

Abstract
Mitochondrial dysfunction causes numerous human diseases and is widely believed to be involved in aging. However, mechanisms through which compromised mitochondrial gene expression elicits the reported variety of cellular defects remain unclear. The amino-terminal domain (ATD) of yeast mitochondrial RNA polymerase is required to couple transcription to translation during expression of mitochondrial DNA-encoded oxidative phosphorylation subunits. Here we report that several ATD mutants exhibit reduced chronological life span. The most severe of these (harboring the rpo41-R129D mutation) displays imbalanced mitochondrial translation, conditional inactivation of respiration, elevated production of reactive oxygen species (ROS), and increased oxidative stress. Reduction of ROS, via overexpression of superoxide dismutase (SOD1 or SOD2 product), not only greatly extends the life span of this mutant but also increases its ability to respire. Another ATD mutant with similarly reduced respiration (rpo41-D152A/D154A) accumulates only intermediate levels of ROS and has a less severe life span defect that is not rescued by SOD. Altogether, our results provide compelling evidence for the “vicious cycle” of mitochondrial ROS production and lead us to propose that the amount of ROS generated depends on the precise nature of the mitochondrial gene expression defect and initiates a downward spiral of oxidative stress only if a critical threshold is crossed.
Mitochondria play an essential role in a wide variety of cellular processes, including ATP production, anabolic and catabolic metabolism, signal transduction, and apoptosis. Mitochondria also contain their own DNA (mtDNA), which encodes a small but essential subset of the oxidative phosphorylation (OXPHOS) machinery responsible for the production of ATP, the remaining subunits of which are encoded by nuclear DNA (26). The nuclear and mitochondrial genomes must therefore coordinate their actions to ensure proper mitochondrial function, and inherited mitochondrial diseases can be the result of mutations in either genome (7, 24, 36). Additionally, mitochondrial dysfunction has long been implicated in age-related pathology and has been linked to a number of increasingly prevalent diseases, including Parkinson's disease, Alzheimer's disease, and diabetes (36). It is therefore clear that an understanding of mitochondrial function and dysfunction is essential to improve human health and possibly to ameliorate aging.
One cost incurred by the use of oxygen for respiration is the production of reactive oxygen species (ROS) such as superoxide, hydrogen peroxide, and hydroxyl radicals. The “mitochondrial theory” of aging posits that ROS produced as by-products of respiration are responsible for cumulative cellular damage and death (20). This theory explains a great number of observations, such as the accumulation with age of mtDNA mutations and deletions, correlating with declines in respiratory capacity and the appearance of morphologically altered and dysfunctional mitochondria (28). However, studies of aging are often correlative and therefore abrogate the separation of cause and effect (2). Notable exceptions include two recent reports of mice with a mitochondrial mutator phenotype, containing a knock-in version of a proofreading-deficient allele of mtDNA polymerase. These animals show accelerated accumulation of mtDNA mutations and remarkably exhibit overt signs of premature aging (16, 33).
An often-cited corollary to the mitochondrial theory of aging is the “vicious cycle” of ROS production and oxidative damage (3). Briefly, it is thought that an inevitable production of low levels of ROS during oxidative metabolism damages mtDNA and/or mitochondrial proteins. This damage is thought to result in faulty oxidative phosphorylation, leading to still more ROS and perpetuation of the cycle. However, like the mitochondrial theory of aging itself, the existence of the vicious cycle has remained a point of controversy (2). Furthermore, despite the common misconception that high levels of respiration will necessarily generate more ROS and decrease life span, the precise relationships among respiration rate, ROS production, and life span are still far from clear (31). Mitochondrial respiration is indeed responsible for generating the majority of endogenous ROS in many cell types (5, 12, 20), but increased respiration has been associated with either enhanced or reduced rates of ROS production (4). Additionally, extension of life span has been attributed to both increases and decreases in respiration (2, 18, 30). Various respiration inhibitors lead to differential effects on the production of ROS, depending on their site and mode of action (17), and it has even been reported that mild uncoupling of electron transport and ATP synthesis is correlated with longer life span in mice (31).
The budding yeast Saccharomyces cerevisiae, which has long served as a model system for understanding mitochondrial gene expression, function, and biogenesis (25, 35), has recently been used to investigate conserved aspects of both chronological and replicative life span (8, 14). Chronological life span is defined as the ability of a nondividing cell to remain viable over time and is measured in yeast by assaying for viability in stationary phase (8). As for metazoans, there exists a substantial body of work on yeast tying ROS and mitochondrial integrity to the determination of life span (1, 14). Two critical factors in the detoxification of ROS in yeast are the superoxide dismutases (SODs): Sod1p, which localizes to the cytoplasm and the mitochondrial intermembrane space (32), and Sod2p, a mitochondrial matrix protein. Both SOD genes are transcriptionally activated by the partially redundant, stress-responsive transcription factors Msn2p and Msn4p (9).
In S. cerevisiae, expression of the mitochondrial genome is initiated with transcription by a dedicated mitochondrial RNA (mtRNA) polymerase encoded by the nuclear RPO41 gene (11). Transcription is functionally coupled to translation at the inner mitochondrial membrane via an association of the amino-terminal domain (ATD) of mtRNA polymerase with the coupling factors Nam1p and Sls1p (6, 22, 24). Though the ATD is dispensable for transcription initiation (38), we previously reported that mutations in this domain generally abolish interaction with Nam1p (22), resulting in a global reduction of mitochondrial translation and hence misassembly of the OXPHOS complexes (23). However, one such mutant (rpo41-R129D, strain GS129) exhibits a phenotype distinct from that of other ATD mutants studied. This mutant has the most severe glycerol growth phenotype (22), uniquely retains the ability to interact with Nam1p (22), and has relatively robust mitochondrial translation (reported as “data not shown” in reference 23). Yet, like other ATD mutants, the steady-state levels of the intron-containing mtDNA-encoded COX1 and COB messages are decreased (22), indicative of a defect in translation, which is required in yeast mitochondria for splicing of introns present in these two mRNAs and hence their stability. The transcript and the glycerol growth defects of GS129 are both partially rescued by the overexpression of Sls1p (6). Altogether, these observations indicate that GS129 has a unique defect in OXPHOS complex assembly due to altered mitochondrial translation.
Given their variable phenotypes (due to mutations that affect the same process), the ATD mutants presented a unique opportunity to uncover mechanisms that underlie the diverse cellular consequences that arise when expression of the mtDNA-encoded subunits of the OXPHOS complexes are perturbed in different ways. Here we describe novel insights into the biological relevance of coupling mitochondrial transcription to translation with regard to respiration, ROS production, oxidative stress, and life span gained through a comprehensive analysis of our ATD mutant collection.
MATERIALS AND METHODS
Yeast strains, plasmids, and media used.
All yeast strains used in this work are derivatives of DBY2006 (MATα his3-Δ200 leu2-3,112 ura3-52 trp1-Δ1 ade2-1). The construction of GS122, GS129, and GS130 has been described previously (22). GS141 is a nam1Δ strain with Nam1p functions complemented by the NAM1 gene on a plasmid and has been described previously (6). The nam1Δ strain that is partially rescued by a plasmid-borne SLS1 gene was constructed by transforming strain GS140 with pRS314-SLS1 and performing plasmid shuffle as described previously (6). Standard yeast media (yeast extract-peptone-dextrose [YPD], yeast extract-peptone-glycerol [YPG], or synthetic dextrose [SD]) containing the necessary nutrient supplements were prepared as described previously (27).
For the SOD overexpression studies, the SOD1 and SOD2 genes were cloned into the yeast/Escherichia coli shuttle vector pRS316 (29) under the control of their endogenous promoters. The SOD1 locus was cloned by PCR using primers that annealed 533 bp upstream of the ATG and 534 bp downstream of the stop codon. SOD2 was similarly cloned using primers that annealed 591 bp upstream of the ATG to 571 bp downstream of the stop codon. XhoI and XbaI sites were added to the 5′ and 3′ ends of the PCR primers, and these sites were used to clone the PCR products into pRS316.
The plasmid pAC48, a derivative of pAC45 (2μ, URA3) (10, 15) from which the EcoRI fragment containing the GAL1 promoter has been excised, was used to make the RTG3 and MSN4 green fluorescent protein fusion constructs. PCR products were generated using the primers that annealed 519 bp and 674 bp upstream of the ATG for RTG3 and MSN4, respectively. The downstream primers annealed at the 3′ end of the RTG3 and MSN4 open reading frames, excluding the stop codons, and contained XhoI sites to facilitate cloning. An EcoRI site was added to the 5′ end of the RTG3 upstream PCR primer, and an XmaI site (with compatible overhangs to EcoRI) was added to the 5′ end of the MSN4 PCR primer.
Mitochondrial translation assays.
Determination of mitochondrial translation was performed exactly as described previously (23).
Chronological life span.
Starter cultures (5 ml) were inoculated from plates and grown overnight in a roller drum at 30°C in SD medium supplemented with the appropriate amino acids. These cultures were then used to inoculate 50 ml of fresh SD medium in 125-ml flasks to an optical density at 600 nm (OD600) of 0.1. Cultures were grown with shaking (200 rpm) at 30°C. After 24 h, these cultures had reached stationary phase, and in all experiments described this time point is designated day 1 stationary phase. Under these conditions, the titers of cultures varied slightly from strain to strain, but the OD600 was always approximately 2.5, which corresponds to ∼5 × 107 cells/ml. Viability was determined over the course of the experiment using either trypan blue staining or quantitative plating experiments or by comparing serial dilutions of the strains. For trypan blue staining, 100 μl of the culture was combined with 100 μl of 0.4% trypan blue solution (Mediatech) and incubated at 30°C for 5 min. The ratio of viable to inviable cells was determined by counting cells using a hemocytometer. Quantitative plating experiments were carried out by making several different dilutions in rich medium (YPD) and spreading these onto YPD plates. Plates that had approximately 500 colonies were counted, and the viable cell number was calculated. For the spotting assay, 10-fold serial dilutions were made in YPD and 10 μl was spotted by hand onto each YPD plate.
Determination of petite formation.
Starter cultures (5 ml) were grown in YPG overnight at 30°C in a roller drum to select for respiration-competent cells. Fresh SD cultures (50 ml) in flasks were then inoculated in 125-ml flasks from these starter cultures to an OD600 of 0.001. The number of petites was then determined immediately after inoculation, after 24 h, and after 5 days. Petite frequency was determined by plating onto selective SD medium and counting colonies. Due to a mutation in the ADE2 gene, our strain backgrounds form red colonies on YPD and SD media when they are respiratory competent and form white colonies when they are petite (25).
Hydrogen peroxide sensitivity.
Starter cultures (5 ml) were inoculated from plates and grown in a roller drum overnight at 30°C in SD medium supplemented with the appropriate amino acids. These cultures were then diluted into 5 ml of SD medium to an OD600 of 0.1. Cultures were grown for an additional 24 h, and then aliquots were removed to serve as untreated controls. H2O2 was then added to a concentration of 25 mM. Cultures were incubated with H2O2 for the specified amount of time and assayed for viability as described above.
Flow cytometry.
Samples (100 μl) were taken from the indicated cultures, and the cells were pelleted in a microcentrifuge. After removal of the media, the cell pellets were resuspended in phosphate-buffered saline (PBS) (100 μl) containing 50 μM dihydroethidium (DHE; Molecular Probes) and incubated at 30°C for 10 min. Cells were then pelleted again and resuspended in PBS (100 μl) without dye. Flow cytometry was carried out on a Becton-Dickinson FACSCalibur model flow cytometer. The DHE fluorescence indicated is the direct output of the FL3 (red fluorescence-detecting) channel without compensation. A total of 25,000 cells were analyzed for each curve.
Fluorescence microscopy.
For green fluorescent protein fluorescence, cells in day 1 stationary phase were pelleted and resuspended in PBS and viewed with a Leica Microstar IV microscope using a standard fluorescein isothiocyanate filter. DHE staining was performed exactly the same for microscopy as described above for flow cytometry, and cells were viewed using a standard rhodamine filter. In both cases, photographs were taken at ×100 magnification with a Yashica 108 camera using Kodak Elite Chrome ASA 400 slide film. The resulting slides were scanned and are reported without manipulation, aside from cropping.
Oxygen consumption assays.
The rate of oxygen consumption was monitored using a standard potentiometric Clark-type electrode (model 5300A; YSI Bioanalytical Products). Five milliliters of culture at the indicated time points was transferred to an airtight chamber maintained at 30°C, and oxygen content was monitored for at least 6 min. To ensure the oxygen consumption observed was due to mitochondrial activity, sodium azide was routinely added to the cultures (final concentration, 0.05% [wt/vol]) and compared to the rate observed without azide.
RESULTS
Defective mitochondrial gene expression in the rpo41-R129D mutant strain GS129 results in imbalanced mitochondrial translation and reduced chronological life span.
We previously reported that mutations in the ATD of mtRNA polymerase (Rpo41p) disrupt the coupling of mitochondrial transcription and translation, leading to aberrant assembly of mitochondrial OXPHOS complexes (6, 22, 23). As discussed in the introduction, one ATD mutation (rpo41-R129D) stood out in our initial studies, in that it has the most severe glycerol (YPG) growth phenotype, presumably due to a unique defect in mitochondrial translation. We undertook a more detailed analysis of this mutant in order to gain a better understanding of how defective mitochondrial gene expression impacts cellular function.
When cultured on YPG agar plates, GS129 forms colonies that are not only smaller than those of the wild type but also are highly heterogeneous in size (Fig. 1A), suggesting the influence of a stochastic process on the growth of this mutant. Its doubling time in liquid YPG medium is also greatly protracted (data not shown). The growth of GS129 in fermentable glucose (SD) media was usually indistinguishable from that of the isogenic wild-type strain. However, we observed that when glucose cultures of GS129 were inoculated from plates that had been left at 4°C for extended periods of time (>30 days), their growth in liquid culture consistently lagged behind wild-type cultures that had been treated identically (Fig. 1B). Surprisingly, growth curves conducted on these cultures clearly showed that the doubling times of the wild-type and mutant strains were indistinguishable. In fact, equations describing these curves are nearly identical, except for the y intercepts, indicating a different number of viable cells were used to inoculate the cultures (Fig. 1B). We reasoned that one possible explanation for this observation is that GS129 is defective in its ability to maintain viability under non-growth conditions (i.e., has a decreased chronological life span). To measure the chronological life span of GS129, the number of viable cells in stationary-phase cultures of the wild-type and GS129 strains was determined over the course of several days using either trypan blue staining or by plating onto rich medium and counting colonies (Fig. 1C and D). Compared to its isogenic wild-type strain (GS122), the GS129 mutant displayed a drastically reduced (>1,000-fold) ability to maintain viability in stationary phase and therefore a shortened chronological life span (8).
FIG. 1.

The mitochondrial RNA polymerase mutant GS129 exhibits a growth defect on the nonfermentable carbon source glycerol and decreased chronological life span. (A) Colonies of wild-type (WT) and rpo41-R129D (GS129) strains after 48 h of growth on glycerol (YPG) agar plates are shown. Samples were spotted from day 1 stationary-phase glucose (SD) cultures (see Materials and Methods). Shown are a 1,000-fold dilution of the wild type and a 100-fold dilution of the GS129 mutant at ×40 magnification. (B) Cultures of the GS129 mutant inoculated from colonies on glucose (SD) agar plates older than ∼30 days exhibited a reproducible lag when grown in liquid glucose (SD) medium. The OD600 of these cultures (y axis) was monitored over time (in hours; x axis) as a measure of growth. The inset equations represent a best-fit line for the curves, where the exponent is inversely proportional to the doubling time. (C) Viability curves of wild-type and GS129 mutant stationary-phase cultures. Viability was determined by trypan blue staining to day 4 and by quantitative plating thereafter (see Materials and Methods). The percentage of survival as a function of days in stationary phase is plotted on a log scale. (D) Relative viability of wild-type and GS129 cultures as determined by plating serial 10-fold dilutions of day 1 and day 10 stationary-phase glucose (SD) cultures onto rich medium (YPD). (E) Mitochondrial translation profiles of wild-type (WT) and GS129 strains. Mitochondrial translation products were specifically labeled by inhibiting cytoplasmic translation, incubating cells with [35S]methionine, and analyzing the products by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and autoradiography. All eight protein products (indicated on the right on the figure) encoded by the Saccharomyces cerevisiae mitochondrial genome are represented.
Given that all other mtRNA polymerase ATD mutant strains have been shown to globally affect mitochondrial translation to various degrees (23), we next examined the mitochondrial translation profile of GS129. The GS129 mutant exhibited robust translation, but the relative synthesis rates of certain components were altered (Fig. 1E). Specifically, translation of Cox1p and Cobp (and, perhaps to a lesser extent, Cox3p) were reduced, and Atp6 and Atp8p were elevated. We conclude that, unlike other ATD mutants, which have globally reduced translation (23), GS129 has efficient yet imbalanced translation of mtDNA-encoded OXPHOS subunits due to the rpo41-R129D point mutation.
GS129 exhibits a conditional and reversible inactivation of respiration in stationary phase.
Given that GS129 exhibits a viability defect in stationary phase (reduced chronological life span; Fig. 1C and D), we next measured the ability of this mutant to respire under these conditions. The rate of mitochondrial oxygen consumption of the wild type and GS129 in stationary phase in either glucose- or glycerol-containing medium was measured. Cells were grown to saturation (5 × 107 to 6 × 107 cells/ml), and oxygen consumption was measured on both the first and second days of stationary phase (see Materials and Methods). In both media, the wild-type strain decreased oxygen consumption from day 1 to day 2 (Fig. 2A), consistent with the decrease in metabolic rate known to occur as cells enter stationary phase (13). In contrast, we found that GS129 displayed a progressive inactivation of respiration in stationary phase when grown in glucose (SD) medium (Fig. 2A). This mutant exhibited a significantly lower rate of oxygen consumption than the wild-type strain at day 1 that dropped still lower on day 2 (Fig. 2A), and it became undetectable by day 3 (data not shown). Surprisingly, the opposite is true of oxygen consumption of the GS129 mutant when grown in glycerol (YPG) medium. Here, oxygen consumption is sustained and actually increases from day 1 to day 2, under conditions where the wild-type strain downregulates respiration (Fig. 2B).
FIG. 2.

GS129 exhibits conditional inactivation of respiration in stationary phase. (A) Oxygen consumption of day 1 and day 2 stationary-phase glucose (SD) cultures of wild type (WT) and GS129. All values were normalized to the OD600 of the culture used, and the mean oxygen consumption/OD600 unit of the wild-type strain was arbitrarily given a value of 1. Each bar represents the average of three independent experiments, with the standard deviations of the three measurements shown. (B) The experiment was the same as that described for panel A but was carried out in glycerol (YPG) medium.
We considered the possibility that the decrease in respiration observed in GS129 could be attributed to an increased percentage of petite cells (i.e., cells unable to respire as a result of mutation of nuclear DNA or mutation or loss of mtDNA) (25). We therefore measured the fraction of petites in stationary phase under the same conditions used in the longevity assays (petite cell formation [± standard deviation] at day 1 stationary-phase wild-type and GS129 cultures was 1.64% ± 0.995% and 6.20% ± 2.04%, respectively). At day 1 of stationary phase, the GS129 strain exhibited an approximately fourfold increase in petite formation compared to the control wild-type strain (GS122), with approximately 6% of the cells being petite. The relative frequencies of petites in these strains at later points in stationary phase remained similar (data not shown). While these data are consistent with slightly elevated damage and mutation to mitochondrial and/or nuclear DNA in GS129, they fail to support the conclusion that the dramatic loss of viability (Fig. 1C and D) and respiration (Fig. 2A) in this mutant is due to petite formation.
The GS129 mutant is sensitive to oxidative stress and contains higher levels of intracellular ROS.
Mitochondria are the main source of cellular ROS, which can damage cellular components and are thought to promote aging (5, 20, 34). Also, yeast cells upregulate stress resistance genes when transferred from a fermentable to a nonfermentable carbon source (9). We hypothesized that the YPG growth phenotype and accelerated aging of GS129 is due to increased sensitivity to oxidative stress. We therefore tested the sensitivity of the wild-type and GS129 mutant strains to H2O2. Indeed, the GS129 mutant exhibits heightened sensitivity to this oxidizing agent compared to the wild-type strain, with approximately 0.1% of the survival of wild type after 150 min of exposure to 25 mM H2O2 (Fig. 3A and B).
FIG. 3.

GS129 displays heightened susceptibility to oxidative stress and aberrant accumulation and localization of ROS. (A) Serial 10-fold dilutions of wild-type (WT) and GS129 day 1 stationary-phase glucose (SD) cultures plated onto rich medium following 150 min of treatment with 25 mM hydrogen peroxide are shown. The left panel shows untreated cultures (−H2O2), and the right panel shows H2O2-treated cultures (+H2O2). (B) Viability curves of the wild type and GS129 following treatment with 25 mM hydrogen peroxide for the time indicated is shown. Percentage survival was determined by plating on rich glucose medium (YPD) and counting colonies and is plotted on a log scale on the y axis. (C) Flow cytometric analysis of endogenous ROS in wild-type (WT) and GS129 cultures stained with 50 μM DHE after 1 day (top panels) or 2 days (bottom panels) in stationary phase. The x axis on each panel is a linear scale of forward scatter (roughly indicative of cell size), and the y axis is a log scale of the intensity of DHE fluorescence. Any fluorescence below 101 is indistinguishable from background and is marked with a horizontal line across each graph. The percentage of cells above this line is indicated in the upper right corner of each panel. (D) Fluorescence-microscopic examination of wild-type (top panels) and GS129 cells (bottom panels) stained with 50 μM DHE at day 1 of stationary phase (left panels) or after treatment with 50 mM H2O2 (right panels).
We next measured intracellular ROS levels by flow cytometry using the ROS-sensitive probe dihydroethidium (DHE) (Fig. 3C). DHE is normally cell permeant and nonfluorescent, but upon oxidation it becomes impermeant and fluorescent. At day 1 of stationary phase (Fig. 3C, top panels), wild-type cells exhibited relatively low levels of fluorescence, with only 31.6% of cells staining above the background range (∼101). In contrast, GS129 cells contained higher overall levels of fluorescence, and 64.2% of cells showed levels of DHE fluorescence above background. By day 2, the profiles of these two strains had diverged even more (Fig. 3C, bottom panels). At this point, the wild-type strain showed two distinct populations of cells, one staining brightly (and therefore containing more ROS, approximately 50.5% of the total population) with the rest of the population staining at background levels. At the same time point, GS129 showed the great majority of cells (89.3%) within the former, brightly staining population. These observations were not due to differences in cell size or autofluorescence of dead cells, as the forward and side-scatter profiles of these cultures were similar (Fig. 3C and data not shown). We also qualitatively confirmed these results using fluorescence microscopy (Fig. 3D). In fact, not only did GS129 cells show an overall greater level of fluorescence but also the pattern of staining within cells was substantially different between this mutant and the wild-type strain. Wild-type cells exhibited staining almost exclusively distributed among intracellular puncta, which are most likely mitochondria. However, GS129 showed a high degree of fluorescence throughout the cell, similar to the pattern of fluorescence exhibited by wild-type cells treated with H2O2 (Fig. 3D).
Overexpression of SOD or Msn4p reduces ROS and rescues the chronological life span of GS129.
Hypothesizing that ROS are actually causative in the decreased chronological life span of GS129, we tested whether introduction of additional exogenous copies of either of the genes encoding superoxide dismutases (SOD1 or SOD2) would restore normal life span to this mutant. Both SOD1 and SOD2 were inserted into low-copy-number plasmids under the control of their endogenous promoters. We found that transformation of GS129 with either of these plasmids significantly increased life span compared to transformation with a control vector (Fig. 4A). It should be pointed out here that the experiments shown in Fig. 4A are the result of quantitative plating assays, which especially in the case of GS129 give considerably different results than trypan blue staining (as reported in Fig. 1C). Specifically, we often find that the number of viable cells at a given time point in GS129 is much higher than the number of CFU. The most likely explanation for this observation is that there are large numbers of cells which are alive but have accumulated sufficient damage as to render them unable to divide (consistent with the vast majority of GS129 cells at day 2 staining brightly with DHE in Fig. 3D).
FIG. 4.

Overexpression of SOD1 or SOD2 extends life span in GS129 and rescues ROS levels and localization. (A) Viability curves as determined by quantitative plating of wild-type (WT) or GS129 strains containing empty vector or a plasmid carrying either SOD1 or SOD2. The number of CFU for each strain at day 1 of stationary phase was given a value of 1, and the fraction of CFU remaining (y axis) over time (x axis) is shown. (B) Glycerol (YPG) growth of the vector control and SOD2-overexpressing strains listed in panel A. (C) Flow cytometric measurement of ROS in GS129 with empty vector or overexpressing SOD2 at day 2 of stationary phase. The fluorescent dye DHE was used as described in the legend to Fig. 3. GS129 plus empty vector is shown as the light gray curve, while GS129 overexpressing SOD2 is dark gray. For comparison, the profile of wild-type cells treated identically is overlaid as the boldface black line. (D) Microscopic analysis of GS129 cells containing either empty vector (left panel) or overexpressing SOD2 (right panel) at day 1 of stationary phase stained with 50 μM DHE.
In addition, SOD1 and SOD2 also substantially ameliorated the glycerol growth defect of GS129, with SOD2 providing better rescue than SOD1 (Fig. 4B and data not shown). However, addition of SOD1 or SOD2 plasmids neither noticeably affected the growth of the wild-type strain on YPG medium nor increased the number of viable cells in stationary phase at the time points we examined (Fig. 4A). Also, overexpression of both SOD1 and SOD2 substantially decreased intracellular ROS as measured by flow cytometry (Fig. 4C) and restored a wild-type punctate staining pattern to GS129 cells stained with DHE (Fig. 4D and data not shown). Finally, we were able to decrease ROS (Fig. 5A) and increase life span (Fig. 5B) in GS129 by overexpressing Msn4p, a transcriptional activator of both SODs and other stress response genes (9).
FIG. 5.

Overexpression of the stress-responsive transcription factor MSN4 extends life span and decreases ROS in GS129. (A) Flow cytometric analysis of stationary-phase wild type (WT) and GS129 containing either empty vector or a high-copy plasmid containing MSN4. Samples were taken at day 2 of stationary phase and are reported as described in the legend to Fig. 3. (B) Viability (at day 5 of stationary phase) of the strains described for panel A, determined by plating 10-fold serial dilutions onto rich medium (YPD).
Reduction of ROS via SOD or Msn4p overexpression restores respiration of GS129 in stationary phase.
We examined whether reducing ROS had any effect on respiration in GS129. The same plasmids described above to overexpress SOD1, SOD2, and MSN4 were tested for their ability to restore respiration in stationary-phase SD cultures. Overexpression of SOD1 or SOD2, which extended the life span of GS129 (Fig. 4A), also substantially increased stationary-phase oxygen consumption (Fig. 6A). Again, it was SOD2 that provided the better rescue of this phenotype. Notably, SOD overexpression had the opposite result on oxygen consumption in the wild-type strain, with both SOD1 and SOD2 significantly decreasing oxygen consumption (Fig. 6A). As with ROS and life span, overexpression of MSN4, like SOD overexpression, increased oxygen consumption in the mutant strain (Fig. 6B). Remarkably, MSN4 restored respiration to virtually wild-type levels (Fig. 6B).
FIG. 6.

Reducing ROS in the GS129 mutant restores stationary-phase respiration. (A) Oxygen consumption of wild-type (WT) and GS129 cultures containing an empty vector or a plasmid carrying either SOD1 or SOD2 at day 1 of stationary phase. Results are plotted as described in the legend to Fig. 2; all measurements were performed in triplicate and were normalized for differences in OD, with the wild type set to 1. (B) Oxygen consumption of day 2 stationary-phase glucose (SD) cultures of wild-type and GS129 strains containing either an empty vector (+vector) or a plasmid that overexpresses MSN4.
Other mtRNA polymerase ATD mutants exhibit reduced respiration but intermediate ROS and life span phenotypes that are not rescued by SOD.
We sought to determine whether the phenotype of the GS129 mutant is specific to this particular point mutation or is shared with other ATD mutants. First, we found that several of the ATD point mutants we have characterized previously (6, 22, 23) indeed have a chronological life span defect, including two deletion mutants, rpo41Δ2 and rpo41Δ3, and one double-point mutant, rpo41-N152A/Y154A (strain GS130). The life spans of these strains, represented here by the GS130 mutant, were virtually indistinguishable from each other (data not shown) and significantly less severe than that of GS129 (Fig. 7A). In examining the phenotypes of the GS130 mutant in more detail, we determined that, as was the case with life span, this mutant exhibited decreased respiration (Fig. 7B), hydrogen peroxide sensitivity (Fig. 7C), and ROS (Fig. 7D) phenotypes intermediate between those of wild-type and GS129 strains. In addition its petite induction rate (1.89% ± 1.45%) was essentially the same as the wild-type strain. However, we found that neither the respiration nor the life span phenotype of GS130 was improved upon overexpression of either SOD gene or MSN4 (Fig. 7A and B and data not shown) and that overexpression of SOD did not reduce DHE fluorescence (Fig. 7E) or improve growth on glycerol medium (data not shown).
FIG. 7.

GS130 exhibits a less severe chronological life span defect than GS129 and is not rescued by overexpression of SOD. (A) Viability of the GS130 mutant plus either empty vector or plasmids that overexpress SOD1 or SOD2, determined as described in the legend to Fig. 4A. The wild-type (WT) and GS129 curves from Fig. 4A (which were carried out in parallel) are shown as dotted lines for comparison. (B) Stationary-phase oxygen consumption of the GS130 strains depicted in panel A. The wild-type result from Fig. 6A is shown for comparison and is given the arbitrary value of 1. (C) Serial 10-fold dilutions of a stationary-phase GS130 culture before (−H2O2) and after (+H2O2) treatment with 25 mM H2O2 for 2.5 h. (D) DHE fluorescence at day 2 of stationary phase of wild type (dark gray line), GS129 (light gray shading), and GS130 (black line). The median and mean fluorescence values, in relative fluorescence units, were the following: wild-type, 44.1 and 78.7; GS130, 59.3 and 89.3; GS129, 99.6 and 125. (E) DHE fluorescence at day 2 of stationary phase of GS130 containing either empty vector (light gray shading) or a plasmid carrying SOD2 (bold gray line), as described in the legend to Fig. 4C. For comparison, the profile of wild-type cells treated identically is overlaid as the thin black line.
DISCUSSION
Defective mitochondrial gene expression can result from either mtDNA mutations or nuclear mutations that impact mtDNA expression or replication. The biological consequences of these defects are highly variable, as illustrated by the complex clinical presentation of mitochondrial disease symptoms in patients. For example, mtDNA mutations in two different tRNA genes can cause drastically different phenotypes and diseases, despite both mutations ultimately decreasing translation of all mtDNA-encoded proteins (7). In fact, the same mtDNA mutation can generate dramatically diverse phenotypes in different patients or even different tissues within the same patient (7). The implication of these observations is that subtle defects in mitochondrial gene expression can differentially impact respiration and in turn lead to a variety of deleterious downstream cellular consequences. To address this issue experimentally, we analyzed in detail two mtRNA polymerase ATD mutants previously shown to be defective in coupling mitochondrial transcription to translation. One of the strains, GS129, has relatively efficient but imbalanced translation (Fig. 1E), whereas the other strain, GS130, exhibits a global reduction in mitochondrial translation that is representative of the majority of ATD mutants (23). Together, these two mutants represent strains with distinct perturbations in mitochondrial gene expression that differentially affect assembly of the OXPHOS system via disruption of the same process. While these two mutants have several phenotypes in common (e.g., decreased respiration and increased ROS production in stationary phase; Fig. 2, 3, and 7), they have extremely different chronological life span defects and responses to overexpression of superoxide dismutase (SOD1 or SOD2; Fig. 1, 4, 5, and 7).
From this study we draw two primary conclusions. The first is that defective mitochondrial gene expression (specifically, defective coupling of transcription to translation) leads to increased ROS production and inhibition of respiration in stationary phase, ultimately limiting chronological life span. The second is that ROS themselves can lead to the complete inactivation of the respiratory chain and a dramatic loss of viability, but only if produced above a certain threshold level. Altogether, our results support the hypothesized “vicious cycle” of mitochondrial ROS production that, once initiated, is thought to lead to the progressive mitochondrial and cellular dysfunction seen in a number of human diseases and aging. The rationale for these conclusions is discussed in detail below.
In our initial characterizations of the ATD of mtRNA polymerase, we examined several point mutations and deletions in this domain. Most of these mutations cause a global reduction in mitochondrial translation due to an inability to properly couple transcription to translation during mitochondrial gene expression (22, 23). One mutation, rpo41-R129D (strain GS129), stood out in these analyses in that it has the most pronounced glycerol growth phenotype (22, 23) (Fig. 1A). Experiments aimed at better understanding this unique ATD mutant were the starting point for this study. We found that GS129 has a severe chronological life span defect (Fig. 1C and D), imbalanced mitochondrial translation (Fig. 1E), severely decreased respiration in stationary-phase glucose (SD) cultures (Fig. 2A), increased ROS production (Fig. 3C and D), and enhanced sensitivity to oxidative stress (Fig. 3A and B). These results clearly establish that disruption of mitochondrial gene expression can limit life span and strongly suggest mitochondrial ROS are responsible for progressive cellular dysfunction (Fig. 8).
FIG. 8.
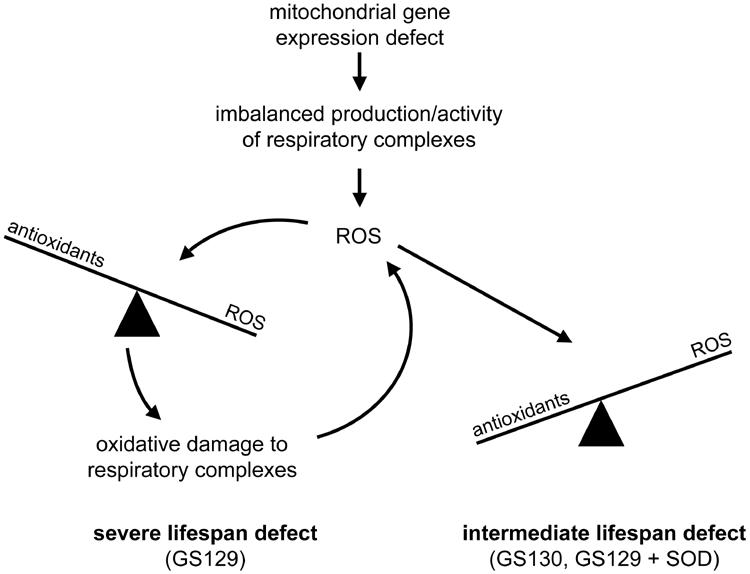
Model depicting how different defects in mitochondrial gene expression differentially affect yeast life span. Depicted is a scenario in which certain types of mitochondrial gene expression defects (e.g., loss of coupling between transcription and translation in this study) result in the imbalanced production/activity of the respiratory chain that initiates increased mitochondrial ROS production (shown at the top of the diagram). Subsequently, the magnitude of the life span reduction depends on the precise nature of the mitochondrial gene expression defect. In the case of the GS130 mutant (rightward branch point), the amount of ROS production is combatted effectively by the normal antioxidant defenses. Nonetheless, the persistent increase in steady-state levels of ROS or decrease in respiration from the underlying mitochondrial gene expression defect causes an intermediate life span decrease. In contrast, in certain circumstances (e.g., GS129 mutant; leftward branch point), the specific manner in which the respiratory chain is disrupted results in enhanced ROS production that damages the OXPHOS complexes and leads to more ROS production (i.e., the vicious cycle of mitochondrial ROS production). Engagement of the vicious cycle overwhelms the antioxidant defenses (irreversibly tipping the balance), leading to a severe life span defect from chronic oxidative stress.
We ruled out the possibility that the stationary-phase respiration defect of GS129 was caused solely by an increased rate of petite mutant formation. Under the conditions in which our life span assays were carried out, the GS129 mutant has a slightly higher rate of petite formation than the wild type (∼6% in GS129 versus ∼1.6% in the wild type at day 1 stationary phase), consistent with increased levels of oxidative stress and potentially greater damage to mtDNA. However, the dramatic decrease in respiration and viability of GS129 in stationary phase (Fig. 1C, D, and 2A) cannot be attributed to such a minimal relative increase in the percentage of petites.
We also point out that the growth rate of the wild-type and GS129 mutant strains are indistinguishable in the SD media used for this assay, and the maximum titers attained by both strains are nearly identical (data not shown), thus discounting the possibility that the differences in respiration observed are due to one strain reaching stationary phase before the other. Importantly, the GS129 mutant is capable of supporting a high level of respiration in stationary phase in glycerol (YPG) medium (Fig. 2B). Though direct comparisons of the wild type and GS129 are more difficult to make in this case (due to differences in growth rates of the two strains in YPG), it can be conservatively concluded that the GS129 mutant mtRNA polymerase can carry out significant gene expression that can support high levels of respiration under at least some conditions. GS129 must therefore be considered a conditional mutant with respect to respiration (i.e., oxygen consumption).
We sought to confirm our hypothesis that the life span defect in GS129 is a direct consequence of its increased ROS production. Therefore, we increased antioxidant defenses in this strain by overexpressing SOD1, SOD2, or the stress-response transcription factor gene MSN4 (Fig. 4 and 5). As predicted, decreasing ROS in GS129 stationary-phase cells by overexpression of SOD1, SOD2, or MSN4 each greatly extended the life span of this mutant (Fig. 4A and 5B). Also, the glycerol growth phenotype of GS129 was greatly ameliorated by SOD overexpression (Fig. 4B), consistent with the interpretation that the heterogeneous colony size and presence of apparently abortive colonies in this mutant (Fig. 1A and 4B) are due to stochastic ROS production and damage. However, perhaps unexpectedly, we found with either SOD or MSN4 overexpression that the inactivation of respiration normally exhibited by GS129 in stationary phase was also reversed (Fig. 6). These observations lead us to the inevitable conclusion that ROS are actually causative in the cessation of respiration in GS129. That is, the respiration block in GS129 cannot be due to a complete lack of expression of an essential OXPHOS component, as reduction of ROS could hardly be imagined to complement this type of defect. Rather, we interpret these data as indicating that, under the nutrient-limited conditions of stationary phase, ROS generated as the result of a partially blocked and aberrant respiration in GS129 overwhelm antioxidant defenses and inactivate the remaining functional complexes, ultimately resulting in death (Fig. 8). Under this scenario, overexpression of antioxidant defenses protects these complexes and prevents the complete inhibition of respiration.
The fact that overexpression of SOD1 and SOD2 both rescue the life span and respiration defects of GS129 is worthy of discussion. The almost complete rescue by SOD2 (a mitochondrial matrix enzyme) strongly suggests that superoxide production or accumulation in the mitochondrial matrix is the major contributor to the observed phenotypes. However, the partial rescue by SOD1 suggests that ROS outside the mitochondrial matrix are also important. Sod1p has been shown to localize to the mitochondrial intermembrane space as well as the cytoplasm, and the loss of Sod1p has been shown to compromise mitochondrial matrix proteins (21, 32, 37). Also, it was recently suggested that mitochondrial superoxide may react with nitric oxide to produce peroxynitrite, a membrane-permeable molecule (19). Therefore, it is possible that superoxide and/or peroxynitrite produced on the outer face of the inner membrane crosses into the matrix, where it can damage OXPHOS components and contribute to the vicious cycle. Also, it is probable that some of the deleterious effects of mitochondria-derived ROS in GS129 are occurring in the cytoplasm or the nucleus, and both Sod1p and Sod2p are capable of detoxifying these ROS before reaching their sites of action. Indeed, this is consistent with the diffuse localization of DHE fluorescence exhibited by GS129 (Fig. 3D) and the restoration of punctate staining by SOD2 overexpression (Fig. 4D).
Having characterized the GS129 mutant in detail, we next compared this revealing mutant to other mtRNA polymerase ATD mutants with similar yet distinct perturbations of mitochondrial gene expression. First, we found that three other ATD mutations (rpo41Δ2, rpo41Δ3, and rpo41-N152A/Y154A) also result in reduced chronological life span. The life span defects in these strains were virtually identical to one another (unpublished observations), and rpo41-N152A/Y154A (strain GS130) was characterized in greater detail as a representative of the group (Fig. 7). While GS130 has a considerable life span defect (e.g., ∼5- to 10-fold loss of viability at day 4), it has a substantially longer life span than GS129, which exhibits a >100-fold loss in viability at the same time point (Fig. 7A). GS130 is also sensitive to hydrogen peroxide (Fig. 7C), has increased ROS levels (Fig. 7D), and carries out substantially decreased respiration in stationary phase (Fig. 7B). Yet, like its life span defect, these phenotypes are less severe than those observed in GS129 (Fig. 2A, 3A, and 7A and D). However, the magnitude of the GS130 respiration defect is arguably on the same order as that observed in GS129 (compare Fig. 2A and 7B).
Perhaps the most noteworthy difference between GS129 and GS130 is the inability of SOD1 or SOD2 overexpression to rescue the life span (Fig. 7A) and respiration (Fig. 7B) phenotypes of GS130. Our interpretation of these results is that the specific block in respiration in GS130 generates more ROS than the isogenic wild-type strain but never in quantities sufficient to overwhelm antioxidant defenses and initiate the vicious cycle (Fig. 8). Therefore, ROS are not causative in either the inactivation of respiration in this mutant or its life span defect. Rather, it is likely that the decrease in the life span of GS130 is due solely to its inability to produce sufficient quantities of OXPHOS components and thus maintain normal amounts of respiration in stationary phase (Fig. 7B). This interpretation is consistent with our results showing that further decreases in respiration in GS130 (via SOD overexpression) shorten its life span (Fig. 7).
Interestingly, GS130 actually carries out lower levels of mitochondrial translation than GS129 (23) (Fig. 1E) and, unlike GS129, its mitochondrial RNA polymerase no longer interacts with the translation-coupling factor Nam1p (22). Based on these differences, it is tempting to speculate that globally reduced translation (in GS130 and the other ATD mutants [23]) represents a less severe defect with regard to ROS production and life span than efficient but imbalanced translation (Fig. 1E, GS129). This interpretation is also consistent with the observation that a Cox1-specific imbalance in mitochondrial translation (in a nam1 null strain that is partially rescued by overexpression of SLS1 [23]) also leads to a stationary-phase respiration defect and dramatically decreased life span (see Fig. S1 in the supplemental material). This implies that perturbations in mitochondrial gene expression (due to mtDNA or nuclear mutations) that cause specific types of imbalanced production or assembly of OXPHOS components lead to extreme cellular dysfunction via a vicious cycle of oxidative stress (Fig. 8). It is tempting to speculate further that the reduced mitochondrial translation of electron transport components of the OXPHOS system, in combination with increased translation of ATP synthase components (i.e., the translation defect observed in GS129 [Fig. 1E]), represents an unusually deleterious imbalance that greatly increases ROS production and aging.
In summary, this study has revealed novel insights into the mechanisms by which defective mitochondrial gene expression contributes to cellular dysfunction and aging. We provide strong evidence that improper assembly of the OXPHOS system (due to loss of coupling of transcription and translation during mitochondrial gene expression) leads to increased ROS production and substantial but conditional decline in respiration. However, the loss of cell viability (and the magnitude of the longevity defect, at least in yeast) resulting from such perturbations depends on the level of mitochondrial ROS production and whether a critical ROS threshold level is reached (Fig. 8). Such a scenario may help explain the variable phenotypic expression (i.e., clinical presentation) of mtDNA mutations in human disease and aging, in that even subtle differences in mitochondrial gene expression can lead to grossly different outcomes depending on the precise nature of the defect. Finally, our results suggest a novel role for SOD in regulating respiration by maintaining proper ROS homeostasis. Increased SOD activity can either increase or decrease respiration depending on genetic background and environmental conditions (Fig. 6A and 7B), implying that ROS may play a role in regulating respiration more complex than simply mediating damage to cellular components. Understanding the dynamic interplay between mitochondrial gene expression, respiration, and ROS in human disease and aging remains fertile ground for future investigation.
Supplementary Material
Acknowledgments
This work was supported by Public Health Service grant HL-59655 from the NHLBI and Army Research Office grant W911NF0510155, awarded to G.S.S.
We thank the following people for insightful discussions, advice, or technical support: Jana Eaton, Sharen McKay, Justin Cotney, Zhibo Wang, Susan Kaech, Ed Rogers, Rusty McGuire, and Anita Corbett.
Footnotes
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Ashrafi, K., D. Sinclair, J. I. Gordon, and L. Guarente. 1999. Passage through stationary phase advances replicative aging in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 96:9100-9105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balaban, R. S., S. Nemoto, and T. Finkel. 2005. Mitochondria, oxidants, and aging. Cell 120:483-495. [DOI] [PubMed] [Google Scholar]
- 3.Bandy, B., and A. J. Davison. 1990. Mitochondrial mutations may increase oxidative stress: implications for carcinogenesis and aging? Free Radic. Biol. Med. 8:523-539. [DOI] [PubMed] [Google Scholar]
- 4.Barros, M. H., B. Bandy, E. B. Tahara, and A. J. Kowaltowski. 2004. Higher respiratory activity decreases mitochondrial reactive oxygen release and increases life span in Saccharomyces cerevisiae. J. Biol. Chem. 279:49883-49888. [DOI] [PubMed] [Google Scholar]
- 5.Boveris, A., N. Oshino, and B. Chance. 1972. The cellular production of hydrogen peroxide. Biochem. J. 128:617-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bryan, A. C., M. S. Rodeheffer, C. M. Wearn, and G. S. Shadel. 2002. Sls1p is a membrane-bound regulator of transcription-coupled processes involved in Saccharomyces cerevisiae mitochondrial gene expression. Genetics 160:75-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DiMauro, S., and E. A. Schon. 2003. Mitochondrial respiratory-chain diseases. N. Engl. J. Med. 348:2656-2668. [DOI] [PubMed] [Google Scholar]
- 8.Fabrizio, P., and V. D. Longo. 2003. The chronological life span of Saccharomyces cerevisiae. Aging Cell 2:73-81. [DOI] [PubMed] [Google Scholar]
- 9.Gorner, W., E. Durchschlag, M. T. Martinez-Pastor, F. Estruch, G. Ammerer, B. Hamilton, H. Ruis, and C. Schuller. 1998. Nuclear localization of the C2H2 zinc finger protein Msn2p is regulated by stress and protein kinase A activity. Genes Dev. 12:586-597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Green, D. M., K. A. Marfatia, E. B. Crafton, X. Zhang, X. Cheng, and A. H. Corbett. 2002. Nab2p is required for poly(A) RNA export in Saccharomyces cerevisiae and is regulated by arginine methylation via Hmt1p. J. Biol. Chem. 277:7752-7760. [DOI] [PubMed] [Google Scholar]
- 11.Greenleaf, A. L., J. L. Kelly, and I. R. Lehman. 1986. Yeast RPO41 gene product is required for transcription and maintenance of the mitochondrial genome. Proc. Natl. Acad. Sci. USA 83:3391-3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harman, D. 1956. Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 11:298-300. [DOI] [PubMed] [Google Scholar]
- 13.Jakubowski, W., T. Bilinski, and G. Bartosz. 2000. Oxidative stress during aging of stationary cultures of the yeast Saccharomyces cerevisiae. Free Radic. Biol. Med. 28:659-664. [DOI] [PubMed] [Google Scholar]
- 14.Jazwinski, S. M. 2005. Yeast longevity and aging-the mitochondrial connection. Mech. Aging Dev. 126:243-248. [DOI] [PubMed] [Google Scholar]
- 15.Kahana, J. A., B. J. Schnapp, and P. A. Silver. 1995. Kinetics of spindle pole body separation in budding yeast. Proc. Natl. Acad. Sci. USA 92:9707-9711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kujoth, G. C., A. Hiona, T. D. Pugh, S. Someya, K. Panzer, S. E. Wohlgemuth, T. Hofer, A. Y. Seo, R. Sullivan, W. A. Jobling, J. D. Morrow, H. Van Remmen, J. M. Sedivy, T. Yamasoba, M. Tanokura, R. Weindruch, C. Leeuwenburgh, and T. A. Prolla. 2005. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309:481-484. [DOI] [PubMed] [Google Scholar]
- 17.Kushnareva, Y., A. N. Murphy, and A. Andreyev. 2002. Complex I-mediated reactive oxygen species generation: modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem. J. 368:545-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin, S. J., M. Kaeberlein, A. A. Andalis, L. A. Sturtz, P. A. Defossez, V. C. Culotta, G. R. Fink, and L. Guarente. 2002. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature 418:344-348. [DOI] [PubMed] [Google Scholar]
- 19.Liochev, S. I., and I. Fridovich. 2005. Cross-compartment protection by SOD1. Free Radic. Biol. Med. 38:146-147. [DOI] [PubMed] [Google Scholar]
- 20.Miquel, J., A. C. Economos, J. Fleming, and J. E. Johnson, Jr. 1980. Mitochondrial role in cell aging. Exp. Gerontol. 15:575-591. [DOI] [PubMed] [Google Scholar]
- 21.O'Brien, K. M., R. Dirmeier, M. Engle, and R. O. Poyton. 2004. Mitochondrial protein oxidation in yeast mutants lacking manganese-(MnSOD) or copper- and zinc-containing superoxide dismutase (CuZnSOD): evidence that MnSOD and CuZnSOD have both unique and overlapping functions in protecting mitochondrial proteins from oxidative damage. J. Biol. Chem. 279:51817-51827. [DOI] [PubMed] [Google Scholar]
- 22.Rodeheffer, M. S., B. E. Boone, A. C. Bryan, and G. S. Shadel. 2001. Nam1p, a protein involved in RNA processing and translation, is coupled to transcription through an interaction with yeast mitochondrial RNA polymerase. J. Biol. Chem. 276:8616-8622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rodeheffer, M. S., and G. S. Shadel. 2003. Multiple interactions involving the amino-terminal domain of yeast mtRNA polymerase determine the efficiency of mitochondrial protein synthesis. J. Biol. Chem. 278:18695-18701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shadel, G. S. 2004. Coupling the mitochondrial transcription machinery to human disease. Trends Genet. 20:513-519. [DOI] [PubMed] [Google Scholar]
- 25.Shadel, G. S. 1999. Yeast as a model for human mtDNA replication. Am. J. Hum. Genet. 65:1230-1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shadel, G. S., and D. A. Clayton. 1997. Mitochondrial DNA maintenance in vertebrates. Annu. Rev. Biochem. 66:409-435. [DOI] [PubMed] [Google Scholar]
- 27.Sherman, F. 1991. Getting started with yeast. Methods Enzymol. 194:3-21. [DOI] [PubMed] [Google Scholar]
- 28.Shigenaga, M. K., T. M. Hagen, and B. N. Ames. 1994. Oxidative damage and mitochondrial decay in aging. Proc. Natl. Acad. Sci. USA 91:10771-10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sikorski, R. S., and P. Hieter. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sohal, R. S., and R. Weindruch. 1996. Oxidative stress, caloric restriction, and aging. Science 273:59-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Speakman, J. R., D. A. Talbot, C. Selman, S. Snart, J. S. McLaren, P. Redman, E. Krol, D. M. Jackson, M. S. Johnson, and M. D. Brand. 2004. Uncoupled and surviving: individual mice with high metabolism have greater mitochondrial uncoupling and live longer. Aging Cell 3:87-95. [DOI] [PubMed] [Google Scholar]
- 32.Sturtz, L. A., K. Diekert, L. T. Jensen, R. Lill, and V. C. Culotta. 2001. A fraction of yeast Cu,Zn-superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria. A physiological role for SOD1 in guarding against mitochondrial oxidative damage. J. Biol. Chem. 276:38084-38089. [DOI] [PubMed] [Google Scholar]
- 33.Trifunovic, A., A. Wredenberg, M. Falkenberg, J. N. Spelbrink, A. T. Rovio, C. E. Bruder, Y. M. Bohlooly, S. Gidlof, A. Oldfors, R. Wibom, J. Tornell, H. T. Jacobs, and N. G. Larsson. 2004. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429:417-423. [DOI] [PubMed] [Google Scholar]
- 34.Turrens, J. F. 1997. Superoxide production by the mitochondrial respiratory chain. Biosci. Rep. 17:3-8. [DOI] [PubMed] [Google Scholar]
- 35.Tzagoloff, A., and A. M. Myers. 1986. Genetics of mitochondrial biogenesis. Annu. Rev. Biochem. 55:249-285. [DOI] [PubMed] [Google Scholar]
- 36.Wallace, D. C. 2005. A Mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer a dawn for evolutionary medicine. Annu. Rev. Genet. 39:359-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wallace, M. A., L. L. Liou, J. Martins, M. H. Clement, S. Bailey, V. D. Longo, J. S. Valentine, and E. B. Gralla. 2004. Superoxide inhibits 4Fe-4S cluster enzymes involved in amino acid biosynthesis. Cross-compartment protection by CuZn-superoxide dismutase. J. Biol. Chem. 279:32055-32062. [DOI] [PubMed] [Google Scholar]
- 38.Wang, Y., and G. S. Shadel. 1999. Stability of the mitochondrial genome requires an amino-terminal domain of yeast mitochondrial RNA polymerase. Proc. Natl. Acad. Sci. USA 96:8046-8051. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.