

Abstract
In this study we developed a new method for accurately determining the pentose phosphate pathway (PPP) split ratio, an important metabolic parameter in the primary metabolism of a cell. This method is based on simultaneous feeding of unlabeled glucose and trace amounts of [U-13C]gluconate, followed by measurement of the mass isotopomers of the intracellular metabolites surrounding the 6-phosphogluconate node. The gluconate tracer method was used with a penicillin G-producing chemostat culture of the filamentous fungus Penicillium chrysogenum. For comparison, a 13C-labeling-based metabolic flux analysis (MFA) was performed for glycolysis and the PPP of P. chrysogenum. For the first time mass isotopomer measurements of 13C-labeled primary metabolites are reported for P. chrysogenum and used for a 13C-based MFA. Estimation of the PPP split ratio of P. chrysogenum at a growth rate of 0.02 h−1 yielded comparable values for the gluconate tracer method and the 13C-based MFA method, 51.8% and 51.1%, respectively. A sensitivity analysis of the estimated PPP split ratios showed that the 95% confidence interval was almost threefold smaller for the gluconate tracer method than for the 13C-based MFA method (40.0 to 63.5% and 46.0 to 56.5%, respectively). From these results we concluded that the gluconate tracer method permits accurate determination of the PPP split ratio but provides no information about the remaining cellular metabolism, while the 13C-based MFA method permits estimation of multiple fluxes but provides a less accurate estimate of the PPP split ratio.
Quantification of primary metabolic fluxes in microorganisms provides researchers with an important tool for a more rational approach to metabolic engineering. A part of the cellular metabolism that has received special attention when possible strain improvement strategies have been examined is the flux distribution around branch points (9, 30, 31). At these metabolic branch points an entering flux diverges in two or more different directions, thus forming a potential target for rerouting fluxes. Under glucose-feeding conditions the first metabolic node encountered by the glucose entering a cell is the node around glucose-6-phosphate (g6p), which results in carbon flux partitioning toward glycolysis, the pentose phosphate pathway (PPP), storage carbohydrates, and in some prokaryotes the Entner-Doudoroff pathway. Each of these pathways has its own unique function in the cell. Glycolysis (combined with the tricarboxylic acid cycle) is the general route for glucose catabolism and energy formation in the cell, while the PPP plays a crucial role in the redox metabolism of the cell. In addition to their catabolic functions these pathways also have anabolic functions as they provide the precursors for the monomers (amino acids, fatty acids, nucleotides, sugar phosphates, etc.) required for growth and product formation. Therefore, accurate determination of the flux distribution around the g6p node provides valuable insight into the functioning of a cell.
An important flux ratio of the g6p node is the fraction of g6p entering the oxidative branch of the PPP in relation to the total uptake of glucose by the cell (referred to as the PPP split ratio below). From an industrial point of view, split ratio determinations are interesting in the quest for strains that overproduce (partially) PPP-originating products, such as phenylalanine and riboflavin. Recently, workers have also revealed the importance of the oxidative branch of the PPP in regulating the cytosolic NADPH levels when specific amino acids (20) and antibiotics are overproduced (35).
One of the earliest documented quantifications of the PPP split ratio dates from 1955, when Katz et al. (18) developed a method to calculate the flux into the oxidative branch of the PPP based on the evolution of 14CO2 in rat liver cells that were consecutively fed [U-14C]glucose, [6-14C]glucose, and [1-14C]glucose. In the ensuing decades the flux of the oxidative branch of the PPP has usually been estimated by feeding cells specifically 13C- or 14C-labeled glucose and then analyzing the 13C or 14C distribution in the CO2 produced or in derivatives of the triose phosphates produced (e.g., lactate, glycerol, and amino acids) (8, 21, 25). Some of these methods are rather laborious, requiring parallel experiments with two differently labeled substrates, while others can be quite costly due to the specifically labeled substrates used. However, the major problem with these methods is that they are based on assumptions concerning the recycling of fructose-6-phosphate (f6p) produced in the PPP, the reversibility (12) and structure (22) of the reactions in the nonoxidative branch of the PPP, the degree of isotopic equilibrium of the hexose monophosphate pool, and the drain on PPP metabolites as precursors for the synthesis of biomass (19). The various assumptions lead to widely different determinations of the PPP split ratio.
Recently, a more robust method was developed by Christensen et al. (3), in which an overall 13C balance is set up for the upper part of glycolysis and the PPP. This method can estimate the PPP flux irrespective of the isotope redistributions arising from f6p recycling and PPP reversibility. However, the method is also based on assumptions that limit the validity of the estimated PPP split ratios. It is assumed that C-5 of g6p is naturally labeled when cells are fed [1-13C]glucose, that the label pattern of glyceraldehyde-3-phosphate is identical to that of serine, and that the labeling of C-1 of serine and the labeling of C-2 of serine are identical, thereby neglecting the possible effect of the enzymes threonine aldolase and glycine decarboxylase on the label distribution in serine.
Nowadays, 13C tracer experiments are increasingly used for simultaneously determining the flux distributions of multiple convergent metabolic nodes. This method, often referred to as 13C-based metabolic flux analysis (MFA), combines 13C labeling data for primary metabolites with uptake and secretion rates and biomass composition to determine the fluxes throughout a metabolic network. The labeling of the primary metabolites can be measured either directly using liquid chromatography-mass spectrometry (LC-MS) (38) or indirectly by measuring the 13C labeling of proteinogenic amino acids using gas chromatography-mass spectrometry (11, 13) and/or nuclear magnetic resonance (26). Until now, however, it has been difficult to accurately determine the PPP split ratio using this method. van Winden et al., for example, showed that the PPP split ratio estimates for Penicillium chrysogenum are highly dependent on the network stoichiometry used (36), while large confidence intervals were found for the PPP split ratios of Saccharomyces cerevisiae and Bacillus subtilis (6, 38).
In this study we developed a new method for determining the PPP split ratio based on simultaneous feeding of unlabeled glucose and trace amounts of [U-13C]gluconate. By measuring the mass isotopomers of the intracellular metabolites around the g6p node, the flux into the oxidative branch of the PPP can be directly calculated. The applicability of the gluconate tracer method was studied by using a penicillin G-producing chemostat culture of the filamentous fungus P. chrysogenum. The sensitivity and accuracy of the estimated PPP split ratio were compared to the sensitivity and accuracy of the PPP split ratio estimated from a 13C-labeling-based MFA of P. chrysogenum grown under identical conditions. For the first time mass isotopomer measurements of 13C-labeled primary metabolites were determined for P. chrysogenum and used for 13C-based MFA.
Theory.
The first step in the PPP is the irreversible dehydrogenation of g6p to 6-phosphoglucono-δ-lactone catalyzed by g6p dehydrogenase, followed by hydrolysis to 6-phosphogluconate (6pg). 6-Phosphogluconate is subsequently catabolized via the Entner-Douderoff pathway or the PPP. Under most growth conditions the irreversibility of these two pathways ensures that 6pg can originate only from g6p, resulting in identical labeling patterns for the two metabolites. An additional inflow into the 6pg pool can be obtained by feeding gluconate to the cells. After uptake, gluconate is directly phosphorylated to 6pg by the enzyme gluconokinase and in this way enters the PPP directly after the g6p branch point. Cofeeding of [U-13C]gluconate to a culture grown on naturally labeled glucose results in a different labeling pattern for g6p and 6pg. The difference in the labeling pattern is directly related to the amount of g6p that enters the oxidative branch of the PPP.
Figure 1 schematically shows the primary metabolism of a cell grown simultaneously on glucose (glc) and gluconate (gln). The flux into the oxidative branch of the PPP (v3) can be calculated by setting up a metabolite balance and a mass isotopomer balance over the metabolite 6pg:
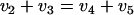 |
(1) |
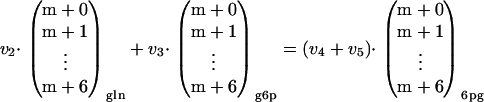 |
(2) |
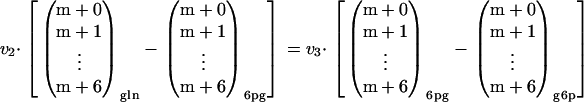
where v2 is the uptake rate of gluconate. Equation 1 shows the mass balance for 6pg, while equation 2 shows the labeling balance in which the fluxes are multiplied by the mass isotopomer distribution vector of the corresponding metabolite. Substituting equation 1 into equation 2 yields the following:
 |
(3) |
By measuring the uptake rate of gluconate and the mass isotopomer distributions of g6p, 6pg, and gluconate an overdetermined system is obtained, from which v3 can be estimated. Since measurement errors occur on both sides of equation 3, a sequential quadratic programming algorithm was used to estimate v3. The PPP split ratio is obtained by dividing v3 by the rate of glucose uptake (v1). Note that for accurate estimation of the PPP split ratio, the measured mass isotopomer distribution vectors in equation 3 should be corrected for naturally occurring isotopes of hydrogen, nitrogen, and oxygen. This was done by inverting the procedure proposed by van Winden et al. (37).
FIG. 1.

Schematic diagram of the metabolic network surrounding the g6p node when cells are growing simultaneously on glucose and gluconate. The PPP split ratio is defined as v3/v1. g3p, glyceraldehyde-3-phosphate; TCA, tricarboxylic acid.
By introducing a labeling difference among the metabolites directly surrounding the entry point of the PPP and by measuring the labeling pattern of these metabolites, the split ratio can be accurately determined without any of the assumptions made in the previously developed methods. Furthermore, direct assessment of the surrounding metabolites rules out any uncertainties introduced by deducing the labeling of primary metabolites from measured labeling patterns of proteinogenic amino acids.
In order to accurately measure the PPP split ratio using the method described above, several conditions have to be met. First, the rate of gluconate uptake should be low, so the overall metabolism of the cell is not disturbed. Thus, only tracer amounts of gluconate should be added to the medium (hence the name gluconate tracer method). Second, the microorganism investigated has to simultaneously metabolize glucose and gluconate. In general, mixtures of carbon sources are taken up simultaneously by a cell only under carbon-limiting conditions (10). Under excess-carbon conditions, the molecular mechanisms of catabolite repression and inducer exclusion ensure that the cell first depletes the energetically more favorable carbon source. Growth on both glucose and gluconate simultaneously has been reported for various microorganisms, such as Corynebacterium glutamicum (24), Bacillus subtilis (7), Escherichia coli (17), and Schizosaccharomyces pombe (2); in this study we focused on P. chrysogenum. The last prerequisite for accurately applying the gluconate tracer method is knowledge about the glucose metabolism of the microorganism studied. Glucose-oxidizing reactions catalyzed by glucose oxidase or glucose dehydrogenase can give rise to an influx of unlabeled carbon (originating from glucose) into the uniformly 13C-labeled gluconate pool, thereby complicating determination of the PPP split ratio. In general, however, these enzymes are expressed only under excess-glucose conditions. Harris et al. (15), for example, showed that the enzyme glucose oxidase could not be detected in a glucose-limited chemostat culture of P. chrysogenum.
MATERIALS AND METHODS
Strain and cultivation conditions.
All cultivations described in this study were performed in a carbon-limited chemostat system operated at a growth rate of 0.02 h−1, using a high-yield industrial P. chrysogenum strain (strain DS17690, kindly donated by DSM Anti-Infectives, Delft, The Netherlands). This strain was a reisolation of the strain previously used in our lab (strain DS12975) (35).
Both the gluconate tracer analysis and the 13C-based MFA for determining the PPP split ratio were carried out using a 1-liter bioreactor (Applikon, Schiedam, The Netherlands). The working volume of the reactor was kept constant at 0.6 liter by means of an overflow system. Effluent was removed discontinuously by pumping out liquid at fixed time intervals (1 min every 10 min). The temperature was controlled at 25°C, while the pH was controlled at 6.50 by automatic titration with 0.5 M NaOH and 0.25 M H2SO4. To keep the dissolved oxygen tension above 50%, the bioreactor was equipped with one Rushton turbine stirrer (600 rpm) and was aerated with pressurized air at a rate of 10 liters/h (0.28 [vol/vol/min]) with an overpressure of 0.1 × 105 Pa. A silicone antifoam agent (BDH, Poole, United Kingdom) was diluted 1:10 (vol/vol) and added to the reactor at a fixed rate of 0.2 ml/h.
Cells were grown on a scaled-down version of the minimal medium described by van Gulik et al. (35) in order to minimize the cost of the labeling experiments. For the 13C-based MFA experiment the minimal medium contained 3.3 g/liter glucose H2O, 0.68 g/liter sodium acetate 3H2O, 0.35 g/liter KH2PO4, 1.54 g/liter (NH4)2SO4, 0.22 g/liter MgSO4 7H2O, 0.53 g/liter phenylacetic acid (PAA), and 0.90 ml/liter trace element solution. The trace element solution contained 75 g/liter Na2-EDTA 2H2O, 2.5 g/liter CuSO4 5H2O, 10 g/liter ZnSO4 7H2O, 10 g/liter MnSO4 H2O, 20 g/liter FeSO4 7H2O, and 2.5 g/liter CaCl2 2H2O. A similar medium was used for the gluconate tracer experiment; the only modification was that 5 Cmol% of glucose was replaced by an equimolar amount of glucono-δ-lactone (0.005 Cmol/liter). The reason for addition of a small amount of sodium acetate to the medium was to introduce an additional inflow of labeled carbon into the metabolism for better estimation of the fluxes in the lower part of the metabolism when the 13C-based MFA method was used.
PAA, the side chain precursor for penicillin G, was added to the medium at a concentration that kept the residual concentration in the chemostat around 3 mM. At this concentration PAA neither limited penicillin G production nor inhibited the growth of P. chrysogenum. For preparation of the minimal medium the appropriate amount of PAA was dissolved in 1 liter of demineralized water, the pH was adjusted to 5.40 using 1 M KOH, and the preparation was autoclaved at 121°C for 40 min. The remaining medium components were dissolved in 4 liters of demineralized water, the pH was adjusted to 5.40 using 1 M KOH, and the preparation was filter sterilized and added to the PAA solution using an Acropak20 filter (PALL, East Hills, NY). After preparation the minimal medium was stirred for at least 12 h with a magnetic stirrer. At a medium pH of 5.40, this incubation for 12 h ensured that all the added glucono-δ-lactone was hydrolyzed to gluconate (32).
Except for the smaller chemostat volume, the procedures for the batch phase and the first part of the chemostat phase were carried out as described by van Gulik et al. (35). During cultivation the batch phase was inoculated with spores from 2.0 g of rice grains. The sole energy source during the batch phase was 3.3 g/liter of glucose H2O. The end of the batch phase was typically reached after 50 h, after which the reactor was switched to the continuous mode using the minimal medium described above. After 2 residence times this medium was replaced by a medium that was chemically identical except that part of the naturally labeled carbon was replaced by 13C. For the 13C-based MFA, 60 Cmol% of the glucose was replaced by specifically labeled [1-13C]glucose (Sigma-Aldrich, St. Louis, MO), 20 Cmol% of the glucose was replaced by [U-13C]glucose (Sigma-Aldrich), and 100 Cmol% of the acetate was replaced by [U-13C]acetate (Sigma-Aldrich). For the gluconate tracer experiment 100 Cmol% of the gluconate (0.005 Cmol/liter) was replaced by [U-13C]glucono-δ-lactone (Omicron, South Bend, IN).
Biomass sampling.
Duplicate 10-ml samples were withdrawn from the bioreactor every second day to determine the biomass dry weight. Samples were filtered over preweighed glass fiber filters (PALL) and dried at 70°C for at least 24 h. The filtrate collected was immediately frozen in liquid nitrogen and used to analyze the extracellular penicillin G and PAA concentrations.
Recently, Mashego et al. (28) observed that the feeding time with 13C-labeled substrate that is necessary in order to obtain an isotopic steady state of the intracellular metabolites is longer than previously anticipated. A possible explanation for the slow replacement of unlabeled metabolites is the turnover of unlabeled storage sugars. To ensure that the intracellular primary metabolites measured in this study were in both chemical and isotopic steady states, the culture was incubated for 3 residence times after the medium was changed to the 13C-labeled medium, before broth samples were harvested to determine the extracellular residual substrate concentrations and the mass isotopomer distributions of intracellular metabolites. Extracellular samples were acquired by rapidly withdrawing 2 ml of broth with a syringe containing precooled stainless steel beads (−18°C), followed immediately by separation of the cells and medium by filtration (27). The samples were stored at −80°C prior to analysis. Samples used for intracellular metabolite determination were obtained by rapidly withdrawing 1 ml of broth from the bioreactor and then injecting the sample directly into 5 ml of a 60% (vol/vol) methanol-water mixture (−40°C) for instantaneous quenching of the cell metabolism (23). A total of 16 1-ml samples of broth were obtained from the bioreactor and processed for metabolite extraction.
Metabolite extraction.
Eight of the 16 samples harvested for metabolite extraction were centrifuged for 5 min at 5,000 × g in a cooled (−20°C) centrifuge equipped with a precooled (−40°C) swing-out rotor with four buckets (Heraeus, Hanau, Germany). After the supernatant was decanted, each pellet was resuspended along with one of the eight remaining samples, which resulted in doubling the amount of biomass in each tube. This step was performed to compensate for the low biomass concentration in the bioreactor (ca. 1 g/liter). The pooled samples were centrifuged for 5 min at 5,000 × g. After the supernatant was decanted, each pellet was resuspended in fresh 60% (vol/vol) methanol-water (−40°C) and centrifuged for 5 min at 5,000 × g. This step ensured removal of any extracellular components (e.g., salts) that would otherwise hamper the metabolite analysis. After this washing step was repeated, the samples were decanted and stored at −40°C in a cryostat. The metabolites were extracted from the pellets using the boiling ethanol procedure described by Lange et al. (23). The boiled extracts were evaporated under a controlled vacuum at a controlled temperature for 45 min in a Rapid-Vap (Labinco, Kansas City, MO) to remove the ethanol and traces of methanol. It was found that within 45 min all of the ethanol and methanol had evaporated from the samples, which left a mixture of metabolites and cell debris suspended in approximately 200 μl of water. The samples were centrifuged for 10 min at 13,000 × g, and the supernatants were stored at −80°C prior to LC-MS analysis.
Sample analysis.
The mass isotopomer distributions of the intracellular metabolites were determined as described by van Winden et al. (38). Metabolites were first separated by high-performance anion-exchange chromatography (Waters, Milford, MA) and then analyzed by mass spectrometry with a Quatro-LC triple quadrupole mass spectrometer (Micromass Ltd., Manchester, United Kingdom) equipped with an electrospray ionization interface. Samples were analyzed for the following intermediates of glycolysis and the PPP: g6p, f6p, 6pg, gln, mannose-6-phosphate, 1,6-fructose bisphosphate (fbp), phosphoenolpyruvate (pep), the combined pool of 2-phosphoglycerate and 3-phosphoglycerate (2/3pg), the combined pool of xylulose-5-phosphate, ribose-5-phosphate, and ribulose-5-phosphate, and sedoheptulose-7-phosphate.
The concentrations of glucose, gluconate, and acetate in the medium and the bioreactor were determined enzymatically (Scil Diagnostics, Viernheim, Germany).
Enzymatic analysis.
The cell extracts used for analysis of glucose oxidase, glucose dehydrogenase, and 6pg phosphatase were prepared as described by Harris et al. (15). The glucose dehydrogenase activities of cell extracts were determined using the assay described by van Dijken et al. (34). The glucose oxidase activities of filtrates and cell extracts were determined using the assay described by Harris et al. (15). 6pg phosphatase activity was determined by incubating (25°C, 30 min) 1 ml of cell extract with 100 μmol of 6pg and enzymatically measuring the production of gluconate (Scil Diagnostics, Viernheim, Germany). Background gluconate levels in the cell extracts were used as blanks.
13C label distribution for glucose and gluconate.
The mass isotopomer distributions of the labeled glucose mixture used for the 13C-labeling-based MFA and the [U-13C]gluconate used for the gluconate tracer method were determined by LC-MS analysis. Since glucose could not be directly measured by LC-MS, a mixture of [1-13C]glucose, [U-13C]glucose, and naturally labeled glucose was first phosphorylated to g6p by incubating 200 μl of medium with 15 μl of 0.25 M ATP (pH 7.0) and 3 μl of hexokinase (1,500 U/ml; Roche Diagnostics, Almere, The Netherlands) for 30 min at 30°C. The enzyme and metabolites were separated by centrifugation at 6,000 × g for 30 min on an Ultrafree-MC 10.000 NMWL spin filter (Millipore, Billerica, MA). The supernatants were stored at −80°C prior to LC-MS analysis.
13C-based metabolic flux analysis.
The metabolic network model of glycolysis used for the 13C-based MFA of P. chrysogenum included all conventional reactions. The reactions of the nonoxidative branch of the PPP were modeled as metabolite-specific, reversible, C2 and C3 fragments producing and consuming half-reactions as proposed by Kleijn et al. (22). For reasons described below, the metabolic network model also included the decarboxylation of oxaloacetate into pep catalyzed by the enzyme pep carboxykinase. The reversible reactions were modeled as separate forward and backward reactions and are referred to as net and exchange fluxes, where
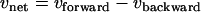 |
(4) |
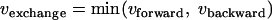 |
(5) |
The flux fitting procedure employed was described in detail by van Winden et al. (38). Briefly, in this procedure the cumomer balances and cumomer-to-isotopomer mapping matrices introduced by Wiechert et al. (39) are used to calculate the isotopomer distributions of metabolites in a predefined metabolic network model for a given flux set. The flux set that gives the best correspondence between the measured and simulated 13C label distributions is determined by nonlinear optimization and is designated the optimal flux fit. All calculations were performed with Matlab 6.1 (The Mathworks Inc., Natick, MA).
RESULTS
Validity of the gluconate tracer method.
Prior to using the gluconate tracer experiment for direct determination of the PPP split ratio, we checked whether P. chrysogenum could simultaneously consume glucose and gluconate under carbon-limiting conditions. Furthermore, we investigated to what extent the metabolism of P. chrysogenum was influenced by replacement of 5 Cmol% of glucose by gluconate. A trial cultivation experiment was performed in which P. chrysogenum was grown for 4.0 residence times on the 13C-based MFA medium containing only glucose and acetate, followed by 4.0 residence times on a gluconate tracer medium with 5 Cmol% of gluconate. As shown in Fig. 2, no significant changes in the biomass dry weight, penicillin G production rate, and PAA consumption rate were observed for the two different media. Throughout cultivation the rate of PAA uptake matched the rate of penicillin G production, indicating that PAA was not catabolized by the cells.
FIG. 2.

Rate of penicillin G production (qPenicillin-G), rate of PAA consumption (qPAA), and biomass concentration during chemostat cultivation of P. chrysogenum performed to validate the gluconate tracer method. After ∼4 residence times the composition of the feed was slightly altered; 5 Cmol% of the original glucose was replaced by an equimolar amount of gluconate. At the same time the chemostat was pulsed with a gluconate solution so that the initial concentration in the chemostat was 0.160 g/liter.
The effect of gluconate addition on intracellular metabolism was investigated by determining the steady-state intracellular metabolite levels before and after the medium was changed to the gluconate-containing medium (Fig. 3). Metabolite levels were determined by isotope dilution mass spectrometry using U-13C-labeled metabolite extracts as internal standards as described by Wu et al. (40). In their study Wu et al. found that for most metabolite levels the maximal standard deviation did not exceed 10%. A t test based on a maximal standard deviation of 10% showed that most of the metabolite levels before and after addition of gluconate were not significantly different; the only exception to this was the level of fbp. The difference in the fbp level observed might have been due to the extremely low concentrations of this metabolite in the sample. The metabolite levels shown in Fig. 3 seemed to be slightly lower after addition of gluconate. However, the lower levels were not necessarily an effect of the gluconate added. The levels of the metabolites examined are biomass specific; therefore, overestimation of the biomass concentration at the time of sampling results in underestimation of all biomass-specific metabolite levels.
FIG. 3.

Steady-state intracellular metabolite concentrations for P. chrysogenum before and after the switch to the gluconate-containing feed. Broth samples used for metabolite analysis were harvested after about 4 and 8 residence times. The standard deviations (indicated by error bars) were fixed at 10%. A metabolite whose concentrations were significantly different before and after addition of gluconate was identified by using a two-tailed equal variance t test and is indicated by an asterisk. A P value less than 0.01 was considered significant. Glyox, glyoxylate; PYR, pyruvate; FUM, fumarate; MAL, malate; a-KG, α-ketoglutarate; G1P, glucose-1-phosphate; G6P, glucose-6-phosphate; F6P, fructose-6-phosphate; M6P, mannose-6-phosphate; T6P, threhalose-6-phosphate; PEP, phosphoenolpyruvate; 2/3PG, combined pool of 2-phosphoglycerate and 3-phosphoglycerate; FBP, 1,6-fructose bisphosphate; 6PG, 6-phosphogluconate.
Immediately after the switch to the gluconate-containing medium, the bioreactor was pulsed with a gluconate solution in order to obtain the same gluconate concentration in the bioreactor as in the medium (∼0.160 g/liter). After the pulse, the decrease in the extracellular residual gluconate concentration in the bioreactor was monitored over time (Fig. 4). Within 0.5 h after the pulse the gluconate concentration in the bioreactor started to decrease. The residual glucose concentration in the bioreactor remained ∼3 mg/liter throughout the experiment. These results clearly demonstrate that P. chrysogenum is capable of simultaneously metabolizing glucose and gluconate under carbon-limiting conditions. The simultaneous uptake was probably triggered by the low glucose concentration during the experiment, which enabled induction of the genes needed for the uptake of other substrates (there was no catabolite repression). The gluconate consumption rate was calculated from the pattern of the extracellular gluconate concentration over time. Figure 4 shows that during the first 12 h the specific rate of uptake of gluconate reached ∼6 × 10−3 mol/Cmol/h, which was followed by a decrease to a steady-state uptake rate of 0.64 × 10−3 mol/Cmol/h. P. chrysogenum clearly needed time to fully adjust to the presence of gluconate, indicating that the transport system required for gluconate and/or the enzyme gluconokinase is induced rather than constitutively expressed.
FIG. 4.

Measured gluconate and glucose concentrations in the chemostat before and after addition of the gluconate pulse (at zero time). The specific rate of gluconate uptake was estimated based on the measured uptake dynamics of gluconate.
Gluconate tracer method and P. chrysogenum.
The gluconate tracer method was used to determine the PPP split ratio in a penicillin G-producing chemostat culture of P. chrysogenum operated at a dilution rate of 0.02 h−1. Table 1 shows the “measured” macroscopic parameters of the cultivation. Apart from an erroneous oxygen consumption rate, identified by rate balancing as described by van der Heijden et al. (33), no major differences between the balanced and “measured” macroscopic rates were found. Note that the rate of gluconate uptake was just less than 5% of the rate of glucose uptake.
TABLE 1.
Macroscopic parameters for the gluconate tracer experiment and the 13C-based MFA experiment
| Parameter | Gluconate tracer expt | 13C-based MFA expt |
|---|---|---|
| Consumption (mmol/Cmol/h) | ||
| Glucose | 8.60 ± 0.40 | 8.37 ± 0.45 |
| Gluconate | 0.42 ± 0.02 | NDa |
| Acetate | 2.67 ± 0.12 | 2.52 ± 0.14 |
| Oxygen | 48.91 ± 2.10 | 32.6 ± 1.3 |
| PAA | 0.50 ± 0.05 | 0.55 ± 0.06 |
| Production | ||
| Carbon dioxide (mmol/Cmol/h) | 31.34 ± 1.57 | 26.01 ± 4.53 |
| Biomass (1/h) | 0.020 ± 0.001 | 0.019 ± 0.001 |
| Penicillin G (mmol/Cmol/h) | 0.51 ± 0.05 | 0.48 ± 0.08 |
| Biomass concn (g/liter) | 1.04 ± 0.05 | 1.07 ± 0.06 |
| Carbon recovery (%) | 104.0 ± 2.2 | 92.2 ± 5.9 |
ND, not determined.
The measured mass isotopomer distribution of the [U-13C]gluconate added to the medium is shown in Table 2. Based on the measured mass fractions, an isotopic purity of 99% (∼0.941/6) was calculated. This value is higher than the 95% isotopic purity specified by the manufacturer, from which an m + 6 mass isotopomer fraction of only 0.74 was predicted for gluconate. Moreover, use of the manufacturer-specified isotopic purity would have resulted in a very different estimate for the PPP split ratio, illustrating the importance of measuring the label distribution of the substrate(s) used.
TABLE 2.
Measured mass isotopomer fractions and standard deviations for the gluconate tracer experiment
| Metabolite | Mass isotopomer | Measured fractiona |
|---|---|---|
| Gluconate in medium | m + 0 | 0.000 ± 0.000 |
| m + 1 | 0.000 ± 0.000 | |
| m + 2 | 0.000 ± 0.000 | |
| m + 3 | 0.000 ± 0.000 | |
| m + 4 | 0.002 ± 0.000 | |
| m + 5 | 0.059 ± 0.001 | |
| m + 6 | 0.939 ± 0.001 | |
| Gluconateb | m + 0 | 0.217 ± 0.006 |
| m + 1 | 0.019 ± 0.001 | |
| m + 2 | 0.003 ± 0.001 | |
| m + 3 | 0.001 ± 0.000 | |
| m + 4 | 0.002 ± 0.001 | |
| m + 5 | 0.040 ± 0.003 | |
| m + 6 | 0.718 ± 0.007 | |
| g6pb | m + 0 | 0.866 ± 0.003 |
| m + 1 | 0.094 ± 0.002 | |
| m + 2 | 0.017 ± 0.002 | |
| m + 3 | 0.015 ± 0.000 | |
| m + 4 | 0.007 ± 0.000 | |
| m + 5 | 0.001 ± 0.000 | |
| m + 6 | 0.001 ± 0.000 | |
| f6pb | m + 0 | 0.852 ± 0.004 |
| m + 1 | 0.094 ± 0.003 | |
| m + 2 | 0.024 ± 0.001 | |
| m + 3 | 0.016 ± 0.001 | |
| m + 4 | 0.012 ± 0.002 | |
| m + 5 | 0.002 ± 0.001 | |
| m + 6 | 0.000 ± 0.000 | |
| 6pgb | m + 0 | 0.786 ± 0.003 |
| m + 1 | 0.082 ± 0.003 | |
| m + 2 | 0.015 ± 0.001 | |
| m + 3 | 0.011 ± 0.000 | |
| m + 4 | 0.016 ± 0.003 | |
| m + 5 | 0.008 ± 0.001 | |
| m + 6 | 0.081 ± 0.003 |
The mass fractions were corrected for the natural isotopes of hydrogen and oxygen.
Intracellular metabolite surrounding the g6p node.
After 3 residence times of feeding on medium containing [U-13C]gluconate, broth was harvested in order to measure the mass isotopomer fractions of 6pg, g6p, f6p, and intracellular gluconate (Table 2). As expected, addition of [U-13C]gluconate led to a distinct m + 6 mass isotopomer fraction for 6pg. No discernible m + 6 mass fractions were observed for g6p and f6p due to the decarboxylation of 6pg in the oxidative branch of the PPP and its carbon redistribution in the nonoxidative branch of the PPP.
Unexpected mass fractions were observed for intracellular gluconate. The unlabeled fraction (m + 0) of the intracellular gluconate was determined to be 22%, while this fraction was not detectable in the medium to which gluconate was added (Table 2). Apparently, an unidentified reaction caused inflow of unlabeled carbon into the otherwise uniformly 13C-labeled gluconate pool. Two possible reactions were hypothesized to explain this phenomenon: oxidation of glucose to gluconate by either glucose oxidase or glucose dehydrogenase and dephosphorylation of intracellular 6pg to gluconate by a phosphatase (Fig. 5). Several aspecific phosphatases have been reported previously for P. chrysogenum (14, 29). Given the competitive inhibition of these phosphatases by inorganic phosphate (ki = 0.42 mM), it seems unlikely that these enzymes were expressed in the presence of the relatively high phosphate concentrations employed in this study. However, the glucose-oxidizing enzymes have the same restriction. As pointed out above, expression of these enzymes is normally triggered by excess glucose, while in this study the chemostat cultures were grown under carbon-limiting conditions.
FIG. 5.

Two candidate reactions for explaining the observed unlabeled mass isotopomer fraction of intracellular gluconate (glnin): dephosphorylation of 6pg (v8) by a phosphatase and oxidation of glucose (v6) by either glucose oxidase or glucose dehydrogenase. glnex, extracellular gluconate.
Enzymatic analysis of cell extracts from the gluconate tracer experiment showed that there was no measurable activity for any of the three proposed enzymes, demonstrating that the levels of enzyme activity were below the detection limit of the assays used. Based on the conceived fluxes for the oxidation of glucose or the dephosphorylation of 6pg (see Table A1 in Appendix A), the minimal specific activities for all three enzymes were estimated to be around 2 × 10−4 μmol/mg protein/min. This value is indeed much lower than the detection limit of the assays (0.01 μmol/mg protein/min). In spite of the fact that the in vitro assays for these enzymes showed that there was no measurable activity, the possibility that the measured m + 0 fractions of intracellular gluconate were due to the activity of one of these enzymes cannot be excluded.
TABLE A1.
Estimated oxidative PPP flux (v3) and corresponding PPP split ratio for a stoichiometric model containing either the glucose oxidation (v6) or 6pg dephosphorylation (v8) flux
| Pathway | Metabolic fluxes (mmol/Cmol/h)
|
PPP split ratio (%) | SSres | |||||
|---|---|---|---|---|---|---|---|---|
| v1 | v2 | v3 | v6 | v7 | v8 | |||
| Oxidation of glucose | 8.60 | 0.42 | 4.30 | 0.13 | 0.55 | 50.8a | 3.48 | |
| Dephosphorylation of 6pg | 8.60 | 0.42 | 4.45 | 0.57 | 0.15 | 51.7 | 2.59 | |
Normalized for the uptake rate of glucose (v1 − v6).
In Appendix A the effects of the two proposed metabolic scenarios on estimation of the PPP split ratio are examined. The oxidation of unlabeled glucose to gluconate caused a small fraction of the unlabeled carbon to flow into the 6pg pool via uptake of gluconate instead of via conventional oxidation of g6p, thereby altering the estimated PPP split ratio. However, due to the small size of the glucose oxidation flux, the change in the PPP split ratio was practically negligible. The dephosphorylation of 6pg had no effect on the label inflow into the 6pg pool. Irrespective of whether the phosphatase was actively expressed, all unlabeled carbon in 6pg originated from g6p, while all 13C label originated from [U-13C]gluconate added to the medium. Hence, an unchanged estimate for the PPP split ratio was obtained. These findings justify the fact that the glucose-oxidizing reaction is neglected in the PPP split ratio estimates presented below.
The mass isotopomers shown in Table 2 and the measured rates of glucose and gluconate uptake were combined with equation 3 to estimate the PPP split ratio in P. chrysogenum. The 95% confidence interval of the PPP split ratio was determined by Monte Carlo simulation in which the added noise was normally distributed with the measured standard deviations of the mass isotopomer fractions. The optimization routine yielded a PPP flux of 4.45 ± 0.36 mmol/Cmol/h, meaning that 51.7% ± 4.2% of the total glucose entering the cell was metabolized via the PPP. Note that for every 1 mol of glucose catabolized in the oxidative branch of the PPP 2 mol of NADPH is produced, while for every 1 mol of gluconate catabolized only 1 mol of NADPH is produced. Since the flux through the PPP is related to the cytosolic NADPH demand of P. chrysogenum (35), the PPP split ratios calculated by the gluconate tracer method are slight overestimates. In the most extreme case of direct proportionality between the PPP flux and the cytosolic NADPH demand, the PPP split ratio should be slightly corrected to 49.5%. However, normalization of this value for the total rate of glucose and gluconate uptake (v1 + v2) increased the PPP split ratio to 51.8%.
Mass isotopomer fractions less than 0.03 were not included in the fitting procedure as we observed that these fractions led to a considerable increase in the variance-weighted sum of squared residuals (SSres). This increase may be explained by the fact that the standard deviations shown in Table 2 are based only on precision limitations of the LC-MS and do not account for any systematic measurement errors. Especially for the small-mass isotopomer fractions these systematic errors can result in large contributions to the minimized sum of squared residuals. A minimal SSres of 2.59 was obtained when we fitted the PPP split ratio. To test whether the SSres is solely caused by normally distributed measurement errors, a χ2 distribution was used, in which the degrees of freedom equaled the number of fitted mass isotopomers fractions minus the number of fitted parameters. Exclusion of the m + 2, m + 3, and m + 4 mass isotopomer fractions of g6p, 6pg, and gluconate left 3 df. Given the probability P(χ2[3]<2.59) = 0.5408, it follows that the fitted PPP split ratio is well within the statistically acceptable range.
13C-based MFA of P. chrysogenum.
By measuring the mass isotopomers of 13C-labeled primary metabolites, the metabolic fluxes in the PPP and glycolysis of P. chrysogenum were estimated for a second chemostat cultivation operated under conditions identical to those used for the gluconate tracer experiment except that labeled glucose instead of gluconate was added to the feed. The macroscopic parameters for the cultivation are shown in Table 1. As expected, similar biomass concentrations and uptake and secretion rates were observed for the gluconate tracer experiment and the 13C-based MFA experiment.
Table 3 shows the measured mass isotopomer fractions of the glucose in the medium and the nine intracellular metabolites after P. chrysogenum was grown for 3 residence times on the 13C-labeled medium. The mass fractions of glucose were used to calculate the isotopic purity of the [U-13C]glucose (98%) and [1-13C]glucose (99%) used in the feed. Both levels of isotope purity matched the manufacturer-specified values.
TABLE 3.
Measured and fitted mass isotopomer fractions for the 13C-based MFA experiment
| Metabolitea | Mass isotopomer | Measured fraction | Fitted fraction | Difference | Metabolitea | Mass isotopomer | Measured fraction | Fitted fraction | Difference | |
|---|---|---|---|---|---|---|---|---|---|---|
| Glucose in medium | m + 0 | 0.196 ± 0.001 | m + 4 | 0.063 ± 0.005 | 0.056 | 0.006 | ||||
| m + 1 | 0.569 ± 0.002 | m + 5 | 0.031 ± 0.003 | 0.027 | 0.004 | |||||
| m + 2 | 0.037 ± 0.000 | m + 6 | 0.081 ± 0.004 | 0.079 | 0.002 | |||||
| m + 3 | 0.012 ± 0.000 | |||||||||
| m + 4 | 0.002 ± 0.000 | fbpb | m + 0 | 0.223 ± 0.014 | 0.223 | 0.000 | ||||
| m + 5 | 0.019 ± 0.000 | m + 1 | 0.345 ± 0.012 | 0.349 | −0.004 | |||||
| m + 6 | 0.166 ± 0.001 | m + 2 | 0.142 ± 0.008 | 0.143 | −0.001 | |||||
| m + 3 | 0.128 ± 0.010 | 0.130 | −0.002 | |||||||
| g6pb | m + 0 | 0.200 ± 0.012 | 0.205 | −0.005 | m + 4 | 0.074 ± 0.004 | 0.068 | 0.006 | ||
| m + 1 | 0.413 ± 0.011 | 0.423 | −0.011 | m + 5 | 0.028 ± 0.003 | 0.029 | 0.000 | |||
| m + 2 | 0.129 ± 0.005 | 0.124 | 0.005 | m + 6 | 0.060 ± 0.006 | 0.056 | 0.004 | |||
| m + 3 | 0.084 ± 0.003 | 0.081 | 0.003 | |||||||
| m + 4 | 0.058 ± 0.004 | 0.053 | 0.005 | 2/3pgb | m + 0 | 0.514 ± 0.010 | 0.501 | 0.013 | ||
| m + 5 | 0.028 ± 0.002 | 0.027 | 0.002 | m + 1 | 0.253 ± 0.006 | 0.254 | −0.001 | |||
| m + 6 | 0.088 ± 0.004 | 0.084 | 0.004 | m + 2 | 0.081 ± 0.013 | 0.097 | −0.015 | |||
| m + 3 | 0.152 ± 0.007 | 0.144 | 0.008 | |||||||
| 6pgb | m + 0 | 0.205 ± 0.008 | 0.205 | 0.000 | ||||||
| m + 1 | 0.382 ± 0.016 | 0.422 | −0.040 | pepb | m + 0 | 0.446 ± 0.009 | 0.456 | −0.010 | ||
| m + 2 | 0.146 ± 0.016 | 0.124 | 0.021 | m + 1 | 0.285 ± 0.007 | 0.277 | 0.008 | |||
| m + 3 | 0.081 ± 0.002 | 0.082 | −0.001 | m + 2 | 0.126 ± 0.006 | 0.123 | 0.003 | |||
| m + 4 | 0.063 ± 0.005 | 0.053 | 0.009 | m + 3 | 0.143 ± 0.005 | 0.139 | 0.003 | |||
| m + 5 | 0.036 ± 0.004 | 0.027 | 0.009 | |||||||
| m + 6 | 0.088 ± 0.003 | 0.084 | 0.004 | p5pb | m + 0 | 0.481 ± 0.017 | 0.486 | −0.005 | ||
| m + 1 | 0.213 ± 0.006 | 0.205 | 0.009 | |||||||
| f6pb | m + 0 | 0.192 ± 0.006 | 0.206 | −0.014 | m + 2 | 0.091 ± 0.016 | 0.108 | −0.017 | ||
| m + 1 | 0.379 ± 0.016 | 0.414 | −0.035 | m + 3 | 0.075 ± 0.005 | 0.081 | −0.006 | |||
| m + 2 | 0.130 ± 0.005 | 0.130 | 0.001 | m + 4 | 0.062 ± 0.009 | 0.052 | 0.010 | |||
| m + 3 | 0.087 ± 0.004 | 0.086 | 0.001 | m + 5 | 0.078 ± 0.014 | 0.066 | 0.012 | |||
| m + 4 | 0.059 ± 0.005 | 0.056 | 0.003 | |||||||
| m + 5 | 0.032 ± 0.003 | 0.027 | 0.005 | s7pb,c | m + 0 | 0.221 ± 0.012 | 0.231 | −0.011 | ||
| m + 6 | 0.121 ± 0.004 | 0.079 | 0.042 | m + 1 | 0.286 ± 0.008 | 0.292 | −0.006 | |||
| m + 2 | 0.192 ± 0.008 | 0.188 | 0.004 | |||||||
| m6pb | m + 0 | 0.205 ± 0.006 | 0.206 | 0.000 | m + 3 | 0.121 ± 0.005 | 0.119 | 0.002 | ||
| m + 1 | 0.393 ± 0.016 | 0.414 | −0.021 | m + 4 | 0.084 ± 0.006 | 0.080 | 0.004 | |||
| m + 2 | 0.137 ± 0.005 | 0.130 | 0.007 | m + 5 | 0.067 ± 0.004 | 0.064 | 0.003 | |||
| m + 3 | 0.090 ± 0.004 | 0.086 | 0.004 | m + 6 | 0.029 ± 0.003 | 0.027 | 0.003 |
m6p, mannose-6-phosphate; p5p, combined pool of xylulose-5-phosphate, ribose-5-phosphate, and ribulose-5-phosphate; s7p, sedoheptulose-7-phosphate.
Intracellular glycolytic or PPP metabolite.
The m + 7 mass fraction of sedoheptulose-7-phosphate was not measured; as a result, the values were normalized for mass fractions m + 0 to m + 6.
Before any calculations were performed, a qualitative analysis of the mass isotopomer data obtained provided additional insight into the primary metabolism of P. chrysogenum under the cultivation conditions used. For example, the nearly identical mass isotopomer fractions for g6p and f6p shown in Table 3 indicate that the glucose isomerase-catalyzed reaction was close to equilibrium. Only a high reversibility of the glucose isomerase-catalyzed reaction could efface the labeling difference in the g6p and f6p pool caused by f6p originating from the nonoxidative branch of the PPP.
The difference in labeling between the 2/3pg and pep data shown in Table 3 suggests that the reaction catalyzed by the enolase is not fully reversible. Furthermore, the difference in labeling also indicates that pep originates not only from 3pg. The most likely candidate for a second pep-producing reaction is the gluconeogenic reaction catalyzed by pep carboxykinase, which converts one molecule of oxaloacetate into pep and CO2 at the expense of one molecule of ATP. One possible explanation for the presence of this enzyme is induction by the cosubstrate acetate. Nevertheless, it is intriguing that a microorganism expresses an energy-consuming gluconeogenic reaction when it can produce all upper glycolytic intermediates from glucose. van Winden et al. (36) have previously shown the importance of including a pep carboxykinase in fitting P. chrysogenum 13C labeling data.
Based on the qualitative analysis of the data in Table 3, a refined metabolic network model of glycolysis and the PPP was constructed for the 13C-based MFA of P. chrysogenum. Flux estimates for the glycolytic and PPP reactions were obtained by fitting the mass isotopomer distribution model to the measured mass isotopomer distributions. The fitted mass isotopomer fractions and the estimated fluxes are shown in Table 3 and Fig. 6, respectively. All fluxes were normalized (on a mole basis) to a glucose influx of 100. Using the 13C-based MFA method, the PPP split ratio was estimated to be 51.1%, which is very similar to the value determined with the gluconate tracer method. Figure 6 also shows that in accordance with previous observations, a flux was fitted for the pep carboxykinase-catalyzed reaction. However, the extent of this flux could not be quantified because only mass isotopomer fractions of glycolytic and PPP metabolites were measured.
FIG. 6.

Flux estimates for 13C-based MFA of P. chrysogenum. Values in gray type represent the drain on the primary metabolites for monomer synthesis (e.g., amino acids, polysaccharides, fatty acids, nucleotides etc.). Fluxes are normalized for the glucose uptake rate. Values not in parentheses indicate the net fluxes, while values in parentheses indicate the exchange fluxes. The arrows with the solid arrowheads indicate the direction of the net flux. e4p, erythrose-4-phosphate; E-C2, glycolaldehyde moiety covalently bound to the thiamine pyrophosphate/transketolase complex; E-C3, dihydroxyacetone moiety covalently bound to the enzyme transaldolase; p5p, combined pool of xylulose-5-phosphate, ribose-5-phosphate, and ribulose-5-phosphate; g3p, glyceraldehyde-3-phosphate; oaa, oxaloacetate; s7p, sedoheptulose-7-phosphate.
The fit shown in Fig. 6 yielded a minimized variance-weighted SSres of 60.2. As described previously, this variable follows a chi-square distribution with, in this case, 30 df (47 independent data points were used for fitting 17 parameters). Given the probability P(χ2[30]<60.2) = 0.999, it follows that with a 95% confidence interval the fit has to be statistically rejected. An analysis showed that the mass isotopomer fractions of f6p and 6pg contributed the most to the total SSres (data not shown).
Accuracy and reproducibility of the estimated PPP split ratios.
The accuracy of the two methods for determining the PPP split ratio was examined by studying the sensitivity of the minimized SSres to changes in the PPP split ratio. The metabolic network model used for the 13C-based MFA is nonlinear, which makes estimation of the confidence interval based on linearization of the model around the optimally fitted fluxes prone to errors. Therefore, the measured mass isotopomer fractions of both methods were refitted at fixed values for the PPP split ratio, ranging from 20% to 80% at a 5% interval. The SSres for each fit is plotted against the fixed PPP split ratio in Fig. 7. By determining the points of intersection of the curve with a horizontal line at the 95% confidence value for SSres (SSres95%) and by subsequently projecting the intersection points onto the x axis, the 95% confidence interval for the PPP split ratio can be obtained. The 95% confidence value for the SSres can be calculated as follows (see Appendix B):
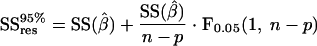 |
(6) |
where SSres,optimal is a constant corresponding to the globally fitted minimum for SSres at the optimal PPP split ratio, n is the number of independent data points, and p is the total number of parameters in the model.
FIG. 7.

Sensitivity analysis of PPP split ratio determination for the 13C-based MFA method (A) and the gluconate tracer method (B). The horizontal dotted lines indicate the 95% cutoff values for SSres, as calculated with equation 5. The vertical dotted lines indicate the upper and lower boundaries for the 95% confidence intervals of the PPP split ratios.
The SSres95% values were calculated to be 68.6 and 16.7 for the 13C-based MFA and the gluconate tracer method, respectively, which corresponded to 95% confidence intervals of 40.0 to 63.5 and 46.0 to 56.5 (Fig. 7). Note that the confidence interval for the gluconate tracer method corresponds well to the confidence interval previously estimated using Monte Carlo simulations (47.7 to 56.1). The results clearly show that PPP split ratio estimation by the gluconate tracer method is more precise.
The reproducibility of the estimated PPP split ratios depends on the consistency of three factors: (i) the chemostat cultivation, (ii) the rapid sampling procedure, and (iii) the sample analysis. The reproducibility of chemostat cultivation is indicated by the data in Table 1, which shows that despite the presence of 5 Cmol% of gluconate, similar macroscopic parameters were obtained for the gluconate tracer method and the 13C-labeling-based MFA method. Furthermore, when the gluconate tracer experiment was performed with naturally labeled gluconate added to the feed medium, similar macroscopic parameters were obtained (results not shown). During each experiment multiple independent samples were taken using the rapid sampling procedure. A one-way analysis of variance showed that the variability between samples was not significantly greater than the variability within a sample (i.e., between multiple injections in the LC-MS analysis). Thus, sample withdrawal from the chemostat was also reproducible. The reproducibility of the sample analysis is indicated by the small standard deviations for the five independent injections in the LC-MS analysis (<1%) (Tables 2 and 3).
DISCUSSION
Replacement of 5 Cmol% of glucose by [U-13C]gluconate in a glucose-fed chemostat culture of P. chrysogenum allowed accurate determination of the PPP split ratio without disturbing the metabolism of the cell. Use of this method with a penicillin G-producing culture of P. chrysogenum resulted in a PPP split ratio of 51.8%. Determination of the PPP split ratio in P. chrysogenum by a 13C-based MFA resulted in a similar PPP split ratio, 51.1%. This is the first report of the use of mass isotopomer measurements of 13C-labeled primary metabolites for determining the metabolic fluxes in P. chrysogenum.
Sensitivity analysis of the two estimated PPP split ratios showed that the gluconate tracer method is more sensitive, as shown by a 95% confidence interval for the PPP split ratio that was threefold smaller. The lower sensitivity of the 13C-based MFA method can be attributed primarily to the nature of the method. While the gluconate tracer method is dedicated to estimation of the PPP split ratio, the 13C-based MFA method aims at determining multiple fluxes in a defined metabolic network model. The high interconnectivity of cellular metabolism allows changes in metabolite labeling (due to, for example, an altered PPP split ratio) to be counteracted by changes in the flux patterns in other parts of the metabolism. As a result, the fitting procedure can produce similar minimized SSres values for dissimilar metabolic flux patterns. The preferred method thus depends on the objective of the experiment. The gluconate tracer method allows accurate determination of only the PPP split ratio, whereas the 13C-based MFA method provides an estimate of the global flux distribution in the cell, although it is not as accurate as the gluconate tracer method.
In principle, the gluconate tracer method can be extended to other parts of the metabolic network. By using route-specific 13C-labeled tracers it is possible to determine flux patterns in parts of the metabolic network which otherwise might be hard to accurately determine using the “holistic” 13C-labeled MFA method. Simultaneous uptake of the tracer and the main carbon source is essential for such tracer studies. Furthermore, the effect of tracer addition on the overall cellular metabolism should be kept to a minimum. Quantification of the flux distribution in a metabolic node requires an influx of the tracer directly after the branch point and measurement of the metabolites directly surrounding the branch point.
Table 4 shows an overview of the previously published PPP split ratios for penicillin-producing chemostat cultivation of P. chrysogenum, together with the measured penicillin production rates, the growth rates used, and the estimation methods used. PPP split ratios ranging from 33% to 75% have been reported for P. chrysogenum. The large variation can be explained in part by different experimental conditions, such as differences in the penicillin production rate (due to strain diversity) and the growth rate used. Nonetheless, the large variations are also a result of assumptions made concerning the underlying stoichiometric model. The difference in the PPP split ratios observed by van Gulik et al. (35) and Henriksen et al. (16), for example, can be explained largely by inclusion of a different cysteine biosynthesis pathway. Furthermore, van Winden et al. (36) showed that different metabolic networks have great effects on the estimated PPP split ratio in a 13C-based MFA.
TABLE 4.
Overview of the previously determined PPP split ratios for penicillin-producing chemostat cultivation of P. chrysogenum
| Reference | PPP split ratio (%) | Specific growth rate (h−1) | Penicillin production (mmol/Cmol/h)b | Method used |
|---|---|---|---|---|
| Henriksen et al. (16) | 61 | 0.1 | 0.66 | MFA |
| van Gulik et al. (35) | 37 | 0.045 | ∼0.55 | MFA |
| Christensen and Nielsen (4) | 75 | 0.08 | 0.72 | 13C-based MFA |
| Christensen et al. (5)a | 70 | 0.06 | 0.31 | 13C-based MFA |
| van Winden et al. (36) | 33 | 0.03 | 0.45 | 13C-based MFA |
| Christensen et al. (3)a | 75 | 0.06 | 0.31 | Algebraic solution |
| This study | 51 | 0.02 | 0.45 | 13C-based MFA |
| This study | 51 | 0.02 | 0.51 | Gluconate tracer |
These studies were based on the same 13C labeling data set, but different methods were used to calculate the PPP split ratio.
The molecular mass of the biomass used for calculating the specific penicillin production rates was assumed to be growth rate independent and to be 28.05 g (dry weight) per Cmol biomass (35).
As described above, the main advantage of the gluconate tracer method is that it does not rely on a metabolic network model but relies merely on accurate determination of the metabolite labeling directly surrounding the g6p pool. The improved sensitivity of the gluconate tracer method compared to the 13C-based MFA method described in this paper increases the credibility of the gluconate tracer method. In this respect, none of the other studies listed in Table 4 mention the confidence interval associated with the estimated PPP split ratio.
Acknowledgments
This work was financially supported by the Dutch EET program (project EETK20002) and by DSM.
Diana Harris (Delft University of Technology) is gratefully acknowledged for her practical help with the enzymatic determinations.
APPENDIX A
Two candidate reactions were hypothesized to explain the difference between the labeling of the gluconate added to the medium and the labeling of the intracellular gluconate in the gluconate tracer experiment (Table 2): oxidation of glucose to gluconate by either glucose oxidase or glucose dehydrogenase and dephosphorylation of intracellular 6pg to gluconate by a phosphatase (Fig. 5). Flux estimates for these reactions were derived by combining the mass and labeling balance around the intracellular gluconate pool, similar to the derivation of equation 3. In the case of a glucose oxidation reaction (v6) the mass balance around the intracellular gluconate pool becomes
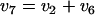 |
(A1) |
Combining equation A1 with the labeling balance yields
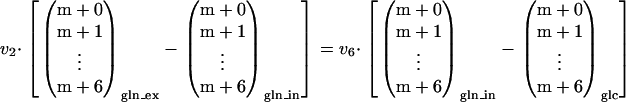 |
(A2) |
where gln_in and gln_ex are the intracellular and extracellular gluconate pools, respectively. In the case of a dephosphorylation reaction (v8) the mass balance around the intracellular gluconate pool becomes
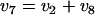 |
(A3) |
Combining equation A3 with the labeling balance gives
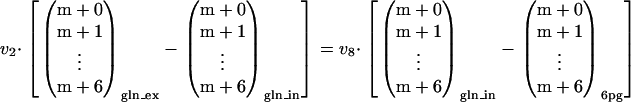 |
(A4) |
Using a sequential quadratic programming algorithm, the fluxes v6 (using equation A2) and v8 (using equation A4) were estimated to be 0.13 and 0.15 mmol/Cmol/h, respectively. The two fits yielded similar SSres values (14.85 and 15.86), which made it impossible to distinguish between the two proposed reactions based on the measured labeling of intracellular gluconate.
The flux estimates for v6 and v8 were subsequently used to estimate the corresponding oxidative PPP flux (v3). Due to the changed mass and labeling balance around the 6pg pool (Fig. 5), equation 3 could not be directly used for estimating v3. A new mass balance around the 6pg pool was set up:
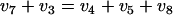 |
(A5) |
Combining equation A5 with the labeling balance around 6pg yielded a new relationship:
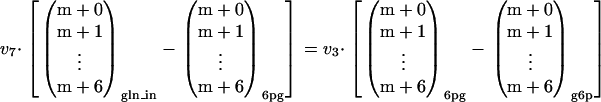 |
(A6) |
Oxidative PPP fluxes (v3) were calculated for the metabolic pathway containing the glucose oxidation reaction and for the metabolic pathway containing the 6pg dephosphorylation reaction by substituting equations A1 and A3 into equation A6, respectively. An overview of the estimated fluxes is shown in Table A1.
The dephosphorylation of 6pg has no effect on the estimated PPP split ratio since it does not alter the origin of the label entering the 6pg pool. Irrespective of an active phosphatase, all unlabeled carbon entering the 6pg pool originates from g6p, and all 13C label enters via the uptake of gluconate. Mathematical proof of this is obtained by substituting equations A3 and A4 in equation A6, which results in the originally derived equation (equation 3) for calculating the PPP split ratio (not shown).
In contrast, the oxidation of unlabeled glucose to gluconate causes flow of unlabeled carbon into the 6pg pool via gluconate uptake instead of via the classical oxidation of g6p. Consequently, a slightly different PPP split ratio was obtained. However, due to the small size of the glucose oxidation flux, the observed effect on the PPP split ratio was marginal.
APPENDIX B
The asymmetric confidence interval parameter (β) in a nonlinear model can be derived by refitting the measurement data for a fixed range of β values. By plotting the minimized sum of squared residuals [SS(β)] for each fit as a function of β, an asymmetrical curve is obtained, from which the confidence interval can be calculated by defining a cutoff value for the SS(β) using the following relationship (1):
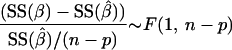 |
(B1) |
where p is the total number of parameters in the model and SS(β̂) is a constant corresponding to the global minimized sum of squared residuals (determined when β is not fixed).
The cutoff value for SSres at a chosen confidence level (α) is obtained by solving equation B1 for SS(β):
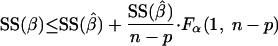 |
(B2) |
Note that the term SS(β̂)/(n − p) corresponds to the estimator for the measurement error (σ̂2).
REFERENCES
- 1.Bates, D. M., and D. G. Watts. 1988. Nonlinear regression analysis and its applications. John Wiley and Sons, Inc., New York, NY.
- 2.Caspari, T., and S. Urlinger. 1996. The activity of the gluconate-H+ symporter of Schizosaccharomyces pombe cells is down-regulated by d-glucose and exogenous cAMP. FEBS Lett. 395:272-276. [DOI] [PubMed] [Google Scholar]
- 3.Christensen, B., T. Christiansen, A. K. Gombert, J. Thykaer, and J. Nielsen. 2001. Simple and robust method for estimation of the split between the oxidative pentose phosphate pathway and the Embden Meyerhof Parnas pathway in microorganisms. Biotechnol. Bioeng. 74:517-523. [DOI] [PubMed] [Google Scholar]
- 4.Christensen, B., and J. Nielsen. 2000. Metabolic network analysis of Penicillium chrysogenum using C-13-labeled glucose. Biotechnol. Bioeng. 68:652-659. [PubMed] [Google Scholar]
- 5.Christensen, B., J. Thykaer, and J. Nielsen. 2000. Metabolic characterization of high- and low-yielding strains of Penicillium chrysogenum. Appl. Microbiol. Biotecnol. 54:212-217. [DOI] [PubMed] [Google Scholar]
- 6.Dauner, M., J. E. Bailey, and U. Sauer. 2001. Metabolic flux analysis with a comprehensive isotopomer model in Bacillus subtilis. Biotechnol. Bioeng. 76:144-156. [DOI] [PubMed] [Google Scholar]
- 7.Dauner, M., M. Sonderegger, M. Hochuli, T. Szyperski, K. Wuthrich, H. P. Hohmann, U. Sauer, and J. E. Bailey. 2002. Intracellular carbon fluxes in riboflavin-producing Bacillus subtilis during growth on two-carbon substrate mixtures. Appl. Environ. Microbiol. 68:1760-1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delgado, T. C., M. M. Castro, C. F. Geraldes, and J. G. Jones. 2004. Quantitation of erythrocyte pentose pathway flux with [2-(13)]glucose and H-1 NMR analysis of the lactate methyl signal. Magnet. Reson. Med. 51:1283-1286. [DOI] [PubMed] [Google Scholar]
- 9.Drysch, A., M. El Massaoudi, W. Wiechert, A. A. de Graaf, and R. Takors. 2004. Serial flux mapping of Corynebacterium glutamicum during fed-batch l-lysine production using the sensor reactor approach. Biotechnol. Bioeng. 85:497-505. [DOI] [PubMed] [Google Scholar]
- 10.Egli, T., U. Lendenmann, and M. Snozzi. 1993. Kinetics of microbial growth with mixtures of carbon sources. Antonie Leeuwenhoek 63:289-298. [DOI] [PubMed] [Google Scholar]
- 11.Fischer, E., and U. Sauer. 2003. Metabolic flux profiling of Escherichia coli mutants in central carbon metabolism using GC-MS. Eur. J. Biochem. 270:880-891. [DOI] [PubMed] [Google Scholar]
- 12.Follstad, B. D., and G. Stephanopoulos. 1998. Effect of reversible reactions on isotope label redistribution—analysis of the pentose phosphate pathway. Eur. J. Biochem. 252:360-371. [DOI] [PubMed] [Google Scholar]
- 13.Gombert, A. K., M. M. dos Santos, B. Christensen, and J. Nielsen. 2001. Network identification and flux quantification in the central metabolism of Saccharomyces cerevisiae under different conditions of glucose repression. J. Bacteriol. 183:1441-1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haas, H., B. Redl, E. Leitner, and G. Stoffler. 1991. Penicillium chrysogenum extracellular acid phosphatase—purification and biochemical characterization. Biochim. Biophys. Acta 1074:392-397. [DOI] [PubMed] [Google Scholar]
- 15.Harris, D. M., J. A. Diderich, Z. A. van der Krogt, M. A. H. Luttik, L. M. Raamsdonk, R. A. L. Bovenberg, W. M. van Gulik, J. P. van Dijken, and J. T. Pronk. 2006. Enzymic analysis of NADPH metabolism in β-lactam-producing Penicillium chrysogenum: presence of a mitochondrial NADPH dehydrogenase. Metab. Eng. 8:91-101. [DOI] [PubMed] [Google Scholar]
- 16.Henriksen, C. M., L. H. Christensen, J. Nielsen, and J. Villadsen. 1996. Growth energetics and metabolic fluxes in continuous cultures of Penicillium chrysogenum. J. Biotechnol. 45:149-164. [DOI] [PubMed] [Google Scholar]
- 17.Izu, H., O. Adachi, and M. Yamada. 1997. Gene organization and transcriptional regulation of the gntRKU operon involved in gluconate uptake and catabolism of Escherichia coli. J. Mol. Biol. 267:778-793. [DOI] [PubMed] [Google Scholar]
- 18.Katz, J., S. Abraham, R. Hill, and I. L. Chaikoff. 1955. The occurrence and mechanism of the hexose monophosphate shunt in rat liver slices. J. Biol. Chem. 214:853-868. [PubMed] [Google Scholar]
- 19.Katz, J., and R. Rognstad. 1967. Labeling of pentose phosphate from glucose-14C and estimation of rates of transaldolase, transketolase, the contribution of the pentose cycle and ribose phosphate synthesis. Biochemistry 6:2227-2247. [DOI] [PubMed] [Google Scholar]
- 20.Kiefer, P., E. Heinzle, O. Zelder, and C. Wittmann. 2004. Comparative metabolic flux analysis of lysine-producing Corynebacterium glutamicum cultured on glucose or fructose. Appl. Environ. Microbiol. 70:229-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kingsley-Hickman, P. B., B. D. Ross, and T. Krick. 1990. Hexose monophosphate shunt measurement in cultured cells with [1-13C]glucose: correction for endogenous carbon sources using [6-13C]glucose. Anal. Biochem. 185:235-237. [DOI] [PubMed] [Google Scholar]
- 22.Kleijn, R. J., W. A. van Winden, W. M. van Gulik, and J. J. Heijnen. 2005. Revisiting the13C-label distribution of the non-oxidative branch of the pentose phosphate pathway based upon kinetic and genetic evidence. FEBS J. 272:4970-4982. [DOI] [PubMed] [Google Scholar]
- 23.Lange, H. C., M. Eman, G. van Zuijlen, D. Visser, J. C. van Dam, J. Frank, M. J. de Mattos, and J. J. Heijnen. 2001. Improved rapid sampling for in vivo kinetics of intracellular metabolites in Saccharomyces cerevisiae. Biotechnol. Bioeng. 75:406-415. [DOI] [PubMed] [Google Scholar]
- 24.Lee, H. W., J. G. Pan, and J. M. Lebeault. 1998. Enhanced l-lysine production in threonine-limited continuous culture of Corynebacterium glutamicum by using gluconate as a secondary carbon source with glucose. Appl. Microbiol. Biotechnol. 49:9-15. [Google Scholar]
- 25.Lee, W. N. P., L. G. Boros, J. Puigjaner, S. Bassilian, S. Lim, and M. Cascante. 1998. Mass isotopomer study of the nonoxidative pathways of the pentose cycle with [1,2-13C2]glucose. Am. J. Physiol. Endoc. Metab. 37:E843-E851. [DOI] [PubMed] [Google Scholar]
- 26.Maaheimo, H., J. Fiaux, Z. P. Cakar, J. E. Bailey, U. Sauer, and T. Szyperski. 2001. Central carbon metabolism of Saccharomyces cerevisiae explored by biosynthetic fractional C-13 labeling of common amino acids. Eur. J. Biochem. 268:2464-2479. [DOI] [PubMed] [Google Scholar]
- 27.Mashego, M. R., W. M. van Gulik, J. L. Vinke, and J. J. Heijnen. 2003. Critical evaluation of sampling techniques for residual glucose determination in carbon-limited chemostat culture of Saccharomyces cerevisiae. Biotechnol. Bioeng. 83:395-399. [DOI] [PubMed] [Google Scholar]
- 28.Mashego, M. R., L. Wu, J. C. van Dam, C. Ras, J. L. Vinke, W. A. van Winden, W. M. van Gulik, and J. J. Heijnen. 2004. MIRACLE: mass isotopomer ratio analysis of U-C-13-labeled extracts. A new method for accurate quantification of changes in concentrations of intracellular metabolites. Biotechnol. Bioeng. 85:620-628. [DOI] [PubMed] [Google Scholar]
- 29.Politino, M., J. Brown, and J. J. Usher. 1996. Purification and characterization of an extracellular alkaline phosphatase from Penicillium chrysogenum. Prep. Biochem. Biotechnol. 26:171-181. [DOI] [PubMed] [Google Scholar]
- 30.Pronk, J. T., H. Y. Steensma, and J. P. vanDijken. 1996. Pyruvate metabolism in Saccharomyces cerevisiae. Yeast 12:1607-1633. [DOI] [PubMed] [Google Scholar]
- 31.Sauer, U., V. Hatzimanikatis, J. E. Bailey, M. Hochuli, T. Szyperski, and K. Wuthrich. 1997. Metabolic fluxes in riboflavin-producing Bacillus subtilis. Nat. Biotechnol. 15:448-452. [DOI] [PubMed] [Google Scholar]
- 32.Sawyer, D. T., and J. B. Bagger. 1959. The lactone acid salt equilibria for d-glucono-delta-lactone and the hydrolysis kinetics for this lactone. J. Am. Chem. Soc. 81:5302-5306. [Google Scholar]
- 33.van der Heijden, R. T. J. M., J. J. Heijnen, C. Hellinga, B. Romein, and K. C. A. M. Luyben. 1994. Linear constraint relations in biochemical reaction systems. 1. Classification of the calculability and the balanceability of conversion rates. Biotechnol. Bioeng. 43:3-10. [DOI] [PubMed] [Google Scholar]
- 34.van Dijken, J. P., A. van Tuijl, M. A. H. Luttik, W. J. Middelhoven, and J. T. Pronk. 2002. Novel pathway for alcoholic fermentation of delta-gluconolactone in the yeast Saccharomyces bulderi. J. Bacteriol. 184:672-678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Gulik, W. M., W. T. de Laat, J. L. Vinke, and J. J. Heijnen. 2000. Application of metabolic flux analysis for the identification of metabolic bottlenecks in the biosynthesis of penicillin-G. Biotechnol. Bioeng. 68:602-618. [DOI] [PubMed] [Google Scholar]
- 36.van Winden, W. A., W. M. van Gulik, D. Schipper, P. J. T. Verheijen, P. Krabben, J. L. Vinke, and J. J. Heijnen. 2003. Metabolic flux and metabolic network analysis of Penicillium chrysogenum using 2D [C-13, H-1] COSYNMR measurements and cumulative Bondomer simulation. Biotechnol. Bioeng. 83:75-92. [DOI] [PubMed] [Google Scholar]
- 37.van Winden, W. A., C. Wittmann, E. Heinzle, and J. J. Heijnen. 2002. Correcting mass isotopomer distributions for naturally occurring isotopes. Biotechnol. Bioeng. 80:477-479. [DOI] [PubMed] [Google Scholar]
- 38.van Winden, W. A., J. C. van Dam, C. Ras, R. J. Kleijn, J. L. Vinke, W. M. van Gulik, and J. J. Heijnen. 2005. Metabolic-flux analysis of Saccharomyces cerevisiae CEN.PK113-7D based on mass isotopomer measurements of 13C-labeled primary metabolites. FEMS Yeast Res. 5:559-568. [DOI] [PubMed] [Google Scholar]
- 39.Wiechert, W., M. Mollney, N. Isermann, M. Wurzel, and A. A. de Graaf. 1999. Bidirectional reaction steps in metabolic networks. III. Explicit solution and analysis of isotopomer labeling systems. Biotechnol. Bioeng. 66:69-85. [PubMed] [Google Scholar]
- 40.Wu, L., M. R. Mashego, J. C. van Dam, A. M. Proell, J. L. Vinke, C. Ras, W. A. van Winden, W. M. van Gulik, and J. J. Heijnen. 2005. Quantitative analysis of the microbial metabolome by isotope dilution mass spectrometry using uniformly C-13-labeled cell extracts as internal standards. Anal. Biochem. 336:164-171. [DOI] [PubMed] [Google Scholar]