

Abstract
This study examined whether Legionella pneumophila is able to thrive on heat-killed microbial cells (necrotrophy) present in biofilms or heat-treated water systems. Quantification by means of plate counting, real-time PCR, and flow cytometry demonstrated necrotrophic growth of L. pneumophila in water after 96 h, when at least 100 dead cells are available to one L. pneumophila cell. Compared to the starting concentration of L. pneumophila, the maximum observed necrotrophic growth was 1.89 log units for real-time PCR and 1.49 log units for plate counting. The average growth was 1.57 ± 0.32 log units (n = 5) for real-time PCR and 1.14 ± 0.35 log units (n = 5) for plate counting. Viability staining and flow cytometry showed that the fraction of living cells in the L. pneumophila population rose from the initial 54% to 82% after 96 h. Growth was measured on heat-killed Pseudomonas putida, Escherichia coli, Acanthamoeba castellanii, Saccharomyces boulardii, and a biofilm sample. Gram-positive organisms did not result in significant growth of L. pneumophila, probably due to their robust cell wall structure. Although necrotrophy showed lower growth yields compared to replication within protozoan hosts, these findings indicate that it may be of major importance in the environmental persistence of L. pneumophila. Techniques aimed at the elimination of protozoa or biofilm from water systems will not necessarily result in a subsequent removal of L. pneumophila unless the formation of dead microbial cells is minimized.
As the causative agent of Legionnaires' disease, Legionella pneumophila poses a substantial human health threat through colonization of water systems worldwide (13, 14, 24). Since person-to-person transmission of L. pneumophila has not yet been observed, measures to prevent the bacterium from spreading have concentrated on the elimination of this (opportunistic) pathogen from water sources (5). Consequently, the analysis of L. pneumophila in complex environmental consortia has become increasingly important (8, 19).
Despite its widespread distribution, L. pneumophila seems to have unusual nutritional requirements when grown in pure culture (8), and information about factors that contribute to the survival or active growth of the pathogen in its (natural) environment is very limited and divergent (5). On the one hand, it is supposed that L. pneumophila grows within protozoan hosts in aquatic environments and that this interaction is central to the pathogenesis and ecology of L. pneumophila (10, 12, 15, 16). Conversely, a number of studies suggested the protozoon-independent survival and growth of L. pneumophila (18, 21, 27, 32), in which case the bacterium uses biofilm matrices as a shelter and source of nutrients. Although scarce, there is evidence that a number of microorganisms in the proximity of L. pneumophila may favor its growth by excreting extracellular compounds as carbon and energy sources (1, 23, 29). Furthermore, sediment (mineral deposits and detritus) has also been shown to improve the survival of L. pneumophila by means of a synergistic effect (20), and recently, it was suggested that the required exogenous supply of amino acids may originate from other microorganisms or from decaying organic matter (5).
The prevention of Legionella multiplication in water systems is currently emulated by a combination of frequent heating of the water and limiting its residence time (stagnation). However, many water systems suffer from secluded spots, such as dead-end plumbing, which hamper treatment efficacy. Furthermore, L. pneumophila has been shown to survive extreme ranges of environmental conditions, including extensive temperatures ranging from 5 to 63°C (9). Considering these facts, it is evident that the complete eradication of Legionella from water systems is hard to achieve, with small numbers of residing bacteria thriving again shortly after water treatment. Recent studies using model warm water systems demonstrated significantly elevated Legionella concentrations (>107 CFU/liter) following heat treatment of the water system (27, 28).
Because biofilm debris can accumulate in the periphery of distribution systems, leading to sediment accumulation and microbial proliferation (26), the hypothesis is that heat treatment induces the formation of sudden loads of dead microbial cells, which become readily available as feed to surviving L. pneumophila, resulting in a fast (re)colonization of the environment. This paper aimed to verify whether L. pneumophila is potentially necrotrophic, thereby using dead microbial cells as a source of nutrients to sustain its persistence and growth. In order to estimate the significance of this necrotrophy, a comparison was made to protozoon-mediated replication of L. pneumophila.
MATERIALS AND METHODS
Strains and cultivation.
Strains used in the course of this study were L. pneumophila sg1 (ATCC 33152), Pseudomonas putida UWC3, Escherichia coli ATCC 4157, Bacillus subtilis LMG 7135, Lactobacillus plantarum LMG 6907, Acanthamoeba castellanii ATCC 30234, and Saccharomyces boulardii MUCL 43341. L. pneumophila was grown aerobically in BYE broth or on BCYE agar plates for 72 h at 37°C (7). P. putida, E. coli, B. subtilis, and S. boulardii were grown aerobically in Luria-Bertani broth (Oxoid, Basingstoke, United Kingdom) for 24 h at 37°C (except for P. putida, which was grown at 28°C). A. castellanii was grown aerobically in ATCC medium 711 for 5 days at 28°C. Cell suspensions for subsequent experiments were prepared by centrifugation of a fully grown culture for 4 min at 5,000 × g (Minispin; Eppendorf, Hamburg, Germany) and suspension of the pellet in 0.22-μm-filter-sterilized tap water. In order to remove all nutrients originating from the growth medium, each suspension was washed twice by centrifugation and again suspended in sterile tap water. Optical density measurements at 610 nm (ISIS9000; Dr. Lange, Berlin, Germany) were performed to obtain the desired starting concentration of each cell suspension, as determined initially by means of plate counting.
Necrotrophic growth experiments.
In order to demonstrate necrotrophic growth of L. pneumophila, a number of growth experiments using mainly heat-killed (30 min at 60°C) P. putida, unless otherwise stated, were performed. Efficiency of the heat treatment was verified throughout the experiments by means of plating 50 μl of the suspension on the appropriate culture medium.
Setup 1.
Setup 1 tackled the possibility of L. pneumophila thriving on dead microbial cells in an aquatic environment. Sterile tubes containing 5 ml of 0.22-μm-filter-sterilized tap water were inoculated with L. pneumophila, to which the following organisms were added: (i) live P. putida, (ii) heat-killed P. putida (concentrations ranging from 103 to 109 CFU/ml), (iii) heat-killed E. coli, (iv) heat-killed B. subtilis, (v) heat-killed Lactobacillus plantarum, (vi) heat-killed A. castellanii, (vii) heat-killed S. boulardii, and (viii) heat-treated biofilm (originating from 22-cm2 polyvinyl chloride coupons installed in a cooling tower water circuit with a 211-mg chemical oxygen demand/cm2). An axenic culture of L. pneumophila in sterile tap water served as a negative control. Cell suspension starting concentrations, as estimated using optical density measurements, were approximately 106 CFU/ml for L. pneumophila, 108 CFU/ml for A. castellanii, and 109 CFU/ml for P. putida, E. coli, B. subtilis, and S. boulardii. All tubes were aerobically incubated at 37°C, with determination of L. pneumophila numbers at 0, 24, and 96 h using real-time PCR and plate counting of 10-fold dilution series on BCYE agar plates. This experiment was performed five times.
Setup 2.
Supplemental to plate counting and real-time PCR, flow cytometry was also applied to demonstrate necrotrophic growth of L. pneumophila. Using a double fluorescent stain, information on the fractions of live and dead L. pneumophila cells, besides their total count, could be obtained. The experimental design was similar to that of setup 1, using 5 ml of filter-sterilized tap water to which the following organisms were added: (i) L. pneumophila, (ii) L. pneumophila and heat-killed P. putida, and (iii) L. pneumophila and live P. putida. Final concentrations were 104 CFU/ml for L. pneumophila and 107 CFU/ml for P. putida. Incubation of the mixture was done at 37°C, with flow cytometry measurements at 0, 24, and 96 h. To detect and quantify the number of live and dead L. pneumophila cells, 1-ml samples were stained for 10 min using the LIVE/DEAD BacLight Bacterial Viability kit (Invitrogen, Merelbeke, Belgium) according to the manufacturer's instructions. Stained samples were analyzed with a Cyan LX flow cytometer (Dakocytomation, Heverlee, Belgium) (3). The threshold trigger was set to sideward scatter, and differences in Syto 16 and propidium iodide fluorescence allowed a clear separation between live and dead L. pneumophila cells.
Setup 3.
To compare necrotrophic growth of L. pneumophila with protozoon-mediated replication, sterile 2.2-liter polyvinyl chloride recipients were filled with 1 liter of filtered (0.22-μm filter) tap water. Inoculation of the recipients was done with L. pneumophila and (i) heat-killed P. putida, (ii) live A. castellanii, or (iii) live A. castellanii and heat-killed P. putida. Again, a recipient containing only L. pneumophila served as a negative control. Starting concentrations for L. pneumophila, P. putida, and A. castellanii were approximately 103 CFU/ml, 106 CFU/ml, and 104 CFU/ml, respectively. Water samples (200 ml) for real-time PCR and denaturing gradient gel electrophoresis (DGGE) analysis were taken after 0, 1, 4, 8, and 14 days at 28°C. This experiment was performed three times.
Setup 4.
As a final indication of necrotrophic growth, a biological oxygen demand (BOD) test was performed, which measured the oxygen consumption during microbial growth. Sterile BOD bottles (500 ml) were filled with 365 ml of sterile tap water supplemented with the following organisms: (i) 105 CFU/ml L. pneumophila, (ii) 105 CFU/ml L. pneumophila and 108 CFU/ml heat-killed P. putida, and (iii) 108 CFU/ml heat-killed P. putida. All bottles were provided with a magnetic stirrer and sealed with a cap containing an electronic pressure indicator (Oxitop; WTW, Weilheim, Germany). All cultures were washed twice using sterile water and incubated overnight to remove all internal and external medium reserves prior to transfer to the autoclaved BOD bottles. A digital oxygen pressure measurement at 37°C was performed on a daily basis during five consecutive days. The BOD value for each sample was calculated according to the instructions of the manufacturer of the Oxitop apparatus (33).
Identification of L. pneumophila.
In order to verify the purity of the L. pneumophila culture used throughout the experiments, a latex agglutination test kit (Oxoid, Basingstoke, United Kingdom) was used according to the manufacturer's instructions. Furthermore, real-time PCR, flow cytometry, and DGGE analyses (4) provided additional certainty on the identity of the strain used.
DNA extraction.
The majority of experiments provided 1-ml samples that were directly suitable for DNA extraction. In the case of setup 3, 200 ml of sample was filtered using sterile filtration units (250 ml; Nalgene, Rochester, N.Y.) equipped with a 0.22-μm filter (47 mm; Millipore, Bedford, Mass.). Subsequent DNA extraction was performed directly on the filter paper based on the protocol described previously by Boon and coworkers (2).
Real-time PCR.
Real-time PCR was based on the protocol described previously by Wellinghausen and coworkers (31) for the specific quantification of L. pneumophila by amplifying the mip gene. PCR was performed in 25-μl reaction mixtures using the qPCR Core kit for SYBR Green I, according to the manufacturer's instructions (Eurogentec, Liège, Belgium), in MicroAmp optical 96-well reaction plates with optical caps (PE Applied Biosystems, Nieuwerkerk a/d IJssel, The Netherlands). The thermal profile was as follows: 50°C for 2 min and 95°C for 10 min followed by 40 cycles of 95°C for 30 s, 57°C for 1 min, and 60°C for 1 min. Amplicon dissociation curves were determined by constant fluorescent measurement during a final heating step at 60°C to 95°C at a 0.1°C/s ramping speed. The template DNA in the reaction mixtures was amplified in triplicate and monitored with an ABI Prism SDS 7000 instrument (PE Applied Biosystems, Nieuwerkerk a/d Ijssel, The Netherlands). Construction of the mip gene standard curve was performed by means of a dilution series of an L. pneumophila culture in sterile tap water. Correlation to the number of CFU and total cell count was determined by plate counting on BCYE medium and flow cytometry, respectively.
PCR-DGGE.
PCR was performed using a Taq polymerase kit (Applied Bio Systems, N.J.). Primers used in this study amplified the V3 region of the 16S rRNA gene (17) and the eukaryotic 18S rRNA gene (6). PCR and subsequent DGGE analysis of the amplicons were performed, as described previously (22), by using the Dcode system (Bio-Rad). Gels were stained in 200 ml of 1× Tris-acetate-EDTA containing 16 μl SYBR Green I nucleic acid gel stain (1:10,000 dilution; FMC BioProducts, Rockland, Maine) and immediately photographed on a UV transillumination table with a video camera module (Vilbert Lourmat, Marne-la Vallé, France). Normalization of the gels was performed using the BioNumerics software package, version 2.50 (Applied-Maths, St.-Martens-Latem, Belgium).
RESULTS
The goal of this study was to verify whether L. pneumophila is capable of surviving and/or growing on dead microbial cells that are presumed to be present after heat treatment of a biofilm-containing water system.
Setup 1.
Because of the use of one L. pneumophila starting suspension and equal volumes in all experiments, only the number of L. pneumophila cells from this starting suspension was determined at 0 h. After 96 h, real-time PCR showed significant necrotrophic growth of L. pneumophila when at least 10 dead P. putida cells were available to one L. pneumophila cell (10:1), whereas plate counts indicated the requirement of a 100:1 ratio (Table 1). Compared to the starting concentration of L. pneumophila, the observed maximum necrotrophic growth was 1.89 log units for real-time PCR and 1.49 log units for plate counting, with an average of 1.57 ± 0.32 log units and 1.14 ± 0.35 log units (n = 5), respectively. Table 1 contains the results at 96 h only, because no significant rise in L. pneumophila numbers was witnessed after 24 h in all experiments (data not shown). When the necrotrophic growth was compared to the axenic L. pneumophila suspension after 96 h, the actual necrotrophic growth surpasses 2 log units because of the slight decay of L. pneumophila in the negative control. No growth stimulation of L. pneumophila by live P. putida cells was observed and could be measured only by real-time PCR because of P. putida overgrowth on the BCYE plates.
TABLE 1.
Culture-dependent and real-time PCR-monitored necrotrophic growth of L. pneumophilaa
| Inoculum |
L. pneumophila CFU/ml (SD)
|
L. pneumophila mip gene copies/ml (SD)
|
||
|---|---|---|---|---|
| 0 h | 96 h | 0 h | 96 h | |
| L. pneumophila (approx 106 cells) | 7.8 × 105 (2.5 × 105) | 2.5 × 105 (1.3 × 105) | 2.4 × 106 (8.6 × 105) | 6.2 × 105 (6.3 × 104) |
| L. pneumophila + 109 live P. putida cells | NA | 3.7 × 106 (1.9 × 106) | ||
| L. pneumophila + 103 dead P. putida cells | 4.2 × 105 (2.6 × 105) | 3.5 × 106 (9.6 × 105) | ||
| L. pneumophila + 104 dead P. putida cells | 3.8 × 105 (9.5 × 104) | 9.8 × 105 (4.5 × 105) | ||
| L. pneumophila + 105 dead P. putida cells | 6.5 × 105 (3.6 × 105) | 3.5 × 106 (1.8 × 106) | ||
| L. pneumophila + 106 dead P. putida cells | 4.5 × 105 (3.1 × 105) | 4.6 × 106 (1.7 × 106)* | ||
| L. pneumophila + 107 dead P. putida cells | 6.9 × 105 (2.6 × 105) | 9.1 × 106 (2.0 × 106)* | ||
| L. pneumophila + 108 dead P. putida cells | 5.3 × 106 (1.1 × 106)* | 4.8 × 107 (9.6 × 106)* | ||
| L. pneumophila + 109 dead P. putida cells | 1.1 × 107 (7.2 × 106)* | 8.9 × 107 (2.0 × 107)* | ||
| L. pneumophila + 109 dead E. coli cells | 6.3 × 106 (2.6 × 106)* | 6.5 × 107 (1.3 × 107)* | ||
| L. pneumophila + 109 dead L. plantarum cells | 6.0 × 105 (4.2 × 105) | 7.5 × 105 (5.2 × 105) | ||
| L. pneumophila + 109 dead B. subtilis cells | NA | 1.8 × 105 (8.6 × 104) | ||
| L. pneumophila + 108 dead A. castellanii cells | 6.8 × 106 (3.0 × 106)* | 4.8 × 107 (1.3 × 107)* | ||
| L. pneumophila + 109 dead S. boulardii cells | 5.5 × 106 (2.8 × 106)* | 1.1 × 107 (8.2 × 106)* | ||
| L. pneumophila + heat-treated biofilm | NA | 2.9 × 107 (7.6 × 106)* | ||
Necrotrophic growth of L. pneumophila in sterile tap water on heat-killed (30 min, 60°C) microbial cells was measured. All L. pneumophila starting concentrations were approximately 106 CFU/ml. Quantification of L. pneumophila cells during 96 h was performed using plate counts on BCYE agar and real-time PCR targeting of the mip gene. Numbers presented are mean values of five repetitive experiments for which each separate experiment was measured in triplicate. An asterisk indicates significant necrotrophic growth of L. pneumophila compared to the starting concentration. NA, not available due to bacterial overgrowth.
Within the same experimental setup, a variety of heat-killed microorganisms as well as a biofilm sample were included in order to determine the specificity of necrotrophic growth. As presented in Table 1, with the exception of the tested gram-positive bacteria, all organisms as well as heat-treated biofilm resulted in a significant growth of L. pneumophila compared to that of the axenic control strain. No large differences between the growth stimulation by E. coli, P. putida, A. castellanii, S. boulardii, and biofilm existed. Based on these results, we decided to perform further experiments using P. putida, an organism frequently encountered in natural biofilms.
Setup 2.
In a separate experiment, flow cytometry was used to demonstrate necrotrophic growth and to acquire information on the ratio of live to dead L. pneumophila cells in each sample. A visual presentation of necrotrophic growth as recorded by flow cytometry is presented in Fig. 1. The applied heat treatment did not result in a complete lysis of the P. putida cells, and the dead P. putida population was clearly separated from both live and dead L. pneumophila cells, thereby facilitating a reliable quantification of the latter. After 96 h, most dead P. putida cells disappeared, and the total number of L. pneumophila cells increased from 2.5 × 104 ± 7.2 × 103 cells/ml to 1.6 × 106 ± 5.5 × 105 cells/ml. In the negative control, L. pneumophila dropped from 2.5 × 104 ± 6.0 × 103 cells/ml to 9.2 × 103 ± 4.2 × 103 cells/ml. By means of viability staining, it was shown that the fraction of living cells in the L. pneumophila population rose from the initial 54% to 82% after 96 h. Between 103 and 108 L. pneumophila cells/ml, total counts by real-time PCR and flow cytometry were similar within 1 log and approximately 10 times higher than that found by plate counting. However, based on the results from live/dead staining using the flow cytometer, it could be concluded that real-time PCR slightly overestimated and plate counting slightly underestimated the number of viable L. pneumophila cells (data not shown). Finally, it was shown that the filter-sterilized tap water used in the experiments is a suitable matrix, with only a minor drop in viable L. pneumophila cells in the negative control.
FIG. 1.

Live/dead analysis of necrotrophic growth. Both plots show Syto 16 fluorescence in the ordinate and propidium iodide fluorescence in the abscissa. Red dots indicate dead P. putida cells, green dots indicate live L. pneumophila cells, and black dots indicate dead L. pneumophila cells (colors are for visualization purposes only and do not correspond to actual fluorescence wavelengths). (A) L. pneumophila plus dead P. putida at 0 h; (B) L. pneumophila plus dead P. putida cells at 96 h.
Setup 3.
One liter of filter-sterilized tap water was used to determine the importance of necrotrophic growth in larger volumes and to compare the growth with A. castellanii-mediated replication of L. pneumophila. Also, in larger volumes, necrotrophic growth of L. pneumophila occurs (ratio of L. pneumophila to P. putida being 1:1,000), with a maximum of 1.42 log units and an average of 1.05 ± 0.37 log units (n = 3) (Fig. 2). After 14 days, the total cell count of L. pneumophila was substantially lower on dead P. putida cells in the case of necrotrophy (6.2 × 105 ± 2.1 × 105 cells/liter) compared to protozoon-mediated replication (1.5 × 108 ± 7.3 × 107 cells/liter). However, during the first 96 h, necrotrophy kept up with protozoon-mediated growth, even demonstrating a head start during the first 48 h. The combination of dead P. putida cells with A. castellanii did not result in a significantly higher L. pneumophila count than that with A. castellanii alone. During all experiments, pro- and eukaryotic DGGE was used to verify the microbial composition of the samples as well as to confirm the axenic nature of the strains used. As an additional means of identification, the band position of L. pneumophila in the samples was compared to that of the type strain, as previously described by Calvo-Bado and coworkers (4) (Fig. 3). Furthermore, it was demonstrated that no protozoa besides the inoculated A. castellanii were present in the filtered tap water (data not shown).
FIG. 2.

Necrotrophic growth of L. pneumophila at 28°C in filter-sterilized tap water on heat-killed (30 min 60°C) P. putida cells and live A. castellanii cells. Quantification of L. pneumophila cells was performed during 2 weeks using real-time PCR targeting the mip gene.
FIG. 3.
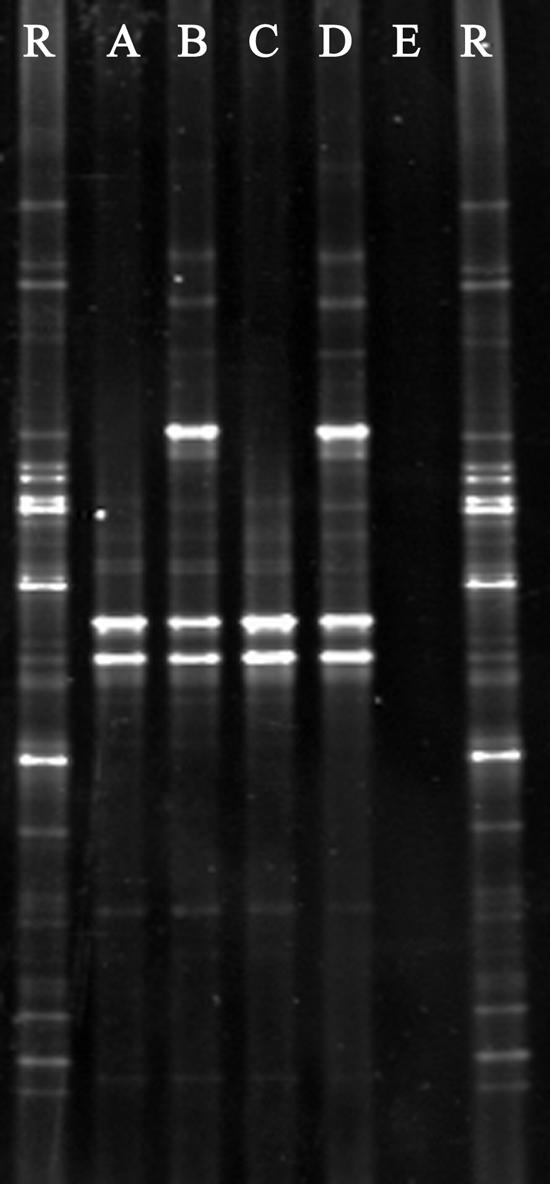
Example of a prokaryotic DGGE gel to verify the bacterial composition of starting samples during a necrotrophic growth experiment in filter-sterilized tap water. R, reference pattern for normalization; A, pure culture of L. pneumophila strain ATCC 33152; B, L. pneumophila plus live P. putida cells; C, L. pneumophila in sterile tap water (negative control); D, L. pneumophila plus heat-killed P. putida cells; E, filter-sterilized tap water as a blank sample.
Setup 4.
Although indirect, a supplemental indication of necrotrophic growth was obtained by measuring oxygen consumption during bacterial growth of L. pneumophila on dead P. putida cells. After removing all residual nutrients originating from the culture medium, it was shown that L. pneumophila was unable to grow actively in sterile tap water when present as an axenic culture. Oxygen consumption remained nearly zero, with a maximum of 2 mg/liter after 5 days. In contrast, L. pneumophila cells supplemented with dead P. putida cells (ratio, 1,000:1) clearly demonstrated active growth, with levels reaching 57 mg/liter at day 5. To verify the influence of decaying organic matter, heat-killed P. putida in sterile tap water was monitored as well, with a maximum BOD value of 29 mg/liter, clearly lower than the value obtained with necrotrophic growth.
DISCUSSION
Despite increasing research efforts, our understanding of the survival and/or growth of L. pneumophila in natural and human-made environments is still limited and divergent (5). Protozoan hosts are supposed to be crucial for replication of L. pneumophila in aquatic environments (10, 12, 15, 16), although alternative pathways have been suggested (5, 18, 20, 21, 27, 32). This paper demonstrates that L. pneumophila is able to thrive on heat-killed microbial cells present in biofilms or heat-treated water systems.
A fivefold repetition of several growth experiments using both culture-dependent and culture-independent quantification techniques clearly demonstrated significant growth of L. pneumophila when at least 100 dead cells are available to one L. pneumophila cell (Table 1). The growth yield was approximately 10%. The inclusion of live P. putida cells in the experiment did not result in any growth of L. pneumophila, indicating that the latter is not capable of actively killing other bacteria. Growth of Legionella on mineral deposits and detritus was previously demonstrated by Stout and coworkers (20), although it required a synergism with other bacteria. To our knowledge, the results presented in this paper are the first to demonstrate that L. pneumophila is able to grow on dead bacterial cells that may occur in the environment (biofilm) or originate from water disinfection measures (25, 26).
The specificity of necrotrophic growth was determined by including a taxonomic range of heat-killed microorganisms in the setup, with the eukaryotic organism A. castellanii inducing a slightly higher growth yield of L. pneumophila, most likely due to their larger cell mass. In contrast, the heat-killed gram-positive organisms B. subtilis and Lactobacillus plantarum did not result in the growth of L. pneumophila, probably because of the robust cell wall structure of these organisms, which prevents nutrients from becoming readily available. In the case of heat-killed Bacillus, growth of this organism was seen on the BCYE plates, indicating insufficient heat treatment or the survival of Bacillus spores. As a representative for more environmental situations, approximately 22 cm2 of biofilm was collected from a cooling tower and added to L. pneumophila after heat treatment. Again, a significant growth of more than 1 log unit was recorded. Given the fact that a biofilm is a constantly renewing entity, containing nearly 50% dead microbial cells (25, 30), this demonstrates how L. pneumophila is able to survive and grow in most water systems in a protozoon-free manner.
Although plate counting and real-time PCR were mainly used to quantify L. pneumophila cells during the experiments, flow cytometry was also applied to validate the methodologies and verify the fractions of live and dead cells within a population. From our results, it was noted that real-time PCR produces counts that are approximately 1 log higher for L. pneumophila because of the detection of mip genes from both live and dead cells. Live/dead analysis using a flow cytometer indicated that only 18% of the L. pneumophila population was dead after 96 h of necrotrophic growth. This implies that real-time PCR overestimates the number of viable L. pneumophila cells but also that plate counting underestimates the number because of a small fraction of L. pneumophila cells being viable but not culturable. Despite these findings, both techniques indicated necrotrophic growth of more than 1 log unit compared that of to their own negative control. This magnitude of growth was also confirmed using flow cytometry, which added the information that after 96 h of necrotrophic growth, 82% of the L. pneumophila cells were still alive. Finally, during the flow cytometric analyses, is was seen that the heat-treated P. putida cells did not lyse completely and that these cells may serve as nutrient packages for L. pneumophila, although the actual mechanism of necrotrophic growth needs to be studied in the future.
As mentioned previously, the major route for L. pneumophila survival and growth in the environment is considered to be replication within protozoan hosts (15). Therefore, another experiment compared necrotrophic growth to protozoon-mediated growth. It was noted that necrotrophic growth of L. pneumophila was also significant in larger volumes of water, as long as the ratio of at least 100 heat-killed P. putida cells to one L. pneumophila cell is maintained. Remarkably, after 24 h and 48 h, necrotrophic growth resulted in higher numbers of L. pneumophila than that found with protozoon-mediated growth. From 72 h on, the latter surpassed the former, resulting in numbers of L. pneumophila that were 1 log higher than those obtained with necrotrophic growth, which came to a halt after 96 h. The combination of A. castellanii and heat-killed P. putida in the same recipient did not significantly increase the numbers of Legionella cells compared to growth mediated by A. castellanii itself. These results indicate that the major route of L. pneumophila replication is by means of protozoa, although necrotrophy is far from negligible. It has to be kept in mind that the setup was batch mode; continuous feeding of the water system with heat-killed P. putida or protozoa may result in other growth yields of L. pneumophila. In this context, batch mode corresponds to sudden loads of dead cells as a result of heat treatment of (biofilm-containing) water systems (26), whereas a continuous supply of dead organics can be found within biofilm itself (25). Although it has been demonstrated that when bursting out of protozoa, L. pneumophila has shifted towards a virulent phenotype (11), from our results, it is not possible to conclude whether this is also the case during necrotrophic growth.
Finally, an experiment was performed to measure oxygen consumption as an indirect indication of bacterial growth. Again, compared to the negative control, L. pneumophila supplemented with dead P. putida cells demonstrated bacterial growth despite the influence of dead P. putida cells on the measurements. Although this experiment by itself does not provide any direct evidence for necrotrophic growth, it further supports the findings of the other experiments with specific monitoring of L. pneumophila.
In conclusion, this paper clearly demonstrates the possibility that L. pneumophila can survive and grow on dead microbial cells, as no other nutrients were sufficiently available in the experiments to achieve growth of over 1 log unit. Although this necrotrophy shows lower growth yields than replication within protozoan hosts, our findings indicate that it may have major importance in the persistence of L. pneumophila in the environment, such as in biofilms. Techniques aimed at the elimination of protozoa or biofilms from water systems will not necessarily result in a subsequent removal of L. pneumophila unless the formation of dead cells is minimized. Necrotrophy, as demonstrated in this paper, may also explain the elevated L. pneumophila concentrations witnessed recently after heat treatment of a water system (27, 28). The concept of necrotrophy by L. pneumophila warrants further research in terms of its environmental importance and its relation to the virulence of L. pneumophila. It also suggests that the eradication of L. pneumophila from water systems may be more difficult than already expected.
Acknowledgments
This work was supported by the Institute for the Promotion of Innovation by Science and Technology in Flanders and the Fund for Scientific Research in Flanders (IWT-Vlaanderen) and by the Bijzonder Onderzoeksfonds (BOF) of Ghent University.
Special thanks to F. Ollevier from the Laboratory of Aquatic Ecology (KULeuven) for supplying the A. castellanii strain and to Joke Geets, Dirk Halet, Tom Van de Wiele, and Lieven Wittebolle for critically reviewing the manuscript.
REFERENCES
- 1.Bohach, G. A., and I. S. Snyder. 1983. Characterization of surfaces involved in adherence of Legionella pneumophila to Fischerella species. Infect. Immun. 42:318-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boon, N., J. Goris, P. De Vos, W. Verstraete, and E. M. Top. 2000. Bioaugmentation of activated sludge by an indigenous 3-chloroaniline-degrading Comamonas testosteroni strain, I2gfp. Appl. Environ. Microbiol. 66:2906-2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boon, N., S. Depuydt, and W. Verstraete. 2006. Evolutionary algorithms and flow cytometry to examine the parameters influencing transconjugant formation. FEMS Microbiol. Ecol. 55:17-27. [DOI] [PubMed]
- 4.Calvo-Bado, L. A., J. A. W. Morgan, M. Sergeant, T. R. Pettitt, and J. M. Whipps. 2003. Molecular characterization of Legionella populations present within slow sand filters used for fungal plant pathogen suppression in horticultural crops. Appl. Environ. Microbiol. 69:533-541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Devos, L., N. Boon, and W. Verstraete. Legionella pneumophila in the environment: occurrence of a fastidious bacterium in oligotrophic conditions. Rev. Environ. Sci. Biotechnol., in press.
- 6.Diez, B., C. Pedros-Alio, T. Marsh, and R. Massana. 2001. Application of denaturing gradient gel electrophoresis (DGGE) to study the diversity of marine picoeukaryotic assemblages and comparison of DGGE with other molecular techniques. Appl. Environ. Microbiol. 67:2942-2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edelstein, P. H. 1981. Improved semiselective medium for isolation of Legionella pneumophila from contaminated clinical and environmental specimens. J. Clin. Microbiol. 14:298-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fields, B., R. Benson, and R. Besser. 2002. Legionella and Legionnaires' disease: 25 years of investigation. Clin. Microbiol. Rev. 15:506-526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fliermans, C. B., W. B. Cherry, L. H. Orrison, S. J. Smith, D. L. Tison, and D. H. Pope. 1981. Ecological distribution of Legionella pneumophila. Appl. Environ. Microbiol. 41:9-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harb, O. S., L. Y. Gao, and Y. Abu Kwaik. 2000. From protozoa to mammalian cells: a new paradigm in the life cycle of intracellular bacterial pathogens. Environ. Microbiol. 2:251-265. [DOI] [PubMed] [Google Scholar]
- 11.Josenhans, C., and S. Suerbaum. 2002. The role of motility as a virulence factor in bacteria. Int. J. Med. Microbiol. 291:605-614. [DOI] [PubMed] [Google Scholar]
- 12.Kuiper, M., B. Wullings, A. Akkermans, R. Beumer, and D. van der Kooij. 2004. Intracellular proliferation of Legionella pneumophila in Hartmannella vermiformis in aquatic biofilms grown on plasticized polyvinyl chloride. Appl. Environ. Microbiol. 70:6826-6833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leoni, E., P. P. Legnani, M. A. B. Sabattini, and F. Righi. 2001. Prevalence of Legionella spp. in swimming pool environment. Water Res. 35:3749-3753. [DOI] [PubMed] [Google Scholar]
- 14.Leoni, E., G. De Luca, P. P. Legnani, S. Sacchetti, S. Stampi, and F. Zanetti. 2005. Legionella waterline colonization: detection of Legionella species in domestic, hotel and hospital water systems. J. Appl. Microbiol. 98:373-379. [DOI] [PubMed] [Google Scholar]
- 15.Molmeret, M., M. Horn, M. Wagner, M. Santic, and Y. Abu Kwaik. 2005. Amoebae as training grounds for intracellular bacterial pathogens. Appl. Environ. Microbiol. 71:20-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murga, R., T. Forster, E. Brown, J. Pruckler, B. Fields, and R. Donlan. 2001. Role of biofilms in the survival of Legionella pneumophila in a model potable-water system. Microbiology 147:3121-3126. [DOI] [PubMed] [Google Scholar]
- 17.Muyzer, G., E. C. de Waal, and A. G. Uitterlinden. 1993. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 59:695-700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rogers, J., and C. W. Keevil. 1992. Immunogold and fluorescein immunolabeling of Legionella pneumophila within an aquatic biofilm visualized by using episcopic differential interference contrast microscopy. Appl. Environ. Microbiol. 58:2326-2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steinert, M., U. Hentschel, and J. Hacker. 2002. Legionella pneumophila: an aquatic microbe goes astray. FEMS Microbiol. Rev. 26:149-162. [DOI] [PubMed] [Google Scholar]
- 20.Stout, J. E., V. L. Yu, and M. G. Best. 1985. Ecology of Legionella pneumophila within water distribution systems. Appl. Environ. Microbiol. 49:221-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Surman, S., G. Morton, B. Keevil, and R. Fitzgeorge. 2002. Legionella pneumophila proliferation is not dependent on intracellular replication, p. 86-89. In R. Marre, Y. Abu Kwaik, C. Bartlett, N. P. Cianciotto, B. Fields, M. Frosch, J. Hacker, and P. C. Luck (ed.), Legionella. ASM Press, Washington, D.C.
- 22.Temmerman, R., I. Scheirlinck, G. Huys, and J. Swings. 2003. Culture-independent analysis of probiotic products using denaturing gradient gel electrophoresis. Appl. Environ. Microbiol. 69:220-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tison, D. L., D. H. Pope, W. B. Cherry, and C. B. Fliermans. 1980. Growth of Legionella pneumophila in association with blue-green algae (cyanobacteria). Appl. Environ. Microbiol. 39:456-459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tobin, J., J. Beare, and M. Dunell. 1980. Legionnaires' disease in a transplant unit: isolation of the causative organism from shower baths. Lancet ii:118-121. [DOI] [PubMed] [Google Scholar]
- 25.Tresse, O., S. Lescob, and D. Rho. 2003. Dynamics of living and dead bacterial cells within a mixed-species biofilm during toluene degradation in a biotrickling filter. J. Appl. Microbiol. 94:849-854. [DOI] [PubMed] [Google Scholar]
- 26.van der Kooij, D., J. S. Vrouwenvelder, and H. R. Veenendaal. 2003. Elucidation and control of biofilm formation processes in water treatment and distribution using the unified biofilm approach. Water Sci. Tech. 47:83-90. [PubMed] [Google Scholar]
- 27.van der Kooij, D., H. Veenendaal, and W. Scheffer. 2005. Biofilm formation and multiplication of Legionella in a model warm water system with pipes of copper, stainless steel and cross-linked polyethylene. Water Res. 39:2789-2798. [DOI] [PubMed] [Google Scholar]
- 28.Vervaeren, H., R. Temmerman, L. Devos, N. Boon, and W. Verstraete. Unpublished data.
- 29.Wadowsky, R. M., and R. B. Yee. 1985. Effect of non-Legionellaceae bacteria on the multiplication of Legionella pneumophila in potable water. Appl. Environ. Microbiol. 49:1206-1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Webb, J. S., L. S. Thompson, S. James, T. Charlton, T. Tolker-Nielsen, B. Koch, M. Givskov, and S. Kjelleberg. 2003. Cell death in Pseudomonas aeruginosa biofilm development. J. Bacteriol. 185:4585-4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wellinghausen, N., C. Frost, and R. Marre. 2001. Detection of legionellae in hospital water samples by quantitative real-time LightCycler PCR. Appl. Environ. Microbiol. 67:3985-3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wright, J. B., I. Ruseka, M. A. Athar, S. Corbett, and J. W. Costerton. 1989. Legionella pneumophila grows adherent to surfaces in vitro and in situ. Infect. Control Hosp. Epidemiol. 10:408-415. [DOI] [PubMed] [Google Scholar]
- 33.WTW. 1997. Bedienungsanleitung System Oxitop Control. Wissenschaftlich-technische Werkstätten, Weilheim, Germany.