

Abstract
Sinorhizobium meliloti genome sequence determination has provided the basis for different approaches of functional genomics for this symbiotic nitrogen-fixing alpha-proteobacterium. One of these approaches is gene disruption with subsequent analysis of mutant phenotypes. This method is efficient for single genes; however, it is laborious and time-consuming if it is used on a large scale. Here, we used a signature-tagged transposon mutagenesis method that allowed analysis of the survival and competitiveness of many mutants in a single experiment. A novel set of signature tags characterized by similar melting temperatures and G+C contents of the tag sequences was developed. The efficiencies of amplification of all tags were expected to be similar. Thus, no preselection of the tags was necessary to create a library of 412 signature-tagged transposons. To achieve high specificity of tag detection, each transposon was bar coded by two signature tags. In order to generate defined, nonredundant sets of signature-tagged S. meliloti mutants for subsequent experiments, 12,000 mutants were constructed, and insertion sites for more than 5,000 mutants were determined. One set consisting of 378 mutants was used in a validation experiment to identify mutants showing altered growth patterns.
Sinorhizobium meliloti is a model organism for studies of plant-microbe interactions. This gram-negative soil bacterium can enter an endosymbiosis with alfalfa plants through the formation of nitrogen-fixing nodules. The availability of the 6.7-Mb S. meliloti genome sequence, which consists of one chromosome (3.65 Mb) and two megaplasmids, pSymA (1.36 Mb) and pSymB (1.68 Mb) (16), has enabled transcriptome (5, 32), proteome (14), and metabolome (6) studies. These approaches focus on the monitoring of RNA, protein, and metabolite levels. Moreover, a library of mobilizable plasmids carrying all open reading frames of this microorganism has been constructed (36). Another step toward a better functional understanding of the S. meliloti genome is the creation of large libraries of defined mutants by site-directed or random mutagenesis. Such mutant libraries can be used to study each mutant's phenotype under defined conditions.
Usually, selection of mutants that can survive under certain conditions is simple and efficient and can be performed using a mixture of different mutants. However, selection of mutants that have an attenuated phenotype in test conditions is problematic, because all mutants have to be checked one by one. A microarray-based signature-tagged mutagenesis (STM) strategy (20; for reviews see references 4, 11, 19, 29, 34, and 38) can overcome this problem.
Signature-tagged mutagenesis is based on a collection of mutants split into sets, in which each mutant is modified by one or more different signature tags. The tags are short DNA segments that are unique for each mutant in a set and can be amplified using invariant (for a review see reference 11) or specific (26) priming sites. Tagged mutants from the same set are pooled prior to an experiment, and each mutant in the mixture can be identified based on the unique tag in its genome. The presence of a particular tag in the mixture can be detected by a tag-specific PCR (26) or by hybridization of amplified products to a dot blot (for a review see reference 11), to a macroarray (for a review see reference 38), or to a microarray (18, 23, 44) containing tag-specific probes. In order to integrate the signature tags into the genome, a PCR targeting strategy (39, 44) or a strategy based on libraries of tag-carrying transposons can be used. Different variants of the latter were used in numerous studies that were aimed at identifying genes important for virulence of pathogenic bacteria (for a review, see reference 4).
In transposon-based STM, the number of mutants that can be pooled in one experiment depends on the number of transposons containing different tags. The largest tagged transposon library reported so far contains 192 transposons (23). In this study, we describe construction of a novel set of 412 mini-Tn5 (mTn5)-based signature-tagged transposons and use of this set for STM of S. meliloti. A large signature-tagged S. meliloti library was generated. Here we describe mapping of insertion sites for more than 5,000 mutants of this library and pilot competition experiments in which a subset of mutants were used in combination with a microarray carrying probes specific for the tag sequences.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Bacterial strains and plasmids are listed in Table 1. Escherichia coli strains were grown at 37°C in Luria-Bertani medium (35). S. meliloti cells were grown at 30°C in tryptone yeast extract (TY) complex medium (8), 2*TY medium (10 g/liter tryptone, 6 g/liter yeast extract, 0.4 g/liter CaCl2), or Vincent minimal medium (VMM) (7, 42). When required, the media were supplemented with kanamycin (50 μg/ml), gentamicin (10 μg/ml), or chloramphenicol (170 mg/ml) for E. coli or with neomycin (120 μg/ml), streptomycin (600 μg/ml), and nalidixic acid (10 μg/ml) for S. meliloti.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristicsa | Source or reference |
|---|---|---|
| S. meliloti Rm2011 | Wild type, Nxr Smr | J. Dénarié, France |
| E. coli strains | ||
| DH5α | F−supE44 ΔlacU169 (φ80lacZΔM15) hsdR17 recA1 endA17 gyrA96 thi-1 relA1 | 35 |
| S17-1 | MM294, RP4-2-Tc::Mu-Km::Tn7, chromosomally integrated | 40 |
| Plasmids | ||
| pBC KS(−) | Cloning vector, Cmr, 3.4 kb | Stratagene |
| pG18Mob2 | pK18Mob derivative, Gmr, 2.8 kb | 24 |
| pCRS530 | Contains mTn5-GNm, Kmr Apr | 31 |
| pG18-STM | pG18Mob2 derivative containing modified mTn5-GNm, Kmr Gmr | This study |
Apr, ampicillin resistance; Cmr, chloramphenicol resistance; Gmr, gentamicin resistance; Kmr, kanamycin resistance; Nxr, nalidixic acid resistance; Smr, streptomycin resistance.
Construction of the mTn5 STM transposon and carrier plasmid pG18-STM.
Plasmid pG18-STM (Fig. 1) carrying the transposase gene tnpA** and a modified mTn5-GNm transposon was constructed by using suicide vector pG18Mob2 (24). Restriction, annealing, ligation, PCR, transformation, and plasmid isolation were performed using standard methods (35).
FIG. 1.

(a) Vector constructed for sequence-tagged mutagenesis of S. meliloti, based on pG18Mob2 containing a modified mTn5-GNm transposon and transposase gene tnpA** devoid of the HindIII restriction site. (b) Detailed diagram of the mTn5-STM transposon. The artificial linker designated AL was inserted into the SfiI restriction site of mTn5-GNm. HindIII and KpnI restriction sites of the artificial linker were used to clone the signature tags (H tag and K tag). P1, P2, P3, and P4 indicate the annealing sites for primers P1_Kpn, P2_Kpn, P3_Hind, and P4_Hind, respectively; Qseq1 indicates the annealing site for the sequencing primer. IE and OE indicate the inside and outside ends required for transposition.
First, the HindIII restriction site situated 1,105 bp downstream of the start codon of the tnpA* gene from pCRS530 (13, 31) was mutated by using the procedure described by Carter (10). The tnpA** gene containing the mutated HindIII restriction site (GAGCTT) was inserted into the SphI and XbaI restriction sites of pG18Mob2. The mini-transposon mTn5-GNm was recovered from pCRS530 using the flanking XbaI and EcoRI restriction sites and was inserted into pG18mob2. Subsequently, the HindIII restriction site in the polylinker of pG18Mob2 was inactivated by treatment of the HindIII sticky ends with the Klenow enzyme and blunt-end ligation.
An artificial linker (AATTCGGCCGCCTAGGCCAAAGGACGTGGTTTACGGGGCACGTAGTTTAAGGAAGTACGGTAAGGTACCG GGG GTG GCG GCA TTC ATA TAGCTGCGTGATTTCATT TTA ACT CCC CTC CGC CGCAAGCTTAGGTGGACCGTCGTAGAGCTAGTAGGG CTC AAT GCA CCA GGA CTA GGCCGCCTAGGCCGAATTC) containing four priming sites (underlined) flanking KpnI and HindIII restriction sites (boldface type) for insertion of variable tag sequences was generated and inserted into the SfiI restriction site of the mTn5-GNm transposon (13, 31), which resulted in transposon mTn5-STM (Fig. 1b). Candidate sequences were designed using the program DNASequenceGenerator (15). For primer design, 87 candidate sequences that fulfilled the following requirements were generated: less than seven identical contiguous nucleotides, a length of 21 bp, a melting temperature of 65°C to 70°C, and no KpnI, HindIII, EcoRI, or SfiI restriction sites. Four of these primer sequences were chosen for construction of the linker sequence. The primer and linker sequences were checked for similarity to the S. meliloti genome using the program vmatch (http://www.vmatch.de) (1). Sequences with the lowest levels of similarity to the S. meliloti genome were chosen. The linker was synthesized as six separate oligonucleotides that were annealed and ligated using the sticky ends of the KpnI and HindIII restriction sites.
Design of tag sequences and cloning into the mTn5-STM transposon.
A total of 1,498 signature tags that were 24 nucleotides long and had melting temperatures between 69.5°C and 70.5°C were designed using the programs DNASequenceGenerator and vmatch and allowing sequence identity of less than eight contiguous nucleotides. One half of the tags had sticky ends for the HindIII restriction site, and the other half had sticky ends for the KpnI restriction site. These tags, which are referred to below as H tags and K tags, respectively, were inserted into the linker cassette of transposon mTn5-STM.
Tags were synthesized as complementary single-stranded oligonucleotides and annealed prior to insertion into the linker. Complementary single-stranded oligonucleotides (50 μM) were mixed in 0.1 M NaCl. After incubation at 95°C for 3 min, samples were slowly cooled to 4°C. For ligation, 3 to 5 ng of plasmid (0.5 μl) was mixed with 7.3 μl of the annealed tag solution, 0.5 μl of polyethylene glycol (nested deletion kit; Amersham), 0.7 μl of T4 DNA ligase (1 U/μl; Roche), and 1 μl of T4 ligase buffer (Roche). Ligation was carried out overnight using a temperature gradient from 16°C to 8°C. Transformation of E. coli strain DH5α was performed as described previously (21). First, the H tags were inserted into the HindIII restriction site of pG18-STM. Subsequently, K tags were cloned into the KpnI restriction site of each plasmid that contained an H tag from the first tag cloning step. Tag-containing clones were verified by PCR using primer pairs for amplification of the tags (primers P1_Kpn [AAAGGACGTGGTTTACGGGGC] and P2_Kpn [TATATGAATGCCGCCACCCCC] in the case of K tags and primers P3_Hind [ATTTTAACTCCCCTCCGCCGC] and P4_Hind [TAGTCCTGGTGCATTGAGCCC] in the case of H tags). To check if the tags were cloned correctly, the linker region of each plasmid was sequenced.
Transposon mutagenesis.
E. coli S17-1-mediated conjugation (40) was used to transfer the plasmids carrying the tagged transposons into S. meliloti strain Rm2011. The transconjugants were selected on 2*TY medium supplemented with nalidixic acid, streptomycin, and neomycin. Clones were picked and rearrayed in sets of mutants.
Mapping of transposon insertion sites.
Transposon insertion sites were mapped by sequencing of the transposon-genome junction region using primer Qseq1 (ATCTAGCCCGCCTAATGAGC) (Fig. 1b). Sequencing was performed by QIAGEN (Hilden, Germany) using genomic DNA as the template. No amplification or cloning steps were carried out.
For transposon insertion site mapping, the GenDB annotation system was used (30). A GenDB extension was used for automated transposon position identification, and the sequences of transposon-genome junction regions were compared to the S. meliloti genome using the BLAST algorithm (3). The transition from the transposon sequence to the genome sequence was identified as the “jump-in” position and was mapped onto the S. meliloti genome.
Contents and layout of the tag microarray.
The mTn5-STM-1 microarray contained 23-mer oligonucleotides carrying 5′ C12-amino modifications. The probes were directed against the variable sequences of signature tags of the 412 mTn5-STM transposons. A microarray slide contained two arrays, each with 4,608 spots in 16 grids consisting of 18 rows and 16 columns. The 16 grids were arrayed in a 4-by-4 pattern. Each oligonucleotide was present in at least four replicates per array. In addition, three 23-mer genomic control sequences were printed in 192 replicates. Spotting was performed as described previously (25). Tag sequences and the layout of the mTn5-STM-1 microarray are described at http://www.cebitec.uni-bielefeld.de/groups/nwt/transcriptomics_facility/services_and_printed_arrays/.
Competition experiments and probe preparation.
Roughly equal quantities of 378 mutants containing different tags were mixed, and the mixture was stored as a glycerol stock. Part of the mutant mixture was stored separately and used as a reference (input pool). In all experiments, three biological replicates were used. Rich (TY) medium and minimal medium (VMM) were inoculated with the glycerol culture stock of the mixture of mutants to obtain an optical density at 600 nm (OD600) of 0.003. For salt- and detergent-induced stress experiments, S. meliloti cells were cultured in TY medium for 6 h after inoculation, and then NaCl was added to a concentration of 400 mM or sodium dodecyl sulfate (SDS) was added to a concentration of 0.87 mM. Cultures reached the stationary growth phase at an OD600 of 14 in TY medium, at an OD600 of 11.3 in VMM, and at an OD600 of 8.5 in stressed cultures. In all conditions, cells were collected in the exponential growth phase (OD600 for TY medium cultures, 7.2; OD600 for VMM cultures, 5.9; OD600 for NaCl-stressed cultures, 4.5; and OD600 for SDS-stressed cultures, 5). Genomic DNA was isolated using a NucleoSpin Tissue kit (Macherey-Nagel). Tags were amplified using primers P1_Kpn and P2_Kpn in the case of K tags and primers P3_Hind and P4_Hind in the case of H tags (Fig. 1b), which were 5′ modified by Cy3 in the case of the experiment and by Cy5 in the case of the input pool. PCR products were purified using a NucleoSpin Extract PCR purification kit (Macherey-Nagel).
Hybridization and image acquisition.
Fluorescently labeled PCR products were lyophilized and resuspended in Easyhyb hybridization solution (Roche Diagnostics). Preprocessing of microarrays was performed as described previously (25). Hybridization was carried out at 36°C for 1 h with an HS4800 hybridization station (Tecan). Before a hybridization sample was applied to a microarray, it was denatured for 3 min at 95°C. Following hybridization, the arrays were washed twice in 2× SSC-0.2% SDS for 5 min at 30°C and then twice in 0.5× SSC for 2 min at 20°C (1× SSC is 0.15 M sodium chloride plus 0.015 M sodium citrate, pH 7.0).
Spot detection, image segmentation, and signal quantification were performed using the ImaGene 6.0 software (Biodiscovery), as described previously (7).
Normalization and statistical analysis of microarray data.
After image processing, the mean intensity (ai value) was calculated for each spot using the formula ai = log2(RiGi)0.5, where Ri = Ich1i − Bgch1i and Gi = Ich2i − Bgch2i, where Ich1i and Ich2i are the intensities of spots in channel 1 and channel 2, respectively, and Bgch1i and Bgch2i are the background intensities of spots in channel 1 and channel 2, respectively. The log2 of the ratio of intensities (mi value) was calculated for each spot using the formula mi = log2(Ri/Gi) (7). LOWESS normalization (45) based on local regression that accounted for intensity and spatial dependence in dye biases was performed for data for the two tags separately. At the next step, the distances between the H and K tags for each mutant (ct) were calculated using the formula 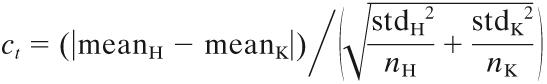 where meanH and meanK, stdH and stdK, and nH and nK are the means, standard deviations, and numbers of mean intensity values, respectively, for a single mutant. Subsequently, 10% of the mutants with the highest ct values and therefore with the greatest difference between the H and K tags were removed.
where meanH and meanK, stdH and stdK, and nH and nK are the means, standard deviations, and numbers of mean intensity values, respectively, for a single mutant. Subsequently, 10% of the mutants with the highest ct values and therefore with the greatest difference between the H and K tags were removed.
For each mutant and experiment the weighted mean of the medians of the m values (meanw) derived from the biological replicates was calculated by combining values for H tags and K tags, using the formula
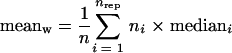 |
where nrep, ni, and mediani are the number of replicates, the number of spots of replicate i, and the median of the m values of replicate i, respectively, and 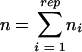 is the overall number of spots.
is the overall number of spots.
Mutants whose meanw values were greater than 0.7 and mutants whose meanw values were less than −0.7 were considered to have induced and attenuated phenotypes, respectively. The data matrix for cluster analysis after filtering of absolute m values consisted of 29 rows (mutants) and four columns (conditions). K-means clustering (28) for group mutants that exhibited similar behaviors in the four growth conditions was performed using K = 8 and a distance based on an uncentered Pearson correlation.
Nucleotide sequence accession number.
The sequence of plasmid pG18-STM has been deposited in the GenBank database under accession no. DQ408591.
RESULTS AND DISCUSSION
Construction of a signature-tagged transposon library and mutagenesis of S. meliloti.
STM was successfully used to study genes important for competitiveness and survival of a number of pathogenic bacteria in the host. Here, we applied a modified STM approach to a symbiotic bacterium and combined it with determination of transposon insertion sites in the mutants. This approach was based on transposons that were each modified by two different short 24-bp signature tags, similar to the method used by Karlyshev et al. (23). The utilization of short tags with similar G+C contents and melting temperatures made it possible to construct a large library of tagged transposons because these tags were amplified with similar efficiencies and therefore no preselection of tags was required (11). A high specificity of tag detection was achieved by bar coding each transposon with two different tags.
The mini-transposon mTn5-GNm (13, 31) used in this study contains the nptII resistance gene and a promotorless gusA reporter gene. mTn5-GNm was additionally modified by an artificial linker containing HindIII and KpnI restriction sites for cloning and priming sites for amplification of the signature tags. pG18-STM carrying the transposon modified by the linker and a transposase gene was constructed as a carrier plasmid for the signature-tagged transposons (Fig. 1).
A total of 1,498 different tags were designed, and 824 of them were synthesized and used to generate a collection of 412 transposons. Each transposon in this set was individually marked by two unique sequence tags. These transposons were used for random mutagenesis of S. meliloti Rm2011 (Fig. 2a). The RP4 mobilizable region (40) of pG18-STM enabled conjugal transfer of plasmids from E. coli donor strain S17-1 into the S. meliloti recipient cells by biparental mating. Mutants were selected based on resistance to neomycin conferred by the nptII gene of the transposon. Twenty-four to 30 clones were picked from each conjugation, resulting in a library of 12,000 tagged mutants. The mutant clones were rearrayed into sets, each containing mutants that differed by their signature tags. A microarray carrying tag-specific probes was constructed and used for detection and quantification of mutants in pilot competition experiments (Fig. 2b).
FIG. 2.

(a) Construction of the transposon mutant library. Nx, nalidixic acid; Nm, neomycin; Sm, streptomycin. (b) Schematic diagram of a competition experiment using signature-tagged mutants. The microarray image shows one of the 16 grids of the mTn5-STM-1 microarray.
Mapping and statistical analysis of the mutant library suggested that the insertion sites were random.
The transposon insertion sites of 5,089 mutants were determined by sequencing the junction using primer Qseq1, which bound 64 bp upstream of the linker with the cloned tags (Fig. 1b). Therefore, it was possible not only to determine the insertion sites of the transposons but also to check the tags in the mutants. Figure 3 summarizes the results of transposon mapping. Mapped transposon insertion sites of the mutants in the test set are shown in Table S3 in the supplemental material. The complete list of mapped mutations is available on the website of the public S. meliloti GenDB Genome Project (http://www.cebitec.uni-bielefeld.de/groups/nwt/sinogate).
FIG. 3.

Distribution of the mTn5-STM transposon insertions in the S. meliloti genome: genes with transposon insertions.
We performed several statistical tests to ensure that the distribution of transposon insertions was random and that the mTn5-STM transposons had no hot spots in the S. meliloti genome. An important parameter that shows the randomness of transposon insertions is the quantity of genes that carry a transposon insertion. A low number of genes hit by the transposon indicates a bias in the pattern of transposition. In order to determine the theoretical number of genes that has to be hit by at least one transposon, the neutral-base-pair model (22) was used. This model allows estimation of the number of gene hits based on the genome length, the number of transposon insertions, and the gene sizes. Applying this model to the library of 5,089 S. meliloti transposon mutants, we predicted that 2,890 genes (standard deviation, 378 genes) had to be mutated. The actual number of genes that were hit by at least one transposon was 2,711 (43.68% of the predicted number in S. meliloti 6207 protein-encoding genes), which was consistent with the expectations based on the neutral-base-pair model.
Furthermore, we performed a genome-wide analysis of all transposon insertion sites in relation to the G+C (A+T) content. Using a 100-bp window centered at the transposon insertion position, we calculated the mean G+C (A+T) content. The differences between the mean G+C and A+T contents within all of these windows and the mean G+C and A+T contents of the whole genome were 0.2% (G+C) and 0.1% (A+T). We also performed a χ2 test to exclude balancing effects in deviations of the G+C (A+T) content mean. The result was very low χ2 scores, 12.8 for the G+C distribution and 14.2 for the A+T distribution with 2,710 degrees of freedom, implying that there was no preference of the mTn5-STM transposons to jump into G+C- or A+T-rich regions.
In order to test the uniform distribution of all transposon insertions, we performed a χ2 test. Using a P value of 0.01 and 29 degrees of freedom per replicon, we found that a uniform distribution was highly improbable. This could have been explained by the existence of essential genes that were not represented in the mutant library. Moreover, mutations that resulted in slow growth of bacteria under the conditions used for selection of the transconjugants in this study resulted in underrepresentation of such mutants in the library. We therefore repeated the χ2 test, assuming that the S. meliloti genome contains essential genes. There is not enough information available about the quantity and position of essential genes in the S. meliloti genome that we could exclude defined groups of genes from the χ2 test. To cope with this problem, we created sets of randomly chosen genes and performed the χ2 test many times, leaving out one of the gene sets each time. The numbers of genes per set ranged from 1 to 100% of all genes that were localized on a certain replicon and did not have transposon insertions.
Such a modified χ2 test showed that the distribution of transposon insertions in the genome was likely to be random. The best χ2 test result for pSymB was a likelihood of 90% for a random distribution of transposons in this replicon. This result was obtained when a set containing 6% of the genes with no transposon insertion was excluded from the test. For pSymA, we observed an 87% likelihood of randomness when 10% of the genes with no hit were left out. In contrast, when all genes that did not have a transposon insertion were excluded from the χ2 test, the probability that the distribution was random was less than 11% for both megaplasmids.
Although the modified χ2 test worked well for the megaplasmids, it failed in the analysis of the transposon insertion distribution throughout the S. meliloti chromosome. We could not find a set of genes whose exclusion from the χ2 test increased the likelihood of randomness to more than 15%. The reason for this might have been a high proportion of essential genes on the chromosome (9) and the great size of the replicon, which led to the large fraction of genes not hit by a transposon. Therefore, a random search to detect essential genes seems to be unsuitable if the number of transposon insertions is not saturating. Nevertheless, based on the data for pSymA and pSymB and the results from the analysis of transposon insertion sites in relation to the G+C (A+T) content, we assumed that there was genome-wide random distribution of transposon insertion sites.
Pilot competition experiments identified signature-tagged mutants with altered growth patterns under different conditions.
In order to validate our STM approach, pilot experiments were carried out using a set of 378 signature-tagged mutants. The test conditions used were growth in rich (TY) medium and minimal medium (VMM), as well as growth in high-osmolarity medium and in medium containing SDS as a detergent. In all cases, the input pool was used as the reference. Each experiment was repeated three times using independent cultures.
The method used for processing the tag microarray data differed from the standard methods used for microarray data analysis due to the use of two signature tags per mutant and because of the comparatively small number of spots on the tag microarray.
After filtering steps performed to exclude technical and nonsignificant variations, 29 mutants were found to have a changed phenotype in at least one of the conditions tested. Using a K-means clustering approach, these mutants were divided into eight clusters corresponding to the pattern of competitiveness (Table 2 and Fig. 4).
TABLE 2.
Characteristics of mutants having altered phenotypes
| Mutant | Mutated genea | meanw for mutant grown in:
|
Gene product | |||
|---|---|---|---|---|---|---|
| TY | VMM | TY+SDS | TY+NaCl | |||
| Cluster 1 (highly competitive under most of the conditions tested) | ||||||
| 2-2E | SMb21633 | 0.39 | 0.55 | 0.67 | 0.88 | PaaG enoyl-coenzyme A hydratase protein |
| 3-5A | SMb20037 | 0.90 | 0.79 | 0.65 | 0.71 | AroE2 shikimate 5-dehydrogenase protein |
| 3-6D | SMc03032 | 0.51 | 0.29 | 0.83 | 0.81 | FlgI flagellar P-ring precursor transmembrane protein |
| Cluster 2 (highly competitive in TY medium and VMM under nonstress conditions) | ||||||
| 1-3G | pSymB.543971 | 1.46 | 0.77 | −0.01 | 0.12 | Intergenic |
| 1-5G | SMa0621 | 1.02 | 0.79 | 0.07 | 0.12 | FixI2 E1-E2-type cation ATPase |
| 3-12B | SMb20476 | 0.92 | 0.61 | −0.09 | 0.14 | Putative ABC transporter periplasmic dipeptide-binding protein |
| Cluster 3 (impaired growth in VMM) | ||||||
| 1-11C | SMc04346 | −0.33 | −1.08 | −0.11 | −0.04 | IlvC ketol-acid reductoisomerase protein |
| 2-9A | SMc02899 | 0.10 | −0.83 | 0.26 | 0.04 | PheA prephenate dehydratase protein |
| 3-1C | SMc01842 | −0.20 | −0.99 | −0.15 | 0.08 | Putative methyltransferase transcription regulator protein |
| 4-12C | SMc01053 | −0.32 | −1.35 | −0.08 | −0.01 | CysG siroheme synthase protein |
| 4-1H | SMc03776 | −0.65 | −1.23 | 0.21 | 0.01 | ProB1 glutamate 5-kinase protein |
| Cluster 4 (highly competitive in stress conditions) | ||||||
| 2-11B | SMc01881 | −0.50 | −0.47 | 2.09 | 2.03 | PanB 3-methyl-2-oxobutanoate hydromethyltransferase protein |
| 2-5H | SMc03164 | −0.18 | −0.08 | 0.77 | 0.43 | XylB xylulose kinase protein, putative |
| 2-7H | pSymB.96435 | 0.28 | 0.11 | 0.82 | 0.43 | Intergenic |
| 3-10H | SMb20360 | −0.36 | −0.22 | 1.67 | 1.50 | Hypothetical protein, putative protease subunit of ATP-dependent Clp protease |
| 3-11B | pSymB.1003846 | 0.19 | 0.43 | 1.95 | 1.85 | Intergenic |
| 3-5B | SMb20931 | 0.12 | 0.21 | 0.94 | 0.76 | Putative sugar uptake ABC transporter periplasmic solute-binding protein precursor |
| Cluster 5 (impaired growth under all conditions tested; strongly impaired growth in VMM) | ||||||
| 2-9E | SMc03782 | −0.40 | −1.01 | −0.37 | −0.52 | Hypothetical signal peptide protein |
| 3-7D | SMc01174 | −0.58 | −0.77 | −0.40 | −0.37 | CysK2 cysteine synthase A protein |
| Cluster 6 (low competitiveness under normal conditions and slightly increased competitiveness under stress conditions) | ||||||
| 3-11D | SMb20377 | −0.67 | −0.90 | 0.46 | 0.46 | Putative translation initiation inhibitor protein |
| 3-12D | SMc00334 | −1.26 | −0.98 | 0.79 | 0.51 | Cmk cytidylate kinase protein |
| Cluster 7 (low competitiveness under normal conditions but not disadvantaged in stress conditions) | ||||||
| 3-12C | SMc00808 | −0.76 | −0.35 | 0.21 | 0.15 | ChrA chromate transport protein |
| 4-10C | SMa0091 | −0.75 | −0.42 | 0.13 | 0.13 | Conserved hypothetical protein |
| 4-11C | SMc02597 | −0.72 | −0.50 | −0.03 | −0.13 | SodC superoxide dismutase Cu-Zn precursor transmembrane protein |
| 4-12D | SMa0070 | −0.75 | −0.46 | 0.13 | 0.08 | ABC transporter permease |
| Cluster 8 (impaired growth in TY medium and partially impaired growth under other conditions) | ||||||
| 2-1H | SMc01219 | −0.64 | −0.35 | −0.74 | −0.19 | Lipopolysaccharide core biosynthesis mannosyltransferase LpsB |
| 3-11H | SMc01700 | −0.72 | −0.31 | −0.26 | −0.24 | Peptidyl-prolyl cis-trans isomerase A (PpiA) |
| 3-8C | SMc02050 | −0.74 | −0.70 | −0.54 | −0.49 | Trigger factor protein Tig, probable |
| 4-12B | SMc02144 | −1.17 | −0.65 | −0.70 | −0.62 | PstC phosphate transport system permease ABC transporter protein |
For mutants carrying a transposon insertion in an intergenic region the exact position of insertion in the replicon is given.
FIG. 4.

K-means cluster analysis of tag microarray data. The normalized m values for the mutants with changed phenotypes are shown. Rows indicate separate mutants, and columns represent specific growth conditions. Green indicates an attenuated phenotype, and red indicates an intensified phenotype.
Cluster 1 contained clones that were highly competitive under most of the conditions tested. In particular, it included an flgI mutant, which was impaired for synthesis of flagella. The fast-growth phenotype of this mutant supports the observation that the synthesis of flagella is energetically disadvantageous (27). Two other mutants in this cluster were paaG (SMb21633) and aroE2 (SMb20037) mutants. paaG encodes a putative enoyl-coenzyme A hydratase/isomerase involved in phenylacetate catabolism, and aroE2 codes for a putative shikimate 5-dehydrogenase protein involved in chorismate metabolism. Both genes have paralogs in the S. meliloti genome.
Cluster 2 contained clones that were highly competitive in TY medium and VMM under nonstress conditions. This cluster consisted of three mutants bearing a transposon insertion in fixI2 (encoding an E1-E2-type cation ATPase), in SMb20476 (coding for a putative ABC transporter periplasmic dipeptide-binding protein), and in the intergenic region between SMb20518 (encoding a putative endohitinase) and SMb20519 (encoding a conserved hypothetical protein), probably influencing transcription of SMb20519.
The growth in VMM of mutants that belonged to cluster 3 was strongly impaired. Characteristically, all mutants in this cluster had a transposon insertion in genes involved in the synthesis of amino acids or cofactors not present in VMM, including isoleucine/valine (ilvC), phenylalanine (pheA), ubiquinone/menaquinone (SMc01842), cysteine (cysG), and proline (proB1). It was previously shown that ilvC mutants of S. meliloti are isoleucine/valine auxotrophs (2) and that cysG mutants of Rhizobium etli are cysteine auxotrophs (41).
Cluster 4 contained six mutants that exhibited high competitiveness in stress conditions but not in nonstress conditions. Two of these mutants had a transposon insertion in the intergenic regions of pSymB, preceding SMb20088 (encoding a conserved hypothetical protein) and upstream of SMb21337 (coding for a putative iron-sulfur-binding protein, probably a subunit of an oxidoreductase-like aldehyde oxidase or xanthine dehydrogenase). This cluster also contained a panB (SMc01881) mutant. In Salmonella enterica, a panB mutation causes auxotrophy for pantothenate (33). We suggest that in S. meliloti the function of PanB can also be performed by another protein, probably by the product of SMb20821, which at the amino acid level exhibits 31% identity with the SMc01881 product and contains a conserved PanB domain. Three other clones in cluster 4 had mutations in xylB (coding for a putative xylulose kinase protein that participates in degradation of d-xylose), SMb20360 (encoding a putative protease subunit of an ATP-dependent Clp protease), and SMb20931 (coding for a putative sugar uptake ABC transporter periplasmic solute-binding protein precursor).
Cluster 5 contained two clones, cysK2 and SMc03782 mutants, whose growth was impaired under all conditions tested and was more strongly impaired in VMM. cysK2 encodes a probable cysteine synthase A (O-acetylserine sulfhydrylase A), whereas the gene product of SMc03782 has similarities to membrane-bound metallopeptidases involved in cell division and chromosome partitioning.
The competitiveness of mutants in clusters 6 and 7 was impaired more strongly under normal conditions than under stress conditions. Such a pattern probably occurred due to the fast growth of nonstressed cultures in the exponential phase compared to the growth of SDS- and salt-stressed cultures. Mutants that grew and divided more slowly than other mutants may have been less competitive in the fast-growing cultures than in stressed slowly growing cultures, if the slow-growth phenotypes were not caused by the stress conditions themselves. Cluster 6 contained mutants with mutations in the cmk gene encoding a putative cytidylate kinase and in SMb20377 encoding a putative translation initiation inhibitor protein. Cluster 7 contained two clones with mutations in transporter genes (chrA and SMa0070), a sodC (coding for a superoxide dismutase) mutant, and an SMa0091 (encoding a hypothetical protein) mutant.
Cluster 8 contained mutants whose growth was impaired in TY medium and was partially impaired under other conditions. This cluster included an lpsB (encoding a lipopolysaccharide core biosynthesis mannosyltransferase) mutant whose competitiveness was weakened in a fast-growing TY medium culture and in a TY medium-SDS culture. Since it was previously shown (12) that lpsB mutants are sensitive to sodium deoxycholate, we expected this mutant to be attenuated in SDS-containing medium as well. The second mutant in the cluster had a transposon insertion in the ppiA gene, which encodes a peptidyl-prolyl isomerase. This enzyme has a chaperone-like activity and facilitates the cis-trans isomerization of peptide bonds N terminal to proline residues within polypeptide chains (37). Interestingly, another mutant in this cluster had a transposon insertion in the tig gene that encoded a peptidyl-prolyl isomerase as well. Trigger factor encoded by tig is a ribosome-bound protein that combines two functions, peptidyl-prolyl isomerization and chaperone-like activities (17), similar to the ppiA gene product. Cluster 8 also contained a pstC mutant, whose slow-growth phenotype was especially noticeable in the fast-growing TY medium culture and was less obvious in VMM and in the stressed cultures. In E. coli, pstC encodes a permease protein of a high-affinity Pi-specific ABC transporter (43). A comparatively high concentration of inorganic phosphate in VMM might have been the reason for the faster growth of the pstC mutant in this medium than in TY medium.
Three mutants that showed altered growth behavior during cultivation of the mutant pool in VMM compared to the growth behavior in TY medium were analyzed individually in competition with the wild type. In these competition experiments the proB mutant (cluster 3) was analyzed in VMM, whereas the chrA mutant (cluster 7) and the tig mutant (cluster 8) were tested in TY medium. In accordance with the competition experiment analyzing the mutant pool by quantification of the signature tags in microarray hybridizations, the three individually tested mutants showed reduced competitiveness compared to the wild type.
Conclusions.
In this study, we used a modified signature-tagged mutagenesis strategy, which for the first time was applied to a nitrogen-fixing symbiotic bacterium. A novel set of tags that does not require preselection of the tags was designed, and a library of 412 different double-tagged transposons was created using this set of tags. In a number of previous studies the workers demonstrated that there was a broad host range for transposition of the mTn5 transposon (for a review see reference 34) for several organisms, including S. enterica serovar Typhimurium, E. coli, Klebsiella pneumoniae, Vibrio cholerae, Proteus mirabilis, Bacillus melitensis, Yersinia pestis, and Citrobacter rodentium. This broad application spectrum in combination with the large number of signature tags and the tag-specific microarray makes the mTn5-STM transposon set a powerful and easy-to-use tool that can be applied to a broad spectrum of bacteria.
An extensive library of transposon mutants containing more than 12,000 clones was created by using the set of tagged transposons. The transposon insertion sites were determined for 42% of the mutants in this library. As a result, 44% coverage of all predicted protein-encoding genes by mapped transposon insertions was achieved. Analysis of the transposon library suggested that the insertion sites of the mTn5-STM transposons were random and that there were no hot spots.
Pilot experiments performed to verify the novel signature-tagged transposon set in combination with a microarray hybridization approach designed to identify and quantify individual mutants in the pool proved the reliability of this system for identification of attenuated mutants. The statistical processing of the tag microarray data comprising normalization and clustering allowed identification of clusters of mutants that had similar growth patterns under different growth conditions. We found that clones carrying similar kinds of mutation were grouped into the same cluster.
In future experiments, sets of mutants can be generated using up to 412 mutants carrying different unique tags. These sets should allow testing of the phenotypes of the mutants in diverse conditions. Of special interest is utilization of the signature-tagged S. meliloti mutants to identify genes important for survival and competitiveness in symbiosis with the host plants.
Supplementary Material
Acknowledgments
We thank Wayne Reeve for providing the mTn5-GNm transposon and Alexander Goesmann for helpful discussions.
This work was funded by grant 031U213D from Bundesministerium für Bildung und Forschung, Germany. N.P. and D.W. were supported by the Graduate School for Bioinformatics and Genome Research, funded by the Ministerium für Wissenschaft und Forschung (MWF), North-Rhine Westphalia, Germany.
Footnotes
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Abouelhoda, M. I., S. Kurtz, and E. Ohlebusch. 2004. Replacing Suffix trees with enhanced Suffix arrays. J. Discrete Algorithms 2:53-86. [Google Scholar]
- 2.Aguilar, O. M., and D. H. Grasso. 1991. The product of the Rhizobium meliloti ilvC gene is required for isoleucine and valine synthesis and nodulation of alfalfa. J. Bacteriol. 173:7756-7764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Altschul, S. F., T. L. Madden, A. A. Schäffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Autret, N., and A. Charbit. 2005. Lessons from signature-tagged mutagenesis on the infectious mechanisms of pathogenic bacteria. FEMS Microbiol. Rev. 29:703-717. [DOI] [PubMed] [Google Scholar]
- 5.Barnett, M. J., C. J. Toman, R. F. Fisher, and S. R. Long. 2004. A dual-genome Symbiosis Chip for coordinate study of signal exchange and development in a prokaryote-host interaction. Proc. Natl. Acad. Sci. USA 101:16636-16641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barsch, A., T. Patschkowski, and K. Niehaus. 2004. Comprehensive metabolite profiling of Sinorhizobium meliloti using gas chromatography-mass spectrometry. Funct. Integr. Genomics 4:219-230. [DOI] [PubMed] [Google Scholar]
- 7.Becker, A., H. Bergès, E. Krol, C. Bruand, S. Rüberg, D. Capela, E. Lauber, E. Meilhoc, F. Ampe, F. J. de Bruijn, J. Fourment, A. Francez-Charlot, D. Kahn, H. Küster, C. Liebe, A. Pühler, S. Weidner, and J. Batut. 2004. Global changes in gene expression in Sinorhizobium meliloti 1021 under microoxic and symbiotic conditions. Mol. Plant-Microbe Interact. 17:292-303. [DOI] [PubMed] [Google Scholar]
- 8.Beringer, J. E. 1974. R factor transfer in Rhizobium leguminosarum. J. Gen. Microbiol. 84:88-98. [DOI] [PubMed] [Google Scholar]
- 9.Capela, D., F. Barloy-Hubler, J. Gouzy, G. Bothe, F. Ampe, J. Batut, P. Boistard, A. Becker, M. Boutry, E. Cadieu, S. Dreano, S. Gloux, T. Godrie, A. Goffeau, D. Kahn, E. Kiss, V. Lelaure, D. Masuy, T. Pohl, D. Portetelle, A. Pühler, B. Purnelle, U. Ramsperger, C. Renard, P. Thebault, M. Vandenbol, S. Weidner, and F. Galibert. 2001. Analysis of the chromosome sequence of the legume symbiont Sinorhizobium meliloti strain 1021. Proc. Natl. Acad. Sci. USA 98:9877-9882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carter, P. 1991. Mutagenesis facilitated by the removal or introduction of unique restriction sites, p. 1-25. In M. J. McPherson (ed.), Directed mutagenesis: a practical approach. Oxford University Press, New York, N.Y.
- 11.Chiang, S. L., J. J. Mekalanos, and D. W. Holden. 1999. In vivo genetic analysis of bacterial virulence. Annu. Rev. Microbiol. 53:129-154. [DOI] [PubMed] [Google Scholar]
- 12.Clover, R. H., J. Kieber, and E. R. Signer. 1989. Lipopolysaccharide mutants of Rhizobium meliloti are not defective in symbiosis. J. Bacteriol. 171:3961-3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Lorenzo, V., M. Herrero, U. Jakubzik, and K. N. Timmis. 1990. Mini-Tn5 transposon derivatives for insertion mutagenesis, promoter probing, and chromosomal insertion of cloned DNA in gram-negative eubacteria. J. Bacteriol. 172:6568-6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Djordjevic, M. A., H. C. Chen, S. Natera, G. Van Noorden, C. Menzel, S. Taylor, C. Renard, O. Geiger, and G. F. Weiller. 2003. A global analysis of protein expression profiles in Sinorhizobium meliloti: discovery of new genes for nodule occupancy and stress adaptation. Mol. Plant-Microbe Interact. 16:508-524. [DOI] [PubMed] [Google Scholar]
- 15.Feldkamp, U., S. Saghafi, W. Banzhaf, and H. Rauhe. 2001. DNA sequence generator: a program for the construction of DNA sequences, p. 23-32. In N. Jonoska and N. C. Seeman (ed.), Proceedings of the Seventh International Workshop on DNA Based Computers (DNA 7). University of South Florida, Tampa, Fla.
- 16.Galibert, F., T. M. Finan, S. R. Long, A. Pühler, P. Abola, F. Ampe, F. Barloy-Hubler, M. J. Barnett, A. Becker, P. Boistard, G. Bothe, M. Boutry, L. Bowser, J. Buhrmester, E. Cadieu, D. Capela, P. Chain, A. Cowie, R. W. Davis, S. Dreano, N. A. Federspiel, R. F. Fisher, S. Gloux, T. Godrie, A. Goffeau, B. Golding, J. Gouzy, M. Gurjal, I. Hernandez-Lucas, A. Hong, L. Huizar, R. W. Hyman, T. Jones, D. Kahn, M. L. Kahn, S. Kalman, D. H. Keating, E. Kiss, C. Komp, V. Lelaure, D. Masuy, C. Palm, M. C. Peck, T. M. Pohl, D. Portetelle, B. Purnelle, U. Ramsperger, R. Surzycki, P. Thébault, M. Vandenbol, F. J. Vorhölter, S. Weidner, D. H. Wells, K. Wong, K. C. Yeh, and J. Batut. 2001. The composite genome of the legume symbiont Sinorhizobium meliloti. Science 293:668-672. [DOI] [PubMed] [Google Scholar]
- 17.Genevaux, P., F. Keppel, F. Schwager, P. S. Langendijk-Genevaux, F. U. Hartl, and C. Georgopoulos. 2004. In vivo analysis of the overlapping functions of DnaK and trigger factor. EMBO Rep. 5:195-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Groh, J. L., Q. Luo, J. D. Ballard, and L. R. Krumholz. 2005. A method adapting microarray technology for signature-tagged mutagenesis of Desulfovibrio desulfuricans G20 and Shewanella oneidensis MR-1 in anaerobic sediment survival experiments. Appl. Environ. Microbiol. 71:7064-7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hayes, F. 2003. Transposon-based strategies for microbial functional genomics and proteomics. Annu. Rev. Genet. 37:3-29. [DOI] [PubMed] [Google Scholar]
- 20.Hensel, M., J. E. Shea, C. Gleeson, M. D. Jones, E. Dalton, and D. W. Holden. 1995. Simultaneous identification of bacterial virulence genes by negative selection. Science 269:400-403. [DOI] [PubMed] [Google Scholar]
- 21.Inoue, H., H. Nojima, and H. Okayama. 1990. High efficiency transformation of Escherichia coli with plasmids. Gene 96:23-28. [DOI] [PubMed] [Google Scholar]
- 22.Jacobs, M. A., A. Alwood, I. Thaipisuttikul, D. Spencer, E. Haugen, S. Ernst, O. Will, R. Kaul, C. Raymond, R. Levy, L. Chun-Rong, D. Guenthner, D. Bovee, M. V. Olson, and C. Manoil. 2003. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 100:14339-14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karlyshev, A. V., P. C. Oyston, K. Williams, G. C. Clark, R. W. Titball, E. A. Winzeler, and B. W. Wren. 2001. Application of high-density array-based signature-tagged mutagenesis to discover novel Yersinia virulence-associated genes. Infect. Immun. 69:7810-7819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kirchner, O., and A. Tauch. 2003. Tools for genetic engineering in the amino acid-producing bacterium Corynebacterium glutamicum. J. Biotechnol. 104:287-299. [DOI] [PubMed] [Google Scholar]
- 25.Krol, E., and A. Becker. 2004. Global transcriptional analysis of the phosphate starvation response in Sinorhizobium meliloti strains 1021 and 2011. Mol. Genet. Genomics 272:1-17. [DOI] [PubMed] [Google Scholar]
- 26.Lehoux, D. E., F. Sanschagrin, and R. C. Levesque. 1999. Defined oligonucleotide tag pools and PCR screening in signature-tagged mutagenesis of essential genes from bacteria. BioTechniques 26:473-480. [DOI] [PubMed] [Google Scholar]
- 27.Macnab, R. M. 1996. Flagella and motility, p. 123-145. In F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, B. Masaganik, W. S. Reznikoff, M. Riley, M. Schaechter, and H. E. Umbarger (ed.), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed. American Society for Microbiology, Washington, D.C.
- 28.MacQueen, J. B. 1967. Some methods for classification and analysis of multivariate observations, p. 281-297. In L. M. Le Cam and J. Neyman (ed.), Proceedings of 5th Berkeley Symposium on Mathematical Statistics and Probability. University of California Press, Berkeley.
- 29.Mecsas, J. 2002. Use of signature-tagged mutagenesis in pathogenesis studies. Curr. Opin. Microbiol. 5:33-37. [DOI] [PubMed] [Google Scholar]
- 30.Meyer, F., A. Goesmann, A. C. McHardy, D. Bartels, T. Bekel, J. Clausen, J. Kalinowski, B. Linke, O. Rupp, R. Giegerich, and A. Pühler. 2003. GenDB—an open source genome annotation system for prokaryote genomes. Nucleic Acids Res. 31:2187-2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reeve, W. G., R. P. Tiwari, P. S. Worsley, M. J. Dilworth, A. R. Glenn, and J. G. Howieson. 1999. Constructs for insertional mutagenesis, transcriptional signal localization and gene regulation studies in root nodule and other bacteria. Microbiology 145:1307-1316. [DOI] [PubMed] [Google Scholar]
- 32.Rüberg, S., Z. X. Tian, E. Krol, B. Linke, F. Meyer, Y. Wang, A. Pühler, S. Weidner, and A. Becker. 2003. Construction and validation of a Sinorhizobium meliloti whole genome DNA microarray: genome-wide profiling of osmoadaptive gene expression. J. Biotechnol. 106:255-268. [DOI] [PubMed] [Google Scholar]
- 33.Rubio, A., and D. M. Downs. 2002. Elevated levels of ketopantoate hydroxymethyltransferase (PanB) lead to a physiologically significant coenzyme A elevation in Salmonella enterica serovar Typhimurium. J. Bacteriol. 184:2827-2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saenz, H. L., and C. Dehio. 2005. Signature-tagged mutagenesis: technical advances in a negative selection method for virulence gene identification. Curr. Opin. Microbiol. 8:612-619. [DOI] [PubMed] [Google Scholar]
- 35.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 36.Schroeder, B. K., B. L. House, M. W. Mortimer, S. N. Yurgel, S. C. Maloney, K. L. Ward, and M. L. Kahn. 2005. Development of a functional genomics platform for Sinorhizobium meliloti: construction of an ORFeome. Appl. Environ. Microbiol. 71:5858-5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shaw, P. E. 2002. Peptidyl-prolyl isomerases: a new twist to transcription. EMBO Rep. 3:521-526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shea, J. E., J. D. Santangelo, and R. G. Feldman. 2000. Signature-tagged mutagenesis in the identification in virulence genes in pathogens. Curr. Opin. Microbiol. 3:451-458. [DOI] [PubMed] [Google Scholar]
- 39.Shoemaker, D. D., D. A. Lashkari, D. Morris, M. Mittman, and R. W. Davis. 1996. Quantitative phenotypic analysis of yeast deletion mutants using a highly parallel molecular bar-coding strategy. Nat. Genet. 14:450-456. [DOI] [PubMed] [Google Scholar]
- 40.Simon, R., U. Priefer, and A. Pühler. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram-negative bacteria. Bio/Technology 1:784-791. [Google Scholar]
- 41.Tate, R., A. Riccio, M. Iaccarino, and E. J. Patriarca. 1997. A cysG mutant strain of Rhizobium etli pleiotropically defective in sulfate and nitrate assimilation. J. Bacteriol. 179:7343-7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vincent, J. M. 1970. A manual for the practical study of root nodule bacteria. IBP handbook no. 15. Blackwell Scientific Publications, Oxford, United Kingdom.
- 43.Wanner, B. L. 1996. Phosphorus assimilation and control of the phosphate regulon, p. 1357-1381. In F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, B. Masaganik, W. S. Reznikoff, M. Riley, M. Schaechter, and H. E. Umbarger (ed.), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed. American Society for Microbiology, Washington, D.C.
- 44.Winzeler, E. A., D. D. Shoemaker, A. Astromoff, H. Liang, K. Anderson, B. Andre, R. Bangham, R. Benito, J. D. Boeke, H. Bussey, A. M. Chu, C. Connelly, K. Davis, F. Dietrich, S. W. Dow, M. El Bakkoury, F. Foury, S. H. Friend, E. Gentalen, G. Giaever, J. H. Hegemann, T. Jones, M. Laub, H. Liao, N. Liebundguth, D. J. Lockhart, A. Lucau-Danila, M. Lussier, N. M'Rabet, P. Menard, M. Mittmann, C. Pai, C. Rebischung, J. L. Revuelta, L. Riles, C. J. Roberts, P. Ross-MacDonald, B. Scherens, M. Snyder, S. Sookhai-Mahadeo, R. K. Storms, S. Veronneau, M. Voet, G. Volckaert, T. R. Ward, R. Wysocki, G. S. Yen, K. Yu, K. Zimmermann, P. Philippsen, M. Johnston, and R. W. Davis. 1999. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285:901-906. [DOI] [PubMed] [Google Scholar]
- 45.Yang, Y. H., S. Dudoit, P. Luu, D. M. Lin, V. Peng, J. Ngai, and T. P. Speed. 2002. Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 30:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.