

Abstract
p53 not only functions as a transcription factor but also has a direct, apoptogenic role at the mitochondria. We have discovered that DNA damage-induced p53-Bcl2 binding is associated with decreased Bcl2-Bax interaction and increased apoptotic cell death in a mechanism regulated by Bcl2's flexible loop regulatory domain (FLD), since purified p53 protein can disrupt the Bcl2/Bax complex by directly binding to a negative regulatory region of the FLD (amino acids [aa] 32 to 68). Deletion of the negative regulatory region (Δ32-68) abolishes Bcl2-p53 binding and enhances Bcl2's antiapoptotic function. Conversely, removal of a positive regulatory region (aa 69 to 87) of the FLD, which contains the Bcl2 phosphorylation site(s) T69, S70, and S87, enhances Bcl2-p53 binding and significantly abrogates Bcl2's survival activity. The phospho-mimetic T69E/S70E/S87E (EEE) but not the nonphosphorylatable T69A/S70A/S87A (AAA) Bcl2 mutant displays a reduced capacity to bind p53 and potently inhibits p53-induced cytochrome c release from isolated mitochondria. Furthermore, the FLD-only aa32-87 and aa32-68 peptides but not the aa69-87 peptide can directly bind p53 in vitro. p53-induced cytochrome c release occurs through a mechanism involving Bax's integral insertion into the outer mitochondrial membrane. Either DNA damage to cells or expression of p53 selectively targeted to the mitochondria results in Bcl2-p53 binding followed by exposure of Bcl2's BH3 domain in association with inactivation of Bcl2's antiapoptotic function, indicating a conformational change in Bcl2 can occur upon direct ligation of p53. Thus, Bcl2's FLD contains both positive and negative regulatory regions which functionally regulate Bcl2's antiapoptotic activity by affecting Bax or p53 binding.
The apoptotic process occurs in three interdependent phases: induction, decision, and execution. The decision phase is largely regulated by the Bcl2 family of apoptotic regulators (1, 21). Bcl2-related proteins share homology in regions designated the Bcl2 homology (BH) domains BH1, BH2, BH3, and BH4 (28). All four homology regions are present in the antiapoptotic family members, including Bcl2, Bcl-XL, and MCL1. The proapoptotic family members can be divided into two subgroups based on the presence of BH domains: the BH123 multidomain proteins (i.e., Bax and Bak) and the BH3-only molecules (5, 29, 39). Recent studies suggest that there are two different subgroups in the BH3-only members. One group, including Bid and Bim, can function both directly to bind and activate Bax as well as indirectly to counteract the inhibition of Bax or Bak by antiapoptotic members, including Bcl2 and Bcl-XL. Other BH3-only proteins (i.e., Bad, Bik, Noxa B, and PUMA) lack the ability to directly activate Bax but can oppose the action of antiapoptotic family members. Thus, both direct and indirect functions of BH3-only proteins may initiate apoptosis via selective interaction of the BH3 domain with a groove on the antiapoptotic Bcl2-like proteins and/or facilitate a conformational change in the multidomain proapoptotic proteins Bax and Bak, which induce a death effect by promoting their insertion into mitochondrial membranes (5, 29, 39). While antiapoptotic Bcl2, the founding member of the family, can suppress cell death induced by a variety of stresses (11), it is still not clear how Bcl2 actually functions to block apoptosis and promote survival. For example, growth factors like interleukin-3 (IL-3) mediate Bcl2 phosphorylation at serine 70 (S70), a site located within the flexible loop regulatory domain (FLD) that lies between the BH4 and BH3 regions to positively regulate its antiapoptotic function (25). The FLD of Bcl2 undergoes posttranslational modification through phosphorylation mediated by several growth factor-activated protein kinases, including the mitogen-activated protein kinases, extracellular signal-regulated kinases 1 and 2, and Jun N-terminal protein kinase 1, as well as protein kinase C (11, 12, 42). In addition, multisite phosphorylation of Bcl2 at S70, threonine 69 (T69), and S87 can also occur when cells are treated with microtubule-disrupting agents, such as paclitaxel and vincristine (46). As revealed using compound phospho-mimetic or nonphosphorylatable Bcl2 mutants, phosphorylation at any of these sites significantly enhances Bcl2's antiapoptotic function, and there is a cumulative advantage for multisite phosphorylation of Bcl2 in survival (9). Bcl2 phosphorylation also stabilizes the Bcl2-Bax interaction and protects Bcl2 from degradation that can occur during apoptosis (11, 15), but the mechanism is not yet clear.
Tumor suppressor activity of the p53 protein has been primarily explained by its ability to induce apoptosis in response to genotoxic stress by activating its transcriptional function that leads to expression of the proapoptotic Bcl2 family members, including Bax, Bid, Noxa, and PUMA (34, 35, 37, 43). However, it has recently been reported that p53 can also induce apoptosis independently of new protein synthesis (8, 33). Since transactivation-deficient p53 mutants can also potently induce apoptotic cell death in certain cell lines (22), this indicates that p53 possesses proapoptotic activity independent of its transcriptional activity. Furthermore, Marchenko et al. reported that a fraction of p53 can localize to mitochondria in tumor cells undergoing DNA damage-induced apoptosis (32). Intriguingly, enhanced apoptotic activity of the P72R p53 mutant has been found to rely in part on its mitochondrial localization (16). Other studies have confirmed that targeting of p53 to the mitochondria can occur in normal lymphocytes induced to die in response to ionizing radiation, suggesting a physiological mechanism (33). p53 accumulation at the mitochondria is rapid following ionizing radiation or treatment with DNA-damaging agents (starting after 1 h) and precedes the early dysfunctional changes in the mitochondria, including cytochrome c (Cyt c) release, that lead to procaspase-3 activation of the intrinsic apoptosis pathway (32). Interestingly, p53, which lacks any BH3 domain, can mimic the BH3-only molecules (Bim or Bid) and trigger the rapid release of Cyt c from isolated mitochondria by activating the membrane permeabilization function of Bax (8, 33). This suggests that the “extranuclear” apoptotic function of p53 may result from a direct effect on a multidomain proapoptotic protein, Bax or Bak, in mitochondria. Finally, a recent report indicates that expression of the p53-inducible, BH3-only death protein PUMA can displace p53 from Bcl-XL, allowing p53 to facilitate activation of Bax with mitochondrial permeabilization (7).
Mitochondria are central death regulators of the intrinsic apoptotic pathway in response to DNA damage, growth factor withdrawal, hypoxia, or oncogene deregulation and are critical for p53-dependent cell death (13, 33, 44). When mitochondria receive a death signal, the outer mitochondrial membrane (OMM) undergoes permeabilization to facilitate release of potent death factors from the intermembranous space into the cytosol that can activate the caspases responsible for apoptosis (19). OMM permeabilization is regulated by the opposing actions of pro- and antiapoptotic Bcl2 family proteins, although the exact mechanism(s) of how these family members regulate OMM permeability is not clear. A popular paradigm holds that antiapoptotic members such as Bcl2 and Bcl-XL, which reside in the OMM, mediate their survival function by preventing the release of death factors from mitochondria. Indeed, overexpression of Bcl2 suppresses both p53-dependent and -independent activation of the intrinsic death pathway. Recent reports indicate that genotoxic stress not only up-regulates p53 transcriptional activity but also leads to translocation of a portion of p53 to the cytoplasm/mitochondria, where it can interact with Bcl-XL or Bcl2 on the OMM and induce mitochondrial dysfunction in a mechanism involving Bax activation (8, 32, 33). Therefore, it is possible that p53 cannot only function directly to activate Bax but may also suppress Bcl2's antiapoptotic function directly by interacting with Bcl2. However, the mechanism by which Bcl2 and p53 bind and how or whether p53 affects Bcl2's survival activity and Bax function remain unclear. Therefore, studies were conducted to determine how p53 and Bcl2 interact and the role of Bcl2's FLD in regulating this interaction.
MATERIALS AND METHODS
Materials.
p53 small interfering RNA (siRNA), anti-Bcl2, Bax, p53, His-probe, prohibitin, and 4′,6′-diamidino-2-phenylindole (DAPI) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Cisplatin and 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate (CHAPS) were obtained from Sigma (St. Louis, MO). The mito-L-p53/pEX-3B construct was obtained from Daiqin Lao (University of Florida). pSilencer 2.1-U6 hygro vector was purchased from Ambion (Austin, TX). Proteinase K was purchased from Invitrogen (Carlsbad, CA). The Bcl2/BH3 domain-specific antibody was obtained from Abgent (San Diego, CA). Purified recombinant p53 protein was purchased from Active Motif (Carlsbad, CA). Purified recombinant wild-type (WT) Bcl2 protein was obtained from Protein X Lab (San Diego, CA). All reagents used were obtained from commercial sources unless otherwise stated.
Plasmids, cDNA, cell lines, and stable transfections.
The phospho-mimetic and nonphosphorylatable Bcl2 mutants were created as described previously (9). For generation of Δ32-68, Δ69-87, Δ32-87, and Δ32-80 Bcl2 loop deletion mutants, the 5′-phosphorylated mutagenic primers for various precise deletion mutants were synthesized as follow: Δ32-68, 5′-CAT TAT AAG CTG TCA CAG AGG GGC TAC GAG TGG GAT ACG TCT CCT CTC AGG CCC CTC GTT GCC ACC GCT GGG-3′; Δ69-87, 5′-GTG CAC CGG GAC ATG GCT GCC AGG CCT GTG CCA CCT GTG GTC CAT CTG-3′; Δ32-87, 5′-CTG TCA CAG AGG GGC TAC GAG TGG GAT CCT GTG CCA CCT GTG GTC CAT CTG ACC-3′; Δ32-80, 5′-CAT TAT AAG CTG TCA CAG AGG GGC TAC GAG TGG GAT CCT GCG CTC AGC CCT GTG CCA CCT GTG GTC CAT CTG-3′. The WT Bcl2/pUC19 construct was used as the target plasmid, which contains a unique NdeI restriction site for selection against the unmutated plasmid. The NdeI selection primer is 5′-GAG TGC ACC ATG GGC GGT GTG AAA-3′. These Bcl2 deletion mutants were created using a site-directed mutagenesis kit (Clontech) according to the manufacturer's instructions. Each single mutant was confirmed by sequencing of the cDNA and was then cloned into the pCIneo (Promega) mammalian expression vector. The pCIneo plasmid containing each Bcl2 mutant cDNA was transfected into IL-3-dependent murine myeloid NSF.H7 or p53-null H1299 cells by electroporation or using Lipofectamine 2000 (Invitrogen). Clones stably expressing WT or mutant Bcl2 were selected in medium containing G418 (0.6 mg/ml). The expression levels of exogenous Bcl2 were compared by Western blot analysis using a Bcl2 antibody. Three separate clones for each mutant expressing similar amounts of exogenous Bcl2 were selected for analysis.
Generation of purified recombinant Bcl2-WT, the phospho-mimetic and nonphosphorylatable FLD-only mutant proteins, or the FLD-only mutant mammalian constructs.
To create various FLD-only (amino acids [aa] 32 to 87) mutants, WT, S70A, and S70E Bcl2 cDNAs in pUC 19 were used as templates. The primers used for PCR were as follows: forward, 5′-AGT GAA TTC CTG ATG GCT GGA GAT GCG GAC GCG-3′; reverse, 5′-CTT GTC GAC TCA GAC CAC AGG TGG CAC AGG-3′. The conditions for PCR were denaturation at 94°C for 2 min, annealing at 58°C for 30 s, and extension at 72°C for 1 min. The samples were run for 30 cycles. PCR products were ligated directly into pCR 2.1-TOPO vector, amplified using TOP10 cells (Invitrogen), and screened by PCR for positive clones. Each FLD-only mutant was confirmed by sequencing of the cDNA. Following digestion with EcoRI and SalI, the cDNA was cloned into pET28a(+) containing a His tag (Novagen) or the pCIneo mammalian expression vector (Promega). The various FLD-only mutants contained in pET28a(+) were transformed into BL21(DE3) RIL-CP bacteria cells from Stratagene (La Jolla, CA). The various His-tagged Bcl2 FLD-only recombinant proteins were purified using a His Bind kit (Novagen) and confirmed by Western blotting using a His antibody.
Preparation of cell lysates.
Cells were washed with 1× phosphate-buffered saline (PBS) and resuspended in either ice-cold 1% CHAPS lysis buffer (1% CHAPS, 50 mM Tris, pH 7.6, 120 mM NaCl, 1 mM EDTA, 1 mM Na3VO4, 50 mM NaF, and 1 mM β-mercaptoethanol) or 0.5% NP-40 EBC lysis buffer (0.5% NP-40, 50 mM Tris, pH 7.6, 120 mM NaCl, 1 mM EDTA, 1 mM Na3VO4, 50 mM NaF, and 1 mM β-mercaptoethanol) with a cocktail of protease inhibitors (Calbiochem). Cells were lysed by sonication and centrifuged at 14,000 × g for 10 min at 4°C. The resulting supernatant was collected as the total cell lysate and used for protein analysis or coimmunoprecipitation as described elsewhere (4, 48).
Metabolic labeling, immunoprecipitation, and Western blot analysis.
Cells were washed with phosphate-free RPMI medium and metabolically labeled with [32P]orthophosphoric acid for 90 min. After agonist or inhibitor addition, cells were washed with ice-cold phosphate-buffered saline and lysed in detergent buffer. Bcl2 was then immunoprecipitated as described previously (11, 25). The samples were subjected to 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to a nitrocellulose membrane, and exposed to Kodak X-Omat film at −80°C. Bcl2 phosphorylation was determined by autoradiography. The same filter was probed by Western blotting using a Bcl2 antibody and developed using an ECL kit (Amersham Biosciences) as described previously (11).
Treatment of isolated, intact mitochondria with proteinase K in vitro.
A total of 2 × 107 H7 cells expressing WT Bcl2 were treated with the DNA-damaging agent cisplatin (20 μM) for 24 h. Cells were washed once with cold 1× PBS and resuspended in isotonic mitochondrial buffer (210 mM mannitol, 70 mM sucrose, 1 mM EGTA, 10 mM HEPES, pH 7.5) containing a cocktail of protease inhibitors. The resuspended cells were homogenized with a Polytron homogenizer operating for four bursts of 10 seconds each at a setting of 5 and then centrifuged at 2,000 × g for 3 min to pellet the nuclei and unbroken cells. The supernatant was centrifuged at 13,000 × g for 10 min to pellet mitochondria as described previously (26). The resulting mitochondria were washed twice with mitochondrial buffer. Purified mitochondria were treated with various concentrations of proteinase K (i.e., 2, 5, 25, 50, and 100 μg/ml) for 25 min on ice. Phenylmethylsulfonyl fluoride was then added to a final concentration of 2 mM, and the samples were incubated for another 10 min on ice as described elsewhere (17). Mitochondria were pelleted by centrifugation at 13,000 × g for 10 min and then washed twice with isotonic mitochondrial buffer. The pellets were resuspended in 1% NP-40 lysis buffer and rocked for 60 min prior to centrifugation at 17,530 × g for 10 min at 4°C. Protein (100 μg) from the resulting supernatant was subjected to SDS-PAGE and analyzed by Western blotting using Bcl2, p53, or prohibitin antibodies, respectively.
Cyt c release from isolated, purified mitochondria.
Intact mitochondria were isolated from p53-null human H1299 cells expressing WT or various Bcl2 mutants and incubated with increasing concentrations of purified p53 protein in MSB buffer (400 mM mannitol, 50 mM Tris, pH 7.2, 10 mM KH2PO4, 5 mg/ml bovine serum albumin) containing a cocktail of protease inhibitors at 30°C for 30 min. Samples were centrifuged at 14,000 rpm for 10 min. The resulting supernatant and pelleted mitochondrial fractions were subjected to SDS-PAGE and analyzed by Western blotting using a Cyt c antibody as described previously (27, 33).
Alkali extraction of Bax peripherally associated with mitochondrial membranes.
Mitochondria were isolated by subcellular fractionation and incubated with increasing concentrations of p53 in MSB buffer containing a cocktail of protease inhibitors at 30°C for 30 min. After washing to remove unbound p53, mitochondria were resuspended in freshly prepared 0.1 M Na2CO3, pH 11, and incubated on ice for 30 min to remove “peripherally” associated but not integral membrane proteins (2, 18). The samples were then centrifuged at 200,000 × g for 30 min, and the alkali-extracted membrane pellet was resuspended with 1% NP-40 lysis buffer and rocked for 60 min and then centrifuged at 17,530 × g for 10 min at 4°C. The lysate was subjected to SDS-PAGE. The alkali-resistant Bax (i.e., nonextractable from or integral to the mitochondrial membranes) was determined by Western blotting using a Bax antibody as described elsewhere (18).
Immunofluorescence.
Cells were grown in a Lab-Tek II chambered slide (Nunc) until they were 80 to 100% confluent. Cells were incubated with prewarmed (37°C) growth medium containing MitoTracker (Molecular Probes, Invitrogen) for 30 min. Cells were then washed with 1× PBS, fixed, permeabilized with ice-cold methanol and acetone, and blocked with 10% rabbit serum. Cells were stained with DAPI or various primary antibodies and fluorescein isothiocyanate (FITC)-conjugated secondary antibody. Cells were washed with 1× PBS and observed under a fluorescence microscope (Zeiss). Pictures were taken and colored with the same exposure setting for each experiment.
Depletion of p53 by RNA interference (RNAi) from H7 cells.
Mouse p53 siRNA (Santa Cruz Biotechnology, Santa Cruz, CA) was transfected into H7 cells expressing WT Bcl2 by using Lipofectamine 2000 according to the manufacturer's instructions. A control siRNA (nonhomologous to any known gene sequence) was employed as a negative control. The levels of p53 expression in the absence or presence of cisplatin were determined by Western blotting using a p53 antibody. Three independent experiments were conducted for specific silencing of the targeted p53 gene.
Vector-based gene silencing of Bax by RNAi.
The human Bax DNA target sequence used for siRNA design is AACTGATCAGAACCATCATGG as determined with Ambion's siRNA target finder. A Bax-specific hairpin siRNA insert (sense-loop-antisense) was determined using a computerized insert design tool based on a target sequence following instructions from Ambion's website. The oligonucleotide encoding the Bax-specific hairpin insert or control hairpin insert (nonhomologous to any known gene sequence) was synthesized and ligated into pSilencer 2.1-U6 hygro vector from Ambion (Austin, TX). The pSilencer 2.1-U6 hygro plasmids bearing Bax hairpin or control hairpin were transfected into H1299 cells using Lipofectamine 2000 according to the manufacturer's instructions. Stable clones persistently expressing Bax siRNA were selected in a medium containing hygromycin (0.8 mg/ml), and Bax expression was analyzed by Western blotting using a Bax antibody.
Cell viability assay.
Apoptotic and viable cells were detected using an ApoAlert annexin-V kit (Clontech) according to the manufacturer's instructions. The percentage of annexin-Vlow (i.e., viable) or annexin-Vhigh (i.e., apoptotic) cells was determined using the data obtained by fluorescence-activated cell sorter analysis as described previously (12). Cell viability was confirmed using the trypan blue dye exclusion method (25).
RESULTS
DNA damage enhances Bcl2-p53 binding, reduces Bcl2-Bax association, and promotes apoptotic cell death.
Recent reports indicate that p53 has an unexpected extranuclear apoptotic effect as the result of targeting mitochondria and stimulating Bax-dependent mitochondrial dysfunction in the absence of new protein synthesis (8, 31, 32). However, the mechanism(s) remains unknown. Bax is a proapoptotic member of the Bcl2 family that is required for mitochondrial p53-induced cell death (8). One popular model holds that the noncovalent heterodimerization of Bcl2 and Bax serves to quench Bax's apoptotic function, potentially by inhibiting a proapoptotic BH3-only family member from activating Bax (5, 29, 38). Therefore, p53 may disrupt the Bcl2/Bax complex to facilitate Bax activation. To test whether a DNA damage-induced p53-Bcl2 interaction could influence Bcl2/Bax heterodimerization in vivo, H7 cells expressing WT Bcl2, p53, and Bax were treated with cisplatin (10 μM) for various times as indicated. Bcl2/Bax and Bcl2/p53 complexes were immunoprecipitated using a Bcl2 or a p53 antibody from cell lysates disrupted in 1% CHAPS lysis buffer, since a previous report indicated that the nonionic detergent NP-40 may alter Bax conformation and potentially affect the Bcl2-Bax interaction (23). Results indicate that cisplatin up-regulates and stimulates a population of p53 to translocate to mitochondria (i.e., up to 40%) in association with increased formation of a Bcl2/p53 complex, decreased Bcl2-Bax binding, and cell death (Fig. 1). To verify purity of the subcellular fractions obtained, fraction-specific proteins were assessed by probing the same filters. Prohibitin, an exclusively mitochondrial protein (24), was detected only in the mitochondrial fraction, while proliferating cell nuclear antigen (PCNA), a nuclear marker (33), was detected exclusively in the nuclear fraction (N) (Fig. 1A). These data reveal that the fractionation procedure does not cause cross-contamination between the fractions. These findings suggest that increased Bcl2-p53 binding is associated with decreased Bcl2/Bax heterodimers, in effect, to liberate Bax from Bcl2 with potential activation of its proapoptotic activity. Parenthetically, similar results were obtained when cells were disrupted in 0.5% NP-40 lysis buffer (data not shown). Thus, the ratio of Bcl2/Bax and Bcl2/p53 complexes may be predictive of cell fate (i.e., survival or death).
FIG. 1.

Treatment of cells with cisplatin results in increased Bcl2-p53 binding and decreased Bcl2-Bax association and promotes apoptotic cell death. (A) H7 myeloid cells expressing p53 and Bcl2 were treated with cisplatin (10 μM) for 48 h. Subcellular fractionation was performed to isolate nuclear (N) and mitochondrial (M) fractions. p53 was analyzed by Western blotting using a p53 antibody. The purity of fractions was confirmed by assessing localization of fraction-specific proteins, including prohibitin (a mitochondrial marker) and PCNA (a nuclear marker). (B) H7 cells expressing WT Bcl2 were treated with cisplatin (10 μM) for various times followed by lysis in 1% CHAPS-containing buffer as described in Materials and Methods. Coimmunoprecipitation was performed using a Bcl2 or p53 antibody, respectively. p53, Bcl2, or Bax was then analyzed by Western blotting using a p53, Bcl2, or Bax antibody as indicated. (C) H7 cells expressing WT Bcl2 were treated with cisplatin (10 μM) for various times. Cell viability was determined by analyzing annexin-V binding on a fluorescence-activated cell sorter. Data represent the mean ± standard deviation of three separate determinations. (D) Mitochondria obtained from H7 cells expressing WT Bcl2 were incubated with increasing concentrations of proteinase K for 25 min on ice as described in Materials and Methods. Mitochondria were washed twice with isotonic mitochondria buffer. A 100-μg aliquot of mitochondrial protein was subjected to SDS-PAGE and analyzed by Western blotting using a Bcl2 or p53 antibody. Prohibitin is used as a marker for the inner mitochondrial membrane.
To identify where Bcl2 and p53 interact in the mitochondria (i.e., outer or inner mitochondrial membranes), studies were conducted using intact mitochondria isolated from H7 cells expressing WT Bcl2 that were treated with cisplatin. Proteinase K treatment selectively removes proteins from the outer but not inner mitochondrial membranes (14, 17). Results indicate that the majority of Bcl2 and p53 is rapidly removed even at the lowest concentration of proteinase K (Fig. 1D). By contrast, prohibitin, an inner mitochondrial marker (24), is not affected even at the highest concentration. Therefore, we can conclude that Bcl2 and p53 colocalize and/or interact on the outer mitochondrial membranes.
Next, to assess the percentage of cisplatin-induced cell death that may result from p53, an RNAi approach was employed. H7 cells expressing WT Bcl2 and p53 were transfected with p53 siRNA or control siRNA using Lipofectamine 2000. After 48 h, cells were treated with cisplatin (10 μM) for up to72 h. Results showed that the p53 siRNA efficiently and specifically “knocked down” p53 expression by more than 95% as assessed by Western blotting in either the absence or presence of cisplatin, while control siRNA had no effect (Fig. 2A). Importantly, the knockdown of p53 expression significantly enhanced the percentage of viable cells following cisplatin treatment (84% versus 40%) (Fig. 2B), indicating that approximately 44% of cisplatin-induced total apoptotic cells (60%) result from p53 under these experimental conditions.
FIG. 2.

Depletion of p53 by RNAi prolongs cell survival following cisplatin treatment. (A and B) p53 siRNA or control siRNA was transfected into WT Bcl2-expressing H7 cells using Lipofectamine 2000. Cells were treated with cisplatin (10 μM) for 72 h, and the levels of p53 expression were analyzed by Western blotting using a p53 antibody. Cell viability was assessed as described in the legend for Fig. 1C. Data represent the mean ± standard deviation of three separate experiments.
p53 disrupts the Bcl2/Bax complex.
To test whether p53 binding to Bcl2 may affect the Bcl2-Bax interaction, p53-null H1299 cells expressing high levels of endogenous Bax were stably transfected with WT Bcl2. The Bcl2/Bax complex was immunoprecipitated using an agarose-conjugated Bcl2 antibody. The immune complex was incubated with purified, recombinant p53 at 4°C for up to 2 h, and proteins released from the complex were identified in the supernatant following centrifugation at 14,000 × g for 5 min. Bcl2-associated p53 (i.e., bound p53), Bcl2-associated Bax (i.e., bound Bax), unbound Bax (i.e., present in the supernatant), and total Bcl2 were analyzed by Western blotting using a p53, Bax, or Bcl2 antibody. Results revealed that p53 can directly disrupt Bcl2/Bax in vitro, since increased levels of Bax were present in the supernatant (Fig. 3).
FIG. 3.

p53 directly disrupts the Bcl2-Bax interaction in vitro. The Bcl2/Bax complex was coimmunoprecipitated from H1299 p53-null cells expressing WT Bcl2 and Bax using an agarose-conjugated Bcl2 antibody and incubated with purified WT p53 (50 ng) at 4°C for 2 h. The samples were centrifuged at 14,000 × g for 5 min. The resulting supernatant and immuno-complex beads were subjected to SDS-PAGE. Bcl2, p53, and Bax that bound to Bcl2 or nonbound Bax present in the supernatant were then analyzed by Western blotting using a Bcl2, p53, or Bax antibody.
Mono- and multisite phospho-mimetic Bcl2 mutants display increased Bax but decreased p53 binding in association with enhanced antiapoptotic activity.
We previously discovered that mono- or multisite phosphorylation of Bcl2 in the FLD enhances its antiapoptotic function, at least in part, by stabilizing the Bcl2-Bax interaction (9). To test whether Bcl2 phosphorylation may affect the association with p53, we tested compound Bcl2 phospho-mimetic and nonphosphorylatable mutants. Results from H7 cells expressing equivalent amounts of the T69E/S70A/S87A (EAA), T69A/S70E/S87A (AEA), T69A/S70A/S87E (AAE), AAA, and EEE mutants were compared. Expression of WT or various Bcl2 mutants does not affect the expression of Bax or p53 in the presence or absence of cisplatin (Fig. 4A and data not shown). However, coimmunoprecipitation indicates that the AEA, AAE, and EEE phospho-mimetic Bcl2 mutants display enhanced interaction with Bax and reduced p53 binding compared with the nonphosphorylatable AAA Bcl2 following treatment of cells with cisplatin. Importantly, this inverse binding relationship is reciprocally associated with increased cell survival (Fig. 4). These data reveal that Bcl2's antiapoptotic function will vary dependent upon its ability to bind Bax or p53 in a mechanism regulated by Bcl2 phosphorylation.
FIG. 4.

Mono- and multisite phosphorylation of Bcl2 enhance the Bcl2-Bax association and reduce Bcl2-p53 binding. (A) WT, EAA, AEA, AAE, AAA, and EEE Bcl2 mutants were stably transfected into H7 cells. Coimmunoprecipitation was performed following treatment with cisplatin (10 μM) for 48 h using a Bcl2 antibody. Total Bcl2, Bax, and p53 as well as Bcl2-associated Bax (bound Bax) and Bcl2-associated p53 (bound p53) were analyzed by Western blotting using a Bcl2, Bax, or p53 antibody. (B) Cells expressing WT or various Bcl2 mutants were treated with cisplatin (10 μM) for various times, and cell viability was determined by analyzing annexin-V binding on a fluorescence-activated cell sorter. Data represent the mean ± standard deviation of three separate experiments.
Mono- and multisite phospho-mimetic Bcl2 mutants directly inhibit mitochondrially targeted p53-induced Cyt c release and apoptosis.
To test whether Bcl2 phosphorylation can affect mitochondrial p53-induced apoptosis, a mito-L-p53/pEX-3B construct containing a mitochondrial targeting sequence was employed to selectively target p53 to mitochondria as described elsewhere (47). The mito-L-p53/pEX-3B plasmid was transfected into p53-null human H1299 cells that do not express endogenous p53 and stably express various Bcl2 mutants and endogenous Bax. Expression of mitochondrial p53 and Bcl2 and cell viability were assessed at 72 h after transfection. Results demonstrated that transfection of the mito-L-p53/pEX-3B construct into H1299 cells results in p53 expression exclusively targeted to mitochondrial membranes. This is characterized by punctuate cytoplasmic p53 staining (green) that colocalizes with MitoTracker staining (red) and appears yellow when merged (Fig. 5A). Intriguingly, mitochondrial targeting of p53 is sufficient to induce apoptosis of these cells in the absence of a genotoxic stress (Fig. 5B and C). This indicates that extranuclear p53 does not require nuclear p53 to kill cells. Furthermore, expression of either mono- or multisite phospho-mimetic Bcl2 mutants but not the nonphosphorylatable AAA Bcl2 more potently inhibits mitochondrially targeted p53-induced apoptosis compared to WT Bcl2 (Fig. 5B and C). Since purified p53 can directly induce Cyt c release from isolated mitochondria (33), we tested whether Bcl2 phosphorylation may also play a role. Mitochondria isolated from p53-null H1299 cells expressing WT, the nonphosphorylatable AAA, or phospho-mimetic EEE Bcl2 were incubated with increasing concentrations of purified recombinant p53. Cyt c release into the supernatant was determined as described in Materials and Methods. In contrast to the WT, expression of the EEE but not the AAA mutant Bcl2 more potently blocks p53-induced Cyt c release from mitochondria (Fig. 5D, E, and F).
FIG.5.


Expression of mono- or multisite phospho-mimetic Bcl2 mutants can block p53-induced mitochondrial dysfunction and apoptosis. (A) The mito-L-p53/pEX-3B plasmid or vector-only control was transfected into p53-null H1299 cells using Lipofectamine 2000. After 72 h, the cells were incubated with prewarmed (37°C) growth medium containing MitoTracker (red) for 30 min. Cells were washed with 1× PBS, fixed, permeabilized with ice-cold methanol and acetone, and then blocked with 10% rabbit serum followed by staining with DAPI and FITC-conjugated p53 antibodies. Images were merged using Open-Lab 3.1.5 software to detect areas of colocalization (yellow). (B and C) The mito-L-p53/pEX-3B plasmid was transfected into p53-null H1299 cells expressing WT and various Bcl2 mutants using Lipofectamine 2000. After 72 h, expression of mitochondrial p53 or Bcl2 was analyzed using a p53 or Bcl2 antibody. Cell viability was assessed at 72 h after transfection using annexin-V binding and a fluorescence-activated cell sorter. Data represent the mean ± standard deviation of three separate experiments. (D, E, and F) Intact mitochondria were isolated from p53-null human H1299 cells expressing the WT, the phospho-mimetic EEE, or the nonphosphorylatable AAA Bcl2 mutant. The mitochondria were then incubated with increasing concentrations of purified p53 protein in MSB buffer at 30°C for 30 min. Samples were centrifuged at 14,000 rpm for 10 min. The resulting supernatant and pelleted mitochondrial fractions were subjected to SDS-PAGE and analyzed by Western blotting using a Cyt c or Bcl2 antibody, respectively.
Depletion of Bax by RNA interference blocks p53-induced Cyt c release from isolated mitochondria and prolongs cell survival following DNA damage, and p53 promotes Bax insertion into mitochondrial membranes.
Genetic studies using Bax homozygous knockout mice reveal that Bax (or Bak) is required for inducing apoptotic cell death via the intrinsic pathway (45). To test whether Bax is also essential for DNA damage-induced and p53-mediated mitochondrial apoptotic cell death, a vector-based stable gene silencing approach was employed to specifically deplete Bax from p53-null H1299 human lung cancer cells. Since H1299 cells express high levels of endogenous Bax but very low to nondetectable levels of Bak (Fig. 6A), this suggests that Bax (but not Bak) may play the major role in mitochondrial p53-induced apoptotic cell death in these cells. The pSilencer 2.1-U6 hygro plasmid containing a Bax hairpin insert was used to transfect cells, and stable clones producing Bax siRNA were selected in medium containing hygromycin. Clones expressing Bax siRNA but not control siRNA were found to have more than 95% reduction in Bax and were significantly more resistant to apoptosis following treatment with cisplatin (Fig. 6A and B).
FIG. 6.

Depletion of Bax by RNA interference blocks p53-induced Cyt c release from isolated mitochondria, and p53 can promote Bax insertion into mitochondrial membranes. (A) The pSilencer 2.1-U6 hygro plasmids bearing the Bax hairpin insert or a control hairpin insert were transfected into H1299 p53-null cells using Lipofectamine 2000. The levels of Bax expression were analyzed by Western blotting using a Bax antibody. (B) H1299 cells expressing Bax siRNA or control siRNA were treated with cisplatin (20 μM) for 48 h. Cell viability was assessed as described in the legend for Fig. 1C. (C) Intact mitochondria were isolated from H1299 cells expressing Bax siRNA or vector control and incubated with increasing concentrations of purified p53 protein in MSB buffer at 30°C for 30 min. Alkali extraction of Bax using 0.1 M Na2CO3 (pH 11.5) was performed as described in Materials and Methods. The alkali-resistant Bax (i.e., nonextractable) was determined by Western blotting using a Bax antibody. (D) Intact mitochondria were isolated from p53-null human H1299 cells expressing Bax siRNA or vector control and incubated with increasing concentrations of purified p53 protein in MSB buffer at 30°C for 30 min. Cyt c release was analyzed as described in the legend for Fig. 5D.
Bax is not only located in the cytosol but also is peripherally associated with the outer mitochondrial membranes in unstimulated cells (18). The stress of a death signal then results in the “translocation” of these nonintegral Bax molecules from the cytosol and/or peripheral location to the mitochondria and insertion into the outer mitochondrial membranes, which induces mitochondrial dysfunction (18, 45). Since the isolated mitochondria contain Bax (Fig. 6C), this indicates that Bax is “peripherally” associated with mitochondrial membranes in H1299 cells during nonstressed growth. To test whether p53 targeting of mitochondria can promote peripherally associated Bax to insert into the mitochondrial membranes, an alkali extraction technique was employed. Alkali extraction of isolated mitochondria will strip any “peripherally” associated proteins from membranes but not affect the integral associated proteins (2, 18). Isolated mitochondria were incubated with purified p53 as described above, washed to remove unbound p53, and then incubated in 0.1 M Na2CO3, pH 11.5, on ice for 30 min. The preparation was centrifuged at 200,000 × g to yield a mitochondrial pellet containing membranes stripped of peripheral proteins. The resulting alkali-extracted mitochondrial membrane pellets were resuspended in 1% NP-40 lysis buffer, and the alkali-resistant Bax (i.e., integral to membranes) was analyzed by Western blotting using a Bax antibody. Results revealed that Bax is peripherally associated with mitochondria isolated from unstimulated cells, since it is completely extracted by alkali (Fig. 6C, lane 1). By contrast, prohibitin, an integral mitochondrial protein (24), was not affected by this treatment (Fig. 6C). Importantly for this mechanism, results indicate that Bax becomes alkali unextractable after incubation with purified recombinant p53 (Fig. 6C, lanes 2 to 4). These findings reveal that p53 can facilitate Bax insertion into the outer mitochondrial membranes. Intriguingly, specific knockdown of Bax expression by RNAi suppresses p53-induced Bax insertion (Fig. 6C, lanes 6 to 8). To assess whether depletion of Bax affects p53-induced Cyt c release, intact mitochondria were isolated from H1299 cells expressing either Bax siRNA or control siRNA and incubated with increasing concentrations of recombinant p53. Results revealed that depletion of Bax potently blocks p53-induced Cyt c release from isolated mitochondria (Fig. 6D). These results suggest that Bax may be required for p53 to induce Bax insertion into mitochondrial membranes as well as Cyt c release.
p53 directly binds to Bcl2's FLD.
The Bcl2 FLD (aa 32 to 87) contains the known phosphorylation sites (i.e., T69, S70, and S87) and a caspase cleavage site (Asp34) that can regulate Bcl2's function (3, 6, 9). Interestingly, deletion of aa 32 to 80 of the FLD, which retains the S87 phosphorylation site, was reported to enhance Bcl2's antiapoptotic function, indicating a potential negative regulatory function for this region of the FLD (3). Bcl2 phosphorylation may potentially induce a conformational change that may inhibit the negative function of the FLD (11). We propose that the FLD may contain both a positive regulatory domain that can be phosphorylated at S70, S87, and T69 (i.e., aa 69 to 87) and a negative regulatory domain (i.e., aa 32 to 68) that binds p53. To test this, a series of loop deletion mutants were created, including the Δ69-87, Δ32-68, Δ32-87, and Δ32-80 mutants. The FLD Bcl2 mutants were stably transfected into IL-3-dependent H7 cells, and clones expressing quantitatively similar levels of the Bcl2 mutants were selected and tested for their sensitivity to killing by cisplatin. Results indicated that following DNA damage and p53 activation, cells expressing Bcl2 mutants that do not contain a phosphorylation site (i.e., Δ69-87 and Δ32-87) have markedly reduced survival compared to WT Bcl2. By contrast, cells expressing the Δ32-68 or Δ32-80 Bcl2 deletion mutants, which retain either all (T69, S70, and S87) or a single (i.e., S87) phosphorylation site but are devoid of the potential D34 caspase site, display increased cell survival compared to WT Bcl2 (Fig. 7). Specifically, the Δ32-68 deletion mutant possesses the most potent antiapoptotic activity, with an order of survival potency for the mutants as follows: Δ32-68 > Δ 32-80 > WT > Δ32-87 > Δ69-87 (Fig. 7). These results indicate that the FLD contains two functional regions with opposite effects on Bcl2's survival activity. One regulatory region (aa 69 to 87) of the FLD enhances and the other (aa 32 to 68) inhibits Bcl2's survival function. Since Bcl2 can serve as the mitochondrial “docking” site for p53, it is possible that p53 may interact with Bcl2 in a mechanism regulated by the FLD. To identify a potential p53 binding site in Bcl2, H7 cells expressing WT, Δ32-68, or Δ69-87 Bcl2 mutants were treated with cisplatin for 48 h. Pull-down of Bcl2 and p53 was accomplished using an agarose-conjugated p53 antibody. Results demonstrated that p53 is able to associate with both WT and Δ69-87 but not with the Δ32-68 loop mutant (Fig. 8A). Therefore, p53 appears to target the negative regulatory region of the FLD (aa 32 to 68). These findings help explain why deletion of this portion of the FLD (i.e., Δ32-68) can significantly enhance Bcl2's antiapoptotic activity, since p53 binding to Bcl2 is lost (Fig. 7 and 8A). Importantly, deletion of the p53 binding site from Bcl2 (i.e., aa 32 to 68) does not affect Bcl2-Bax binding, while removal of aa 69 to 87, which contains the phosphorylation sites, markedly dampens the Bcl2-Bax interaction (Fig. 8B). In addition, Bryostatin-1 (Bryo), a potent protein kinase C activator, can still induce phosphorylation of the Δ32-68 but not the Δ69-87 Bcl2 mutant, indicating that removal of the p53 binding site contained in aa 32 to 68 fails to affect its phosphorylation capacity (Fig. 8C). Importantly, Bryo can prolong survival of cells expressing WT or the Δ32-68 mutant but not those cells expressing the Δ69-87 Bcl2 mutant when exposed to cisplatin (Fig. 8D). Also, expression of the Δ32-68 but not the Δ69-87 Bcl2 mutant can more potently block p53-induced Bax insertion into mitochondrial membranes, Cyt c release, and mitochondrial p53-induced apoptosis compared to WT Bcl2 (Fig. 8E to H). These findings indicate that DNA damage-induced binding of p53 to Bcl2's negative regulatory region in the FLD can function by inhibiting Bcl2's antiapoptotic function. However, this inhibition can be overcome by Bcl2 phosphorylation, since phospho-mimetic Bcl2 blocks this interaction and preserves its antiapoptotic activity (Fig. 4).
FIG. 7.

Effects of expression of various Bcl2 loop deletion mutants on its survival function. (A) Schematic representation of phosphorylation sites in the Bcl2 flexible loop domain. (B) WT and various loop deletion Bcl2 mutant cDNAs were stably transfected into H7 cells. Bcl2 protein expression levels were determined by Western blotting using a Bcl2 antibody. (C) Cells expressing WT or various loop deletion Bcl2 mutants were treated with cisplatin (20 μM) for 48 h. Cell viability was determined by analyzing annexin-V binding on a fluorescence-activated cell sorter. Data represent the mean ± standard deviation of three determinations.
FIG.8.
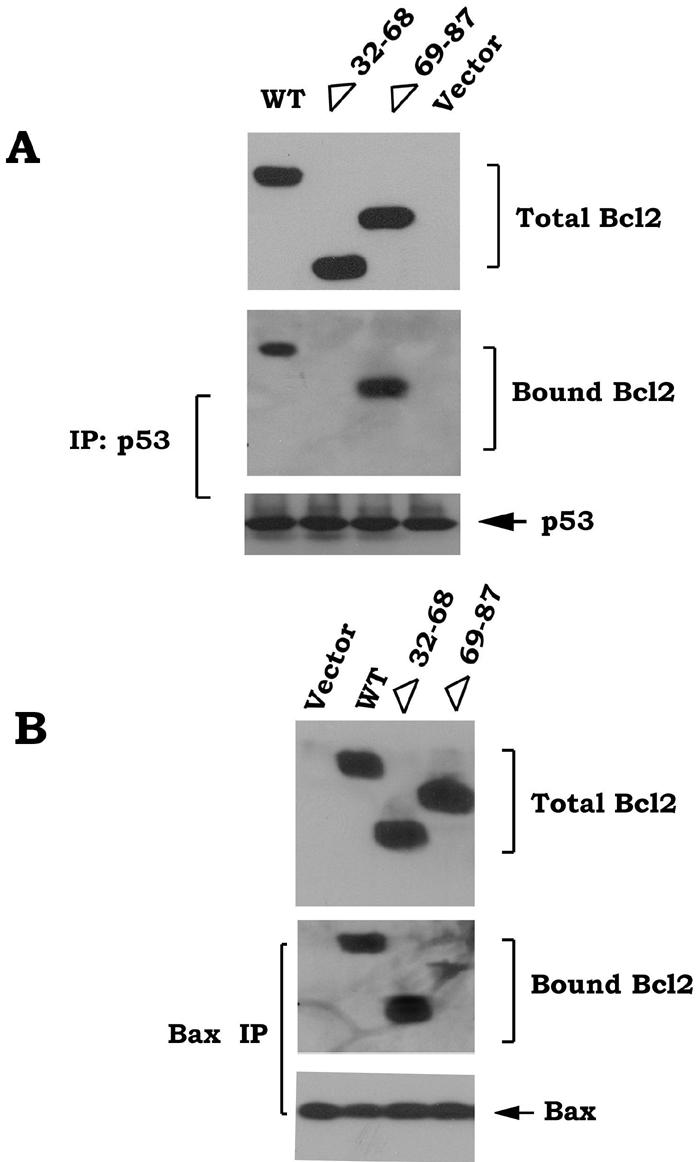


p53 interacts with Bcl2's negative regulatory region in the FLD. (A and B) H7 cells expressing WT or Bcl2 deletion mutants (i.e., Δ32-68 or Δ69-87) were treated with cisplatin (10 μM) for 48 h. Coimmunoprecipitation was carried out using an agarose-conjugated p53 or Bax antibody. Bcl2, p53, Bax, and bound Bcl2 were then analyzed by Western blotting. (C) H7 cells expressing WT or Bcl2 deletion mutants (i.e., Δ32-68 or Δ69-87) were metabolically labeled with [32P]orthophosphoric acid and treated with Bryo for 30 min. Bcl2 was immunoprecipitated using an agarose-conjugated Bcl2 antibody. Phosphorylation of Bcl2 was determined by autoradiography (upper). Western blot analysis was performed to confirm and quantify Bcl2 protein (lower). (D) H7 cells expressing WT or Bcl2 deletion mutants (i.e., Δ32-68 or Δ69-87) were treated with cisplatin in the absence or presence of Bryo for 48 h. Cell viability was analyzed by detecting annexin-V binding by using a fluorescence-activated cell sorter. Data represent the mean ± standard deviation of three separate experiments. (E) Intact mitochondria were isolated from p53-null human H1299 cells expressing the WT or Δ32-68 or Δ69-87 Bcl2 mutants and incubated with increasing concentrations of purified p53 protein in MSB buffer at 30°C for 30 min. Alkali extraction of Bax using 0.1 M Na2CO3 (pH 11.5) was performed as described in Materials and Methods. Alkali-resistant Bax (i.e., nonextractable) was determined by Western blotting using a Bax antibody. (F) Intact mitochondria were isolated from p53-null human H1299 cells expressing the WT or Δ32-68 or Δ69-87 Bcl2 mutants and incubated with increasing concentrations of purified p53 protein in MSB buffer at 30°C for 30 min. Cyt c release was analyzed using a Cyt c antibody. (G and H) The mito-L-p53/pEX-3B plasmid was transfected into H1299 p53-null cells expressing various Bcl2 mutants using Lipofectamine 2000. After 72 h, expression of mito-p53 was analyzed by Western blotting using a p53 antibody. Cell viability was also assessed using annexin-V binding and a fluorescence-activated cell sorter. Data represent the mean ± standard deviation of three separate experiments.
A purified, full-length Bcl2 loop-only protein (aa 32 to 87) directly binds to p53 and inhibits the Bcl2-p53 interaction in vitro.
Our data provide strong evidence that p53 potentially binds to the FLD of Bcl2 following DNA damage (Fig. 8A). To characterize the mechanism responsible for regulating p53 binding to Bcl2's FLD, various purified, recombinant, full-length His-tagged “FLD-only” (i.e., aa 32 to 87) proteins, including an unmodified (WT), a nonphosphorylatable (S70A), and a phospho-mimetic (S70E) FLD mutant, were produced, purified, and used in an in vitro binding pull-down assay. Results revealed that “FLD-only” proteins can directly interact with p53 and that the S70A but not the S70E “FLD-only” protein more efficiently binds to purified p53 (Fig. 9A). These findings help to explain the observation that phosphorylation of Bcl2 impairs p53 binding (Fig. 4). To further test which “FLD-only” protein may affect the p53-Bcl2 interaction, purified Bcl2 was incubated with purified p53 in the absence or presence of increasing concentrations of WT, S70A, or S70E FLD-only proteins. p53-associated Bcl2 (i.e., bound Bcl2) was pulled down with an agarose-conjugated p53 antibody. Bound Bcl2 or p53 was assessed by Western blotting. Results revealed that the nonphosphorylatable S70A but not the phospho-mimetic S70E FLD-only protein can more potently inhibit the Bcl2-p53 interaction compared to the WT FLD-only protein in a competitive binding mechanism (Fig. 9B). These findings support the notion that physiologically phosphorylated Bcl2 at S70 is capable of disrupting/blocking Bcl2-p53 binding. The results also help explain how the positive regulatory region (i.e., aa 69 to 87) of the FLD can affect the activity of the negative regulatory region (i.e., aa 32 to 68).
FIG. 9.

A purified Bcl2 full-length FLD-only protein (aa 32 to 87) can directly bind to p53 and competitively inhibit the Bcl2-p53 interaction. (A) His-tagged Bcl2 loop-only (aa32-87) proteins including WT, S70A, and S70E were produced and purified. Purified p53 was incubated with His-tagged WT, S70A, or S70E Bcl2 FLD-only proteins at 4°C for 2 h. Coimmunoprecipitation was carried out using an agarose-conjugated p53 antibody. The p53-associated Bcl2 FLD and p53 were analyzed by Western blotting using an anti-His probe or p53 antibody, respectively. (B) Purified p53 was incubated with full-length WT Bcl2 in the absence or presence of increasing concentrations of WT, S70A, or S70E FLD-only proteins, and coimmunoprecipitation was carried out using an agarose-conjugated p53 antibody as above. The p53-associated Bcl2 (bound Bcl2) and p53 were analyzed by Western blotting using a Bcl2 or p53 antibody, respectively. (C) Purified p53 was incubated with His-tagged aa32-87, aa32-68, or aa69-87 Bcl2 FLD-only peptides in 1% CHAPS lysis buffer at 4°C for 2 h. Coimmunoprecipitation was carried out using an anti-His antibody. The FLD-associated p53 and various His-tagged Bcl2 FLD-only proteins were analyzed by Western blotting using an anti-p53 or anti-His antibody.
Since deletion of aa 32 to 68 in the FLD results in loss of Bcl2's ability to associate with p53 (Fig. 8A), this suggests that the aa32-68 region of the FLD may be a potential p53 binding site. To directly test this, purified p53 was incubated with purified recombinant His-tagged aa32-87, aa32-68, or aa69-87 FLD peptides in 1% CHAPS lysis buffer at 4°C for 2 h. Coimmunoprecipitation was carried out using an anti-His antibody. Results indicated that the aa32-87 or aa32-68 but not the aa69-87 FLD peptide can directly bind p53 (Fig. 9C). These findings provide strong, direct evidence that aa 32 to 68 in Bcl2's FLD is the p53 binding site.
Expression of the Bcl2 FLD-only peptide in cells suppresses cisplatin-induced Bcl2-p53 binding in association with increased cell survival.
Our data show that the Bcl2 FLD-only peptide can directly bind to p53 and competitively suppress the Bcl2-p53 interaction in vitro (Fig. 9). To test a functional role of the FLD peptide in vivo, the “FLD-only” (i.e., aa 32 to 87)/pCIneo mammalian constructs, including unmodified (WT), nonphosphorylatable (S70A), and phospho-mimetic (S70E) FLD mutants, were created. The constructs were transfected into H7 cells expressing WT Bcl2. After transfection, cells were treated with cisplatin (10 μM) for up to72 h. The Bcl2/p53 complex was immunoprecipitated using an agarose-conjugated Bcl2 antibody. Results revealed that WT and S70A but not S70E FLD-only mutants potently inhibit cisplatin-stimulated Bcl2-p53 binding and prolong cell survival (Fig. 10). Intriguingly, the S70A FLD mutant is more potent than the WT FLD, a result consistent with the in vitro results reported above (compare Fig. 9B and 10).
FIG. 10.

Expression of Bcl2 FLD-only proteins inhibits cisplatin-induced Bcl2-p53 binding and prolongs cell survival. (A) The full-length Bcl2 FLD-only (aa 32 to 87) or point mutant cDNAs, including nonmutated WT and S70A and S70E mutants, were cloned into the pCIneo mammalian expression vector as described in Materials and Methods. These constructs were transfected into WT Bcl2-expressing H7 cells. After transfection, cells were treated with cisplatin for 72 h and lysed in 1% CHAPS lysis buffer. Coimmunoprecipitation was carried out using an agarose-conjugated Bcl2 antibody as above. Bcl2-associated p53 and total Bcl2 were analyzed by Western blotting using a p53 or Bcl2 antibody. (B) H7 cells expressing WT Bcl2 were transfected with WT, S70A, and S70E FLD-only constructs and treated with cisplatin for 72 h as above. Cell viability was assessed using annexin-V binding by fluorescence-activated cell sorting. Data represent the mean ± standard deviation of three separate experiments.
DNA damage-induced p53-Bcl2 binding, expression of mitochondrially targeted p53, or binding of p53 to Bcl2 in a cell-free system results in a conformational change in Bcl2.
Bax and Bak undergo conformational changes in association with their conversion from a latent to an active proapoptotic protein (20, 36). A recent report indicates that the interaction between the nuclear orphan receptor Nur77/TR3 and Bcl2 can induce a conformational change in Bcl2 that induces the “exposure” of its own BH3 domain (30). To test whether DNA damage-induced p53-Bcl2 binding is also associated with a Bcl2 conformational change, we used Bcl2 antibodies that recognize different Bcl2 epitopes. Antibody binding to Bcl2 was measured by immunofluorescence or immunoprecipitation. It has been reported that the Bcl2 BH3 domain-specific antibody can detect a conformational change in Bcl2's BH3 domain (30). H460 human lung cancer cells expressing endogenous Bcl2 and p53 were treated with cisplatin (20 μM) for various times. Immunoprecipitation of Bcl2 was performed using a Bcl2/BH3 domain-specific or pan-Bcl2 antibody, and Bcl2 was analyzed by Western blotting using a pan-Bcl2 antibody. Results indicated that treatment of cells with cisplatin potently enhances the ability of the Bcl2/BH3 domain-specific antibody to immunoprecipitate Bcl2 compared to the pan-Bcl2 antibody (Fig. 11A). These findings suggest that DNA damage-induced p53-Bcl2 binding can alter the conformation of Bcl2's BH3 domain epitope in association with increased susceptibility of cells to apoptosis. Thus, we propose that DNA damage-induced p53-Bcl2 binding induces a conformational change in Bcl2 which results in impairment of its antiapoptotic function, perhaps by facilitating a BH3-only proapoptotic protein to bind and activate Bax.
FIG. 11.

Treatment of H460 cells with cisplatin or expression of mitochondria-targeted p53 in p53-null H1299 cells results in a conformational change in Bcl2 with exposure of the Bcl2 BH3 domain. (A) Human lung cancer H460 cells expressing WT p53 and Bcl2 were treated with cisplatin (20 μM) for 48 h. Coimmunoprecipitation was carried out using a Bcl2 BH3 or full-length Bcl2 antibody, respectively. Bcl2 was analyzed by Western blotting using a Bcl2 antibody. (B) The mito-L-p53/pEX-3B or vector-only control plasmid was transfected into p53-null H1299 cells expressing WT Bcl2 by using Lipofectamine 2000. Cells were incubated with prewarmed (37°C) growth medium containing MitoTracker (red) for 30 min. Cells were then washed with 1× PBS, fixed, permeabilized with ice-cold methanol and acetone, and then blocked with 10% rabbit serum followed by staining with Bcl2 BH3 domain primary and FITC-conjugated (green) secondary antibodies. Images were merged using Open-Lab 3.1.5 software to detect areas of colocalization (yellow). (C) The mito-L-p53/pEX-3B plasmid was transfected into p53-null H1299 cells expressing WT Bcl2 by using Lipofectamine 2000. Coimmunoprecipitation was carried out with a Bcl2 BH3 or full-length Bcl2 antibody as described above. Bcl2 was analyzed by Western blotting using a Bcl2 antibody. (D) Purified Bcl2 protein was incubated with increasing concentrations of purified p53 in 1% CHAPS lysis buffer at 4°C for 2 h. Coimmunoprecipitation was carried out using a Bcl2 BH3 or full-length Bcl2 antibody, respectively. Bcl2 was analyzed by Western blotting.
To test whether direct targeting of p53 to mitochondria and binding to Bcl2 can also induce a Bcl2 conformational change, the mito-L-p53/pEX-3B construct was transfected into H1299 p53-null cells that express exogenous WT Bcl2. Cells were incubated with prewarmed (37°C) growth medium containing MitoTracker for 30 min, fixed, and permeabilized with ice-cold methanol and acetone. Fixed cells were blocked with 10% rabbit serum and then immunostained with the Bcl2 BH3 domain primary and FITC-conjugated secondary antibodies. While Bcl2 immunofluorescence was low or undetectable in vector-only cells, it increased significantly in cells transfected with the mito-L-p53/pEX-3B construct (Fig. 11B). Furthermore, immunoprecipitation of Bcl2 with the anti-Bcl2/BH3 domain or a pan-Bcl2 antibody confirmed that expression of mito-p53 alters Bcl2's conformation (Fig. 11C) but does not affect the expression level of Bcl2 (Fig. 11C). Collectively then, these findings provide strong evidence that targeting p53 to mitochondria can lead to Bcl2 binding with induction of a conformational change that potentially inactivates Bcl2.
To directly test whether the binding of p53 to Bcl2 can induce this type of Bcl2 conformational change, an in vitro cell-free assay was employed. Purified p53 was added to Bcl2, followed by immunoprecipitation using the anti-Bcl2/BH3 domain antibody. Results indicated that addition of purified p53 can enhance the ability of the Bcl2/BH3 domain-specific antibody to bind Bcl2, and this occurs in a p53 dose-dependent manner (Fig. 11D). These findings would appear to provide direct evidence for the notion that binding of p53 and Bcl2 in a cell-free system changes Bcl2's conformation involving its BH3 domain.
DISCUSSION
p53 not only functions in the nucleus to induce cell cycle arrest and apoptosis following a genotoxic stress but also may have an “extranuclear,” proapoptotic function as a result of binding to Bcl-XL or Bcl2 located in the outer mitochondrial membrane (32, 33) (Fig. 1). Here we found that DNA damage induces a significant population of endogenous p53 (i.e., 40%) to translocate to mitochondria and which directly interacts with Bcl2 in association with reduced Bcl2/Bax heterodimerization and enhanced apoptosis (Fig. 1). The nonionic detergent NP-40 has been reported to cause a conformational change in Bax, which may artifactually influence/promote the Bcl2-Bax interaction (23). However, a recent report demonstrated that Bcl2/Bax complexes may indeed exist at the mitochondria and that mitochondrial Bax could be coimmunoprecipitated with Bcl2 when detergent lysis was carried out with CHAPS (48). To rule out the potential artifactual effect of NP-40 on Bcl2-Bax or Bcl2-p53 binding, we used 1% CHAPS lysis buffer and compared it with 0.5% NP-40 lysis buffer in coimmunoprecipitation experiments. Interestingly, similar results were obtained (Fig. 1B and data not shown).
Mechanistically, DNA damage-enhanced p53-Bcl2 binding appears to disrupt Bcl2-Bax binding and thereby promotes “free” Bax to be potentially activated by a BH3-only proapoptotic protein. Because heterodimerization of Bcl2/Bax is thought to quench Bax's apoptotic function (38), the mechanism(s) involving disruption of the Bcl2/Bax complex by p53 becomes highly relevant. Here we found that p53 can directly disrupt the Bcl2/Bax complex by binding to a novel regulatory region of Bcl2's FLD (i.e., aa 32 to 68) that we have identified as a “negative” regulatory domain in Bcl2. The consequence of this interaction is the release of proapoptotic Bax that is “peripherally” associated with mitochondria to become integrally associated in the OMM, leading to Cyt c release with activation of the cytosolic caspase cascade and apoptosis.
Phosphorylation in the FLD can positively regulate Bcl2's antiapoptotic function, but the mechanism is not clear (9). Since single-site S70 and multisite phospho-mimetic Bcl2 mutants (i.e., AEA and EEE) display more efficient binding to Bax with a decreased capacity to associate with p53, while the nonphosphorylatable AAA Bcl2 mutant has the opposite effect (Fig. 4), this suggests that phosphorylation enhances Bcl2's antiapoptotic function potentially through a mechanism involving its ability to simultaneously associate with Bax and block the p53-Bcl2 interaction. These data extend our previous finding that the phosphorylation-negative Bcl2 mutant S70A associates poorly with Bax following IL-3 withdrawal compared to WT Bcl2 (11). Furthermore, the stability of the Bcl2-Bax association is also significantly reduced when Bcl2 phosphorylation is suppressed by treatment of cells with known Bcl2 kinase inhibitors, either PD98059 or staurosporine (11). While the binding of p53 to Bcl2 may impair Bcl2's survival activity, Bcl2 phosphorylation prevents p53 binding. This is clear from results which demonstrate that expression of the phospho-mimetic Bcl2 mutants can more potently inhibit mitochondrial-targeted p53-induced apoptosis (Fig. 5). It was recently reported that p53 plays a direct apoptogenic role at the mitochondria because purified p53 was able to induce Cyt c release from isolated mitochondria (33). In contrast to the WT, we found that expression of the phospho-mimetic EEE but not the nonphosphorylatable AAA Bcl2 mutant in isolated, intact mitochondria from nonstressed H1299 cells more potently blocks purified p53-induced Cyt c release (Fig. 5). This indicates that phosphorylation of Bcl2 can directly inhibit p53's “extranuclear” apoptogenic role at the mitochondria.
Bax is not only located in the cytosol but also “peripherally” associated with the mitochondrial membranes during normal cell growth. Bax's proapoptotic effect will be revealed when it becomes integrally inserted into the mitochondrial membranes after a death signal, which results in mitochondrial dysfunction and apoptosis (18). Thus, the integral insertion of Bax into the outer mitochondrial membranes is essential for Bax's proapoptotic activity (36). Our findings reveal that treatment of isolated mitochondria from unstimulated H1299 cells that lack endogenous p53 but express high levels of endogenous Bax (but very low levels of Bak) with purified p53 can facilitate mitochondria-associated Bax insertion into mitochondrial membranes, leading to Cyt c release (Fig. 6). This may be a mechanism by which p53 activates Bax and induces mitochondrial dysfunction. Intriguingly, depletion of Bax expression by RNAi potently blocks p53-induced Cyt c release and thereby prolongs cell survival following DNA damage (Fig. 6). This indicates that Bax may be required for p53-induced mitochondrial dysfunction and apoptosis.
It is well known that Bcl2's FLD can regulate its antiapoptotic function. Specifically, the FLD contains multiple phosphorylation sites and a potential Asp34 caspase cleavage site (3, 6, 9). Phosphorylation of Bcl2 at one or more sites in the FLD is reported to enhance Bcl2's antiapoptotic function (9), while caspase-mediated cleavage at D34 has been reported to convert Bcl2 to a proapoptotic molecule (6). Recent studies using nuclear magnetic resonance spectroscopy showed that the p53 binding site on Bcl-XL consists of the carboxy terminus of the first α-helix, the loop between α3 and α4, and the loop between α5 and α6 of Bcl-XL (40). However, the binding site of p53 on Bcl2 remains unclear. Our findings demonstrate that the FLD of Bcl2 can be further divided into a negative regulatory domain (i.e., aa 32 to 68) which binds p53 and contains the D34 caspase cleavage site and a positive regulatory domain (i.e., aa 69 to 87) which contains the phosphorylation sites, including T69, S70, and S87. These distinct regulatory regions of the FLD function in opposition to each other. DNA damage facilitates the direct targeting of p53 to Bcl2 by binding to Bcl2's negative regulatory domain (i.e., aa 32 to 68) of the FLD (Fig. 8 and 9). Deletion of the negative regulatory domain (Δ32-68) results in a Bcl2 mutant that retains its phosphorylation capacity and has enhanced antiapoptotic function, while removal of the positive regulatory domain (Δ69-87) results in a nonphosphorylatable Bcl2 and antiapoptotic activity is reduced (Fig. 7 and 8). Since purified His-tagged aa32-87 and aa32-68 but not aa69-87 FLD peptides can directly bind to p53 in vitro (Fig. 9C), this confirms that the aa32-68 region of the FLD is sufficient to bind p53. Intriguingly, phosphorylation of Bcl2 at one or more sites in the positive regulatory region (i.e., aa 69 to 87) can inhibit p53 binding (Fig. 4). With the discovery of these “yin” and “yang” regulatory regions of the FLD in Bcl2, our findings support the notion that p53-Bcl2 binding is likely a dynamic process regulated by “on-off” binding to p53, depending upon Bcl2's phosphorylation status, which is dynamic (10). In addition to a loss of p53 binding, deletion of aa 32 to 68 from Bcl2 also potently enhances Bcl2's ability to interact with Bax (Fig. 8A and B). This may more efficiently suppress Bax's proapoptotic function by “holding” Bax in an inactive form. Importantly, however, phosphorylation of Bcl2 at one or more sites in the positive regulatory domain of the FLD can prevent p53 from binding to Bcl2 and thereby block direct p53-mediated inactivation of Bcl2 during genotoxic stress. Our findings reveal a novel mechanism by which Bcl2 can regulate p53 binding and cell survival even during a genotoxic stress which leads to p53 up-regulation and translocation to the mitochondria.
We previously discovered that S70 is the physiological phosphorylation site of Bcl2 that is required for Bcl2's antiapoptotic function (25). Our data now show that the FLD-only domain (aa 32 to 87) can directly interact with p53 in vitro and specifically that the S70A protein but not the S70E FLD-only protein more efficiently binds to purified p53 (Fig. 9). Functionally, expression of WT or the S70A but not S70E FLD-only protein significantly enhances cell survival following cisplatin treatment in a mechanism involving inhibition of Bcl2-p53 binding (Fig. 10). These findings indicate a potential mechanism and suggest a target for development of novel antineoplastic therapies that can induce the p53-Bcl2 interaction.
Bcl2's FLD is located between the BH3 (i.e., death) and BH4 (i.e., survival) domains (1, 41). Based on our data, we propose that p53 binding to the negative region of the FLD will alter Bcl2's conformation such that its BH3 domain becomes exposed. The Bcl2 family members are divided into either anti- or proapoptotic molecules (21). Interestingly, the antiapoptotic members can be converted into proapoptotic molecules under certain situations. For example, the orphan nuclear receptor Nur77/TR3 can be translocated from the nucleus to mitochondria in response to specific cell death stimuli, where it can also interact with the FLD of Bcl2 to induce a Bcl2 conformational change that “exposes” its BH3 domain, resulting in conversion to a killer molecule (30). Our findings demonstrate that the potent tumor suppressor p53 directly binds Bcl2 through its negative regulatory region of the FLD and induces a BH3 “conformational” change that results in inactivation of Bcl2's antiapoptotic function (Fig. 11). Either DNA damage-induced p53-Bcl2 binding or expression of mitochondrial-targeted p53 induces this conformational change in Bcl2 which facilitates dissociation of Bcl2/Bax and further favors apoptosis. Thus, p53 may mediate this functional change by acting as an allosteric regulator of Bcl2 to induce a conformational reorganization of its hydrophobic cleft that results in exposure of its BH3 domain. In our model, such a conformational change would disrupt high-affinity Bcl2-Bax binding, which not only inactivates Bcl2 but also may promote Bax's proapoptotic function.
In summary, our results have identified a novel mechanism by which p53 can directly inactivate Bcl2 by binding to a newly recognized negative regulatory domain (aa 32 to 68) of Bcl2's FLD. DNA damage-induced p53 interaction with Bcl2 disrupts Bcl2/Bax heterodimerization and thereby enhances Bax-dependent cell death. p53 apparently binds the negative regulatory region of Bcl2's FLD to induce a conformational change in Bcl2 that leads to exposure of its proapoptotic BH3 domain and subsequent disruption of Bax binding with functional inactivation of Bcl2. Phosphorylation of Bcl2 at one or more sites in the positive regulatory region (aa 69 to 87) of the FLD can block p53 binding and preserve or rescue Bcl2's survival function. Thus, therapeutic manipulation of Bcl2-p53 binding by inhibiting the positive regulatory region or stimulating the function of the negative regulatory region of the Bcl2 FLD may represent a novel antineoplastic approach.
Acknowledgments
This work was supported by National Institutes of Health grants RO1 CA44649 (to W.S.M.) and RO1 CA112183 (to X.D.).
We are grateful to Daiqin Liao (University of Florida) for kindly providing the mito-L-p53/pEX-3B construct.
REFERENCES
- 1.Adams, J., and S. Cory. 1998. The Bcl2 protein family: arbitraters of cell survival. Science 281:1322-1326. [DOI] [PubMed] [Google Scholar]
- 2.Cao, X., X. Deng, and W. S. May. 2003. Cleavage of Bax to p18 Bax accelerates stress-induced apoptosis, and a cathepsin-like protease may rapidly degrade p18 Bax. Blood 102:2605-2614. [DOI] [PubMed] [Google Scholar]
- 3.Chang, B., A. Minn, S. Muchmore, S. Fesik, and C. Thompson. 1997. Identification of a novel regulatory domain in Bcl-XL and Bcl2. EMBO J. 16:968-977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen, G., and E. Lee. 1996. The product of the ATM gene is a 370-kDa nuclear phosphoprotein. J. Biol. Chem. 271:33693-33697. [DOI] [PubMed] [Google Scholar]
- 5.Chen, L., S. Willis, A. Wei, B. Smith, J. Fletcher, M. Hinds, P. Colman, C. Day, J. Adams, and D. Huang. 2005. Differential targeting of prosurvival Bcl2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell 17:393-403. [DOI] [PubMed] [Google Scholar]
- 6.Cheng, E., D. Kirsch, R. Clem, R. Ravi, M. Kastan, A. Bedi, K. Ueno, and J. Hardwick. 1997. Conversion of Bcl-2 to a Bax-like death effector by caspases. Science 278:1966-1968. [DOI] [PubMed] [Google Scholar]
- 7.Chipuk, J., L. Hayes, T. Kuwana, D. Newmeyer, and D. Green. 2005. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science 309:1732-1735. [DOI] [PubMed] [Google Scholar]
- 8.Chipuk, J., T. Kuwana, L. Hayes, N. Droin, D. Newmeyer, M. Schuler, and D. Green. 2004. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 303:1010-1014. [DOI] [PubMed] [Google Scholar]
- 9.Deng, X., F. Gao, T. Flagg, and W. S. May. 2004. Mono- and multi-site phosphorylation enhances Bcl2's anti-apoptotic function and inhibition of cell cycle entry functions. Proc. Natl. Acad. Sci. USA 101:153-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deng, X., T. Ito, B. Carr, M. Mumby, and W. S. May. 1998. Reversible phosphorylation of Bcl2 following interleukin 3 or bryostatin 1 is mediated by direct interaction with protein phosphatase 2A. J. Biol. Chem. 273:34157-34163. [DOI] [PubMed] [Google Scholar]
- 11.Deng, X., P. Ruvolo, B. Carr, and W. S. May. 2000. Survival function of ERK1/2 as IL-3-activated, staurosporine-resistant Bcl2 kinases. Proc. Natl. Acad. Sci. USA 97:1578-1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deng, X., L. Xiao, W. Lang, F. Gao, P. Ruvolo, and W. S. May. 2001. Novel role for JNK as a stress-activated Bcl2 kinase. J. Biol. Chem. 276:23681-23688. [DOI] [PubMed] [Google Scholar]
- 13.Desai, B., B. Myers, and S. Schreiber. 2002. FKBP12-rapamycin-associated protein associates with mitochondria and senses osmotic stress via mitochondrial dysfunction. Proc. Natl. Acad. Sci. USA 99:4319-4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diekert, K., G. Kispal, B. Guiard, and R. Lill. 1999. An internal targeting signal directing proteins into the mitochondrial intermembrane space. Proc. Natl. Acad. Sci. USA 96:11752-11757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dimmeler, S., K. Breitschopf, J. Haendeler, and A. Zeiher. 1999. Dephosphorylation targets Bcl2 for ubiquitin-dependent degradation: a link between the apoptosome and the proteasome pathway. J. Exp. Med. 189:1815-1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dumont, P., J. Leu, A. Pietra III, D. George, and M. Murphy. 2003. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat. Genet. 33:357-365. [DOI] [PubMed] [Google Scholar]
- 17.Gao, S., J. Chen, S. Brodsky, H. Huang, S. Adler, J. Lee, N. Dhadwal, L. Gould, S. Gross, and M. Goligorsky. 2004. Docking of endothelial nitric oxide synthase (eNOS) to the mitochondrial outer membrane. J. Biol. Chem. 279:15968-15974. [DOI] [PubMed] [Google Scholar]
- 18.Goping, I., A. Gross, J. Lavoie, M. Nguyen, R. Jemmerson, K. Roth, S. Korsmeyer, and G. Shore. 1998. Regulated targeting of Bax to mitochondria. J. Cell Biol. 143:207-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Green, D., and G. Evan. 2002. A matter of life and death. Cancer Cell 1:19-30. [DOI] [PubMed] [Google Scholar]
- 20.Griffiths, G., L. Dubrez, C. Morgan, N. Jones, J. Whitehouse, B. Corfe, C. Dive, A. John, and J. Hickman. 1999. Cell damage-induced conformational changes of the proapoptotic protein Bak in vivo precede the onset of apoptosis. J. Cell Biol. 144:903-914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gross, A., J. McDonnell, and J. Korsmeyer. 1999. Bcl2 family members and the mitochondria in apoptosis. Gene Dev. 13:1899-1911. [DOI] [PubMed] [Google Scholar]
- 22.Haupt, Y., S. Rowan, E. Shaulian, K. Vousden, and M. Oren. 1995. Induction of apoptosis in HeLa cells by transactivation-deficient p53. Genes Dev. 9:2170-2183. [DOI] [PubMed] [Google Scholar]
- 23.Hsu, Y., and R. Youle. 1997. Nonionic detergents induce dimerization among members of the Bcl-2 family. J. Biol. Chem. 272:13829-13834. [DOI] [PubMed] [Google Scholar]
- 24.Ikonen, E., K. Fiedler, R. Parton, and K. Simons. 1995. Prohibitin, an antiproliferative protein, is localized to mitochondria. FEBS Lett. 358:273-277. [DOI] [PubMed] [Google Scholar]
- 25.Ito, T., X. Deng, B. Carr, and W. S. May. 1997. Bcl-2 phosphorylation required for anti-apoptosis function. J. Biol. Chem. 272:11671-11673. [DOI] [PubMed] [Google Scholar]
- 26.Jin, Z., F. Gao, T. Flagg, and X. Deng. 2004. Tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone promotes functional cooperation of Bcl2 and c-Myc through phosphorylation in regulating cell survival and proliferation. J. Biol. Chem. 279:40209-40219. [DOI] [PubMed] [Google Scholar]
- 27.Jürgensmeier, J. M., Z. Xie, Q. Deveraux, L. Ellerby, D. Bredesen, and J. C. Reed. 1998. Bax directly induces release of cytochrome c from isolated mitochondria. Proc. Natl. Acad. Sci. USA 95:4997-5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kelekar, A., and C. Thompson. 1998. Bcl2-family proteins: the role of the BH3 domain in apoptosis. Trends Cell Biol. 8:324-330. [DOI] [PubMed] [Google Scholar]
- 29.Kuwana, T., L. Bouchier-Hayes, J. Chipuk, C. Bonzon, B. Sullivan, D. Green, and D. Newmeyer. 2005. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell 17:525-535. [DOI] [PubMed] [Google Scholar]
- 30.Lin, B., S. Kumar, F. Lin, W. Liu, Y. Han, X. Cao, M. Dawson, J. Reed, and X. Zhang. 2004. Conversion of Bcl2 from protector to killer by interaction with nuclear orphan receptor Nur77/TR3. Cell 116:527-540. [DOI] [PubMed] [Google Scholar]
- 31.Manfredi, J. J. 2003. p53 and apoptosis: it's not just in the nucleus anymore. Mol. Cell 11:552-554. [DOI] [PubMed] [Google Scholar]
- 32.Marchenko, N., A. Zaika, and U. M. Moll. 2000. Death signal-induced localization of p53 protein to mitochondria. J. Biol. Chem. 275:16202-16212. [DOI] [PubMed] [Google Scholar]
- 33.Mihara, M., S. Erster, A. Zaika, O. Petrenko, T. Chittenden, P. Pancoska, and U. M. Moll. 2003. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell 11:577-590. [DOI] [PubMed] [Google Scholar]
- 34.Miyashita, T., and J. C. Reed. 1995. Tumor suppressor p53 is a direct transcriptional activator of the human Bax gene. Cell 80:293-299. [DOI] [PubMed] [Google Scholar]
- 35.Nakano, K., and K. Vousden. 2001. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 7:683-694. [DOI] [PubMed] [Google Scholar]
- 36.Nechushtan, A., C. L. Smith, Y. Hsu, and R. J. Youle. 1999. Confirmation of the Bax C-terminus regulates subcellular location and cell death. EMBO J. 18:2330-2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oda, E., R. Ohki, H. Murasawa, J. Nemoto, T. Shibue, T. Yamashita, T. Tokino, T. Taniguchi, and N. Tanaka. 2000. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288:1053-1058. [DOI] [PubMed] [Google Scholar]
- 38.Oltvai, Z., C. Milliman, and S. Korsmeyer. 1993. Bcl2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 74:609-619. [DOI] [PubMed] [Google Scholar]
- 39.Pagliari, L., T. Kuwana, C. Bonzon, D. Newmeyer, S. Tu, H. Beere, and D. Green. 2005. The multidomain proapoptotic molecules Bax and Bak are directly activated by heat. Proc. Natl. Acad. Sci. USA 102:17975-17980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petros, A., A. Gunasekera, N. Xu, E. Olejniczak, and S. Fesik. 2004. Defining the p53 DNA-binding domain/Bcl-XL-binding interface using NMR. FEBS Lett. 559:171-174. [DOI] [PubMed] [Google Scholar]
- 41.Petros, A., A. Medek, D. Nettesheim, D. Kim, H. Yoon, K. Swift, E. Matayoshi, T. Oltersdorf, and S. Fesik. 2001. Solution structure of the antiapoptotic protein bcl2. Proc. Natl. Acad. Sci. USA 98:3012-3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruvolo, P., X. Deng, B. Carr, and W. S. May. 1998. A functional role for mitochondrial protein kinase Cα in Bcl2 phosphorylation and suppression of apoptosis. J. Biol. Chem. 273:25436-25442. [DOI] [PubMed] [Google Scholar]
- 43.Sax, J., P. Fei, M. Murphy, E. Bernhard, S. Korsmeyer, and W. El-Deiry. 2002. BID regulation by p53 contributes to chemosensitivity. Nat. Cell Biol. 4:842-849. [DOI] [PubMed] [Google Scholar]
- 44.Wang, X. 2001. The expanding role of mitochondria in apoptosis. Genes Dev. 15:2922-2933. [PubMed] [Google Scholar]
- 45.Wei, M., W. Zong, E. Cheng, T. Lindsten, V. Panoutsakopoulou, A. Ross, K. Roth, G. MacGregor, C. Thompson, and S. Korsmeyer. 2001. Proapoptotic Bax and Bak: a requisite gateway to mitochondrial dysfunction and death. Science 292:727-730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamamoto, K., H. Ichijo, and S. Korsmeyer. 1999. Bcl2 is phosphorylated and inactivated by an ASK/Jun N-terminal protein kinase pathway normally activated at G2/M. Mol. Cell. Biol. 19:8469-8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao, L., and D. Liao. 2003. Sequestration of p53 in the cytoplasm by adenovirus type 12 E1M 55-kilodalton oncoprotein is required for inhibition of p53-mediated apoptosis. J. Virol. 77:13171-13181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou, H., Q. Hou, Y. Chai, and Y. Hsu. 2005. Distinct domains of Bcl-XL are involved in Bax and Bad antagonism and in apoptosis inhibition. Exp. Cell Res. 309:316-328. [DOI] [PubMed] [Google Scholar]