

Abstract
A straightforward mechanism for eliciting transcriptional repression would be to simply block the DNA binding site for activators. Such passive repression is often mediated by transcription factors that lack an intrinsic repressor activity. MafG is a bidirectional regulator of transcription, a repressor in its homodimeric state but an activator when heterodimerized with p45. Here, we report that MafG is conjugated to SUMO-2/3 in vivo. To clarify the possible physiological role(s) for sumoylation in regulating MafG activity, we evaluated mutant and wild-type MafG in transgenic mice and cultured cells. Whereas sumoylation-deficient MafG activated p45-dependent transcription normally and did not affect heterodimer activity, repression by the sumoylation-deficient MafG mutant was severely compromised in vivo. Furthermore, the SUMO-dependent repression activity of MafG was sensitive to histone deacetylase inhibition. Thus, repression by MafG is not achieved through simple passive repression by competing for the activator binding site but requires sumoylation, which then mediates transcriptional repression through recruitment of a repressor complex containing histone deacetylase activity.
Transcriptional repressors are categorized into two classes: passive and active repressors (33). Passive repressors lack intrinsic repressor activity, thereby neutralizing the function of activators by impairing their DNA-protein or protein-protein interactions. Active repressors possess intrinsic repressor activity that modulates chromatin organization. The small Maf proteins, MafG, MafK, MafF (5, 11), and MafT (32), possess a basic region for DNA recognition and a leucine zipper for dimer formation. The small Mafs bind to Maf recognition elements (MAREs) as either homodimers or heterodimers with CNC and Bach family proteins (21). Small Maf proteins lack any canonical transcriptional effector domains and are obligatory partners for CNC and Bach proteins, which contain recognizable functional motifs but cannot bind to DNA as monomers or homodimers. Small Maf homodimers compete with these activating heterodimers for DNA binding and block their activities, a feature characteristic of passive repression (20, 22).
Genetic and biochemical studies have revealed that the small Maf-p45 heterodimer, which is also known as NF-E2, serves as an indispensable transcriptional activator for terminal megakaryocyte differentiation (17, 24, 29, 31). The megakaryocytes of p45-null mutant mice proliferate and differentiate in response to thrombopoietin, without practically any proplatelet generation. Similarly, mafG-null mutant mice and, with increased severity, mafG−/−::mafK+/− compound mutant mice display defective proplatelet formation (PPF). Expression of the thromboxane synthetase (TXS) gene, a well-characterized megakaryocytic MARE-dependent target gene (36), was dramatically suppressed in megakaryocytes of p45-null mice (4), as well as in small maf mutant mice (20). Thus, a synergy between p45 and small Mafs was strongly implicated from these studies.
The repressor activity of small Mafs is observed when its relative abundance surpasses that of p45 (20, 22). Megakaryocytes recovered from MafK- or MafG-overexpressing transgenic mice exhibited reduced PPF and lowered TXS levels, demonstrating that an excess of small Maf proteins represses MARE-dependent transcription, presumably by competing with the small Maf-p45 heterodimer for DNA binding. When these same transgenic mice were crossed into a mafG-null mutant background (thereby diminishing total small Maf protein to approximately normal diploid equivalence), PPF and TXS levels in the compound mutant megakaryocytes were restored. These results provide direct in vivo evidence that small Maf proteins function as quantity-dependent bidirectional transcriptional effectors (20).
In this report, we show that MafG, the most abundant small Maf protein in the bone marrow, is conjugated to SUMO, particularly SUMO-2/3. SUMO is covalently attached to the lysine residues of target proteins via an isopeptide linkage in a multistep process analogous to ubiquitination (18, 28). SUMO conjugation is carried out through the consecutive action of three enzymes: an activating (E1) enzyme (Aos1/Uba2), a conjugating (E2) enzyme (Ubc9), and an E3 ligase (e.g., PIAS family proteins). The importance of SUMO modification has emerged from analyses of a variety of cellular processes, including transcription, mitosis, and nuclear/cytoplasmic transport (35). An increasing number of transcription factors have been reported to be substrates for sumoylation, but many of those studies were based on in vitro or in transfecto experiments, requiring further validation to reveal the genuine in vivo contribution of sumoylation to their activities.
Following the initial discovery that MafG is sumoylated in vivo, we adopted a transgenic complementation rescue approach in parallel with reporter gene transfection assays to clarify the significance of SUMO conjugation to small Maf activity. We generated transgenic mice expressing wild-type and sumoylation-defective MafG in megakaryocytes and then analyzed their activities in a mafG-null mutant background. Mutant or wild-type MafG were capable of rescuing megakaryocyte PPF and TXS expression in the absence of endogenous MafG, indicating that sumoylation is not required for transcriptional activation by MafG. However, unlike the wild type, transgenic sumoylation-defective MafG was unable to repress PPF or TXS expression, demonstrating that sumoylation is required for MafG-mediated transcriptional repression. Thus, a simple increase in the abundance of sumoylation-defective MafG is not sufficient to antagonize MafG-p45 heterodimer activity.
MATERIALS AND METHODS
Transgenes and transgenic mice.
The generation of transgenic mice bearing G1HRD-MafG and G1HRD-MafK has been previously described (20). To construct G1HRD-His-MafG, six histidine residues were inserted between the first methionine and the second threonine residues of MafG, and the resultant coding sequence was ligated to the G1HRD vector. To construct G1HRD-His-MafG K14R, MafG cDNA was first cloned into pET15b (Novagen) between NdeI and BamH I (pET15b-MafG). The BglII-BamHI fragment of pET15b-MafG was cloned into the NotI-NotI vector fragment of pIM-lacZ (19) to generate pIM-His-MafG BB. Lysine 14 of MafG was mutated to arginine to generate pIM-His-MafG K14R BB. The KpnI-XbaI fragment of pIM-His-MafG K14R BB was replaced with the KpnI-NotI fragment from IE3.9int-LacZ (23) to generate G1HRD-His-MafG K14R. Transgenic mice were generated as previously described (19). PCR primer sequences for identification of the transgenes are available upon request. Each G1HRD construct was coinjected with G1HRD-green fluorescent protein (GFP) (20). To obtain mafG−/−::TgHis-MafG and mafG−/−::TgHis-MafG K14R mice, homozygous mafG mutant mice were crossed with G1HRD-His-MafG and G1HRD-His-MafG K14R transgenic mice, respectively. MafG+/−::TgHis-MafG or mafG+/−::TgHis-MafG K14R mice were crossed with mafG homozygous mutant mice, and the resultant litters were used for analysis (see Fig. 7A). MafG mutant and wild-type alleles were determined by PCR (24).
FIG. 7.

Generation of G1HRD-His-MafG and G1HRD-His-MafG K14R transgenic mouse lines. (A) The mating strategy for the transgenic complementation rescue experiment. (B and C) Immunoblot analyses of whole-cell lysates from the bone marrows of transgenic mice carrying G1HRD-His MafG (B) or G1HRD-His-MafG K14R (C) using anti-small Maf antibody. Three independent lines (lines 8, 11, and 25) and four independent lines (lines 11, 18, 45, and 71) were established for G1HRD-His-MafG and G1HRD-His-MafG K14R transgenic mice, respectively. The band intensities of the transgene products relative to that of endogenous MafG are shown at the bottom. (D) Immunoblot analysis of bone marrow lysates from the compound mutant mice. Samples were prepared from G1HRD-His-MafG (lanes 3 and 4) or G1HRD-His-MafG K14R (lanes 5 and 6) mice, both in a mafG-null background, or wild-type (lane 1) or mafG-null (lane 2) mice. White and black arrowheads indicate MafG with or without sumoylation, respectively.
Immunoblotting and nickel bead purification.
For immunoblotting, whole-cell extracts were prepared from the bone marrow cells of 4- to 12-week-old mice from each line of transgenic mouse. MafG and its mutant derivatives were detected using an anti-MafG antibody, which was raised in rabbits that were immunized with purified mouse MafG protein. For the nickel bead pull-down, bone marrow cells were prepared from wild-type and TgHis-MafG mice (line 11) and lysed in guanidinium lysis buffer (6 M guanidine hydrochloride, 20 mM sodium phosphate, pH 7.8, and 500 mM sodium chloride) with 20 mM imidazole. The cell lysates were incubated with Probond resin (Invitrogen) at 4°C for 1 h. Bound proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), blotted onto a polyvinylidene difluoride membrane, and reacted with anti-MafG antibody (described above), anti-SUMO-1 antibody (Alexis), and anti-SUMO-2/3 antibody (a kind gift from H. Saitoh). Cell lysates prepared after transfection were similarly subjected to immunoblot analysis using anti-His antibody (G-18; Santa Cruz), anti-MafG antibody, and anti-SUMO-2/3 antibody.
Transfection analyses.
Plasmids pcDNA3-T7-PIAS1, pcDNA3-T7-PIAS3, pcDNA3-T7-PIASxα, pcDNA3-T7-PIASxβ, and pcDNA3-T7-PIASy were kind gifts from H. Ariga. Plasmids pBOS-HA-SUMO-1 and pBOS-HA-SUMO-2 were kind gifts from J. Mimura. pRBGP2, a reporter plasmid carrying the firefly luciferase gene driven by triplicated MAREs, and pEF-p45 were previously described (9). Mouse Ubc9 cDNA was cloned and inserted into the NotI site of p3xFLAG-CMV-10 (Sigma). pIM-His-MafG XB was generated by inserting the XbaI-BamH I fragment of pET15b-MafG into the NotI-NotI vector fragment of pIM-lacZ. pIM-His-MafG K14R XB and pIM-His-MafG E16A XB were generated from pIM-His-MafG XB by mutating lysine 14 of MafG to arginine and glutamic acid 16 to alanine, respectively. pIM-His-SUMO-2-MafG K14R XB was generated from pIM-His-MafG K14R XB by substituting its NdeI-BstEII fragment with a SUMO-2-coding fragment generated by PCR. The plasmids were transfected into 293T cells using FuGENE 6 (Roche). Trichostatin A (Wako) was added 8 h after transfection to give a final concentration of 330 nM. Twenty-four hours after transfection, cells were harvested in 1× SDS gel-loading buffer (50 mM Tris-HCl, pH 6.8, 100 mM dithiothreitol, 2% SDS, and 10% glycerol) for the immunoblot analysis or in passive lysis buffer (Promega) for the reporter assay. The expression of both firefly and sea pansy luciferases was quantified using the dual-luciferase reporter assay system (Promega). For transfection efficiency, firefly luciferase activity was normalized to the cotransfected sea pansy luciferase activity.
PPF assay.
Megakaryocytes were isolated from the bone marrows of 1- to 4-month-old mice, and the projection of proplatelets was observed as described previously (24). Only GFP-positive cells were counted for calculating the PPF ratios.
Quantification of TXS mRNA.
CD41-positive cells were isolated from bone marrows by MACS (Miltenyi Biotec). Bone marrow cells were collected as previously described (24), reacted with fluorescein isothiocyanate-labeled anti-mouse CD41 antibody (Pharmingen), and mixed with anti-fluorescein isothiocyanate microbeads (Miltenyi Biotec). The associated cells were separated by a large cell separation column (Miltenyi Biotec). CD41-positive cells were pooled from three independent animals from each line. Total RNA was extracted from the cells using Isogen (Nippon Gene, Tokyo) and reverse transcribed by Superscript II (Invitrogen). TXS mRNA was quantified using real-time PCR (ABI PRISM 7700 sequence detection system) as previously described (20).
Electrophoretic mobility shift assay (EMSA).
To produce sumoylated MafG in Escherichia coli, pET15b-MafG1-123 (16) was introduced into E. coli BL21 (DE3) with or without pT-E1E2S2 (34). The bacterial lysates were incubated with Probond resin (Invitrogen) at 4°C for 1 h. The associated proteins were eluted by 500 mM imidazole. The efficiency of SUMO conjugation was checked by SDS-PAGE and ranged between approximately 5 and 50%. His-MafG, His-MafG K14R, and His-SUMO-2-MafG K14R were translated in vitro using a TNT coupled wheat germ extract system (Promega). Double-stranded MARE oligonucleotide probe #25 (10) was radiolabeled with 32P, and incubation of the probe and proteins was carried out as described previously (10). Antibodies against the His6 tag (G-18; Santa Cruz) and p45 (C-19; Santa Cruz) were added for the supershift. The protein-DNA complexes and free probe were resolved by electrophoresis on a 5% polyacrylamide (79:1) gel in 1× Tris-borate-EDTA buffer.
MBP pull-down.
Maltose binding protein (MBP)-p45 (9) was purified with amylose resin (NEB). The mixture of sumoylated and nonsumoylated His-MafG1-123 or purified His-MafG1-76 (15) was incubated with MBP-p45 in binding buffer (20 mM Tris-HCl, pH 8.0, 500 mM sodium chloride, 10% glycerol, and 10 mM 2-mercaptoethanol) at 4°C for 6 h. The protein mixture was then incubated with amylose resin at 4°C for 1 h. Bound proteins were separated by SDS-PAGE, blotted onto a polyvinylidene difluoride membrane, and reacted with anti-His antibody (G-18; Santa Cruz) and anti-MBP antibody (Santa Cruz, C-18).
Biotinylated DNA pull-down.
Biotinylated double-stranded DNA probes were prepared by annealing 5′-bio-TTT GGG GAA CCT GTG CTG AGT CAC TGG AG-3′ to 5′-CCT CCA GTG ACT CAG CAC AGG TTC CCC-3′ (PBGD probe) and 5′-bio-TTT GGG GAA CCT GCA ATT CGT CAA TTG AG-3′ to 5′-CCT CAA TTG ACG AAT TGC AGG TTC CCC-3′ (mutated probe). The mixture of sumoylated and nonsumoylated His-MafG1-123 and/or MBP-p45 was incubated with either of the biotinylated DNA probes in binding buffer (20 mM HEPES, pH 7.6, 200 mM sodium chloride, 4 mM magnesium chloride, 1 mM EDTA, and 0.005% Tween 20) at 37°C for 30 min. In the same buffer, the protein-DNA complex was incubated with TetraLink avidin resin (Promega) at room temperature for 30 min. Bound proteins were detected in the same way as described above. Bound DNAs were separated by electrophoresis in a 15% acrylamide gel and detected by ethidium bromide staining.
RESULTS
Small Maf proteins are conjugated with SUMO-2/3 in bone marrow cells.
We previously generated transgenic mice bearing either G1HRD-MafK or G1HRD-MafG, expressing MafK or MafG, respectively, under the regulation of the Gata1 gene hematopoietic regulatory domain (G1HRD) (20). These mice were crossed with mafG-null mutant animals to recover compound mutant mice whose small Maf expression was complemented by the transgene in erythroid cells and megakaryocytes but not in other cell lineages. During the course of analyzing these mice, we noted the possibility that small Mafs might be posttranslationally modified. Immunoblot analysis of bone marrow cells showed that small Maf was least abundant in mafG-null mutant mice, intermediate in wild-type and compound mutant mice, and most abundant in transgenic mice (Fig. 1A). There were additional larger bands of approximately 40 kDa, with intensities that correlated directly with those of the original 23-kDa (MafG) bands (Fig. 1A). We initially postulated that the 40-kDa bands might represent small Maf proteins bearing posttranslational modification.
FIG. 1.

SUMO-2/3 is conjugated to MafG in the bone marrow. (A) Immunoblot analysis of whole-cell lysates from bone marrows reacted with anti-small Maf antibody. The bone marrow samples were prepared from mafG-null mutant mice (lanes 1 and 5), wild-type mice (lanes 2 and 6), G1HRD-MafG transgenic mice in a mafG-null mutant background (lane 3) or wild-type background (lane 4), and G1HRD-MafK transgenic mice in a mafG-null background (lane 7) or wild-type background (lane 8). (B) Alignments of the N-terminal halves of small Maf proteins found in human, mouse, chicken, and zebra fish. Amino acids comprising the extended homology region (EHR) and basic region are boxed. The conserved sumoylation consensus site is also boxed. (C to E) Nickel pull-down samples of the bone marrows from wild-type mouse and G1HRD-His-MafG mouse (line 11) reacted with anti-MafG antibody (C, lanes 1 and 2), anti-SUMO-1 antibody (D, lanes 4 and 5), or anti-SUMO-2/3 antibody (E, lanes 4 and 5). The 239T cell lysate and those containing overexpressed hemagglutinin (HA)-tagged SUMO-1 and SUMO-2 were used to confirm antibody specificity (lanes 1 to 3 in panels D and E). White and black arrowheads indicate small Maf with or without modification, respectively (A, C, and E). Asterisks indicate nonspecific signals (A).
A previous study reported that acetylation of lysine residues clustered in the basic region of MafG stimulates DNA binding of the MafG-p45 heterodimer (8). However, the size difference of the two bands (Fig. 1A) did not correspond to the addition of an acetyl group. When we aligned the small Maf sequences of vertebrates, we found a consensus sequence for SUMO conjugation that is completely conserved in the N-terminal regions of all the small Maf proteins (Fig. 1B, green box). Hence, we explored the possibility that the 40-kDa band (Fig. 1A) represents a sumoylated small Maf molecule.
To address this hypothesis, we generated transgenic mice carrying G1HRD-His-MafG that expresses His6-tagged MafG under the transcriptional control of Gata1 (see above and Fig. 7B). Since MafG is the major small Maf protein expressed in bone marrow cells, we employed MafG in all subsequent studies. Bone marrow lysates were prepared from one of the high-expressor lines (Line 11) under denaturing conditions, and all proteins interacting with nickel beads were isolated. In this protein fraction, two major bands were detected on Western blots using an anti-MafG antibody; the slow- and fast-migrating bands represent MafG with and without modification, respectively (Fig. 1C). While the anti-SUMO-1 antibody did not react with the nickel-purified protein sample, an anti-SUMO-2/3 antibody recognized the lower-mobility band (Fig. 1D and E). Thus, MafG is conjugated to SUMO-2 or -3 in vivo.
PIASy efficiently promotes MafG lysine-14 sumoylation.
To establish a system for analyzing the possible physiological consequences of MafG sumoylation, we sought to establish conditions for studying MafG sumoylation in cultured cells. His-MafG was transiently expressed in 293T cells with SUMO E2 conjugating enzyme Ubc9, SUMO, and PIAS family proteins, which are well-characterized SUMO E3 ligases (18). Whereas all five PIAS family proteins generated sumoylated MafG when SUMO-1 was added (Fig. 2A), PIASy in particular enhanced the intensity of the more slowly migrating band when SUMO-2 was added (Fig. 2B).
FIG. 2.

PIASy promotes SUMO-2 conjugation to MafG through the lysine 14 residue. (A and B) The effect of PIAS family proteins on the sumoylation of His-MafG. His-MafG was transiently expressed in 293T cells, and sumoylation was examined by immunoblot analysis with anti-His antibody. SUMO-1 and SUMO-2 were expressed in panels A and B, respectively. (C and D) His-MafG K14R and His-MafG E16A were transiently expressed in 293T cells, and their sumoylation was examined in the absence or presence of PIASy. White and black arrowheads indicate MafG with or without sumoylation, respectively (A to D).
To identify the SUMO acceptor site in MafG, we mutated the sumoylation consensus site. PIASy, which turned out to be effective for SUMO-2 conjugation to MafG, was used to examine the efficiency of sumoylation of the MafG mutants. MafG K14R, generated by substituting the lysine residue in the consensus motif with arginine, was not converted into a larger form (Fig. 2C), indicating that K14 is necessary for SUMO conjugation. An alternative mutant form of MafG, MafG E16A, generated by substituting the glutamate residue in the consensus motif with alanine, was also an ineffective sumoylation substrate (Fig. 2D), indicating that E16 is also necessary for modification. The requirement of both residues strongly implicates lysine 14 as the SUMO acceptor site of MafG.
Dimer formation and DNA recognition are not significantly altered by MafG sumoylation in vitro.
We next asked whether sumoylation affects dimer formation or DNA recognition by MafG. For this purpose, we generated sumoylated proteins in E. coli (34). The His6-tagged DNA binding domain of MafG containing the N-terminal region (His-MafG 1-123) (16) was forcibly expressed with Aos1, Uba2, Ubc9, and SUMO-2 in E. coli and then purified using nickel beads. This resulted in the recovery of a mixed preparation (approximately 1:1 molar ratio) of sumoylated and nonsumoylated MafG (Fig. 3A and B).
FIG. 3.

Analysis of DNA binding and dimerization of sumoylated MafG. (A) SUMO conjugation to His-MafG 1-123 in E. coli. Total lysates from bacteria harboring pET15b-MafG1-123 (lane 1) and pET15b-MafG1-123 plus pT-E1E2S2 (lane 2) were incubated with nickel beads. The associated proteins were analyzed by SDS-PAGE followed by Coomassie blue staining. (B and C) DNA binding ability of bacterially synthesized His-MafG 1-123 and its sumoylated form. EMSA was performed with a MARE-containing probe (C). The incubated proteins are shown in the immunoblot reacted with anti-His antibody (B). The lane numbers in panel B correspond to those in panel C. No protein was added to the reaction mixture in lane 0 in panel C. (D) Heterodimer formation between sumoylated or nonsumoylated His-MafG 1-123 and MBP-p45. The mixture of sumoylated and nonsumoylated His-MafG 1-123 was incubated with MBP-p45 (lanes 1 and 3). His-MafG 1-76 was incubated with MBP-p45 as a control (lanes 2 and 4). Input proteins and pull-down samples were reacted with anti-His antibody. (E) DNA binding ability of His-MafG 1-123 or its sumoylated form in the presence of MBP-p45. Constant amounts of His-MafG 1-123 (lanes 2 to 5) and the mixture of His-MafG 1-123 and its sumoylated form (lanes 6 to 9) were incubated with a MARE-containing probe and increasing amounts of MBP-p45. No protein was added to the reaction mixture in lane 1, and only MBP-p45 was added to lane 10. (F) DNA binding of heterodimers containing MafG and p45. The mixture of sumoylated and nonsumoylated His-MafG 1-123 (lane 9) and/or MBP-p45 was incubated with biotinylated PBGD probe (lanes 1 to 4) or a control mutated probe (lanes 5 to 8). A protein interaction was observed only when both the MafG mixture and MBP-p45 were added to the PBGD probe (lane 4). The pull-down efficiency was monitored by the quantity of DNA probe interacting with the avidin resins. The percentages shown at the tops of the panels indicate the approximate molar ratios of sumoylated His-MafG 1-123 to the total of sumoylated and nonsumoylated His-MafG 1-123 used in the reactions (B, C, and E). White and black arrowheads indicate His-MafG 1-123 with or without sumoylation, respectively (A, B, and D), or a shifted band generated by His-MafG 1-123 and the one containing sumoylated His-MafG 1-123, respectively (C and E). An asterisk indicates a shifted band representing heterodimer binding (E).
Homodimer formation and DNA binding were monitored by EMSA using a double-stranded oligonucleotide containing a consensus MARE (#25 [10]) as the radiolabeled probe (Fig. 3C). When nonsumoylated MafG was incubated with MARE, a single major band was observed (Fig. 3C, lanes 1 to 3). The intensities of the shifted bands paralleled the incubated amount of protein (Fig. 3B, lanes 1 to 3). When the equimolar mixture containing both modified and nonsumoylated MafG was used (Fig. 3B, lanes 4 to 6), one additional band of slower mobility appeared (Fig. 3C, lanes 4 to 6). When MafG and the equimolar mixture were combined in a 1:1 ratio, the proportion of the sumoylated form was approximately 25% of the resultant mixture (Fig. 3B, lane 8) and can be seen as the slower-migrating band of intermediate intensity (Fig. 3C, compare lane 8 with lanes 7 and 9). These results indicate that the faster-mobility band represents a homodimer of the nonsumoylated MafG, whereas the slower band contained sumoylated MafG, although we did not determine whether the lower-mobility band represented a homodimer of sumoylated MafG or a heterodimer of sumoylated and nonsumoylated moieties. We conclude that both nonsumoylated and sumoylated MafG proteins are able to bind to DNA and that this modification does not alter the structure that mediates DNA recognition.
Next, we examined the physical properties of MafG complexes as heterodimers with p45. When the mixture of sumoylated and nonsumoylated MafG was incubated with an MBP-p45 bacterially derived fusion protein, both forms were bound by amylose resin (Fig. 3D, lane 3). In contrast, MafG1-76 (lacking the leucine zipper domain) (15) did not bind (lane 4). This result indicates that sumoylation does not inhibit the interaction between MafG and p45 and that the dimerization surface with p45 is maintained irrespective of sumoylation.
To examine the DNA binding ability of each heterodimer, we performed EMSA using increasing amounts of MBP-p45 in association with a constant amount of MafG or with the equimolar mixture of unmodified and sumoylated MafG. The more MBP-p45 added, the less intense was the shifted band composed of MafG and the more intense was the band representing a heterodimer of MBP-p45 and MafG (Fig. 3E, lanes 3 to 5). The two bands generated by the mixed preparation of sumoylated and nonsumoylated MafG similarly decreased in intensities in parallel to the presence of MBP-p45 (lanes 7 to 9), implying that MafG was recruited to the heterodimer complex irrespective of the SUMO conjugation status.
We adopted a pull-down assay using biotinylated double-stranded DNA and further confirmed the DNA binding of a heterodimer containing sumoylated MafG. To eliminate binding of the homodimer, a MARE exclusively bound by the small Maf-p45 heterodimer (a MARE found in the promoter region of the porphobilinogen deaminase gene) (9) was used as a “PBGD probe” along with a control sequence containing a mutated MARE as a “mutated probe.” When the biotinylated PBGD probe was incubated with the mixed MafG preparation and MBP-p45 and pulled down with avidin resins, sumoylated and nonsumoylated MafG were detected in a ratio similar to that found in the input mixture (Fig. 3F, compare lanes 4 and 9). No apparent interaction was detected with the control probe (Fig. 3F, lane 8), confirming the sequence specificity of the interaction between the PBGD probe and the proteins. From this result, we concluded that sumoylation per se does not alter DNA binding of the activating heterodimer formed between MafG and p45.
MafG sumoylation inhibits collaborative MafG-p45 transcriptional activation.
We next examined the effect of sumoylation on the function of MafG in MARE-dependent transcription using reporter gene assays in the absence or presence of PIASy. A reporter plasmid (pRBGP2) bearing a firefly luciferase gene directed by triplicated MAREs and a plasmid that expresses p45 were transiently introduced into 293T cells along with various amounts of MafG expression vector (Fig. 4A, left panel). In the absence of PIASy, the reporter gene was activated and then repressed depending on the abundance of cotransfected MafG (Fig. 4A, left panel) (22). In the presence of PIASy, MafG was unable to activate the reporter gene (Fig. 4A, left panel).
FIG. 4.

Sumoylated MafG behaves as a transcriptional repressor. (A) The effect of PIASy on the function of MafG and its mutants in MARE-dependent transcription. An increasing amount of His-MafG (left panel), His-MafG K14R (middle panel), or MafG E16A (right panel) expression vector was transiently introduced into 293T cells with pRBGP2 and p45 expression vectors in the absence or presence of PIASy. (B and C) Protein accumulation of His-MafG, His-MafG K14R, and His-MafG E16A transiently expressed in 293T cells. Whole-cell extracts were examined by immunoblot analysis using an anti-His antibody. The expression vector for p45 was cotransfected and used as a control for monitoring the transfection efficiency.
To examine whether this effect of coexpressed PIASy was a consequence of MafG sumoylation, we next assayed MARE-dependent transcription in the presence of the sumoylation-defective mutant molecules, MafG K14R and MafG E16A. When MafG K14R was transfected instead of MafG, efficient reporter gene activation was observed in both the absence and presence of PIASy (Fig. 4A, middle panel). MafG E16A yielded essentially the same result (Fig. 4A, right panel). The expressed levels of MafG, MafG K14R, and MafG E16A were all comparable (Fig. 4B and C). Hence, we conclude that both MafG K14R and MafG E16A are insensitive to the negative regulatory effect of PIASy on MARE-mediated transcription. Since PIASy promotes MafG sumoylation that is dependent on the presence of both lysine 14 and glutamate 16 (see Fig. 2C and D), we strongly suspect that SUMO-2 conjugation inhibits MafG from participating in transcriptional activation.
To address the question of whether the presence of the SUMO moiety was sufficient to deprive MafG of its collaborative activator function when associated with p45, we generated a protein in which SUMO-2 and MafG K14R were joined into a single polypeptide and then examined the effect of this MafG-SUMO-2 fusion protein on MARE-dependent transcription. The SUMO-2 polypeptide was inserted between the His6 tag and MafG K14R to generate His-SUMO-2-MafG K14R (Fig. 5A). When transiently expressed in 293T cells, the abundance of His-SUMO-2-MafG K14R was comparable to that of His-MafG or His-MafG K14R (Fig. 5B). The in vitro-translated proteins, His-MafG, His-MafG K14R, and His-SUMO-2-MafG K14R (Fig. 5D), showed comparable DNA binding activities as homodimers and heterodimers with p45 when examined in EMSA (Fig. 5C). Each heterodimer binding appeared as a doublet (Fig. 5C, lanes 3, 8, and 13), probably due to the presence of two isoforms of p45 (31), which is consistent with our previous result showing a doublet MARE-binding activity composed of p45 and small Maf in megakaryocytes (20).
FIG. 5.

Forced conjugation with the SUMO-2 moiety is sufficient to convert MafG into a transcriptional repressor. (A) The generation of His-SUMO-2-MafG K14R was confirmed by immunoblot analysis using antibodies recognizing SUMO-2 (lanes 1 to 3) and MafG (lanes 4 to 6). His-SUMO-2-MafG K14R was transiently expressed in 293T cells, and whole-cell lysates were analyzed (lanes 2 and 6). Mock transfectants were used as negative controls (lanes 1 and 4), and cells transfected with vectors expressing HA-SUMO-2 (lane 3) and His-MafG (lane 5) were used as positive controls for the respective antibodies. (B) Protein accumulation of His-MafG, His-MafG K14R, and His-SUMO-2-MafG K14R transiently overexpressed in 293T cells. Whole-cell extracts were examined by immunoblot analysis using an anti-His antibody. The p45 expression vector was cotransfected and used as a control for monitoring the transfection efficiency. (C and D) Comparison of DNA binding and dimerization abilities of His-MafG, His-MafG K14R, and His-SUMO-2-MafG K14R. (C) EMSA using a MARE probe (no. 25) (10). His-MafG (lanes 1 to 5), His-MafG K14R (lanes 6 to 10), and His-SUMO-2-MafG K14R (lanes 11 to 15) were incubated with the probe. Binding of the homodimers were observed as broader bands (lanes 1, 6, and 11), which were partially supershifted and partially suppressed by anti-His antibody (lanes 2, 7, and 12). Binding of the p45 heterodimers was observed as doublets (lanes 3, 8, and 13), which were supershifted by anti-His antibody (lanes 4, 9, and 14) and by anti-p45 antibody (lanes 5, 10, and 15). (D) Immunoblot analysis with the anti-His antibody showing comparable expression levels of in vitro-translated His-MafG (lane 2), His-MafG K14R (lane 3), and His-SUMO-2-MafG K14R (lane 4) used in EMSA (C). A reaction sample with no template was used as a negative control (lane 1). (E and F) Effects of His-MafG, His-MafG K14R, and His-SUMO-2-MafG K14R on MARE-dependent transcription. To examine their abilities to activate the reporter gene pRBGP2, small amounts of each expression vector (0.1 and 0.3 μg) were transiently introduced into 293T cells along with 0.15 μg of p45 expression vector (E). To examine their abilities to repress the reporter gene, higher abundances of each expression vector (0.6 and 1.3 μg) were added to the state where pRBGP2 was activated by p45 and His-MafG expression vectors (0.15 μg each) (F). The relative luciferase activities are shown. The amounts of transfected plasmids are indicated. Error bars indicate the standard deviations of samples analyzed in triplicate. The results are representative of three independent experiments.
MafG and p45 synergistically activate the MARE-dependent reporter gene when the expression level of MafG is relatively low (Fig. 5E), but reporter transcription is repressed when the abundance of MafG is excessive (Fig. 5F). When MafG K14R was substituted for MafG, the reporter gene was only activated and not repressed, even when MafG K14R was added to excessive levels (Fig. 5E and F). When SUMO-2-MafG K14R was cotransfected with the reporter gene plus p45, no transcriptional activation was observed, regardless of its abundance (Fig. 5E and F), indicating that conjugation of the SUMO-2 moiety alone can convert MafG into a transcriptional repressor. Since SUMO-2-MafG K14R forms a heterodimer with p45 and binds to the MARE (Fig. 5C, lane 13), we surmise that the SUMO-2 moiety antagonizes the function of the transcriptional activation domain carried by p45. These results imply that sumoylation converts MafG into a transcriptional repressor, that nonsumoylated MafG is the obligatory partner of p45 in generating a transcriptional activator, and thus that the bidirectional regulatory capacity of MafG is intimately linked to sumoylation.
MafG-mediated repression is sensitive to HDAC activity.
Based on reports that the differential recruitment of histone deacetylases (HDACs) serves as one of the mechanisms underlying the transcriptional repression directed by sumoylated transcription factors, cofactors, or histones (6, 30, 38), we assessed whether or not MafG-mediated repression was sensitive to trichostatin A (TSA), a potent inhibitor of HDACs (Fig. 6A). In the absence of TSA, excessive MafG repressed reporter gene expression (Fig. 6A, left panel), but TSA treatment abolished this repression (Fig. 6A, right panel). In contrast, MafG K14R and MafG E16A enhanced reporter gene expression regardless of TSA treatment (Fig. 6A). MafG showed the highest derepression ratio compared to the sumoylation-defective mutants (Fig. 6B). This result implicates a direct contribution of HDAC activity to MafG-mediated repression and suggests that sumoylated MafG can recruit a protein complex containing HDAC activity.
FIG. 6.

MafG-mediated transcriptional repression is sensitive to TSA treatment. (A) The effect of TSA treatment on MafG-mediated transcriptional repression. His-MafG, His-MafG K14R, or His-MafG E16A expression vector was transiently introduced into 293T cells with pRBGP2, p45, and His-MafG expression vectors. The relative luciferase activities with (right panel) or without (left panel) TSA treatment are shown. The relative amounts of the transfected plasmids are indicated. Error bars indicate the standard deviations for samples analyzed in triplicate. These results are representative of three independent experiments. (B) Relative derepression caused by TSA treatment. The derepression ratios of MafG and its mutants were calculated, from the result are shown in panel A.
Sumoylation-defective MafG mutants were more effective in activating transcription with p45 (see Fig. 4A, Fig. 5E, and F, and Fig. 6A). This suggests that the activating heterodimer composed of nonsumoylated MafG and p45 is constantly confronted with the negative effect of corepressors recruited by competitive dimers containing sumoylated MafG. In the case of sumoylation-defective MafG, all the available MafG would participate in the activating heterodimer without generating repressive competitors, thereby resulting in the increased net transcriptional activity.
Lysine 14 is necessary for MafG to repress transcription in megakaryocytes.
With the intention of clarifying the functional significance of MafG sumoylation in vivo, we adopted a transgenic complementation rescue assay (Fig. 7A). We prepared transgenic mouse lines expressing His6-tagged MafG or His6-tagged MafG K14R under the control of G1HRD. Three and four independent lines were established for G1HRD-His-MafG and G1HRD-His-MafG K14R transgenic mice, respectively. Immunoblot analysis of bone marrow lysates using anti-MafG antibody confirmed the accumulation of MafG and MafG K14R in those animals (Fig. 7B and C). The relative abundance of the transgene product was approximately 10-fold higher in each line than endogenous MafG, except for G1HRD-His-MafG line 25. Note that while G1HRD-His-MafG lines 8 and 11 displayed an intense SUMO-conjugated band (Fig. 7B and C), none of the G1HRD-His-MafG K14R mice did (Fig. 7C). This observation demonstrates that lysine 14 of MafG is the acceptor residue for SUMO conjugation in vivo.
We examined PPF to elucidate how sumoylation affects MafG activity, since it appears to be one of the most sensitive parameters of MARE-dependent transcriptional activity in vivo (17, 20, 24). Megakaryocytes were purified from the bone marrows of transgenic mice, and the PPF ratios were examined. For this purpose, all of the transgenic mice were coinjected with G1HRD-GFP for monitoring transgene-expressing cells by green fluorescence (20). We also prepared a pseudo-wild-type transgenic line expressing only the G1HRD-GFP transgene. PPF ratios were calculated by counting the GFP-positive megakaryocytes (Fig. 8A to C). Approximately 40% of the megakaryocytes expressing GFP alone generated proplatelets in this assay, whereas PPF was dramatically repressed in MafG-expressing megakaryocytes (Fig. 8D), consistent with our previous observations (20). In contrast, MafG K14R-expressing megakaryocytes displayed completely normal PPF ratios.
FIG. 8.

Analysis of megakaryocytes isolated from the bone marrows of G1HRD-His-MafG and G1HRD-His-MafG K14R transgenic mouse lines. (A to C) PPF was analyzed in bone marrow megakaryocytes. Representative megakaryocytes from G1HRD-GFP mice (A), G1HRD-His-MafG mice (B), or G1HRD-His-MafG K14R (C) mice are shown. Megakaryocytes displaying proplatelets, arrays of beaded filamentous cell projections, are indicated by white arrows. To monitor the transgene expression, G1HRD-GFP was coinjected when G1HRD-His-MafG and G1HRD-His-MafG K14R mice were being generated. GFP-positive megakaryocytes were counted for calculation of PPF ratios. (D and E) PPF incidence in megakaryocytes purified from bone marrows. Transgenic mice expressing His-MafG or His-MafG K14R were examined with control mice expressing GFP (D). Compound mutant mice expressing His-MafG or His-MafG K14R in a mafG-null background were examined with control mice expressing GFP in a mafG-null background (E). The average incidence of more than three independent mice from each line is shown. Error bars indicate the standard deviations. (F and G) TXS gene expression level quantified by real time PCR. Megakaryocytes isolated from G1HRD-His-MafG mice and G1HRD-His-MafG K14R mice were examined in panel F. Megakaryocytes isolated from the mafG−/−::G1HRD-His-MafG and mafG−/−::G1HRD-His-MafG K14R compound mutant mice were examined in panel G. Error bars indicate the standard deviations for samples performed in triplicate.
To directly examine the effects of MafG transgenes on MARE-dependent transcription, we quantified mRNA encoding TXS, a well-characterized target gene of the p45-MafG heterodimer (36). The TXS expression levels correlated well with the PPF ratios, indicating that MafG represses MARE-dependent transcription but that MafG K14R does not (Fig. 8F). Therefore, lysine 14 is a critical residue for MafG-mediated transcriptional repression in vivo.
Sumoylation is dispensable for MafG transcriptional activation in megakaryocytes.
We next asked whether MafG K14R activates transcription by heterodimerizing with p45. This positive regulatory activity can be monitored through the rescue of PPF in the absence of endogenous MafG (20). We purified megakaryocytes from compound mutant mice and examined the PPF ratios (Fig. 8E). Consistent with our previous analyses, PPF was severely defective in the absence of MafG, whereas MafG supplied by transgenic complementation fully rescued PPF. Importantly, we found that MafG K14R also fully rescued PPF in mafG-null megakaryocytes. The TXS expression levels in the megakaryocytes of these compound mutant mice were higher than in mafG-null megakaryocytes, indicating that transgene-derived MafG and MafG K14R both functionally activated MARE-dependent transcription with p45 (Fig. 8G). These results demonstrate that MafG K14R is able to contribute to transcriptional activation in megakaryocytes and that MafG sumoylation is not required for MARE-dependent transcriptional activation.
We note that, for unknown reasons, transgene-derived MafG in the mafG-null background was not efficiently sumoylated (Fig. 1A, lanes 3 and 7, and Fig. 7D, lanes 3 and 4). This may explain the recovery of PPF and TXS expression levels in mafG−/−::G1HRD-His-MafG mice in spite of the higher expression levels of MafG compared to those for wild-type mice. This observation also supports the contention that nonsumoylated MafG cannot antagonize the activity of the MafG-p45 heterodimer, even when it is much more abundantly expressed than endogenous MafG.
DISCUSSION
This study has shown that in addition to the effects elicited by altering the quantitative balance between the small Mafs and their heterodimeric partners, sumoylation is a critical determinant in the acquisition of the bidirectional (positive and negative) regulatory capacity of MafG. We found that MafG is specifically modified by SUMO-2/3 in mouse bone marrow. Transgenic analysis demonstrated that sumoylation-defective MafG loses its repressor activity. In contrast, transgenic complementation rescue analyses demonstrated that sumoylation is dispensable for the participation of MafG in transcriptional activation. These results suggest that SUMO modification promotes the incorporation of MafG into a transcriptional repressor complex, likely through the recruitment of an HDAC-containing moiety.
Reports on the sumoylation of transcription factors have shown that such modification can promote either activation or repression depending on the acceptor proteins involved (7). However, most of the previous work was performed using transient overexpression in cultured cells, and thus, the biological significance of those phenomena was not entirely clear. Two genetic studies with Caenorhabditis elegans have demonstrated that the sumoylation of polycomb group proteins is essential for their physiological repression of Hox genes (39) and that the sumoylation of LIN-11, a LIM homeobox protein, is important for its function in uterine/vulval morphogenesis (2).
To address the basic mechanistic concern regarding the in vivo consequences of sumoylation, the status of sumoylation was examined in mice. As far as we are aware, MafG is the first example of a transcription factor modified specifically by SUMO-2/3 in vivo. The effects of MafG sumoylation on transcriptional activity were examined with mice by comparing MafG to the nonsumoylated MafG mutant, MafG K14R. This represents one of the rare trials for testing the in vivo significance of SUMO modification on the function of mammalian transcription factors. However, these data also indicate that the link between sumoylation and its functional consequences should be further explored by manipulating the sumoylation efficiency of MafG in megakaryocytes (see below).
A consequence of small Maf sumoylation seemed to reside in the differential recruitment of larger transcriptional complexes. Based on the result that MafG-mediated repression was sensitive to TSA (see Fig. 6) and a report that MafK, another small Maf protein, associated with HDAC1 and HDAC2 in undifferentiated MEL cells (1), we envisage that sumoylated MafG achieves transcriptional repression by recruiting a corepressor complex containing HDAC(s) (Fig. 9, bottom panel). A surprising finding was that MafG K14R satisfied the prerequisites for competitive passive repression (i.e., as an inactive homodimer), namely, being sufficient in protein abundance and having proper nuclear localization (H. Motohashi, unpublished observation) and comparable DNA binding (Fig. 5C, lanes 1 to 10), and yet the mutant molecule did not antagonize the activity of MafG-p45. An intriguing notion that emerges from this observation is that the inhibition of MafG-p45 activity by excessive MafG is not attained by a simple competition with the MafG homodimer but likely involves changes at the structural chromatin level. The permissive chromatin environment, once generated by the MafG-p45 heterodimer with a coactivator complex (Fig. 9, top panel), might be maintained even when a homodimer of nonsumoylated MafG competes with the activating heterodimer for the target site (Fig. 9, middle panel). While we originally thought that MafG behaved as a typical passive repressor, the data presented here strongly suggest that MafG becomes qualified as an active repressor via sumoylation.
FIG. 9.
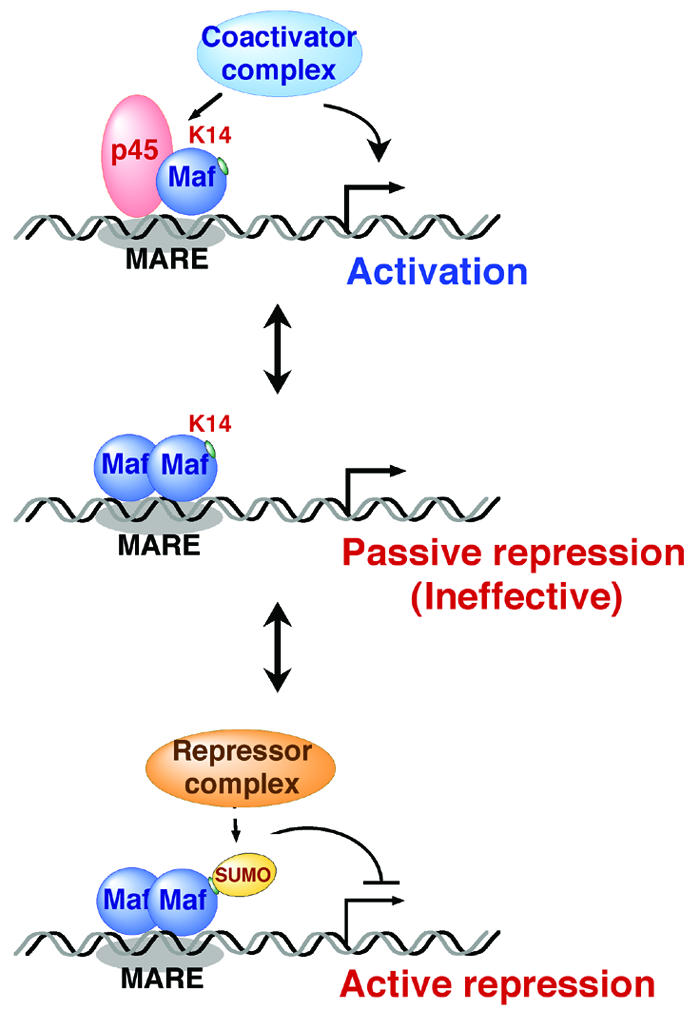
Model of the functional conversion of MafG by sumoylation. MafG activates transcription by forming a heterodimer with p45, thereby recruiting a coactivator complex containing CBP (top panel). MafG homodimer competes with the heterodimer for DNA binding (middle and bottom panels). Simple occupancy of the MARE (passive repression, middle panel) is ineffective, but recruitment of a corepressor complex by sumoylated MafG may be required for achieving transcriptional repression (active repression, bottom panel). Sumoylation is a critical determinant of MafG in its acquisition of a bidirectional regulatory capacity.
A series of in vitro analyses revealed that MafG sumoylation did not dramatically affect the DNA binding or formation of a homodimer or a heterodimer with p45. The DNA binding of homodimers with or without SUMO modification was easily resolved in EMSA. On the contrary, EMSA did not provide conclusive data on the heterodimer binding, since the shifted band of sumoylated heterodimer displayed a mobility similar to that of the nonsumoylated heterodimer, which is often the case with larger proteins (14). Alternatively, a DNA pull-down assay successfully demonstrated the comparable DNA binding of two species of heterodimer. Our next critical question was whether sumoylated MafG binds to MARE as a homodimer or as a heterodimer when it exerts transcriptional repression in vivo. Our preliminary result indicated that the efficiency of MafG sumoylation by PIASy seemed to be decreased in the presence of p45 (Motohashi, unpublished observation), which suggested that MafG is preferentially sumoylated in the form of a homodimer, thereby acquiring repressor activity. Therefore, we contemplate that sumoylated MafG has little chance of existing in the form of a heterodimer with p45 but rather preferentially exists in the form of a homodimer in vivo.
Identification of the SUMO E3 ligase for MafG operating in vivo is an important future objective, since it will help to elucidate the mechanism of MafG sumoylation. This will also provide a strategy for inhibiting the modification of MafG, which would constitute another convincing method for investigating its significance. A recent report demonstrated a rhythmic sumoylation of BMAL1 synchronizing with the circadian cycle (3), which implies an interesting regulatory mechanism for sumoylation. It would be significant if the ratio of sumoylated to nonsumoylated MafG was regulated in a tissue-specific manner and/or in response to various stimuli through differential activation of E3 ligase. PIASy was originally identified as an interacting partner of LEF1 (27) and found to be effective in sumoylating MafG in cultured cells. Therefore, we expect that examination of the status of MafG sumoylation in the megakaryocytes of PIASy-deficient mice (26) may reveal an important clue that could lend credence to this proposed mechanism.
An essential concept requiring further clarification is the physiological role that sumoylation serves in the function of MafG. There are few proteins whose functional alteration by sumoylation has been assigned a specific biological role. Sp3 and Elk1 are good examples of proteins that activate or repress transcription depending on their biological context, and for each protein it was clearly shown that SUMO modification is important for their repressive effects (25, 37). In the case of MafG, our present data seem to suggest that the frequency of PPF is finely tuned by positive and negative MARE-dependent regulation exerted by nonsumoylated and sumoylated MafG, respectively, since the PPF ratios observed in mafG−/−::G1HRD-His-MafG K14R mice (lines 11, 45, and 71) were slightly higher than those of mafG−/−::G1HRD-His-MafG mice (Fig. 8, panel E). We surmise that the higher PPF ratios observed in megakaryocytes containing exclusively nonsumoylatable MafG could result from the loss of negative regulation normally exerted by the sumoylated population of MafG. Our preliminary data also imply that MafG represses the expression of megakaryocyte-specific MARE-dependent genes in immature hematopoietic cells through sumoylation (Motohashi, unpublished observation). A comparison between immature cells in mafG-null mutant bone marrow supplemented with MafG or MafG K14R would be a good starting point for demonstrating the physiological contribution of MafG sumoylation.
In summary, we previously found that small Maf proteins are regulated quite specifically at the transcriptional level (12, 13, 19) and that their abundance is a critical determinant of positive and negative gene regulation through MARE (20, 22). This study revealed that the sumoylation of MafG is an indispensable posttranslational modification that controls the negative regulatory capacity of MafG. We envisage that small Maf sumoylation may be mechanistically utilized in a similar context where CNC partner molecules other than p45 are involved. Thus, another layer of complexity and specificity in transcriptional control by the network of heterodimeric CNC-small Maf and homodimeric small Maf interactions is achieved through the effect of this posttranslational modification of small Maf proteins.
Acknowledgments
We thank Y. Tamagawa, M. Kimura, and R. Kawai for experimental assistance. We also thank H. Ariga and J. Mimura for their kind supplying of reagents.
This work was supported by grants from the NIH (CA80088; H.M., F.K., and J.D.E.), ERATO-JST (M.Y.), Ministry of Education, Science, Sports and Culture (H.M. and M.Y.), Yamanouchi Foundation (H.M.), Uehara Memorial Foundation (H.M.), and Special Coordination Fund for Promoting Science and Technology (H.M.). F.K. is a JSPS Research Fellow.
REFERENCES
- 1.Brand, M., J. A. Ranish, N. T. Kummer, J. Hamilton, K. Igarashi, C. Francastel, T. H. Chi, G. R. Crabtree, R. Aebersold, and M. Groudine. 2004. Dynamic change in transcription factor complexes during erythroid differentiation revealed by quantitative proteomics. Nat. Struct. Mol. Biol. 1:73-80. [DOI] [PubMed] [Google Scholar]
- 2.Broday, L., I. Kolotuev, C. Didier, A. Bhoumik, B. P. Gupta, P. W. Sternberg, B. Podbilewicz, and Z. Ronai. 2004. The small ubiquitin-like modifier (SUMO) is required for gonadal and uterine-vulval morphogenesis in Caenorhabditis elegans. Genes Dev. 18:2380-2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cardone, L., J. Hirayama, F. Giordano, T. Tamaru, J. J. Palvimo, and P. Sassone-Corsi. 2005. Circadian clock control by SUMOylation of BMAL1. Science 309:1390-1394. [DOI] [PubMed] [Google Scholar]
- 4.Deveaux, S., S. Cohen-Kaminsky, R. A. Shivdasani, N. C. Andrews, A. Filipe, I. Kuzniak, S. H. Orkin, P.-H. Romeo, and V. Mignotte. 1997. p45 NF-E2 regulates expression of thromboxane synthase in megakaryocytes. EMBO J. 16:5654-5661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fujiwara, K. T., K. Kataoka, and M. Nishizawa. 1993. Two new members of the maf oncogene family, mafK and mafF, encode nuclear b-Zip proteins lacking putative trans-activator domain. Oncogene 8:2371-2380. [PubMed] [Google Scholar]
- 6.Girdwood, D., D. Bumpass, O. A. Vaughan, A. Thain, L. A. Anderson, A. W. Snowden, E. Garcia-Wilson, N. D. Perkins, and R. T. Hay. 2003. P300 transcriptional repression is mediated by SUMO modification. Mol. Cell 11:1043-1054. [DOI] [PubMed] [Google Scholar]
- 7.Girdwood, D. W., M. H. Tatham, and R. T. Hay. 2004. SUMO and transcriptional regulation. Semin. Cell Dev. Biol. 15:201-210. [DOI] [PubMed] [Google Scholar]
- 8.Hung, H.-L., A. Y. Kim, W. Hong, C. Rakowski, and G. A. Blobel. 2001. Stimulation of NF-E2 DNA binding by CREB-binding protein (CBP)-mediated acetylation. J. Biol. Chem. 276:10715-10721. [DOI] [PubMed] [Google Scholar]
- 9.Igarashi, K., K. Kataoka, K. Itoh, N. Hayashi, M. Nishizawa, and M. Yamamoto. 1994. Regulation of transcription by dimerization of erythroid factor NF-E2 p45 with small Maf proteins. Nature 367:568-572. [DOI] [PubMed] [Google Scholar]
- 10.Kataoka, K., M. Noda, and M. Nishizawa. 1994. Maf nuclear oncoprotein recognizes sequences related to an AP-1 site and forms heterodimers with both Fos and Jun. Mol. Cell. Biol. 14:700-712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kataoka, K., K. Igarashi, K. Itoh, K. T. Fujiwara, M. Noda, M. Yamamoto, and M. Nishizawa. 1995. Small Maf proteins heterodimerize with Fos and potentially act as competitive repressors of NF-E2 transcription factor. Mol. Cell. Biol. 15:2180-2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katsuoka, F., H. Motohashi, K. Onodera, N. Suwabe, J. D. Engel, and M. Yamamoto. 2000. One enhancer mediates mafK transcriptional activation in both hematopoietic and cardiac muscle cells. EMBO J. 19:2980-2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Katsuoka, F., H. Motohashi, J. D. Engel, and M. Yamamoto. 2005. Nrf2 transcriptionally activates the mafG gene through an antioxidant response element. J. Biol. Chem. 280:4483-4490. [DOI] [PubMed] [Google Scholar]
- 14.Komatsu, T., H. Mizusaki, T. Mukai, H. Ogawa, D. Baba, M. Shirakawa, S. Hatakeyama, K. I. Nakayama, H. Yamamoto, A. Kikuchi, and K. Morohashi. 2005. Small ubiquitin-like modifier 1 (SUMO-1) modification of the synergy control motif of Ad4 binding protein/steroidogenic factor 1 (Ad4BP/SF-1) regulates synergistic transcription between Ad4BP/SF-1 and Sox9. Mol. Endocrionol. 118:2451-2462. [DOI] [PubMed] [Google Scholar]
- 15.Kusunoki, H., H. Motohashi, F. Katsuoka, A. Morohashi, M. Yamamoto, and T. Tanaka. 2002. Solution structure of the DNA-binding domain of MafG. Nat. Struct. Biol. 9:252-256. [DOI] [PubMed] [Google Scholar]
- 16.Kyo, M., T. Yamamoto, H. Motohashi, T. Kamiya, T. Kuroita, T. Tanaka, J. D. Engel, B. Kawakami, and M. Yamamoto. 2004. Evaluation of MafG interaction with Maf recognition element arrays by surface plasmon resonance imaging technique. Genes Cells 9:153-164. [DOI] [PubMed] [Google Scholar]
- 17.Lecine, P., J. L. Villeval, P. Vyas, B. Swencki, Y. Xu, and R. A. Shivdasani. 1998. Mice lacking transcription factor NF-E2 provide in vivo validation of the proplatelet model of thrombocytopoiesis and show a platelet production defect that is intrinsic to megakaryocytes. Blood 92:1608-1616. [PubMed] [Google Scholar]
- 18.Melchior, F., M. Schergaut, and A. Pichler. 2003. SUMO: ligases, isopeptidases and nuclear pores. Trends Biochem. Sci. 28:612-618. [DOI] [PubMed] [Google Scholar]
- 19.Motohashi, H., K. Igarashi, K. Onodera, S. Takahashi, H. Ohtani, M. Nakafuku, M. Nishizawa, J. D. Engel, and M. Yamamoto. 1996. Mesodermal- vs. neuronal-specific expression of MafK is elicited by different promoters. Genes Cells 1:223-238. [DOI] [PubMed] [Google Scholar]
- 20.Motohashi, H., F. Katsuoka, J. A. Shavit, J. D. Engel, and M. Yamamoto. 2000. Positive or negative MARE-dependent transcriptional regulation is determined by the abundance of small Maf proteins. Cell 103:865-875. [DOI] [PubMed] [Google Scholar]
- 21.Motohashi, H., T. O'Connora, F. Katsuoka, D. J. Engel, and M. Yamamoto. 2002. Integration and diversity of the regulatory network composed of Maf and CNC families of transcription factors. Gene 294:1-12. [DOI] [PubMed] [Google Scholar]
- 22.Nagai, T., K. Igarashi, J. Akasaka, K. Furuyama, H. Fujita, N. Hayashi, M. Yamamoto, and S. Sassa. 1998. Regulation of NF-E2 activity in erythroleukemia cell differentiation. J. Biol. Chem. 273:5358-5365. [DOI] [PubMed] [Google Scholar]
- 23.Onodera, K., S. Takahashi, S. Nishimura, J. Ohta, H. Motohashi, K. Yomogida, N. Hayashi, J. D. Engel, and M. Yamamoto. 1997. GATA-1 transcription is controlled by distinct regulatory mechanisms during primitive and definitive erythropoiesis. Proc. Natl. Acad. Sci. USA 94:4487-4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Onodera, K., J. A. Shavit, H. Motohashi, M. Yamamoto, and D. J. Engel. 2000. Perinatal synthetic lethality and hematopoietic defects in compound mafG::mafK mutant mice. EMBO J. 19:1335-1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ross, S., J. L. Best, L. I. Zon, and G. Gill. 2002. SUMO-1 modification represses Sp3 transcriptional activation and modulates its subnuclear localization. Mol. Cell 10:831-842. [DOI] [PubMed] [Google Scholar]
- 26.Roth, W., C. Sustmann, M. Kieslinger, A. Gilmozzi, D. Irmer, E. Kremmer, C. Turck, and R. Grosschedl. 2004. PIASy-deficient mice display modest defects in IFN and Wnt signaling. J. Immunol. 173:6189-6199. [DOI] [PubMed] [Google Scholar]
- 27.Sachdev, S., L. Bruhn, H. Sieber, A. Pichler, F. Melchior, and R. Grosschedl. 2001. PIASy, a nuclear matrix-associated SUMO E3 ligase, represses LEF1 activity by sequestration into nuclear bodies. Genes Dev. 15:3088-3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saitoh, H. 2004. Unraveling the SUMO2/3 conjugation and deconjugation pathways, p. 175-207. In V. G. Wilson (ed.), Sumoylation, molecular biology and biochemistry. Horizon Bioscience, Norfolk, United Kingdom.
- 29.Shavit, J. A., H. Motohashi, K. Onodera, J. Akasaka, M. Yamamoto, and J. D. Engel. 1998. Impaired megakaryopoiesis and behavioral defects in mafG-null mutant mice. Genes Dev. 12:2164-2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shiio, Y., and R. N. Eisenman. 2003. Histone sumoylation is associated with transcriptional repression. Proc. Natl. Acad. Sci. USA 23:13225-13230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shivdasani, R. A., M. F. Rosenblatt, D. Zucker-Franklin, C. W. Jackson, P. Hunt, C. J. M. Saris, and S. H. Orkin. 1995. Transcription factor NF-E2 is required for platelet formation independent of the actions of thrombopoietin/MGDF in megakaryocyte development. Cell 81:695-704. [DOI] [PubMed] [Google Scholar]
- 32.Takagi, Y., M. Kobayashi, L. Li, T. Suzuki, K. Nishikawa, and M. Yamamoto. 2004. MafT, a new member of the small Maf protein family in zebrafish. Biochem. Biophys. Res. Commun. 320:62-69. [DOI] [PubMed] [Google Scholar]
- 33.Thiel, G., M. Lietz, and M. Hohl. 2004. How mammalian transcriptional repressors work. Eur. J. Biochem. 271:2855-2862. [DOI] [PubMed] [Google Scholar]
- 34.Uchimura, Y., M. Nakamura, K. Sugasawa, M. Nakao, and H. Saitoh. 2004. Overproduction of eukaryotic SUMO-1- and SUMO-2-conjugated proteins in Escherichia coli. Anal. Biochem. 331:204-206. [DOI] [PubMed] [Google Scholar]
- 35.Verger, A., J. Perdomo, and M. Crossley. 2002. Modification with SUMO. EMBO Rep. 4:137-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yaekashiwa, M., and L. H. Wang. 2002. Transcriptional control of the human thromboxane synthase gene in vivo and in vitro. J. Biol. Chem. 277:22497-22508. [DOI] [PubMed] [Google Scholar]
- 37.Yang, S. H., E. Jaffray, R. T. Hay, and A. D. Sharrocks. 2003. Dynamic interplay of the SUMO and ERK pathways in regulating Elk-1 transcriptional activity. Mol. Cell 12:63-74. [DOI] [PubMed] [Google Scholar]
- 38.Yang, S. H., and A. D. Sharrocks. 2004. SUMO promotes HDAC-mediated transcriptional repression. Mol. Cell 13:611-617. [DOI] [PubMed] [Google Scholar]
- 39.Zhang, H., G. A. Smolen, R. Palmer, A. Christoforou, S. van den Heuvel, and D. A. Haber. 2004. SUMO modification is required for in vivo Hox gene regulation by the Caenorhabditis elegans Polycomb group protein SOP-2. Nat. Genet. 36:507-511. [DOI] [PubMed] [Google Scholar]