, at least in part, through an extracellular, paracrine mechanism.
The suppressed response to activin A or TGF-β1 in KCP-expressing cells was further confirmed by Western blotting with antibodies specific for P-Smad2/3 serine 465/467 (Fig. 2). By use of stably expressing KCP and parental 3T3 cells, whole-cell lysates were prepared 1 h after increasing amounts of TGF-β1 or activin A were added. With increasing amounts of TGF-β1, the KCP-expressing cells still responded, although there was little increase at higher concentrations and the overall levels of P-Smad2/3 were decreased compared to levels for the parental line (Fig. 2A). With increasing amounts of activin A, there was little induction of P-Smad2/3 in KCP-expressing cells even at high concentrations (Fig. 2B). To exclude any variations due to selection of stably expressing clones, we transfected KCP transiently into HEK293 cells and added TGF-β1 or activin 48 h posttransfection (Fig. 2C). For these transient experiments, we utilized the Sec-Tag vector containing an immunoglobulin κ leader sequence to improve secretion. Cells expressing Sec-Tag KCP showed a marked reduction of P-Smad2/3 upon addition of either TGF-β1 or activin A compared to control cells. Duplicate blots were probed for total Smad2 protein to reveal nearly equal amounts in all lysates.
FIG. 2.

KCP expression suppresses Smad2/3 phosphorylation. Increasing concentrations of TGF-β1(A) or activin A (B) were added to 3T3 cells or KCP stably transformed 3T3 cells, and protein lysates were prepared 1 h after addition. Proteins were separated by SDS-PAGE and probed with the indicated antibodies via Western blotting. (C) Transient expression of KCP in HEK293 cells. Cells were transfected with Sec-Tag KCP 48 h prior to addition of TGF-β1 or activin, as indicated. Lysates were prepared 1 h after addition of ligands and analyzed by Western blotting. (D) Activation of the BMP pathway does not significantly affect the activin A response by use of 3TP-Lux. Control NIH 3T3 cells were transiently transfected with 3TP-Lux, and factors were added as indicated.
Given that KCP expression enhances the response to BMPs in transiently and stably expressing cells (11), it is possible that increased basal BMP signaling and Smad1 activation, in the absence of exogenous ligands, may suppress the TGF-β/activin response. In order to rule out this possibility, we tested potential cross talk between the pathways by using the 3TP-Lux reporter and activin A (Fig. 2D). Under these conditions, stimulating the BMP/Smad1 pathway with 10 to 100 ng/ml BMP7 did not suppress activin A-dependent 3TP-Lux activity. Thus, it is unlikely that KCP-mediated suppression of Smad2/3 phosphorylation is related to increased activity of the BMP pathways.
The KCP protein contains multiple cysteine-rich domains that can bind directly to ligands of the TGF-β superfamily. Thus, we tested for direct physical interactions between KCP and TGF-β1 or activin A by using biotinylated KCP or poly-His-tagged KCP from conditioned media (Fig. 3). Total extracellular proteins from KCP-expressing cell lines and control cells were biotinylated and used to capture recombinant TGF-β1 (Fig. 3A). Biotinylated proteins from KCP-expressing cells, but not from control cells, were able to pull down TGF-β by use of neutravidin beads. Furthermore, an independent method utilized poly-His-tagged KCP protein, and ligands were captured on nickel affinity beads. In these assays, both recombinant TGF-β1 (Fig. 3B) and activin A (Fig. 3C) were able to interact with the His-tagged KCP protein, as evidenced by the coprecipitation with nickel agarose. Thus, the data suggest a physical interaction between these ligands and KCP or with another KCP-associated protein. This interaction could act to attenuate the signaling responses observed with the Smad2/3 reporter by partially sequestering ligands from their receptors.
FIG. 3.
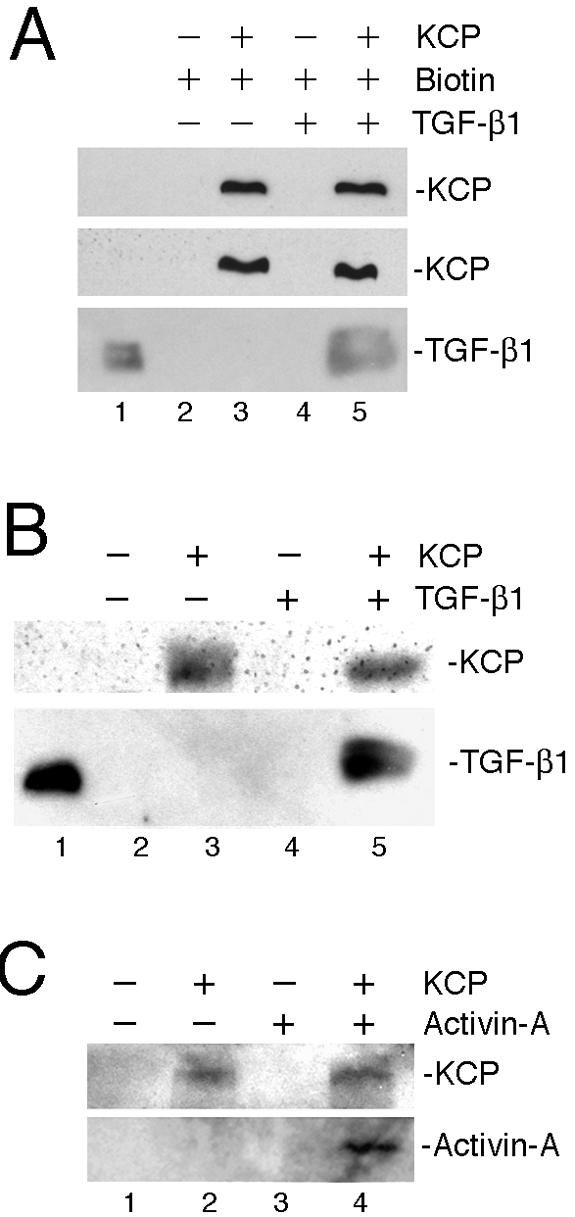
KCP binds to activin A and TGF-β1. (A) Total extracellular proteins from control and KCP-expressing cells were biotinylated and used to pull down recombinant TGF-β1. Note that lysates from control cells do not capture TGF-β1 on neutravidin beads. (B and C) The concentrated conditioned media from NIH 3T3 parental and KCP-myc-His stable cells were incubated with Ni-nitrilotriacetic acid agarose resin and washed under native conditions. The pulled-down proteins were then incubated with either TGF-β1 (B) or activin A (C) overnight and then washed with PBS three times and eluted in 2× SDS-PAGE sample buffer. Immobilized proteins were detected with anti-TGF-β1 or anti-activin A as indicated. Lane 1 contains recombinant TGF-β1 as a positive control for Western blotting.
Mice homozygous for a KCP null allele showed increased sensitivity to developing renal fibrotic disease (11). The ability of proximal tubules to recover from injury was also attenuated in KCP null mice. In the kidney, TGF-β is a profibrotic cytokine. Thus, KCP null mice could be more susceptible to fibrotic disease because lack of KCP protein results in TGF-β hypersensitivity. To test this mechanism, we isolated primary renal epithelial cells from wild-type and KCP mutant adult kidneys. Cells were prepared by collagenase digestion and use of Percoll gradients and cultured in a defined serum-free medium. Primary renal cells were then exposed to increasing amounts of TGF-β and scored for various indicators. In vitro, TGF-β promotes the dissociation of cell-cell junctions, increases motility, and can initiate apoptosis. After 48 h of TGF-β treatment, significant differences between wild-type and KCP null cells were observed. At 2 ng/ml of TGF-β, wild-type cells showed little cell dissociation and cell death at the light microscopic level, whereas KCP null cells were partially dissociated, with many cells floating off the plastic (Fig. 4A). At 10 ng/ml, both cell types showed a robust response, though KCP null cells were even fewer. To better quantify the response, we stained cells for E-cadherin and anti-SMA. At the highest doses, both cell types showed few E-cadherin-positive clusters and a significant increase in SMA-positive cells compared to untreated cells (Fig. 4B). However, at low doses (0.5 ng/ml), few wild-type cells were dissociated from their neighbors, while KCP null cells already showed increased dissociation of E-cadherin-based cell junctions and strong SMA expression. The dose-response curves were quantified by counting fields of cells at various concentrations of TGF-β after 48 h in culture (Fig. 5A and B). KCP null primary cells consistently showed more cell dissociation and increased numbers of SMA-positive cells at low doses of TGF-β. To directly measure TGF-β sensitivity, we examined P-Smad2 levels after 1 h with ligands (Fig. 5C). Similarly to what we observed with the cell morphology assays, KCP null cells showed increased levels of P-Smad2/3 at 0.1 and 0.5 ng/ml. These increased levels were evident despite having lower levels of total Smad2/3 protein present. To confirm the increased cell death observed with KCP mutant cells, we examined caspase-3 activation after 48 h in culture. Even at low doses of TGF-β, KCP null cells showed increased activation of caspase-3, as evidenced by the cleaved 17-kDa active form. Wild-type cells showed measurable amounts of active caspase-3 only at high doses of TGF-β, and this was reversible upon cotreatment with BMP7. Consistent with our previous results, KCP null cells did not show suppression of caspase-3 activation when TGF-β and BMP7 were added together. Our data indicate that KCP can suppress the TGF-β response in renal epithelial cells and help maintain the epithelial phenotype.
FIG. 4.

KCP mutant renal epithelial cells are hypersensitive to TGF-β1. (A) Primary cells were isolated from normal adult kidneys or from KCP−/− adult kidneys and grown on plastic in defined media. Cells were incubated with increasing amounts of TGF-β1 and photographed after 48 h. Note the loss of cell monolayers at 2.0 ng/ml of TGF-β with KCP−/− cells. (B) Immunostaining of cells after 48 h in culture with the indicated amounts of TGF-β1. E-cadherin is shown in green (all panels), smooth muscle actin is in red (right two panels only), and the cell nuclei are blue (all panels). At 10 ng/ml, both cell populations show dissociation and some SMA expression. However, at low doses, only KCP−/− cells show significant dissociation and strong SMA-expressing cells.
FIG. 5.

Quantitative analysis of TGF-β1 responses in KCP mutant cells. Primary cells were isolated from wild-type and KCP null kidneys and cultured with increasing amounts of TGF-β1. (A) After 48 h, cells were stained with anti-SMA and counted. The percentage of SMA-positive cells per field is given for 20 randomly selected areas. (B) Cell scattering was detected after staining with anti-E-cadherin. The percent scattering is the number of E-cadherin-negative cells with no visible junctions to neighboring cells, as counted in 20 randomly selected fields of view. (C) Levels of P-Smad2/3 were detected by Western blotting 1 h after addition of increasing amounts of TGF-β1 to primary renal cells from wild-type and KCP null kidneys. Note the increased levels of P-Smad2/3 in KCP null cells at a low concentration of 0.1 ng/ml of TGF-β1. This increase was evident despite significantly lower levels of total Smad2/3 present. (D) KCP−/− cells were more sensitive to TGF-β-mediated apoptosis. Activation of caspase-3 was measured by Western blotting of total cell lysates after 48 h of culture with increasing amounts of TGF-β. Concentrations are as follows: lane 1, 0 ng/ml; lane 2, 0.1 ng/ml; lane 3, 0.5 ng/ml; lane 4, 2.0 ng/ml; lane 5, 10 ng/ml; lane 6, 2 ng/ml TGF-β and 50 ng/ml BMP7. Proteins from equal numbers of cells were loaded. β-tub., β-tubulin. Arrow, cleaved caspase-3.
In animal models of renal disease, TGF-β is a profibrotic cytokine that is thought to mediate the transition of epithelial cells to mesenchyme (15, 23). Previously, we showed that KCP null mice were hypersensitive to developing renal fibrosis by either the ATN model or the UUO model (11). To address the sensitivity of KCP null mice to TGF-β, we analyzed protein lysates from kidneys after administration of folic acid to induce ATN (Fig. 6). Western blots of proteins were probed for total and phosphorylated Smad2 and analyzed by quantitative chemiluminescence. In the ATN model, levels of P-Smad2 were not significantly different at 2 days after injection of folic acid. However, KCP null kidneys showed more than a 10-fold increase in P-Smad2 levels 7 days postinjury. The increase in P-Smad2 levels seen for KCP null kidneys persisted, as evidenced by a more-than-fivefold difference at 28 days post-folic acid injection. These data are consistent with the increased scarring in KCP null kidneys observed to occur after recovery from ATN.
FIG. 6.

Analysis of Smad2 after acute tubular necrosis. (A) Western blotting with anti-Smad2 or anti-P-Smad2 antibodies of total protein lysates from kidneys. Control animals were untreated, and kidneys were analyzed 2, 7, or 28 days after induction of ATN by folic acid. Genotypes are wild type (+/+) or KCP null (−/−). (B) Quantitative analysis of the Smad2 Western blots by use of an Agfa Arcus scanner and Bio-Rad Multi-Analyst software. (C) Quantitative analysis of the P-Smad2 Western blots. Statistically significant differences are noted by P values. Con, control.
We also determined P-Smad2 levels in an independent model of renal injury, the UUO model of fibrosis. Obstructed and contralateral kidneys from wild-type and KCP null animals were examined for P-Smad levels after UUO (Fig. 7). The increase in P-Smad2 levels in the kidneys of KCP mice after obstruction was modest but reproducible. After 28 days, obstructed kidneys from KCP null mice exhibited approximately 30% more P-Smad2. Contralateral kidneys of KCP null mice, which we had previously shown to exhibit a chronic fibrotic phenotype at 28 days (11) also showed a twofold increase in P-Smad2 levels at 28 days. These data suggest that increased TGF-β signaling may contribute to the phenotype observed after renal injury in KCP mutant kidneys.
FIG. 7.

Analysis of Smad2 after UUO. (A) Western blotting with anti-Smad2 or anti-P-Smad2 antibodies of total protein lysates from kidneys after UUO. Controls are untreated kidneys. Contralateral (C) and obstructed (UUO) kidneys were taken from wild-type and KCP null animals at 7 or 28 days postobstruction. (B) Quantitative analysis of total Smad2 levels. (C) Quantitative analysis of P-Smad2 levels. Statistically significant differences are noted by P values of less than 0.05.
DISCUSSION
The KCP protein has 18 CR domains and is capable of interacting with many ligands of the TGF-β superfamily. Previously, we have shown that KCP binds BMP7 and enhances receptor-ligand interactions to increase the sensitivity of BMP signaling responses (11). As indicated in this report, the ability of KCP to enhance signaling is limited to the BMP family, since both TGF-β and activin signaling are suppressed by KCP. Suppression is mediated by a paracrine mechanism, suggesting that the secreted form of KCP is able to sequester ligands from receptors. Binding of KCP to both activin A and TGF-β complexes supports this interpretation.
KCP is not the first extracellular CR domain protein that can differentially regulate the TGF-β superfamily. Initial characterization of the prototype CR proteins Chordin and Short gastrulation (Sog) revealed inhibition of BMP and Decapentaplegic (Dpp) signaling during axis formation in frogs and flies, respectively. More recently, however, sog was shown to limit dpp activity, in cooperation with twisted gastrulation, and to allow diffusion of a latent complex that can be activated by tolloid (4, 20, 21). These modifiers thus transform a gradient of ligand into a stepwise response with a distinct boundary of Smad activation. CTGF, another CR domain protein, also has dual roles in regulating the TGF-β family members. Similarly to KCP, the CTGF protein binds to both BMPs and TGF-βs (1), but its effect on signaling is opposite that of KCP in that it inhibits BMP-mediated signaling while enhancing the TGF-β response. Thus, CR domain proteins are not exclusive inhibitors but are able to both positively and negatively modulate the signals received from different branches of the TGF-β superfamily of ligands to achieve a balance of outputs.
The mechanism of TGF-β and activin inhibition remains to be clearly defined. While KCP binds to BMPs and can enhance the interaction of BMPs with the type I receptor, we have not been able to assay the effects of KCP on the TGF-β receptors for several technical reasons. Given the size and cysteine-rich nature of KCP, it has not been possible to make a soluble recombinant form of the protein in bacteria, thus precluding any quantitative biochemical studies in vitro. However, we have been able to show binding of KCP to TGF-β and activin in conditioned media, though it is possible that this interaction may be mediated by other proteins. Whether the interaction of KCP with these two ligands prevents receptor-ligand interactions remains to be determined. Given the size and complexity of KCP, sequestration of ligands from the receptor complex certainly is a possibility that needs to be addressed.
Mice homozygous for a targeted allele of KCP show no gross developmental defects but are sensitive to developing renal fibrotic disease (11). Renal fibrosis is due to an increase in interstitial fibroblasts and increased deposition of basement membrane components. Currently, TGF-β1 is regarded as the predominant profibrotic stimulus in the kidney and many other tissues (3, 17, 22). Consistent with this hypothesis, deletion of the downstream effector Smad3 protects against fibrotic disease in the kidney after obstruction (15). TGF-β1 induces cell hypertrophy in the early phase of fibrotic disease and may promote epithelial cell-to-mesenchyme transition and apoptosis in later stages of fibrotic diseases (9). It appears that BMP-7 counteracts this TGF-β1-driven fibrotic response (13, 23, 24). Though KCP expression in adult tissues is undetectable, we have observed the induction of KCP expression in renal fibrosis and renal injury models (11). Furthermore, KCP mutants show decreased levels of P-Smad1 after injury, indicating a decrease in BMP responsiveness. This alone may underlie the increased sensitivity to developing renal fibrosis. However, the potential for KCP mutants to have increased TGF-β responses, due to the absence of KCP-mediated inhibition, may also contribute to the phenotype. The increased P-Smad2 levels observed with both the ATN and the UUO model are consistent with this hypothesis, though the increased P-Samd2 levels are significantly less than the decreased P-Smad1 levels observed with KCP null kidneys. Nevertheless, the increased sensitivity of KCP mutants to developing renal fibrotic disease may be due to a combination of decreased BMP signaling and increased TGF-β signaling. Given the importance of TGF-βs and BMPs in mediation of injury, repair, and fibrosis, the potential role for KCP to affect these processes clearly merits further investigation.
The roles of TGF-β in renal injury are likely to be complex. In addition to stimulating epithelial cell-to-mesenchyme transition, TGF-β can promote cell death in vitro and in vivo (16). Activation of caspase-3 by low doses of TGF-β in primary renal epithelial cells from KCP mutant mice suggests that apoptosis is an important component of the stress response to either obstruction or acute tubular necrosis. Indeed, we have observed significant differences in mortality after folic acid induction of ATN in KCP null mice compared to mortality in wild-type mice (11), suggesting that the initial amount of cell death may be increased.
In summary, KCP is the first enhancer of BMP signaling described to date that suppresses TGF-β and activin signaling and as such may play an important role in mediating the signal specificity between competing inputs in the initiation and progression of renal disease.
Acknowledgments
We thank Anita Roberts for the 3TP-Lux reporter, X. Cheng for help with the UUO model, and Eric Fearon and Ken Cadigan for critical reading of the manuscript.
This work is supported by NIH grants DK062914 to G.R.D and DK002803 to S.R.P.
REFERENCES
- 1.Abreu, J. G., N. I. Ketpura, B. Reversade, and E. M. De Robertis. 2002. Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-beta. Nat. Cell Biol. 4:599-604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Annes, J. P., J. S. Munger, and D. B. Rifkin. 2003. Making sense of latent TGFbeta activation. J. Cell Sci. 116:217-224. [DOI] [PubMed] [Google Scholar]
- 3.Chatziantoniou, C., and J. C. Dussaule. 2005. Insights into the mechanisms of renal fibrosis: is it possible to achieve regression? Am. J. Physiol. Renal Physiol. 289:F227-F234. [DOI] [PubMed] [Google Scholar]
- 4.Eldar, A., R. Dorfman, D. Weiss, H. Ashe, B. Z. Shilo, and N. Barkai. 2002. Robustness of the BMP morphogen gradient in Drosophila embryonic patterning. Nature 419:304-308. [DOI] [PubMed] [Google Scholar]
- 5.Garcia Abreu, J., C. Coffinier, J. Larrain, M. Oelgeschlager, and E. M. De Robertis. 2002. Chordin-like CR domains and the regulation of evolutionarily conserved extracellular signaling systems. Gene 287:39-47. [DOI] [PubMed] [Google Scholar]
- 6.Gumienny, T. L., and R. W. Padgett. 2002. The other side of TGF-beta superfamily signal regulation: thinking outside the cell. Trends Endocrinol. Metab. 13:295-299. [DOI] [PubMed] [Google Scholar]
- 7.Humes, H. D., W. H. Fissell, W. F. Weitzel, D. A. Buffington, A. J. Westover, S. M. MacKay, and J. M. Gutierrez. 2002. Metabolic replacement of kidney function in uremic animals with a bioartificial kidney containing human cells. Am. J. Kidney Dis. 39:1078-1087. [DOI] [PubMed] [Google Scholar]
- 8.Imgrund, M., E. Grone, H. J. Grone, M. Kretzler, L. Holzman, D. Schlondorff, and U. W. Rothenpieler. 1999. Re-expression of the developmental gene Pax-2 during experimental acute tubular necrosis in mice 1. Kidney Int. 56:1423-1431. [DOI] [PubMed] [Google Scholar]
- 9.Kalluri, R., and E. G. Neilson. 2003. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 112:1776-1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larrain, J., D. Bachiller, B. Lu, E. Agius, S. Piccolo, and E. M. De Robertis. 2000. BMP-binding modules in chordin: a model for signalling regulation in the extracellular space. Development 127:821-830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin, J., S. R. Patel, X. Cheng, E. A. Cho, I. Levitan, M. Ullenbruch, S. H. Phan, J. M. Park, and G. R. Dressler. 2005. Kielin/chordin-like protein, a novel enhancer of BMP signaling, attenuates renal fibrotic disease. Nat. Med. 11:387-393. [DOI] [PubMed] [Google Scholar]
- 12.Manning, G., D. B. Whyte, R. Martinez, T. Hunter, and S. Sudarsanam. 2002. The protein kinase complement of the human genome. Science 298:1912-1934. [DOI] [PubMed] [Google Scholar]
- 13.Patel, S. R., and G. R. Dressler. 2005. BMP7 signaling in renal development and disease. Trends Mol. Med. 11:512-518. [DOI] [PubMed] [Google Scholar]
- 14.Piek, E., W. J. Ju, J. Heyer, D. Escalante-Alcalde, C. L. Stewart, M. Weinstein, C. Deng, R. Kucherlapati, E. P. Bottinger, and A. B. Roberts. 2001. Functional characterization of transforming growth factor beta signaling in Smad2- and Smad3-deficient fibroblasts. J. Biol. Chem. 276:19945-19953. [DOI] [PubMed] [Google Scholar]
- 15.Sato, M., Y. Muragaki, S. Saika, A. B. Roberts, and A. Ooshima. 2003. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J. Clin. Investig. 112:1486-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schiffer, M., M. Bitzer, I. S. Roberts, J. B. Kopp, P. ten Dijke, P. Mundel, and E. P. Bottinger. 2001. Apoptosis in podocytes induced by TGF-beta and Smad7. J. Clin. Investig. 108:807-816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schnaper, H. W., T. Hayashida, S. C. Hubchak, and A. C. Poncelet. 2003. TGF-beta signal transduction and mesangial cell fibrogenesis. Am. J. Physiol. Renal Physiol. 284:F243-F252. [DOI] [PubMed] [Google Scholar]
- 18.Sens, D. A., C. J. Detrisac, M. A. Sens, M. R. Rossi, S. L. Wenger, and J. H. Todd. 1999. Tissue culture of human renal epithelial cells using a defined serum-free growth formulation. Exp. Nephrol. 7:344-352. [DOI] [PubMed] [Google Scholar]
- 19.Shi, Y., and J. Massague. 2003. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 113:685-700. [DOI] [PubMed] [Google Scholar]
- 20.Shimmi, O., and M. B. O'Connor. 2003. Physical properties of Tld, Sog, Tsg and Dpp protein interactions are predicted to help create a sharp boundary in Bmp signals during dorsoventral patterning of the Drosophila embryo. Development 130:4673-4682. [DOI] [PubMed] [Google Scholar]
- 21.Sutherland, D. J., M. Li, X. Q. Liu, R. Stefancsik, and L. A. Raftery. 2003. Stepwise formation of a SMAD activity gradient during dorsal-ventral patterning of the Drosophila embryo. Development 130:5705-5716. [DOI] [PubMed] [Google Scholar]
- 22.Tamaki, K., and S. Okuda. 2003. Role of TGF-beta in the progression of renal fibrosis. Contrib. Nephrol. 139:44-65. [DOI] [PubMed] [Google Scholar]
- 23.Zeisberg, M., J. Hanai, H. Sugimoto, T. Mammoto, D. Charytan, F. Strutz, and R. Kalluri. 2003. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 9:964-968. [DOI] [PubMed] [Google Scholar]
- 24.Zeisberg, M., A. A. Shah, and R. Kalluri. 2005. Bone morphogenic protein-7 induces mesenchymal to epithelial transition in adult renal fibroblasts and facilitates regeneration of injured kidney. J. Biol. Chem. 280:8094-8100. [DOI] [PubMed] [Google Scholar]
- 25.Zwijsen, A., K. Verschueren, and D. Huylebroeck. 2003. New intracellular components of bone morphogenetic protein/Smad signaling cascades. FEBS Lett. 546:133-139. [DOI] [PubMed] [Google Scholar]