

Abstract
Focal adhesion kinase (FAK) has been implicated to be a point of convergence of integrin and growth factor signaling pathways. Here we report that FAK directly interacts with the hepatocyte growth factor receptor c-Met. Phosphorylation of c-Met at Tyr-1349 and, to a lesser extent, Tyr-1356 is required for its interaction with the band 4.1 and ezrin/radixin/moesin homology domain (FERM domain) of FAK. The F2 subdomain of the FAK FERM domain alone is sufficient for Met binding, in which a patch of basic residues (216KAKTLRK222) are critical for the interaction. Met-FAK interaction leads to FAK activation and subsequent contribution to hepatocyte growth factor-induced cell motility and cell invasion. Our results provide evidence that constitutive Met-FAK interaction may be a critical determinant for tumor cells to acquire invasive potential.
Focal adhesion kinase (FAK), a 125-kDa nonreceptor tyrosine kinase localized in focal adhesions, is known for its pivotal role in the control of integrin-mediated cell functions, including cell migration, cell cycle progression, and cell survival (29, 32). It rapidly becomes tyrosine phosphorylated and activated following cell adhesion to extracellular matrix proteins. Increasing evidence also suggests that FAK may be a point of convergence of integrin and growth factor signaling pathways. In addition to cell adhesion, a number of growth factors have been reported to stimulate the phosphorylation of FAK (8, 18, 28, 37). However, the mechanisms for FAK activation in integrin signaling and growth factor signaling remain obscure.
FAK contains a central tyrosine kinase domain flanked by large NH2- and COOH-terminal regions. The COOH terminus contains a focal adhesion targeting domain responsible for FAK localization in focal adhesions (19). The NH2 terminus contains a region of sequence homology with band 4.1 and ezrin/radixin/moesin (ERM) proteins, termed a FERM domain. The FERM domain was found in some membrane-targeted proteins and thought to mediate protein-protein and/or protein-phosphoinositide interactions (2, 27). The crystal structure of the FAK FERM domain, which reveals a trilobed architecture (F1, F2, and F3) similar to those of ERM family members (17, 33, 44), has recently been determined (6). The FERM domain of FAK has been described to be involved in interactions with other proteins, including the cytoplasmic region of integrins (9, 42), the FERM domain of ezrin (36), the pleckstrin homology domain of the Tec family kinase Etk (10), and the receptors for platelet-derived growth factor (PDGF) and epidermal growth factor (EGF) (43). It was also proposed that intramolecular interaction of the FAK FERM domain with the kinase domain suppresses the catalytic activity of FAK (13).
Hepatocyte growth factor (HGF), also known as scatter factor, is a mesenchymally derived factor that elicits multiple cellular responses, including proliferation, migration, and morphogenesis, on various types of cells (1, 3, 4). The diverse biological functions of HGF are transmitted through activation of its transmembrane receptor, encoded by the c-met proto-oncogene (5). Inappropriate activation of the HGF/c-Met signaling pathway has been implicated in the etiology of a number of human tumors and has been shown to confer invasive and metastatic properties to cancer cells (3, 4). The Met receptor is a heterodimer composed of a 45-kDa α chain that remains entirely extracellular and a 145-kDa β chain that traverses the plasma membrane and contains the intracellular tyrosine kinase domain (16, 38). Upon HGF binding, the intrinsic tyrosine kinase of the receptor is activated, resulting in autophosphorylation at specific tyrosine residues in the β chain (15, 30). Two tyrosine residues in the COOH terminus of the β chain (Tyr-1349 and Tyr-1356) are required for all biological activities of the receptor (48, 50) and serve as docking sites for the Grb2-associated binder 1 (Gab1) docking protein (40, 47) and multiple Src homology 2 (SH2) domain- and phosphotyrosine binding domain-containing proteins (34, 35).
The c-met proto-oncogene was originally identified as an oncogene activated in vitro after treatment of a human cell line with a chemical carcinogen (12). Under such conditions, activation of the Met proto-oncogene occurred via a chromosomal rearrangement between chromosome 1 and chromosome 7 (31). This rearrangement creates a hybrid gene, Tpr-Met, with its upstream region derived from the Tpr (translocated promoter region) locus fused to downstream sequences encoding the Met kinase. In the fusion, the 5′ region of the Met gene is replaced by Tpr, which provides two leucine zipper domains. These motifs mimic the effect of the ligand, leading to a constitutively dimerized and therefore activated Met kinase. The dimerization domains are essential for Tpr-Met oncogene transforming activity. The activated Tpr-Met oncogene expresses a 5.0-kb hybrid RNA encoding a 65-kDa fusion protein (39). Tyr-482 and Tyr-489 of Tpr-Met, corresponding to Tyr-1349 and Tyr-1356 in c-Met, are responsible for the association with SH2 domain-containing transducers (1, 35). The Tpr-Met rearrangement has been detected in a number of human cell lines derived from human gastric carcinomas (45) and in biopsy samples derived from gastric tumors (46). The Tpr-Met oncogenic potential has been evaluated both with fibroblasts in vitro and with transgenic mice, where it leads to the development of mammary tumors (25, 39).
We have previously demonstrated that FAK is involved in HGF-induced cell motility and that its elevated expression renders epithelial cells susceptible to transformation by HGF stimulation (7, 24). In this study, we report a direct interaction between FAK and Met. This interaction occurs through the F2 subdomain of the FAK FERM domain and the phosphorylated Tyr-1349 and Tyr-1356 of c-Met. A patch of basic residues (216KAKTLRK222) in the F2 subdomain of the FERM domain are crucial for the Met-FAK interaction. We show that direct interaction of FAK with Met is required for FAK to promote HGF-induced cell invasion. Our results implicate that Met-FAK interaction may confer invasive potential to tumor cells and serve as a target for therapeutic purposes.
MATERIALS AND METHODS
Antibodies and reagents.
Polyclonal anti-Met (C-28), anti-Gab1 (H-198), and anti-FAK (A-17) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Polyclonal anti-phospho-Met (Tyr-1234/Tyr-1235) was purchased from Upstate Biotechnology (Lake Placid, NY). Polyclonal anti-phospho-Met (Tyr-1349) was from Cell Signaling Technology (Beverly, MA). Monoclonal anti-FAK (clone 77) and antiphosphotyrosine (PY20) were from BD Transduction Laboratories (Lexington, KY). Polyclonal anti-phospho-FAKs (pY397, pY407, pY576, pY577, pY861, and pY925) were purchased from BioSource International, Inc. (Camarillo, CA). Monoclonal anti-Src (clone 327) was from Oncogene Research Products (San Diego, CA). Monoclonal antihemagglutinin (anti-HA) epitope was purchased from Roche. Recombinant human HGF, protein A-Sepharose beads, and glutathione agarose beads were purchased from Sigma-Aldrich. Lipofectamine and Oligofectamine were purchased from Invitrogen Life Technologies. Alkaline phosphatase was from New England Biolabs (Beverly, MA). G418 sulfate was from Calbiochem (San Diego, CA). Cultrex basement membrane extract was purchased from R&D Systems (Minneapolis, MN). The 24-well transwell chamber used for the invasion assay was purchased from Costar (Cambridge, MA). The Met-binding sequence peptide (NH2-FGMQVPPPAHMGFRSS-COOH) and the phosphopeptide (NH2-IGEHPYVHVNATPYVNVK-COOH) and its unphosphorylated counterpart were purchased from United Biochemical Research, Inc. (Seattle, WA). The small interfering RNA (siRNA) duplex (GGGACAUCGAGUGUAGAGACUdTdT) (where dT is deoxyribosylthymine) used for the Gab-1 knockdown was purchased from Dharmacon, Inc. (Lafayette, CO).
Plasmids.
Plasmid pcDNA3-HA-Gab1 was kindly provided by T. Hirano (Osaka University, Japan). Chicken FAK cDNA was kindly provided by J. T. Parsons (University of Virginia, VA). Human c-Met cDNA was kindly provided by G. Vande Woude (Van Andel Research Institute, MI). Plasmids pMT2-Tpr-Met and pMT2-Tpr-Met Y482F/Y489F were kindly provided by P. M. Comoglio (University of Torino, Italy). Plasmids pXM-Tpr-Met K241A, pXM-Tpr-Met Y482F, and pXM-Tpr-Met Y489F were kindly provided by M. Park (McGill University, Canada). The following plasmids were constructed in our laboratory: the pGEX-Tpr-Met series, pET-21d-Tpr-Met, pcDNA3.1-HA-FAK (amino acids [aa] 1 to 1053), pcDNA3.1-HA-FAK-NH2 (aa 1 to 391), pKH3-FAK-FERM (aa 30 to 377), pKH3-FAK-COOH (aa 693 to 1053), pET-21a-FAK-NH2 (aa 1 to 363), pGEX2T-FAK-NH2 (aa 1 to 391), pGEX2T-FAK-NH2-ΔF1 (aa 126 to 391), pGEX2T-FAK-NH2-ΔF3 (aa 1 to 253), pGEX2T-FAK-F1 (aa 1 to 126), pGEX2T-FAK-F2 (aa 126 to 253), pGEX2T-FAK-F3 (aa 246 to 391), and pGEX1-FAK (aa 378 to 406). Mutagenesis was carried out using a QuikChange site-directed mutagenesis kit (Stratagene). The mutagenic primers used were Met Y1349F, 5′-TTTCATTGGGGAGCACTTTGTCCATGTGAACGCTA-3′; Met Y1356F, 5′-TCCATGTGAACGCTACTTTTGTGAACGTAAAATGTGTCG-3′; FAK K216A, 5′-GAGTTTGCTAGATTCAGTGGCGGCCAAAACACTACGAAAAT-3′; FAK K218A, 5′-GCTAGATTCAGTGAAGGCCGCAACACTACGAAAATTAATCCAA-3′; and FAK K222A, 5′-GAAGGCCAAAACACTACGAGCATTAATCCAACAGACATTTCGA-3′. The underlining indicates the positions of substituted codons. The desired mutations were confirmed by dideoxy DNA sequencing, a service provided by the Biotechnology Center of National Chung Hsing University, Taiwan.
Cell culture and transfections.
FAK+/+ and FAK−/− mouse embryo fibroblasts (MEF) were kindly provided by D. Ilic (University of California at San Francisco, CA) and were described previously (20). They were maintained in Dulbecco's modified Eagle's medium (Invitrogen Life Technologies) supplemented with 10% fetal bovine serum, 100 μM nonessential amino acids, 1 mM sodium pyruvate, and 55 μM 2-mercaptoethanol and cultured at 37°C in a humidified atmosphere of 5% CO2 and 95% air. Gab1+/+ MEF, Gab1−/− MEF, SYF (src−/− yes−/− fyn−/−) cells, and human embryonic kidney (HEK) 293 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. Gab1+/+ and Gab1−/− MEF were provided by T. Hirano (Osaka University, Japan) and were described previously (21). SYF cells were provided by P. Soriano (Fred Hutchinson Cancer Research Center, WA) and were described previously (22). Lung cancer cell lines CL1-0, CL1-1, and CL1-5 were obtained from P. C. Yang (National Taiwan University, Taiwan) and were described previously (11). Lung cancer cell lines A549, H1355, H23, and H838 were purchased from the American Type Culture Collection. All lung cancer cell lines used in this study were grown in RPMI 1640 medium (Invitrogen Life Technologies) supplemented with 10% fetal bovine serum. For transient transfections, 5 × 105 cells were seeded on a 6-cm culture dish. Eighteen hours later, the cells were incubated with plasmid (1 to 2 μg) and Lipofectamine (10 μl) for 5 h; cells were lysed in 1% NP-40 lysis buffer 24 h after incubation with plasmid. For HGF stimulation, cells were serum starved for 18 h and treated with 20 ng/ml of HGF for 15 min. To generate Madin-Darby canine kidney (MDCK) cells stably expressing HA-tagged FAK and its K222A mutant, MDCK cells were grown on 60-mm dishes and transfected with 2 μg of pcDNA3.1-HA-FAK or pcDNA3.1-HA-K222A by using 10 μl of Lipofectamine according to the manufacturer's instructions. The cells were selected in growth medium containing 0.5 mg/ml G418 and screened for HA-tagged FAK expression by immunoblotting with anti-HA.
Immunoprecipitations and immunoblotting.
Cells were lysed in 1% Nonidet P-40 lysis buffer (1% Nonidet P-40, 20 mM Tris-HCl, pH 8.0, 137 mM NaCl, 10% glycerol, and 1 mM Na3VO4) containing protease inhibitors (1 mM phenylmethylsulfonyl fluoride, 0.2 trypsin inhibitory units/ml aprotinin, and 20 μg/ml leupeptin). The lysates were centrifuged for 10 min at 4°C to remove debris, and the protein concentrations were determined by using a Bio-Rad protein assay (Hercules, CA). For immunoprecipitation, aliquots of lysates were incubated with l μg antibody for 1.5 h at 4°C. Immunocomplexes were collected on protein A-Sepharose beads. For monoclonal antibodies, protein A-Sepharose beads were coupled with rabbit anti-mouse immunoglobulin G (1 μg) before use. The beads were washed three times with 1% NP-40 lysis buffer, boiled for 3 min in sodium dodecyl sulfate (SDS) sample buffer, subjected to SDS-polyacrylamide gel electrophoresis, and transferred to nitrocellulose (Schleicher and Schuell, Inc., Keene, NH). Immunoblotting was performed with appropriate antibodies using an Amersham Pharmacia Biotech enhanced chemiluminescence system for detection.
In vitro protein kinase assay.
To perform in vitro kinase assays, the immunoprecipitates by anti-Met, anti-Src, or anti-FAK were washed three times with 1% NP-40 lysis buffer and once in 20 mM Tris buffer. Kinase reactions were carried out in 40 μl of kinase buffer (50 mM Tris-HCl, pH 7.5, 10 mM MnCl2) containing 10 μCi [r-32P]ATP (3,000 Ci mmol−1; NEN) and 0.5 μg of purified glutathione S-transferase (GST) or GST-FAK (aa 378 to 406) fusion proteins for 20 min at 25°C. Reactions were terminated by the addition of SDS sample buffer, and proteins were resolved by SDS-polyacrylamide gel electrophoresis. To prepare soluble GST fusion proteins, immobilized GST fusion proteins on glutathione-agarose beads were eluted by 10 mM reduced glutathione in 50 mM Tris buffer (pH 8.0).
Protein purification.
Recombinant baculoviruses for expressing HA-tagged FAK or its kinase-deficient (kd) mutant were kindly provided by J.-L. Guan (Cornell University, NY) and were described previously (49). To purify recombinant FAK proteins, Sf21 insect cells were infected with the recombinant baculoviruses for 72 h and lysed in 1% Nonidet P-40 lysis buffer. The recombinant FAK proteins were immobilized on protein A-Sepharose with polyclonal anti-FAK, eluted in 0.1 M citric acid (pH 3.0), and neutralized in Tris-HCl buffer. Histidine-tagged fusion proteins (His-FAK-NH2 domain and His-Tpr-Met) were purified by immobilized metal ion affinity chromatography according to the supplier's instructions (Amersham Pharmacia).
In vitro pull-down assay.
GST fusion proteins were immobilized on glutathione-agarose beads and then incubated with baculovirus-expressed recombinant FAK proteins or the HEK 293 cell lysates containing Tpr-Met in 1% Nonidet P-40 lysis buffer for 1 h at 4°C. The complexes were washed four times with 1% Nonidet P-40 lysis buffer, resolved by SDS-polyacrylamide gel electrophoresis, and analyzed by immunoblotting with anti-HA or anti-Met.
Cell scatter assay.
MDCK cells were allowed to grow as discrete colonies by seeding at 2 × 103 cells per 60-mm dish. When the majority of colonies contained between 20 to 40 cells (60 to 72 h after seeding), the medium was replaced by fresh medium containing 5% serum and 20 ng/ml HGF. Six hours later, the effect of HGF on scatter of MDCK cells was photographed under a phase-contrast microscope at ×100 magnification. When half of the cells in a given colony had lost contact with their neighbors and exhibited a fibroblast-like phenotype, the colony was judged a “scattered” colony. The percentage of scattered colonies in the total 50 colonies was measured.
Cell migration assay.
Cell migration assays were carried out with a Neuro Probe 48-well chemotaxis chamber (Cabin John, MD). The medium containing 10 μg/ml collagen with or without 10 ng/ml HGF was added to the lower chamber. The lower and upper chambers were separated by a polycarbonate membrane (8 μm pore size; Poretics, Livermore, CA). Cells were allowed to migrate for 6 h at 37°C in a humidified atmosphere containing 5% CO2. The membrane was fixed in methanol for 10 min and stained with modified Giemsa stain for 1 h. Cells on the upper side of the membrane were removed by cotton swabs. Cells on the lower side of the membrane were counted under a light microscope. Each experiment was performed in triplicate.
Matrigel invasion assay.
Cells (104) in 250 μl of serum-free medium were added to an inner cup of the 24-well transwell chamber that had been coated with 150 μl of Cultrex basement membrane extract (∼8 mg/ml). Medium (750 μl) supplemented with 10% serum was added to the outer cup. After 24 h, cells that had migrated through Matrigel and the filter membrane with 8-μm pores were fixed, stained, and counted under a light microscope. Each experiment was performed in triplicate.
Statistics.
Statistical analyses were performed with Student's t test. Differences were considered to be statistically significant at P of <0.05.
RESULTS
Tyr-1349 and, to a lesser extent, Tyr-1356 of c-Met are required for its association with FAK in intact cells.
Although we have previously shown that FAK engages in HGF signaling (7, 8, 24), it is unclear whether FAK physically associates with c-Met in response to HGF stimulation. To examine this possibility, coimmunoprecipitation of FAK and c-Met was examined with HEK 293 cells. Upon HGF stimulation, c-Met was phosphorylated at Tyr-1234 and Tyr-1235 in the activation loop within the catalytic domain and began to form complexes with FAK (Fig. 1A). Upon HGF stimulation, approximately 5% of total FAK proteins in the cell were estimated to associate with c-Met and, conversely, approximately 20% of total Met proteins in the cell were estimated to associate with FAK (data not shown). Overexpression of c-Met (fourfold greater than endogenous level) led to its constitutive activation and association with FAK independently of HGF stimulation (Fig. 1A). Increased expression of c-Met was correlated with its increased association with FAK (Fig. 1B). Mutation of c-Met at Tyr-1349 and, to a lesser extent, Tyr-1356 abolished its interaction with FAK (Fig. 1C). Tyr-1349 and Tyr-1356 of c-Met are equivalent to Tyr-482 and Tyr-489 of Tpr-Met, an activated cytoplasmic form of c-Met (31). Accordingly, mutations at Tyr-482 and Tyr-489 of Tpr-Met abolished its interaction with FAK (Fig. 1D).
FIG. 1.

Tyr-1349 and, to a lesser extent, Tyr-1356 of Met are required for its interaction with FAK. (A) Met-FAK interaction was examined by coimmunoprecipitation (IP) of both molecules in HEK 293 cells with (+) or without (−) HGF stimulation. To measure the activation of c-Met, whole-cell lysates (WCL) were analyzed by immunoblotting (IB) with a phospho-specific antibody (anti [α]-Met-Y1234/1235-P). (B) Increasing amounts of c-Met were transiently expressed in HEK 293 cells, and its coprecipitation with FAK was examined. (C) c-Met or its mutants were transiently expressed in HEK 293 cells, and their coprecipitations with FAK were examined. Phosphorylation of c-Met at Tyr-1349 was analyzed by immunoblotting with a phospho- specific antibody (α-Met-Y1349-P). (D) HA-tagged FAK was transiently coexpressed with Tpr-Met or its mutants in HEK 293 cells, and their coprecipitations were analyzed. IgG, immunoglobulin G. Molecular size markers (in kilodaltons) are noted at the left of blots.
Gab1 is not required for Met-FAK interaction.
Since FAK lacks a module, such as the SH2 domain, for binding to phosphotyrosine, the results shown in Fig. 1C and D are somewhat unexpected. We suspected that the interaction between FAK and Met might be indirect through some other cellular proteins. To examine the potential involvement of the major Met-docking protein Gab1 in this interaction, the effect of siRNA-mediated knockdown of Gab1 on Met-FAK interaction was analyzed with HEK 293 cells. The extent of Gab1 knockdown was approximately 60%, which had no effect on Met-FAK interaction (Fig. 2A). To further exclude the involvement of Gab1 in this event, Met-FAK interaction was analyzed with Gab1-null (Gab1−/−) cells and their wild-type (wt) counterparts. The extent of Met-FAK interaction in Gab1−/− cells was same as that in Gab1+/+ cells (Fig. 2B). Moreover, previous studies had identified a 13-amino-acid sequence, GMQVPPPAHMGFR, within the Met-binding domain of Gab1 as critical for Met-Gab1 interaction (26, 41). We found that a 16-amino-acid peptide containing the Met-binding sequence of Gab1 was capable of blocking Met-Gab1 interaction but had little effect on Met-FAK interaction (Fig. 2C). These results together exclude the possibility of Gab1 mediating Met-FAK interaction in intact cells.
FIG. 2.
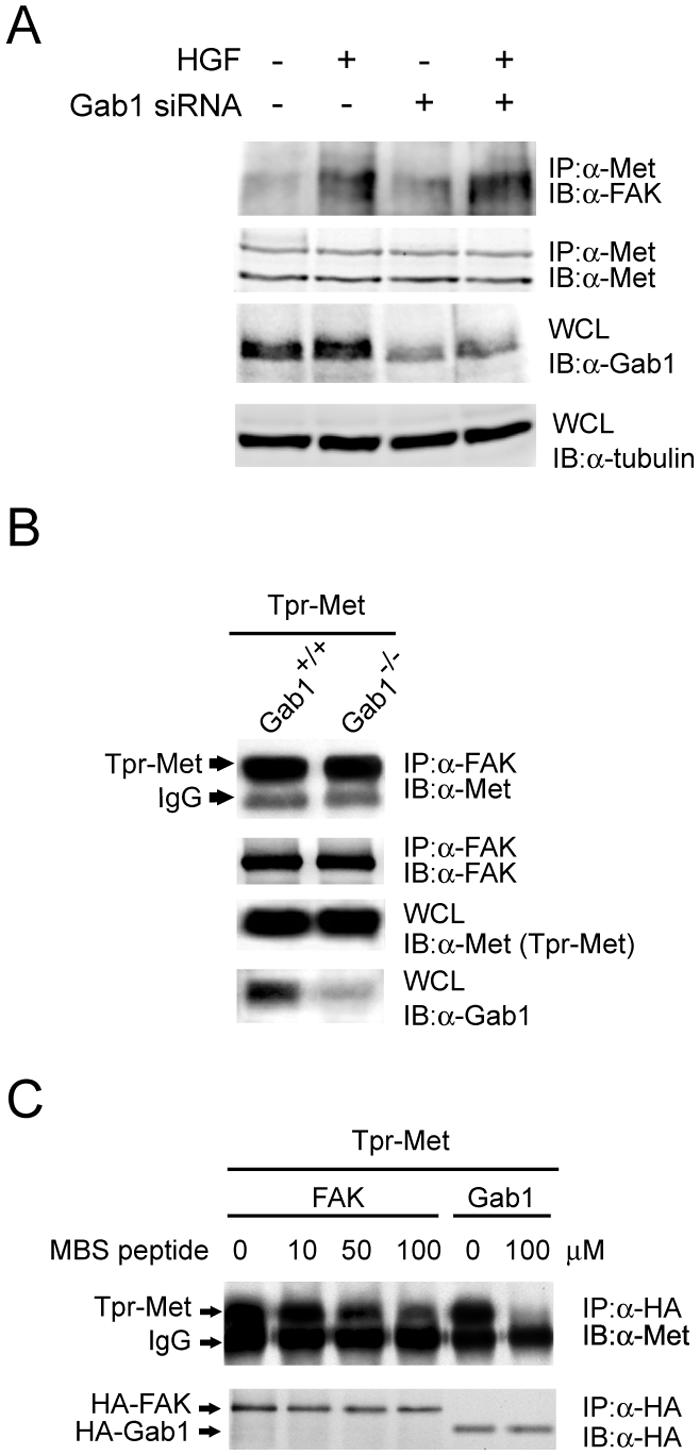
Gab1 does not mediate the interaction between Met and FAK. (A) The siRNA duplex (400 nM) for the Gab1 knockdown was delivered into HEK 293 cells by Oligofectamine. Sixty hours later, the cells were serum starved for 12 h and stimulated with or without HGF for 20 min before lysis. To examine the expression level of Gab1, whole-cell lysates (WCL) were analyzed by immunoblotting (IB) with anti-Gab1 (α-Gab1). The coimmunoprecipitation (IP) of FAK and MET was analyzed. (B) Tpr-Met was transiently expressed in Gab1+/+ cells or Gab1−/− cells, and its coprecipitation with FAK was examined. (C) HEK 293 cell lysates containing HA-tagged FAK or HA-tagged Gab1 were incubated with the lysates containing Tpr-Met in the presence or absence of a 16-amino-acid peptide containing the Met-binding sequence (MBS) of Gab1. One hour later, coprecipitation of Tpr-Met with HA-tagged FAK or Gab1 was examined. IgG, immunoglobulin G.
Direct Met-FAK interaction requires phosphorylation of Met at Tyr-1349 and Tyr-1356.
To examine whether FAK directly interacts with Met, Met-FAK interaction was reconstituted in vitro. GST fusion proteins encoding Tpr-Met and its various mutants were expressed in bacteria and then subjected to an in vitro pull-down assay to analyze their abilities to bind to purified baculovirus-expressed recombinant FAK proteins (Fig. 3A). Except for the kd mutant, GST-Tpr-Met was apparently tyrosine phosphorylated, indicating that Tpr-Met was active and underwent autophosphorylation in bacteria (Fig. 3B). The phosphorylated GST-Tpr-Met bound to recombinant FAK in vitro, and, in contrast, the GST-Tpr-Met kd mutant which failed to phosphorylate itself was unable to interact with recombinant FAK (Fig. 3B). To examine whether phosphorylation is indeed the key determinant for Met to bind to FAK, GST-Tpr-Met was pretreated with alkaline phosphatase in vitro to remove its phosphate groups. The dephosphorylated GST-Tpr-Met decreased its association with recombinant FAK (Fig. 3C). In accordance with the results shown in Fig. 1D, mutation at Tyr-482 or Tyr-489 impaired the ability of GST-Tpr-Met to bind to recombinant FAK (Fig. 3B). To further examine whether the phosphorylation of c-Met at Tyr-1349 and Tyr-1356 is important for its interaction with FAK, a synthetic phosphopeptide (IGEHPY1349VHVNATPY1356VNVK) corresponding to the sequence surrounding Tyr-1349 and Tyr-1356 was used as a competitor for Tpr-Met to bind to recombinant FAK proteins in vitro (Fig. 3D). Apparently, the phosphopeptide, but not its unphosphorylated counterpart, inhibited the in vitro association of Tpr-Met with FAK, suggesting that a short stretch of c-Met covering its phosphorylated Tyr-1349 and Tyr-1356 is involved in FAK binding. Taken together, these results indicate that Met directly interacts with FAK via its phosphorylated Tyr-1349 and Tyr-1356.
FIG. 3.

Direct interaction between Met and FAK. (A) Baculovirus-expressed recombinant FAK proteins (bac-FAK) were purified from insect cell lysates by affinity chromatography. The crude insect cell lysates, the protein A-Sepharose-bound proteins (on beads), and the eluted proteins (eluted) were fractionated by SDS-polyacrylamide gel electrophoresis and visualized by Coomassie blue staining. (B) Immobilized GST fusion proteins encoding Tpr-Met or its mutants were incubated with purified baculovirus-expressed FAK proteins, and the bound proteins were analyzed by immunoblotting (IB) with anti-FAK (α-FAK). Total tyrosine phosphorylation of GST-Tpr-Met and its specific phosphorylation at Tyr-482 (equivalent to Tyr-1349 of c-Met) were analyzed by immunoblotting with antiphosphotyrosine (α-PY) and anti-Met-Y1349-P, respectively. The GST fusion proteins on the nitrocellulose membrane were visualized by staining with Ponceau S solution. (C) Dephosphorylation of Tpr-Met decreases its association with FAK in vitro. Immobilized GST-Tpr-Met was treated with 10 U of alkaline phosphatase (AP) in the presence or absence of 2 mM sodium vanadate (Na3VO4) for 1 h and then incubated with baculovirus-expressed recombinant FAK. The bound recombinant FAK proteins were analyzed by immunoblotting with anti-FAK. The tyrosine phosphorylation of GST-Tpr-Met was analyzed by immunoblotting with anti-PY. (D) HEK 293 cell lysates containing Tpr-Met were incubated with baculovirus-expressed recombinant FAK proteins in the presence of different amounts of the phosphopeptide or its unphosphorylated counterpart. One hour later, coprecipitation of Tpr-Met and baculovirus-expressed recombinant FAK was analyzed. Molecular size markers (in kilodaltons) are noted at the left of blots. IP, immunoprecipitation; IgG, immunoglobulin G.
The FERM domain of FAK directly binds to Met.
To determine the region of FAK responsible for Met binding, the FERM domain (aa 30 to 377) and the COOH-terminal domain (aa 693 to 1053) of FAK were transiently coexpressed with c-Met in HEK 293 cells. The results showed that the FERM domain of FAK, but not its COOH-terminal domain, was sufficient to form stable complexes with c-Met in intact cells (Fig. 4A). Like full-length FAK, the FERM domain did not bind to the Tpr-Met mutant with mutations at Tyr-482 and Tyr-489 (Fig. 4B). The purified histidine-tagged FAK NH2 domain (His-FAK-N) was sufficient to bind to GST-Tpr-Met but not the FF mutant (Fig. 4C), supporting a direct interaction between both molecules. Like the FAK-Met interaction, the FERM domain-Met interaction was blocked by the phosphopeptide IGEHPY1349VHVNATPY1356VNVK (Fig. 4D). Moreover, the FERM domain of FAK was capable of competing with the full-length FAK for Met binding in vitro (Fig. 4E). These results clearly indicate that FAK directly interacts with Met via its FERM domain.
FIG. 4.

The FERM domain of FAK binds to Met. (A) HA-tagged full-length (FL) FAK or its FERM domain (aa 30 to 377) or COOH domain (aa 693 to 1053) was transiently coexpressed with c-Met in HEK 293 cells. Coimmunoprecipitation (IP) of HA-tagged FAK proteins and c-Met was analyzed. (B) The HA-tagged FAK FERM domain was transiently coexpressed with Tpr-Met or its mutants in HEK 293 cells, and their coprecipitation was analyzed. (C) Purified GST-Tpr-Met and the histidine-tagged NH2 domain of FAK (His-FAK-N) were analyzed by SDS-polyacrylamide gel electrophoresis and visualized by Coomassie blue staining (left). Soluble GST-Tpr-Met or its FF mutant was incubated with purified His-FAK-N. One hour later, glutathione agarose beads were added to the mixture and the bound proteins were analyzed by immunoblotting (IB) with an anti-FAK antibody (A-17) specific to the NH2 domain of FAK (α-FAK-N) (right). Molecular size markers (in kilodaltons) are noted at the left of blots. (D) HEK 293 cell lysates containing Tpr-Met and those containing the HA-tagged FAK FERM domain were mixed and incubated with (+) or without (−) 200 μM of the phosphopeptide IGEHPY1349VHVNATPY1356VNVK or its unphosphorylated peptide. One hour later, coprecipitation of Tpr-Met and the HA-tagged FAK FERM domain was analyzed. (E) The FAK FERM domain competes with the full-length FAK for Tpr-Met binding in vitro. HEK 293 cell lysates containing Tpr-Met were incubated with baculovirus-expressed FAK (bac-FAK) in the presence of increasing amounts of HEK 293 cell lysates containing the HA-tagged FAK FERM domain. Coprecipitation of Tpr-Met and HA-tagged proteins was analyzed. IgG, immunoglobulin G; WCL, whole-cell lysates.
A basic patch (216KAKTLRK222) in the F2 subdomain of the FAK FERM domain is critical for Met binding.
The FAK FERM domain contains three subdomains, F1, F2, and F3 (6). To identify which of these subdomains are involved in Met binding, GST fusion proteins encoding the FAK FERM domain or its truncated forms were generated. The F2 subdomain (aa 126 to 253) alone was sufficient to bind to Tpr-Met from HEK 293 cells (Fig. 5A) or purified histidine-tagged Tpr-Met from bacteria (Fig. 5B). The FERM domain with deletion of the F1 or F3 subdomain retained its ability to bind to Tpr-Met (Fig. 5A). Moreover, the F2 subdomain contains a patch of basic residues (216KAKTLRK222) that are highly conserved in the FAK family and play a role in activation of FAK upon cell adhesion (14). To explore the role of those basic residues in Met-FAK interaction, a series of the FERM domain mutants were generated. A single mutation of the FERM domain at Lys-216, Lys-218, or Lys-222 severely impaired its interaction with Tpr-Met both in vivo (Fig. 5C) and in vitro (Fig. 5D). For the full-length FAK, a single mutation at Lys-218 or Lys-222 had no effect on its tyrosine phosphorylation, catalytic activity, or ability to promote haptotaxis (see Fig. S1 in the supplemental material). However, mutation at Lys-218 and, in particular, Lys-222 of the full-length FAK significantly inhibited its interaction with c-Met in intact cells (Fig. 5E). These results suggest that the basic patch 216KAKTLRK222 in the F2 subdomain of the FAK FERM domain may be involved in the interaction with phosphorylated Tyr-1349 and Tyr-1356 of c-Met.
FIG. 5.

The basic residues in the F2 subdomain of the FAK FERM domain are critical for FAK to interact with Met. (A) Immobilized GST fusion proteins encoding the FAK FERM domain or its truncated mutants were incubated with HEK 293 cell lysates containing Tpr-Met. The bound proteins were analyzed by immunoblotting (IB) with anti-Met (α-Met). The GST fusion proteins on the nitrocellulose membrane were visualized by staining with Ponceau S solution. The intact GST fusion proteins on the membrane are marked by circles. (B) Immobilized GST fusion proteins encoding the F1, F2, or F3 subdomain of the FAK FERM domain were incubated with purified histidine-tagged Tpr-Met (His-Tpr-Met). The bound proteins were resolved by SDS-polyacrylamide gel electrophoresis and visualized by Coomassie blue staining (left) or subjected to immunoblotting with anti-Met (right). (C) Tpr-Met was transiently coexpressed with the HA-tagged FAK NH2 domain or its mutants in HEK 293 cells. Coprecipitation of Tpr-Met and the HA-tagged FAK NH2 domain was analyzed. (D) Immobilized GST-Tpr-Met was incubated with HEK 293 cell lysates containing the HA-tagged FAK NH2 domain or its mutants. The bound proteins were analyzed by immunoblotting with anti-HA. (E) c-Met was transiently coexpressed with HA-tagged full-length (FL) FAK or its mutants in HEK 293 cells. Coprecipitation of c-Met with HA-tagged FAK was analyzed. Molecular size markers (in kilodaltons) are noted at the left of blots. IgG, immunoglobulin G; IP, immunoprecipitation; WCL, whole-cell lysates.
Met-FAK interaction leads to FAK activation.
To examine whether Met-FAK interaction leads to FAK activation, the tyrosine phosphorylation and catalytic activity of FAK were measured. Elevated expression of c-Met led to an increase not only in Met-FAK interaction but also in the tyrosine phosphorylation of FAK (Fig. 6A). Whether Tpr-Met was transiently expressed in HEK 293 cells or stably expressed in NIH 3T3 cells, it prominently stimulated the tyrosine phosphorylation of FAK (Fig. 6B). In contrast, the K241A (kd) mutant and the Y482F/Y489F mutant of Tpr-Met had no such effect (Fig. 6B). Moreover, the FAK K222A mutant deficient in Met binding was refractory to phosphorylation by Tpr-Met (Fig. 6C), indicating that direct Met-FAK interaction is essential for Met to phosphorylate FAK. To measure the catalytic activity of FAK, GST-FAK (aa 378 to 406), which encodes 29 residues flanking the FAK autophosphorylation site Tyr-397, was used as a substrate in an in vitro kinase assay. This substrate was phosphorylated only by FAK, not by Tpr-Met or Src (Fig. 6D); therefore, its phosphorylation could faithfully reflect the catalytic activity of FAK. Our results showed that Tpr-Met, but not its Y482F/Y489F mutant, stimulated the catalytic activity of FAK in NIH 3T3 cells (Fig. 6E). In addition, when Tpr-Met was expressed in FAK−/− cells, the catalytic activity of exogenous FAK was activated by Tpr-Met; in contrast, the FAK K222A mutant was refractory to stimulation by Tpr-Met (Fig. 6F). To demonstrate that Met-FAK interaction activates FAK in vitro, purified GST-Tpr-Met or its FF mutant was allowed to interact with purified baculovirus-expressed FAK and then the effect on the catalytic activity of baculovirus-expressed FAK was measured. The results (Fig. 6G) showed that the catalytic activity of FAK was apparently (15-fold) activated by Tpr-Met but activated only slightly by the Tpr-Met FF mutant. Taken together, these results indicate that Met-FAK interaction is a prerequisite for efficient activation of FAK by Met.
FIG. 6.

Met-FAK interaction leads to FAK activation. (A) The tyrosine phosphorylation of endogenous FAK was analyzed with HEK 293 cells transiently expressing increasing amounts of c-Met. To measure the phosphorylation of c-Met, whole-cell lysates (WCL) were analyzed by immunoblotting (IB) with phospho-specific antibodies. (B) v-Src or Tpr-Met was transiently expressed in HEK 293 cells or stably expressed in NIH 3T3 cells. The tyrosine phosphorylation of FAK in those cells was analyzed. (C) HA-tagged FAK or its K222A mutant was transiently coexpressed with (+) or without (−) Tpr-Met in FAK−/− cells. The tyrosine phosphorylation of HA-tagged FAK was analyzed. (D) In vitro kinase assays for Src, Tpr-Met, and FAK, with GST-FAK (aa 378 to 406) as a substrate. Note that GST-FAK (aa 378 to 406) is phosphorylated only by FAK but not by Src, Tpr-Met, or the FAK kd mutant. (E) The kinase activity of endogenous FAK in NIH 3T3 cells stably expressing Tpr-Met or its Y482F/Y489F mutant was analyzed by an in vitro kinase assay using GST-FAK (aa 378 to 406) as a substrate. The results are the averages of two experiments. (F) The kinase activity of HA-tagged FAK or its K222A mutant expressed with or without Tpr-Met in FAK−/− cells was analyzed by an in vitro kinase assay using GST-FAK (aa 378 to 406) as a substrate. The results are the averages of two experiments. (G) Purified baculovirus-expressed FAK (bac-FAK) was incubated with soluble GST-Tpr-Met or its FF mutant in the presence of unlabeled ATP (30 μM). One hour later, the baculovirus-expressed FAK was immunoprecipitated (IP) by anti-HA (α-HA) and subjected to an in vitro kinase assay using GST-FAK (aa 378 to 406) as a substrate in the presence of [γ-32P]ATP. Molecular size markers (in kilodaltons) are noted at the left of blots. α-PY, antiphosphotyrosine; IgG, immunoglobulin G.
Met directly phosphorylates FAK.
We have previously shown that Src is involved in HGF-induced phosphorylation of FAK (8). To examine the necessity of Src in Met-stimulated FAK phosphorylation, the effect of Met on the tyrosine phosphorylation of FAK was analyzed with SYF (src−/− yes−/− fyn−/−) cells. Tpr-Met, but not its FF mutant, stimulated the tyrosine phosphorylation of FAK in SYF cells (Fig. 7A), indicating that the Src family kinases are not required for Met to stimulate FAK phosphorylation. Interestingly, the tyrosine residues known to be phosphorylated by Src were also susceptible to phosphorylation by Tpr-Met (Fig. 7B). Next, to examine whether increased phosphorylation of FAK in Tpr-Met-expressed cells resulted from its autophosphorylation, FAK or its two mutants, the kd mutant and the Y397F mutant, were transiently expressed in the FAK−/− cells expressing Tpr-Met. The results showed that Tpr-Met stimulated the phosphorylation of both FAK kd and Y397F mutants to the same level as that of wt FAK (Fig. 7C), indicating that the catalytic activity and the autophosphorylation site at Tyr-397 of FAK were not required for Tpr-Met to stimulate FAK phosphorylation. Except for Tyr-397, the phosphorylation sites for the FAK Y397F mutant for Tpr-Met were the same as those for wt FAK (see Fig. S2 in the supplemental material). Finally, to examine whether Met could directly phosphorylate FAK in vitro, the purified baculovirus-expressed FAK kd mutant was used as a substrate for Tpr-Met in an in vitro kinase assay. Indeed, Tpr-Met was able to phosphorylate recombinant FAK kd proteins in vitro (Fig. 7D), with a Km of approximately 0.12 μM (data not shown). The in vitro phosphorylation sites of recombinant FAK kd proteins for Tpr-Met were the same as those identified in vivo (Fig. 7E).
FIG. 7.

Met directly phosphorylates FAK. (A) The tyrosine phosphorylation of endogenous FAK in the SYF cells transiently expressing v-Src, Tpr-Met, or its mutant was examined. (B) The phosphorylation of FAK was analyzed by using antibodies specific to phosphorylated FAK at indicated tyrosine residues. (C) HA-tagged FAK or its mutants were transiently coexpressed with Tpr-Met in FAK−/− cells. The tyrosine phosphorylation of HA-tagged FAK was analyzed. (D) In vitro kinase assays for v-Src and Tpr-Met were performed using the baculovirus-expressed FAK kd mutant (bac-FAK-kd) as a substrate in the presence or absence of unlabeled ATP. The mixture of the immunocomplexes and the substrate was analyzed by immunoblotting (IB) with antiphosphotyrosine (α-PY). Molecular size markers (in kilodaltons) are noted at the left of blots. (E) The purified baculovirus-expressed FAK kd mutant was incubated with soluble GST-Tpr-Met or its FF mutant in the presence of unlabeled ATP. One hour later, the baculovirus-expressed FAK kd mutant was immunoprecipitated (IP) with anti-HA and the immunocomplexes were analyzed by immunoblotting with antibodies specific to phosphorylated FAK at indicated tyrosine residues. WCL, whole-cell lysates.
Met-FAK interaction is essential for FAK to promote HGF-stimulated cell invasion.
We have previously shown that FAK overexpression promotes HGF-induced cell motility and Matrigel invasion (7, 24). To examine whether Met-FAK interaction is essential for FAK to promote both events, wt FAK and its K222A mutant were stably expressed (twofold greater than endogenous levels) in MDCK cells. As expected, wt FAK, but not its K222A mutant, was associated with the phosphorylated c-Met upon HGF stimulation (Fig. 8A). The ability of the K222A mutant to promote HGF-induced cell scattering (Fig. 8B) and cell migration (Fig. 8C) was approximately 50% lower than that of wt FAK, indicating that Met-FAK interaction is important, but not absolutely necessary, for FAK to promote HGF-induced cell motility. However, the K222A mutant failed to promote HGF-induced Matrigel invasion (Fig. 8D), suggesting that the ability of FAK to promote HGF-induced invasion may rely largely on its interaction with c-Met.
FIG. 8.

Met-FAK interaction is required for FAK to promote HGF-stimulated cell invasion. (A) MDCK cells stably overexpressing HA-tagged FAK or its K222A mutant deficient in Met binding were established. Equal amounts of cell lysates were incubated with anti-HA (α-HA), and the immunocomplexes were analyzed by immunoblotting (IB) with anti-FAK or anti-phospho-Met (α-Met-Y1234/1235-P). (B) Control MDCK cells and those overexpressing FAK or its K222A mutant were subjected to an HGF-induced scattering assay. Data (means ± standard errors) are from three experiments.  , P < 0.05, compared with the control MDCK cells. (C) The MDCK cells were subjected to a cell migration assay. , P < 0.05, compared with the control MDCK cells in the presence of HGF. (D) The MDCK cells were subjected to a Matrigel invasion assay. , P < 0.05, compared with the control MDCK cells in the presence of HGF. (E) Met-FAK interaction was analyzed with a series of lung cancer cell lines. The ability of those cells to invade through Matrigel was measured. , P < 0.05, compared with A549 cells. (F) CL1-5 lung cancer cells stably expressing the FAK NH2 domain were established. The inhibitory effect of the FAK NH2 domain on Met-FAK interaction was examined. (G) CL1-5 cells and those stably expressing the FAK NH2 domain were subjected to a Matrigel invasion assay. IP, immunoprecipitation; WCL, whole-cell lysates; IgG, immunoglobulin G.
, P < 0.05, compared with the control MDCK cells. (C) The MDCK cells were subjected to a cell migration assay. , P < 0.05, compared with the control MDCK cells in the presence of HGF. (D) The MDCK cells were subjected to a Matrigel invasion assay. , P < 0.05, compared with the control MDCK cells in the presence of HGF. (E) Met-FAK interaction was analyzed with a series of lung cancer cell lines. The ability of those cells to invade through Matrigel was measured. , P < 0.05, compared with A549 cells. (F) CL1-5 lung cancer cells stably expressing the FAK NH2 domain were established. The inhibitory effect of the FAK NH2 domain on Met-FAK interaction was examined. (G) CL1-5 cells and those stably expressing the FAK NH2 domain were subjected to a Matrigel invasion assay. IP, immunoprecipitation; WCL, whole-cell lysates; IgG, immunoglobulin G.
To examine the significance of Met-FAK interaction in the invasiveness of tumor cells, the correlation between Met-FAK interaction and invasiveness was analyzed with lung cancer cells. Met-FAK interaction was detected in some of the lung cancer cell lines (CL1-5, H23, and H838), correlated with their invasiveness (Fig. 8E). To further demonstrate that Met-FAK interaction is critical for tumor cells to acquire invasive potential, the NH2-terminal domain of FAK was stably expressed in CL1-5 lung cancer cells to compete with endogenous FAK for c-Met binding in intact cells. The expression of the NH2-terminal domain of FAK in CL1-5 cells did not interfere with the integrin-mediated signaling (see Fig. S3 in the supplemental material). However, it successfully blocked Met-FAK interaction in CL1-5 cells (Fig. 8F) and, more importantly, it significantly suppressed the invasiveness of the cells (Fig. 8G). These results together implicate that Met-FAK interaction may be a critical determinant for at least certain types of cancer cells to acquire invasive potential.
DISCUSSION
FAK is already known for its pivotal role in the regulation of integrin-mediated cell functions. In this study, we report that upon HGF stimulation FAK is recruited by phosphorylated c-Met, leading to its activation and subsequent contribution to HGF-induced cell migration and invasion. We have identified that the F2 subdomain (aa 126 to 253) of the FERM domain of FAK is responsible for its interaction with c-Met. Since the F2 subdomain lacks obvious sequence similarity to other SH2 and phosphotyrosine binding domains, it may represent a novel type of phosphotyrosine binding domain. Moreover, we have identified lysine residues in a basic patch (216KAKTLRK222) of the F2 subdomain as critical for Met-FAK interaction. Since the sequence of the basic patch has no similarity to the Met-binding sequence (GMQVPPPAHMGFR) of Gab1 (26, 41), the mode of FAK to interact with Met is likely to be different from that of Gab1. This notion is supported by our finding that a Met mutant (the Y1349E mutant) with a substitution of glutamic acid for Tyr-1349 constitutively forms complexes with Gab1 but fails to interact with FAK (data not shown). This finding also indicates that the negative charge at residue 1349 is not the only determinant for c-Met to interact with FAK.
Although the FERM domain of FAK was reported to interact with the PDGF receptor and the EGF receptor (43), the modes for their interactions remain to be elucidated. In this study, we show that phosphorylation of c-Met at Tyr-1349 and, to a lesser extent, Tyr-1356 is essential for its interaction with FAK. Moreover, we demonstrate that the F2 subdomain of the FAK FERM domain by itself is sufficient to bind to phosphorylated Met and that the lysine residues in the 216KAKTLRK222 patch of the F2 subdomain are critical for the interaction. Experiments are in progress to examine whether those basic residues in the F2 subdomain also mediate the interaction of FAK with both PDGF and EGF receptors. In addition, the FAK FERM domain has been reported to interact with the cytoplasmic region of integrins (9, 42), the FERM domain of ezrin (36), and the pleckstrin homology domain of the Tec family kinase Etk (9). It will be of interest to dissect the modes by which the FAK FERM domain interacts with those molecules.
The overall three-lobed architecture of ERM family FERM domains is preserved in FAK (6), whose F2 lobe is composed of all α-helices, with a core four-helix bundle similar to that found in the acyl-coenzyme A binding protein (23). However, the 216KAKTLRK222 patch, which is present in the α3 bundle of the F2 lobe, is unique and well conserved in the FAK family. The positive charges of all four basic residues in the patch are exposed to the surface of the structure and form a basic region at the tip of the F2 lobe. Interestingly, the residues of ezrin, radixin, moesin, and merlin, corresponding to Lys-218, Arg-221, and Lys-222 of FAK, are all acidic. The distance between Lys-216 and Lys-222 of FAK is estimated to be approximately 16 to 17 Å, close to that between Tyr-1349 and Tyr-1356 of c-Met. Nevertheless, the physical nature of the interaction remains to be determined.
It was proposed that the intramolecular interaction of the FAK FERM domain with the kinase domain suppresses the catalytic activity of FAK (13). Since Met-FAK interaction leads to FAK activation, it is possible that the FERM domain-mediated Met interaction may alleviate the intramolecular inhibitory interaction of FAK, thereby leading to its activation. Moreover, Met-mediated FAK phosphorylation could further activate FAK. Indeed, we found that Met phosphorylates FAK at its known phosphorylation sites, including Tyr-576 and Tyr-577, both of which are located in the activating loop within the catalytic domain (Fig. 7B). In addition, we found that Met phosphorylates the NH2-terminal domain of FAK (unpublished data). The functional significance of the phosphorylation at the FAK NH2-terminal domain is under investigation.
Dunty et al. (14) reported that the conserved basic residues in the 216KAKTLRK222 patch are critical for the FAK FERM domain to interact with another FAK molecule and play a role in adhesion-dependent activation of FAK. They thought that this intermolecular interaction could be indirectly mediated by an unknown protein. To our knowledge, c-Met is the first protein to be reported for its direct interaction with the 216KAKTLRK222 patch of the FERM domain. It is not clear whether c-Met could mediate the interaction of the FERM domain with another full-length FAK under any circumstances. Of course, it remains possible that the 216KAKTLRK222 patch could interact with other cellular proteins which then serve as a link for the FERM-FAK interaction. Moreover, it was reported that triple mutations at Lys-216, Lys-218, and Arg-221 of FAK had little effect on its catalytic activity in vitro but that they caused FAK to be defective in adhesion-dependent tyrosine phosphorylation, Src family binding, and ability to promote haptotaxis (14). However, we found in this study that a single mutation at Lys-218 or Lys-222 of FAK has no inhibitory effect on its catalytic activity, tyrosine phosphorylation, or ability to promote haptotaxis (see Fig. S1 in the supplemental material). Although the reason for this discrepancy is unknown, it is possible that the intermolecular interaction of the FERM domain with another FAK molecule may be disturbed only by triple mutations at Lys-216, Lys-218, and Arg-221, but not by a single mutation at Lys-222.
We have previously shown that Src participates in HGF-induced FAK phosphorylation (8). In this study, we show that Met stimulates FAK phosphorylation in SYF cells (Fig. 7A and B), indicating that the Src family kinases are not required for Met to phosphorylate FAK. However, the contribution of Src to HGF-stimulated FAK phosphorylation cannot be excluded. It is possible that the maximal activation of FAK upon HGF stimulation may be reached only when both Met-FAK and Met-Src-FAK pathways are activated (Fig. 9). Our results show that the FAK K222A mutant deficient in c-Met binding can still promote HGF-induced MDCK cell motility to a level approximately 50% of that promoted by wt FAK (Fig. 8B and C), suggesting that HGF-stimulated activation of FAK through the Met-FAK pathway and the Met-Src-FAK pathway may be equally important for FAK to promote HGF-stimulated cell motility. However, the results shown in Fig. 8D indicate that the FAK K222A mutant fails to promote HGF-induced invasion, indicating that the ability of FAK to promote cell invasion depends largely on its direct interaction with c-Met. These results together suggest that the signals transmitted through the Met-FAK pathway might be somewhat different from those transmitted through the Met-Src-FAK pathway. This assumption also underscores that signals propagated by reciprocal regulation between Met and FAK may be necessary for cells to acquire invasive potential.
FIG. 9.

Met activates FAK through both the Met-Src-FAK pathway (our previous studies) and the Met-FAK pathway (this study). The activated FAK functions as a signaling platform to potentiate HGF-elicited signals, thereby contributing to HGF-stimulated cell motility and cell invasion. Note that the ability of FAK to promote HGF-stimulated invasion relies largely on its direct interaction with Met (see Discussion for details). P, phosphorylation.
We demonstrate here that Met-FAK interaction is correlated with the invasiveness of lung cancer cells (Fig. 8E). Moreover, blockade of Met-FAK interaction by expression of the FAK NH2 domain in highly invasive lung cancer cells significantly suppressed their invasiveness (Fig. 8G). It is worth noting that in the absence of HGF stimulation Met-FAK interaction was detected only in highly invasive cancer cells but not in less invasive cancer cells (Fig. 8E). These results suggest that under certain circumstances, for example, when c-Met is overexpressed and/or constitutively activated, Met-FAK interaction could become constitutive independently of HGF stimulation. In fact, we have found that Met-FAK interaction can be detected only in tumor lesions of specimens from lung cancer patients, not in paired nontumor lesions (unpublished data). Therefore, our results implicate that Met-FAK interaction may play a role in tumorigenesis and serve as a potential target for therapeutic purposes.
Supplementary Material
Acknowledgments
We thank D. Ilic for FAK-null MEF, T. Hirano for Gab1-null MEF, P. Soriano for SYF cells, P.-C. Yang for lung cancer cell lines, J.-L. Guan for recombinant baculoviruses, G. Vande Woude for c-Met cDNA, P. M. Comoglio and M. Park for the Tpr-Met constructs, J. T. Parsons for FAK cDNA, and W.-M. Yang for the pcDNA3.1-HA vector. We are grateful to P.-C. Chan, C.-M. Chen, and T.-H. Chen for assistance with plasmid construction and to Y.-C. Li for purification of the histidine-tagged FAK NH2 domain.
This work was supported by grants NSC-93-2311-B-005-012, NSC-94-2320-B005-003, NSC95-3112-B005-001, and TCVGH-NCHU947610 to H.-C.C.
Footnotes
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Bardelli, A., L. Pugliese, and P. M. Comoglio. 1997. “Invasive-growth” signaling by the Met/HGF receptor: the hereditary renal carcinoma connection. Biochim. Biophys. Acta 1333:M41-M51. [DOI] [PubMed] [Google Scholar]
- 2.Barret, C., C. Roy, P. Montcourrier, P. Mangeat, and V. Niggli. 2000. Mutagenesis of the phosphatidylinositol 4,5-bisphosphate (PIP(2)) binding site in the NH(2)-terminal domain of ezrin correlates with its altered cellular distribution. J. Cell Biol. 151:1067-1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Birchmeier, C., W. Birchmeier, E. Gherardi, and G. F. Vande Woude. 2003. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 4:915-925. [DOI] [PubMed] [Google Scholar]
- 4.Birchmeier, C., and E. Gherardi. 1998. Developmental roles of HGF/SF and its receptor, the c-Met tyrosine kinase. Trends Cell Biol. 8:404-410. [DOI] [PubMed] [Google Scholar]
- 5.Bottaro, D. P., J. S. Rubin, D. L. Faletto, A. M. Chan, T. E. Kmiecik, G. F. Vande Woude, and S. A. Aaronson. 1991. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 251:802-804. [DOI] [PubMed] [Google Scholar]
- 6.Ceccarelli, D. F., H. K. Song, F. Poy, M. D. Schaller, and M. J. Eck. 2006. Crystal structure of the FERM domain of focal adhesion kinase. J. Biol. Chem. 281:252-259. [DOI] [PubMed] [Google Scholar]
- 7.Chan, P. C., C. C. Liang, K. C. Yu, M. C. Chang, W. L. Ho, B. H. Chen, and H. C. Chen. 2002. Synergistic effect of focal adhesion kinase overexpression and hepatocyte growth factor stimulation on cell transformation. J. Biol. Chem. 277:50373-50379. [DOI] [PubMed] [Google Scholar]
- 8.Chen, H. C., P. C. Chan, M. J. Tang, C. H. Cheng, and T. J. Chang. 1998. Tyrosine phosphorylation of focal adhesion kinase stimulated by hepatocyte growth factor leads to mitogen-activated protein kinase activation. J. Biol. Chem. 273:25777-25782. [DOI] [PubMed] [Google Scholar]
- 9.Chen, L. M., D. Bailey, and C. Fernandez-Valle. 2000. Association of beta 1 integrin with focal adhesion kinase and paxillin in differentiating Schwann cells. J. Neurosci. 20:3776-3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen, R., O. Kim, M. Li, X. Xiong, J. L. Guan, H. J. Kung, H. Chen, Y. Shimizu, and Y. Qiu. 2001. Regulation of the PH-domain-containing tyrosine kinase Etk by focal adhesion kinase through the FERM domain. Nat. Cell Biol. 3:439-444. [DOI] [PubMed] [Google Scholar]
- 11.Chu, Y. W., P. C. Yang, S. C. Yang, Y. C. Shyu, M. J. Hendrix, R. Wu, and C. W. Wu. 1997. Selection of invasive and metastatic subpopulations from a human lung adenocarcinoma cell line. Am. J. Respir. Cell Mol. Biol. 17:353-360. [DOI] [PubMed] [Google Scholar]
- 12.Cooper, C. S., M. Park, D. G. Blair, M. A. Tainsky, K. Huebner, C. M. Croce, and G. F. Vande Woude. 1984. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature 311:29-33. [DOI] [PubMed] [Google Scholar]
- 13.Cooper, L. A., T. L. Shen, and J. L. Guan. 2003. Regulation of focal adhesion kinase by its amino-terminal domain through an autoinhibitory interaction. Mol. Cell. Biol. 23:8030-8041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dunty, J. M., V. Gabarra-Niecko, M. L. King, D. F. Ceccarelli, M. J. Eck, and M. D. Schaller. 2004. FERM domain interaction promotes FAK signaling. Mol. Cell. Biol. 24:5353-5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferracini, R., P. Longati, L. Naldini, E. Vigna, and P. M. Comoglio. 1991. Identification of the major autophosphorylation site of the Met/hepatocyte growth factor receptor tyrosine kinase. J. Biol. Chem. 266:19558-19564. [PubMed] [Google Scholar]
- 16.Giordano, S., C. Ponzetto, M. F. Di Renzo, C. S. Cooper, and P. M. Comoglio. 1989. Tyrosine kinase receptor indistinguishable from the c-met protein. Nature 339:155-156. [DOI] [PubMed] [Google Scholar]
- 17.Hamada, K., T. Shimizu, T. Matsui, S. Tsukita, and T. Hakoshima. 2000. Structural basis of the membrane-targeting and unmasking mechanisms of the radixin FERM domain. EMBO J. 19:4449-4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hatai, M., H. Hashi, A. Mogi, H. Soga, J. Yokota, and Y. Yaoi. 1994. Stimulation of tyrosine- and serine-phosphorylation of focal adhesion kinase in mouse 3T3 cells by fibronectin and fibroblast growth factor. FEBS Lett. 350:113-116. [DOI] [PubMed] [Google Scholar]
- 19.Hildebrand, J. D., M. D. Schaller, and J. T. Parsons. 1993. Identification of sequences required for the efficient localization of the focal adhesion kinase, pp125FAK, to cellular focal adhesions. J. Cell Biol. 123:993-1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ilic, D., Y. Furuta, S. Kanazawa, N. Takeda, K. Sobue, N. Nakatsuji, S. Nomura, J. Fujimoto, M. Okada, and T. Yamamoto. 1995. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature 377:539-544. [DOI] [PubMed] [Google Scholar]
- 21.Itoh, M., Y. Yoshida, K. Nishida, M. Narimatsu, M. Hibi, and T. Hirano. 2000. Role of Gab1 in heart, placenta, and skin development and growth factor- and cytokine-induced extracellular signal-regulated kinase mitogen-activated protein kinase activation. Mol. Cell. Biol. 20:3695-3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klinghoffer, R. A., C. Sachsenmaier, J. A. Cooper, and P. Soriano. 1999. Src family kinases are required for integrin but not PDGFR signal transduction. EMBO J. 18:2459-2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kragelund, B. B., K. V. Andersen, J. C. Madsen, J. Knudsen, and F. M. Poulsen. 1993. Three-dimensional structure of the complex between acyl-coenzyme A binding protein and palmitoyl-coenzyme A. J. Mol. Biol. 230:1260-1277. [DOI] [PubMed] [Google Scholar]
- 24.Lai, J. F., S. C. Kao, S. T. Jiang, M. J. Tang, P. C. Chan, and H. C. Chen. 2000. Involvement of focal adhesion kinase in hepatocyte growth factor-induced scatter of Madin-Darby canine kidney cells. J. Biol. Chem. 275:7474-7480. [DOI] [PubMed] [Google Scholar]
- 25.Liang, T. J., A. E. Reid, R. Xavier, R. D. Cardiff, and T. C. Wang. 1996. Transgenic expression of tpr-met oncogene leads to development of mammary hyperplasia and tumors. J. Clin. Investig. 97:2872-2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lock, L. S., M. M. Frigault, C. Saucier, and M. Park. 2003. Grb2-independent recruitment of Gab1 requires the C-terminal lobe and structural integrity of the Met receptor kinase domain. J. Biol. Chem. 278:30083-30090. [DOI] [PubMed] [Google Scholar]
- 27.Mangeat, P., C. Roy, and M. Martin. 1999. ERM proteins in cell adhesion and membrane dynamics. Trends Cell Biol. 9:187-192. [DOI] [PubMed] [Google Scholar]
- 28.Matsumoto, K., K. Matsumoto, T. Nakamura, and R. H. Kramer. 1994. Hepatocyte growth factor/scatter factor induces tyrosine phosphorylation of focal adhesion kinase (p125FAK) and promotes migration and invasion by oral squamous cell carcinoma cells. J. Biol. Chem. 269:31807-31813. [PubMed] [Google Scholar]
- 29.Mitra, S. K., D. A. Hanson, and D. D. Schlaepfer. 2005. Focal adhesion kinase: in command and control of cell motility. Nat. Rev. Mol. Cell Biol. 6:56-68. [DOI] [PubMed] [Google Scholar]
- 30.Naldini, L., E. Vigna, R. P. Narsimhan, G. Gaudino, R. Zarnegar, G. K. Michalopoulos, and P. M. Comoglio. 1991. Hepatocyte growth factor (HGF) stimulates the tyrosine kinase activity of the receptor encoded by the proto-oncogene c-MET. Oncogene 6:501-504. [PubMed] [Google Scholar]
- 31.Park, M., M. Dean, C. S. Cooper, M. Schmidt, S. J. O'Brien, D. G. Blair, and G. F. Vande Woude. 1986. Mechanism of met oncogene activation. Cell 45:895-904. [DOI] [PubMed] [Google Scholar]
- 32.Parsons, J. T. 2003. Focal adhesion kinase: the first ten years. J. Cell Sci. 116:1409-1416. [DOI] [PubMed] [Google Scholar]
- 33.Pearson, M. A., D. Reczek, A. Bretscher, and P. A. Karplus. 2000. Structure of the ERM protein moesin reveals the FERM domain fold masked by an extended actin binding tail domain. Cell 101:259-270. [DOI] [PubMed] [Google Scholar]
- 34.Pelicci, G., S. Giordano, Z. Zhen, A. E. Salcini, L. Lanfrancone, A. Bardelli, G. Panayotou, M. D. Waterfield, C. Ponzetto, and P. G. Pelicci. 1995. The motogenic and mitogenic responses to HGF are amplified by the Shc adaptor protein. Oncogene 10:1631-1638. [PubMed] [Google Scholar]
- 35.Ponzetto, C., A. Bardelli, Z. Zhen, F. Maina, P. dalla Zonca, S. Giordano, A. Graziani, G. Panayotou, and P. M. Comoglio. 1994. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell 77:261-271. [DOI] [PubMed] [Google Scholar]
- 36.Poullet, P., A. Gautreau, G. Kadare, J. A. Girault, D. Louvard, and M. Arpin. 2001. Ezrin interacts with focal adhesion kinase and induces its activation independently of cell-matrix adhesion. J. Biol. Chem. 276:37686-37691. [DOI] [PubMed] [Google Scholar]
- 37.Rankin, S., and E. Rozengurt. 1994. Platelet-derived growth factor modulation of focal adhesion kinase (p125FAK) and paxillin tyrosine phosphorylation in Swiss 3T3 cells. Bell-shaped dose response and cross-talk with bombesin. J. Biol. Chem. 269:704-710. [PubMed] [Google Scholar]
- 38.Rodrigues, G. A., M. A. Naujokas, and M. Park. 1991. Alternative splicing generates isoforms of the Met receptor tyrosine kinase which undergo differential processing. Mol. Cell. Biol. 11:2962-2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodrigues, G. A., and M. Park. 1994. Autophosphorylation modulates the kinase activity and oncogenic potential of the Met receptor tyrosine kinase. Oncogene 9:2019-2027. [PubMed] [Google Scholar]
- 40.Sachs, M., H. Brohmann, D. Zechner, T. Muller, J. Hulsken, I. Walther, U. Schaeper, C. Birchmeier, and W. Birchmeier. 2000. Essential role of Gab1 for signaling by the c-Met receptor in vivo. J. Cell Biol. 150:1375-1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schaeper, U., N. H. Gehring, K. P. Fuchs, M. Sachs, B. Kempkes, and W. Birchmeier. 2000. Coupling of Gab1 to c-Met, Grb2, and Shp2 mediates biological responses. J. Cell Biol. 149:1419-1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schaller, M. D., C. A. Otey, J. D. Hildebrand, and J. T. Parsons. 1995. Focal adhesion kinase and paxillin bind to peptides mimicking beta integrin cytoplasmic domains. J. Cell Biol. 130:1181-1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sieg, D. J., C. R. Hauck, D. Ilic, C. K. Klingbeil, E. Schaefer, C. H. Damsky, and D. D. Schlaepfer. 2000. FAK integrates growth-factor and integrin signals to promote cell migration. Nat. Cell Biol. 2:249-256. [DOI] [PubMed] [Google Scholar]
- 44.Smith, W. J., N. Nassar, A. Bretscher, R. A. Cerione, and P. A. Karplus. 2003. Structure of the active N-terminal domain of Ezrin. Conformational and mobility changes identify keystone interactions. J. Biol. Chem. 278:4949-4956. [DOI] [PubMed] [Google Scholar]
- 45.Soman, N. R., G. N. Wogan, and J. S. Rhim. 1990. TPR-MET oncogenic rearrangement: detection by polymerase chain reaction amplification of the transcript and expression in human tumor cell lines. Proc. Natl. Acad. Sci. USA 87:738-742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Soman, N. R., P. Correa, B. A. Ruiz, and G. N. Wogan. 1991. The TPR-MET oncogenic rearrangement is present and expressed in human gastric carcinoma and precursor lesions. Proc. Natl. Acad. Sci. USA 88:4892-4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weidner, K. M., C. S. Di, M. Sachs, V. Brinkmann, J. Behrens, and W. Birchmeier. 1996. Interaction between Gab1 and the c-Met receptor tyrosine kinase is responsible for epithelial morphogenesis. Nature 384:173-176. [DOI] [PubMed] [Google Scholar]
- 48.Weidner, K. M., M. Sachs, and W. Birchmeier. 1993. The Met receptor tyrosine kinase transduces motility, proliferation, and morphogenic signals of scatter factor/hepatocyte growth factor in epithelial cells. J. Cell Biol. 121:145-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xing, Z., H. C. Chen, J. K. Nowlen, S. J. Taylor, D. Shalloway, and J. L. Guan. 1994. Direct interaction of v-Src with the focal adhesion kinase mediated by the Src SH2 domain. Mol. Biol. Cell 5:413-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu, H., M. A. Naujokas, E. D. Fixman, K. Torossian, and M. Park. 1994. Tyrosine 1356 in the carboxyl-terminal tail of the HGF/SF receptor is essential for the transduction of signals for cell motility and morphogenesis. J. Biol. Chem. 269:29943-29948. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.