

Abstract
Cellular O2 sensing enables physiological adjustments to variations in tissue pO2. Under basal conditions, cells are adjusted to an O2 environment biologically read as normoxia. Any sharp departure from that state of normoxia triggers O2-sensitive biological responses. The stabilization of hypoxia-inducible factor (HIF) signifies a robust biological read-out of hypoxia. In the presence of sufficient O2, HIF is hydroxylated and degraded. HIF prolyl hydroxylation is catalyzed by prolyl hydroxylase isoenzymes PHD1, 2 and 3. Using HT22 neurons stably transfected with a HIF reporter construct, we tested a novel hypothesis postulating that biological cells are capable of resetting their normoxic set-point by O2-sensitive changes in PHD expression. Results of this study show that the pO2 of the mouse brain cortex was 35 mm Hg or 5% O2. Exposure of HT22, adjusted to growing in 20% O2, to 5% O2 resulted in HIF-driven transcription. However, cells adjusted to growing in 5% O2 did not report hypoxia. Cells adjusted to growing in 30% O2 reported hypoxia when acutely exposed to room air culture conditions. When grown under high O2 conditions, cells reset their normoxic set-point upwards by down-regulating the expression of PHD1–3. When grown under low O2 conditions, cells reset their normoxic set-point downwards by inducing the expression of PHD1–3. Exposure of mice in vivo to a hypoxic 10% O2 environment lowered blood as well as brain pO2. Such hypoxic exposure induced PHD1–3. Exposure of mice to a hyperoxic 50% O2 ambience repressed the expression of PHD1–3 indicating that O2-sensitive regulation of PHD expression is effective in the brain in vivo. siRNA dependent knock-down of PHD expression revealed that O2-sensitive regulation of PHD may contribute to tuning the normoxic set-point in biological cells.
INTRODUCTION
Cellular O2 homeostasis is tightly maintained within a narrow range (perceived as “normoxia”) due to the risk of oxidative damage from excess O2 (hyperoxia), and of metabolic demise from insufficient O2 (hypoxia) [1]. pO2 ranges from 90 to below 3 Torr in mammalian organs under normoxic conditions with arterial pO2 of about 100 Torr or ~14%O2 [2]. Thus, “normoxia” for cells may be viewed as a variable that is dependent, for example, on the specific localization of the cell in organs and functional status of the specific tissue. Any sharp departure from that state of normoxia, both hypoxia as well as hyperoxia, triggers O2-sensitive signaling and functional biological responses [3–6]. Re-oxygenation associated with reperfusion of an ischemic site causes hyperoxic challenge [3, 4]. On the other hand, focal occlusions in the vasculature, for example, may compromise the local state of oxygenation such that the pO2 is lower than normoxia causing hypoxia. Hypoxia may induce specific molecular responses causing functional changes in the tissue [7]. Because sharp deviations from the normoxic set-point seems to have a significant impact on tissue biology, it is important to understand the principles that influence a cell’s decision to accept any given pO2 as normoxia.
The activation of hypoxia-inducible transcription factor (HIF) has emerged as a robust biological read-out of cellular hypoxia [8]. While pO2 serves as an accurate physical measure of the state of oxygenation, lowering of pO2 coupled with activation of HIF is viewed as biologically effective hypoxia. Once stabilized in the presence of O2 [8], HIF binds to the hypoxia response element to induce the expression of a number of HIF-regulated genes. HIF-alpha activity is regulated by a series of oxygen-dependent enzymatic hydroxylations at specific prolyl and asparaginyl residues. HIF prolyl hydroxylation is performed by a closely related set of prolyl hydroxylase (PHD) isoenzymes [9]. In this study, we sought to test the hypothesis that cells are capable of resetting their normoxic set-point by O2-sensitive changes in PHD expression.
MATERIAL AND METHODS
Materials
Dulbecco’s Modified Eagle Medium, fetal calf serum, hygromycin B, penicillin and streptomycin were obtained from Invitrogen Corporation, Carlsbad, CA. Cell culture dishes were from Nunc, Denmark. The PHD1 expression plasmid was a kind gift from Dr. P. J. Ratcliffe of the University of Oxford, UK.
Cell culture
Stable cell lines expressing an HRE-luciferase reporter were obtained by transfection of HT22 cells with an enolase 1 promoter-reporter construct as described previously [10, 11]. Cells were maintained in Dulbecco's modified Eagle's medium with high glucose, L-glutamine, pyridoxine hydrochloride supplemented with 10% fetal calf serum, 0.1 mg/ml hygromycin B, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C in humidified culture incubators where desired O2 ambience was maintained using an electronic OxyCycler (BioSpherix, Redfield, NY).
Determination of cell viability
The viability was assessed by measuring lactate dehydrogenase (LDH) leakage from cells to media following oxygen exposure (0.5%, 20% or 30%) for different time interval as mentioned in figures using an in vitro toxicology assay kit from Sigma Chemical Co. (St. Louis, MO, USA). In brief, LDH leakage was determined using the following equation: LDH leakage = LDH activity in the cell culture media/total LDH activity (i.e., LDH activity of cells in monolayer + LDH activity in the cell culture media) [12].
pO2 of culture media
Dissolved oxygen of cell culture media was determined using the OxyLite E Tissue Oxygenation and Temperature Monitor System (Oxford Optronix Ltd., Oxford, UK).
Luciferase assay
For luciferase assay cells were seeded in 35 mm plates at a density of 0.2×106 cells per plate in 2 ml media. Cells were maintained in the specified O2 environments for the designated time intervals. When required, cells were extracted for the measurement of luciferase activity using a commercial assay kit (Stratagene, La Jolla, CA) according to the manufacturer’s protocol. Luminescence was measured using a LB 9507 luminometer (EG and G Berthold, Bad Wildbad, Germany). Results were expressed as percentage change in relative light units (RLU)/mg of total protein.
Expression of PHD1 in cells
Following 18 h of seeding, HT22 cells were transiently transfected with either the empty expression plasmid pcDNA3 or the expression plasmid PHD1 (EGLN2) using Lipofectamine2000 reagent (Invitrogen Corporation, Carlsbad, CA) according to manufacturer’s instructions. The cells were maintained in regular culture condition for 24 h to allow for protein expression.
Immunoblot analyses
For PHD1 immunoblot, cytosolic protein extract of cells transiently transfected with pcDNA 3 or PHD1 plasmid were separated on a 10% SDS-polyacrylamide gel under reducing conditions, transferred to PVDF membrane, and probed with anti-PHD1 antibody (1:1000 dilution; Bethyl laboratories, Inc. Montgomery, TX). For HIF1 α immunoblot, cytosolic protein extract (50μg/sample) of cells were separated on a 10% SDS-polyacrylamide gel under reducing conditions, transferred to PVDF membrane, and probed with anti-HIF-1 alpha polyclonal antibody (1:1000 dilution; Novus Biologicals, Inc. Littleton, CO).
siRNA delivery
Cells (0.15×106 cells/well in 12-well plate) were seeded in antibiotic-free medium for 24h prior to transfection. Lipofectamine2000 reagent (Invitrogen Corporation, Carlsbad, CA) was used to transfect cells with 100nM siRNA pool (Dharmacon RNA technologies, Lafayette, CO) for 48h. For control siControl non-targeting siRNA pool (mixture of 4 siRNA, designed to have ≥4 mismatches with known mouse genes) was used.
mRNA quantification
For RNA isolation in in vitro experiments, cells were seeded in 100 mm plates at a density of 1.5×106 cells per plate in 8 ml media. The cells were maintained in the specified O2 environment described in the pertinent figure legends for 24h. After 24h, totalRNA was isolated from cells using Absolutely RNA® Miniprep Kit (Stratagene, La Jolla, CA) according to the manufacturer’s protocol. The abundance of mRNA for PHD1–3 was quantitated using real-time PCR. The double-stranded DNA binding dye SYBR green-I was used. The following primer sets were used:
m_GAPDH F: 5′- ATG ACC ACA GTC CAT GCC ATC ACT –3′
m_GAPDH R: 5′- TGT TGA AGT CGC AGG AGA CAA CCT -3′
m_PHD1_EGLN2 F: 5′- GCG TCT TCG TGA TGG GCA ACT A -3′
m_PHD1_EGLN2 R: 5′- TGC GCC CAT TGA CGT AGT T -3′
m_PHD2_EGLN1 F: 5′- GGA GAT GGA AGA TGC GTG ACA TGT -3′
m_PHD2_EGLN1 R: 5′- TGC TGG CTG TAC TTC ATG AGG GTT A -3′
m_PHD3_EGLN3 F: 5′- TCG GCT TCT GCT ACC TGG ACA A -3′
m_PHD3_EGLN3 R: 5′- GTC GAT GAG GGA CAG GAG GAA GTT -3′
m_HIF-1α F: 5′- GCG ACA CCA TCA TCT CTC TGG ATT -3′
m_HIF-1α R: 5′- GGG CAT GGT AAA AGA AAG TCC CAG T -3′
Note that EGLN1, 2, 3 represents PHD2, 1 and 3, respectively.
In vivo studies
Adult male C57BL/6 mice (Harlan, Indianapolis, IN, USA) were randomly divided into two groups to be held in an environmental chamber: room air (RA) and hypoxia (10% O2). After 24 h of exposure to either room air or hypoxia, brain samples were collected and total RNA was isolated using Trizol. RNA, thus obtained, was cleaned up using Absolutely RNA® Miniprep Kit (Stratagene, La Jolla, CA) according to the manufacturer’s protocol. Real-time PCR was performed as described above for HT22 cells.
Mice in the hypoxia and hyperoxia groups were held in a 10% or 50% O2 environmental chamber, respectively. O2 concentration in the chamber was monitored and automatically adjusted by electronic oxygen controllers (Pro-Ox, BioSpherix, Redfield, NY). Chamber CO2 (<0.03%) and humidity (40–50%) was maintained using CO2-absorbant soda lime and hygroscopic silica gel, respectively. The chamber is fitted with a fan to facilitate the mixing of gas and dissipation of heat. Within the environmental chamber, mice were individually housed in standard cages supported with food and water. Control mice were housed under similar conditions in the chamber containing room air. The custom designed chamber (glove-box) was fitted with two pairs of gloves and an antechamber. This design allows the replacement of cages and handling of mice for blood collection without any transient change in environmental pO2 of the main compartment. The chamber allows the insertion of cables (e.g. O2 sensing OxyLite, Oxford Optronix) while the mice are held in the chamber at the designated O2 ambience. During all experiments, mice were provided with standard breeding rodent chow and water ad libitum. All animal protocols were approved by the Institutional Laboratory Animal Care and Use Committee (ILACUC) of the Ohio State University, Columbus, OH, USA.
Tissue pO2
Mouse brain cortical pO2 was determined using the Oxylite E Tissue Oxygenation and Temperature Monitor System (Oxford Optronix Ltd., Oxford, UK). The brain pO2 of control mice maintained in ambient room oxygen (n=4) was compared to that of mice (n=4) maintained in 10% O2 ambience for 24 h. Control mice were anesthetized with halothane in room air (21% O2) while hypoxic mice were anesthetized with halothane in 10% O2. While under anesthesia, the skull was exposed via a small incision and a 2mm burr hole was drilled over the right hemisphere of the cortex just distal to the bregma suture line, keeping the dura in tact. The fiber-optic probe was then passed through the dura into the cortex a distance of 3mm and then pulled back 0.5mm to relieve any pressure the probe may be exerting on tissue. For the detection of blood pO2, mice were bled from the retro-orbital vein while in the environmental chamber.
Data presentation
Results of at least three experiments are expressed as Mean ± SD. Difference between means was tested by the Student t-test.
RESULTS AND DISCUSSION
Cells in culture are typically maintained at 20% O2, i.e. room air containing 5% CO2. Such ambient pO2 is, in most cases, much higher than the pO2 of the organ represented by the cell. For example, the pO2 of the heart is 35 mm Hg or roughly 5% O2 [3]. We have recently demonstrated that cells isolated from the heart and cultured under conditions of four-fold excess ambient O2, i.e. 20% O2 in room air, demonstrate striking functional changes that may be prevented substantially if cells were grown under iso-pO2 conditions [3, 4]. Such O2-sensitive changes are typically not observed in cell lines grown at 20% O2 because the surviving and propagating cells in the line are those that have adjusted their normoxic set-point to match the room air conditions. We have observed that the pO2 of the mouse brain cortex is approximately 35 mm Hg or roughly 5% O2 (Fig. 6B). This finding is consistent with previous estimates reported using the electron paramagnetic resonance spectroscopic approach [14]. The study of HT22 murine brain neurons demonstrate, however, that exposure to 5% O2 ambience is biologically read as hypoxia as evident by HRE-driven luciferase expression (Fig. 1). Exposure of the same cells to 0.5% O2 ambience further induced HIF activity (Fig. 1A). Of note, if the cells were maintained in 5% O2 on a long-term basis, they no longer responded to that pO2 as detected by HIF activity. When these cells, adjusted to grow in 5% O2, were exposed to 0.5% O2 ambience the resulting induction of HIF activity was less severe than that observed in cells adjusted to grow in 20% O2 room air condition (Fig. 1A). Results from the reporter assay were consistent with Western blot detecting stabilized HIF1α (Fig. 1B). These observations indicate that the biological response to any given pO2 is not tightly dependent on the physical pO2 itself but on the conditioning of the cells. There are HIF1α-independent mechanisms to upregulate HRE-luciferase activity [15]. Thus, we sought to characterize whether HIF1α was indeed responsible for the HRE-luciferase activity detected in our experimental system.
Figure 6. Prolyl hydroxylase (PHD) gene expression in the brain of mice maintained in room air, hypoxic or hyperoxic ambience.
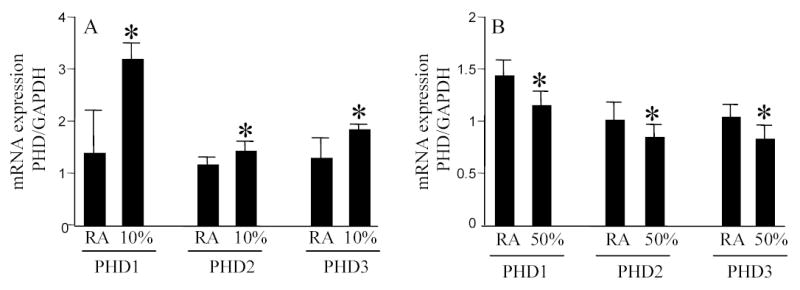
Total RNA was collected from the cortical region of mice brain after 24h in either room air, 10% O2 (A) or 50% O2 (B) ambience. PHD1–3 expression was quantitated using real-time PCR. *, p<0.05 higher (A) or lower (B) compared to the corresponding group maintained in room air. Results are mean ± S.D.
Figure 1. HIF activity in response to changes in ambient O2 environment in cells maintained in either room air (20%) or 5% O2.
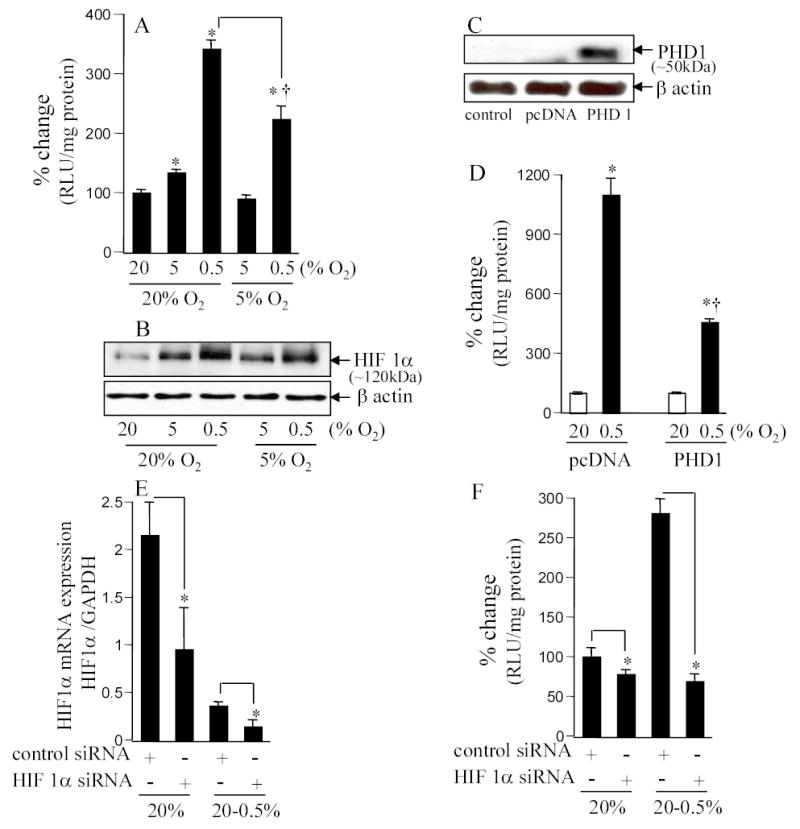
A&B. HT22 cells stably transfected with HRE-luciferase reporter constructs were maintained in 20% or 5% oxygen for 4 weeks before start of experiments. For experiments, cells were plated and maintained in the basal O2 ambience (i.e. either in 20% or 5%) for 24h. After this period, cells were either moved (hypoxic exposure) or not (control) to the lower (5 or 0.5%, as indicated) O2 ambience. Luciferase activity was determined as a measure of HIF-driven transcription (A). Stabilized HIF1a protein was detected by Western Blot (B). *, p<0.05; higher compared to the corresponding control group maintained in 20% O2. †, p<0.05; lower compared to the 20%→0.5% group. Results are mean ± S.D. C&D. PHD over-expression (B) decreased hypoxia-induced HRE-luciferase activity (C). Cells maintained in 20% O2 were transfected with control plasmid (pcDNA 3) or PHD1 plasmid using Lipofectamine 2000. One day (24h) later, cells were moved to 0.5% O2 ambience for 24h and then lucifersase activity was measured. *, p<0.05; higher compared to the corresponding control group maintained in 20% O2; †, lower compared to the pcDNA 0.5% group. E&F. Knock-down of HIF1α (D) abrogated HIF activity (E) observed in A & C. Cells maintained in 20% O2 ambience were plated using antibiotic-free culture media for 24h before transfection. Cells were transfected with 100 nM per sample with control siRNA (non-targeting siRNA pool) or HIF1α mRNA targeting siRNA pool as indicated. Cells were cultured for 48h after transfection and then exposed to hypoxia (E) for 24h before being harvested for mRNA quantitation or determination of luciferase activity. *, p<0.05; lower compared to the corresponding control group as shown.
A major advance in our comprehension of how hypoxia is sensed by mammalian cells came forth with the discovery of a family of oxygen-dependent enzymes responsible for the modulation of HIF-1 stability [16–18]. Degradation of HIF-1α under normoxic conditions is triggered by post-translational O2-dependent enzymatic hydroxylations at specific prolyl and asparaginyl residues within a polypeptide segment known as the oxygen-dependent degradation domain [19]. HIF prolyl hydroxylation is performed by a closely related set of isoenzymes (PHD1–3). Hydroxylation of either human HIF-1α Pro402 or Pro564 promotes interaction with the von Hippel-Lindau tumour suppressor protein (pVHL). In oxygenated cells, this process targets HIF-α for rapid proteasomal destruction [9]. In our effort to determine the significance of HIF in hypoxia-induced HRE reporting observed in Fig. 1A, we over-expressed PHD1 in the cells (Fig. 1C). Hypoxia induced HRE-luciferase reporting was significantly blunted in PHD1 over-expressing cells (Fig. 1D) indicating that HIF is responsible for the observed hypoxia-induced HRE-luciferase activity as evident in Fig. 1A. To confirm the specific role of HIF1α in our experimental system, siRNA based HIF1α knock-down was performed. Delivery of HIF1α siRNA to cells significantly lowered HIF1α gene expression (Fig. 1E). In such HIF1α knock-down cells, the ability of 0.5% O2 ambience failed to induce HRE-luciferase activity (Fig. 1F). Taken together, the results indicated that in our experimental system HIF1α was implicated in enhancing HRE-luciferase activity in response to exposure to hypoxic ambience.
We tested the hypothesis that the normoxic set-point in biological cells is floating and adjustable. To test this hypothesis experiments were conducted where HT22 neurons, stably transfected with the HRE-luciferase construct, were exposed to varying O2 ambience. Exposure of the culture dishes to different O2 ambience clearly influenced the dissolved O2 concentration in the culture media (Fig. 2A). Compared to cells grown under standard room air conditions, the culture media of cells grown in 0.5% O2 ambience contained over ten-fold lower pO2 (Fig. 2A). Exposure to 0.5% O2 ambience did not influence cell viability (Fig. 2B). When cells adjusted to growing in room air condition was moved to 0.5% O2 ambience, potent HIF activity was recorded (Fig. 2C). Continuous exposure of the cells to the same O2 ambience clearly, over time, blunted the biological response reflecting cellular adjustment which may be viewed as a lowering of the normoxic set-point (Fig. 2B). This observation is consistent with our finding reported in Figure 1 demonstrating that cells maintained for 4 weeks in 5% O2 ambience do not report HIF activity. While HRE-driven luciferase activity decreased as a function of time (Fig. 2C), total protein content in the cells maintained at 0.5% O2 ambience did not decline (Fig. 2D).
Figure 2. Culture media pO2 and HIF activity in response to changes in ambient O2.
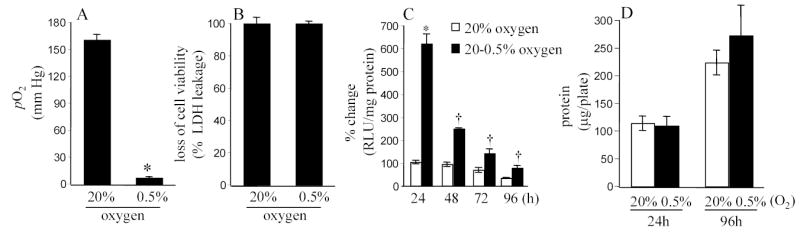
Plates containing cells in culture were moved from 20% O2 ambience to 0.5% O2 ambience. Dissolved O2 concentration in the culture media was determined after 24h of maintenance in 20% or 0.5% as indicated (A). B. The hypoxic exposure did not influence cell viability. C&D, Cells maintained in 20% O2 ambience were plated and moved to 0.5% O2 ambience where they were cultured for the specified duration. HIF activity subsided over time (C) while total protein content (D) in the cells were not affected. *, p<0.05. lower (A) or higher (C) compared to the paired control group maintained in 20% O2. †, p<0.05; lower compared to the HIF activity in the 0.5% group of the immediately previous time point. Results are mean ± S.D.
To further test the hypothesis that the normoxic set-point in biological cells is adjustable we tried another line of investigation testing whether the cells could be made to biologically report hypoxia by adjusting them to an O2 ambience above 20%. Exposure of the cells to 50% O2 resulted in overt growth arrest and some toxicity (not shown). Based on pilot studies, we identified that at 30% O2 ambience HT22 cells grew well and free of O2 toxicity (Fig. 3C). We sought to adjust the putative normoxic set-point upwards by maintaining a colony of cells in 30% O2 ambience. Passage-matched cells were maintained under standard 20% O2 culture conditions. Acute exposure of cells maintained in 30% O2 to 20% O2 ambience resulted in increased HIF activity (Fig. 3A). The observation that exposure to room air ambience was biologically read by the cells as a hypoxic exposure suggests that maintenance of cells in 30% O2 was effective in resetting the normoxic set point upwards. This finding was also consistent with our observation that the exposure of 30% O2 was not toxic to the cells (Fig. 3B).
Figure 3. Inducible HIF activity is dependent on the O2 ambience to which the cells are adjusted to and not on absolute pO2.
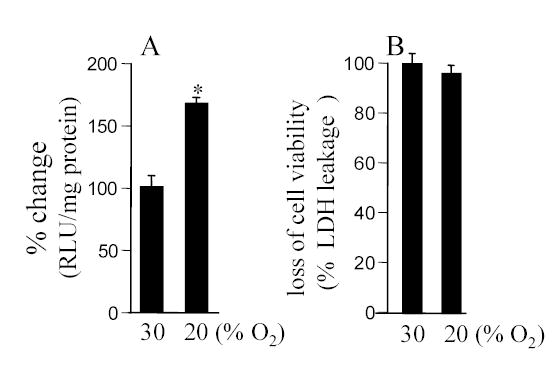
A. Cells were plated and maintained in 30% O2 ambience for 24h. After this period, cells were either moved to 20% or 30% ambience as indicated. After another 24h of culture, luciferase activity was measured. B. The said changes in O2 ambience did not influence cell viability (also see Fig. 2B). Results are mean ± S.D. *, p<0.05.
In the presence of sufficient pO2, HIF-1α is destabilized by post-translational hydroxylation of Pro-564 and Pro-402 by a family of oxygen-sensitive dioxygenases catalyzing prolyl hydroxylation, a specific modification that provides recognition for the E3 ubiquitin ligase complex containing the von Hippel-Lindau tumour suppressor protein. Three HIF prolyl-hydroxylases (PHD1, 2 and 3) were identified recently in mammals and shown to hydroxylate HIF-α subunits. In HT22 cells maintained under standard room air (20% O2) conditions, acute changes in O2 ambience significantly influenced the expression of PHD1–3. Exposure of such cells to lower pO2 increased PHD expression while exposure to higher pO2 decreased expression of PHD 1–3 (Fig. 4, upper row). In cells adjusted to growing in 30% O2 ambience, we had noted an upward shift in the normoxic set-point (Fig. 3). Studies of PHD1–3 mRNA expression revealed that compared to cells grown in 20% O2, cells maintained in 30% O2 ambience had significantly lower PHD1 expression (Fig. 4). In these cells, grown in 30% O2 ambience, acute exposure to 0.5% significantly increased the expression of PHD1–3 (Fig. 4). Next, we sought to examine whether this O2-sensitive expression of PHD1–3 is effective in vivo. Mice were maintained in an environmental chamber either containing standard room air or 10% O2. Exposure to the 10% O2 environment decreased both peripheral blood as well as brain pO2 (Fig. 5). Under physiological conditions while tissue hypoxia is typically associated with ischemic disorders, reoxygenation may cause hyperoxic insult [3, 4, 25]. Consistent with the findings with HT22 cells hypoxia and hyperoxia in vivo significantly increased and decreased the expression of PHD 1–3, respectively (Fig. 6). To test the significance of PHD expression with respect to HIF transactivation, we employed the siRNA approach to specifically knock-down PHD1, 2 or 3. Knock-down of PHD resulted in HRE-luciferase reporting even under conditions of room air (Fig. 7). These observations indicate that changes in PHD expression may be effective in resetting the normoxic set-point of cells. Thus, PHD-targeted approaches may be effective in inducing or silencing HIF-dependent gene expression under a given condition of pO2.
Figure 4. Expression of prolyl hydroxylase (PHD) 1–3 in response to changes in O2 ambience.
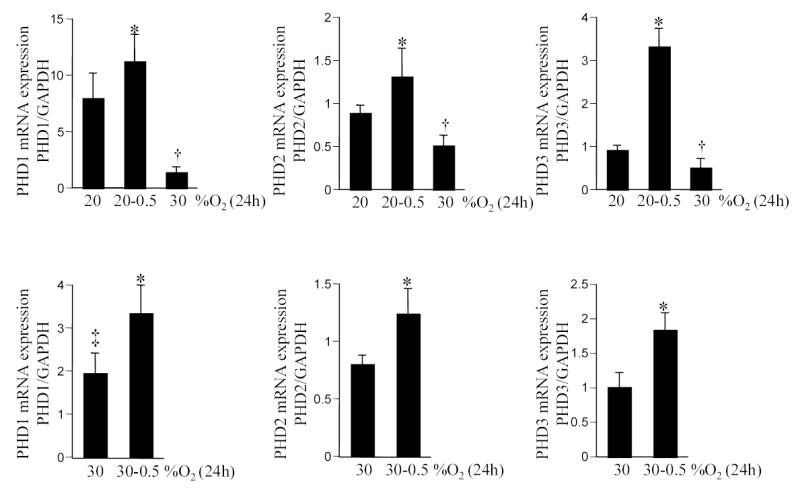
Cells were maintained in 20% (upper row) or 30% (lower row) O2 ambience for 4 weeks. For experiments, cells were plated and maintained in the corresponding O2 ambience for 24h. After that period, cells were moved to either a lower (0.5%) or higher (30%) O2 ambience as indicated. PHD mRNA levels were quantitated by real-time PCR. *or †, p<0.05 compared to the corresponding bar on the extreme left of each panel. ‡, lower compared to PHD1 expression in cells adjusted to growing in 20% O2 ambience (i.e. bar vertically above). Results are mean ± S.D.
Figure 5. Blood (A) and brain (B) pO2 of mice in response to changes in O2 ambience.
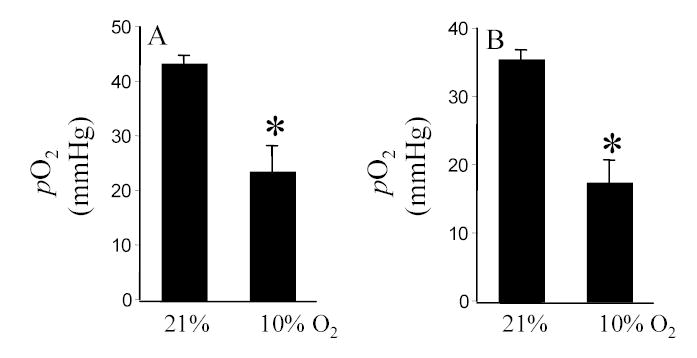
*, p<0.05. Results are mean ± S.D.
Figure 7. Knock-down of prolyl hydroxylase (PHD) 1–3 expression elevates HIF activity.
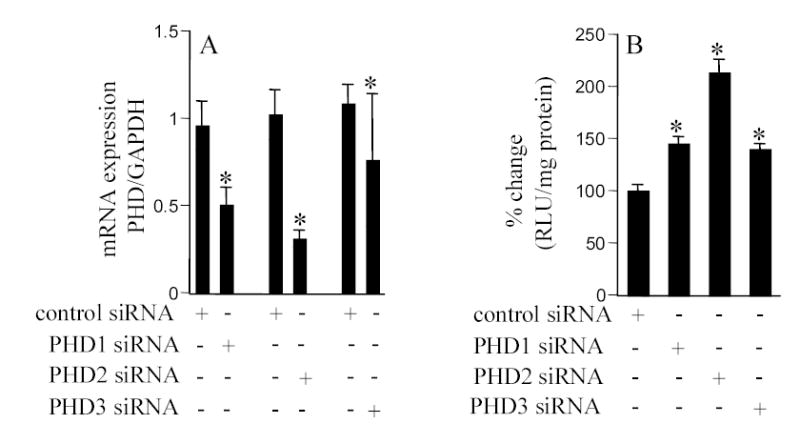
Cells maintained in 20% O2 ambience were plated in antibiotic-free culture media for 24h before transfection. Cells were transfected with 100 nM per sample with control siRNA (non-targeting siRNA pool) or PHD1/PHD2/PHD3 siRNA targeting siRNA pool as indicated. Cells were maintained for 48h after transfection before being harvested for mRNA quantitation (A) or determination of HRE-luciferase activity (B). PHD (1–3) mRNA levels were quantitated by real-time PCR. p<0.05, *, lower (A) or higher (B) compared to the corresponding group treated with control siRNA. Results are mean ± S.D.
The current study provides first evidence demonstrating that PHD expression is down-regulated at higher pO2. Thus, levels of PHD expression are subject to dual regulation by ambient pO2. Using a small interference RNA approach it has been recently demonstrated that each of the three PHD isoforms contributes in a non-redundant manner to the regulation of both HIF-1α and HIF-2α subunits. Of importance, it has been reported that the contribution of each PHD under particular culture conditions is strongly dependent on the abundance of the enzyme [26]. Taken together, this work presents first direct evidence supporting that the normoxic set-point in cells is adjustable and that O2-sensitive PHD expression may be implicated in fine tuning that set-point.
Acknowledgments
Supported by NIH NS42617, HL73087 and GM 69589 to CKS.
References
- 1.Semenza GL. HIF-1, O(2), and the 3 PHDs: how animal cells signal hypoxia to the nucleus. Cell. 2001;107:1–3. doi: 10.1016/s0092-8674(01)00518-9. [DOI] [PubMed] [Google Scholar]
- 2.Porwol T, Ehleben W, Brand V, Acker H. Tissue oxygen sensor function of NADPH oxidase isoforms, an unusual cytochrome aa3 and reactive oxygen species. Respiration Physiology. 2001;128:331–348. doi: 10.1016/s0034-5687(01)00310-3. [DOI] [PubMed] [Google Scholar]
- 3.Roy S, Khanna S, Bickerstaff AA, Subramanian SV, Atalay M, Bierl M, Pendyala S, Levy D, Sharma N, Venojarvi M, Strauch A, Orosz CG, Sen CK. Oxygen sensing by primary cardiac fibroblasts: a key role of p21(Waf1/Cip1/Sdi1) Circ Res. 2003;92:264–271. doi: 10.1161/01.res.0000056770.30922.e6. [DOI] [PubMed] [Google Scholar]
- 4.Roy S, Khanna S, Wallace WA, Lappalainen J, Rink C, Cardounel AJ, Zweier JL, Sen CK. Characterization of perceived hyperoxia in isolated primary cardiac fibroblasts and in the reoxygenated heart. Journal of Biological Chemistry. 2003;278:47129–47135. doi: 10.1074/jbc.M308703200. [DOI] [PubMed] [Google Scholar]
- 5.Covello KL, Simon MC. HIFs, hypoxia, and vascular development. Curr Top Dev Biol. 2004;62:37–54. doi: 10.1016/S0070-2153(04)62002-3. [DOI] [PubMed] [Google Scholar]
- 6.Michiels C. Physiological and pathological responses to hypoxia. Am J Pathol. 2004;164:1875–1882. doi: 10.1016/S0002-9440(10)63747-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, Garcia JG, Semenza GL. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood. 2005;105:659–669. doi: 10.1182/blood-2004-07-2958. [DOI] [PubMed] [Google Scholar]
- 8.Semenza GL. Hydroxylation of HIF-1: oxygen sensing at the molecular level. Physiology (Bethesda) 2004;19:176–182. doi: 10.1152/physiol.00001.2004. [DOI] [PubMed] [Google Scholar]
- 9.Metzen E, Ratcliffe PJ. HIF hydroxylation and cellular oxygen sensing. Biol Chem. 2004;385:223–230. doi: 10.1515/BC.2004.016. [DOI] [PubMed] [Google Scholar]
- 10.Aminova LR, Chavez JC, Lee J, Ryu H, Kung A, Lamanna JC, Ratan RR. Prosurvival and prodeath effects of hypoxia-inducible factor-1alpha stabilization in a murine hippocampal cell line. J Biol Chem. 2005;280:3996–4003. doi: 10.1074/jbc.M409223200. [DOI] [PubMed] [Google Scholar]
- 11.Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, Giallongo A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem. 1996;271:32529–32537. doi: 10.1074/jbc.271.51.32529. [DOI] [PubMed] [Google Scholar]
- 12.Khanna S, Roy S, Ryu H, Bahadduri P, Swaan PW, Ratan RR, Sen CK. Molecular basis of vitamin E action. Tocotrienol modulates 12-lipoxygenase, a key mediator of glutamate-induced neurodegeneration. Journal of Biological Chemistry. 2003;278:43508–43515. doi: 10.1074/jbc.M307075200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sen CK, Khanna S, Babior BM, Hunt TK, Ellison EC, Roy S. Oxidant-induced vascular endothelial growth factor expression in human keratinocytes and cutaneous wound healing. J Biol Chem. 2002;277:33284–33290. doi: 10.1074/jbc.M203391200. [DOI] [PubMed] [Google Scholar]
- 14.O'Hara JA, Khan N, Hou H, Wilmo CM, Demidenko E, Dunn JF, Swartz HM. Comparison of EPR oximetry and Eppendorf polarographic electrode assessments of rat brain PtO2. Physiol Meas. 2004;25:1413–1423. doi: 10.1088/0967-3334/25/6/007. [DOI] [PubMed] [Google Scholar]
- 15.Zaman K, Ryu H, Hall D, O'Donovan K, Lin KI, Miller MP, Marquis JC, Baraban JM, Semenza GL, Ratan RR. Protection from oxidative stress-induced apoptosis in cortical neuronal cultures by iron chelators is associated with enhanced DNA binding of hypoxia-inducible factor-1 and ATF-1/CREB and increased expression of glycolytic enzymes, p21(waf1/cip1), and erythropoietin. J Neurosci. 1999;19:9821–9830. doi: 10.1523/JNEUROSCI.19-22-09821.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 17.Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O'Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 18.Jaakkola P. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 19.Semenza GL. HIF-1 and tumor progression: pathophysiology and therapeutics. Trends in Molecular Medicine. 2002;8:S62–67. doi: 10.1016/s1471-4914(02)02317-1. [DOI] [PubMed] [Google Scholar]
- 20.Dulak J, Jozkowicz A. Regulation of vascular endothelial growth factor synthesis by nitric oxide: facts and controversies. Antioxid Redox Signal. 2003;5:123–132. doi: 10.1089/152308603321223612. [DOI] [PubMed] [Google Scholar]
- 21.Mukhopadhyay D, Datta K. Multiple regulatory pathways of vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) expression in tumors. Semin Cancer Biol. 2004;14:123–130. doi: 10.1016/j.semcancer.2003.09.019. [DOI] [PubMed] [Google Scholar]
- 22.Hopf HW, Gibson JJ, Angeles AP, Constant JS, Feng JJ, Rollins MD, Zamirul Hussain M, Hunt TK. Hyperoxia and angiogenesis. Wound Repair Regen. 2005;13:558–564. doi: 10.1111/j.1524-475X.2005.00078.x. [DOI] [PubMed] [Google Scholar]
- 23.Patel V, Chivukala I, Roy S, Khanna S, He G, Ojha N, Mehrotra A, Dias LM, Hunt TK, Sen CK. Oxygen: from the benefits of inducing VEGF expression to managing the risk of hyperbaric stress. Antioxid Redox Signal. 2005;7:1377–1387. doi: 10.1089/ars.2005.7.1377. [DOI] [PubMed] [Google Scholar]
- 24.Sheikh AY, Gibson JJ, Rollins MD, Hopf HW, Hussain Z, Hunt TK. Effect of hyperoxia on vascular endothelial growth factor levels in a wound model. Arch Surg. 2000;135:1293–1297. doi: 10.1001/archsurg.135.11.1293. [DOI] [PubMed] [Google Scholar]
- 25.Roy S, Khanna S, Sen CK. Perceived hyperoxia: oxygen-regulated signal transduction pathways in the heart. Methods in Enzymology. 2004;381:133–139. doi: 10.1016/S0076-6879(04)81008-5. [DOI] [PubMed] [Google Scholar]
- 26.Appelhoff RJ, Tian YM, Raval RR, Turley H, Harris AL, Pugh CW, Ratcliffe PJ, Gleadle JM. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279:38458–38465. doi: 10.1074/jbc.M406026200. [DOI] [PubMed] [Google Scholar]