

Abstract
We developed a novel quantitative real-time PCR (Q-PCR) method for the soil actinomycete Rhodococcus equi, an important horse pathogen and emerging human pathogen. Species-specific quantification was achieved by targeting the chromosomal monocopy gene choE, universally conserved in R. equi. The choE Q-PCR included an internal amplification control (IAC) for identification of false negatives. A second Q-PCR targeted the virulence plasmid gene vapA, carried by most horse isolates but infrequently found in isolates from other sources. The choE-IAC and vapA assays were 100% sensitive and specific as determined using 178 R. equi isolates, 77 nontarget bacteria, and a panel of 60 R. equi isolates with known vapA+ and vapA-negative (including vapB+) plasmid genotypes. The vapA+ frequency among isolate types was as follows: horse, 85%; human, 20%; bovine and pig, 0%; others, 27%. The choE-IAC Q-PCR could detect up to one genome equivalent using R. equi DNA or 100 bacteria/ml using DNA extracted from artificially contaminated horse bronchoalveolar lavage (BAL) fluid. Quantification was linear over a 6-log dynamic range down to ≈10 target molecules (or 1,000 CFU/ml BAL fluid) with PCR efficiency E of >0.94. The vapA assay had similar performance but appeared unsuitable for accurate (vapA+) R. equi quantification due to variability in target gene or plasmid copy number (1 to 9). The dual-reaction Q-PCR system here reported offers a useful tool to both medical and veterinary diagnostic laboratories for the quantitative detection of R. equi and (optional) vapA+ “horse-pathogenic” genotype determination.
Rhodococcus equi is a soil-dwelling actinomycete of the mycolata group that causes pyogranulomatous infections in the lungs and other different body locations in a variety of animal hosts. This facultative intracellular parasite is well known in veterinary medicine as the causal agent of foal pneumonia, a severe purulent bronchopneumonic infection with high case-fatality rates. The disease is recognized in many countries as the leading cause of mortality in foals and is a cause of serious concern to the equine industry as it can become endemic in stud farms and there is no effective vaccine for its prevention (9, 23). In recent years R. equi has emerged as an opportunistic human pathogen, especially in individuals infected with human immunodeficiency virus. In the human host the infection presents usually as tuberculosis-like cavitary pneumonia or bacteremia (2, 36). R. equi is also being increasingly reported in other animal species, mainly associated with extrapulmonary, purulent, caseating infections (5, 33). In cattle the organism is typically isolated from chronic retropharyngeal, bronchial, or mediastinal pyogranulomatous lymphadenitis (6) and in pigs from submaxillary lymph nodes (18).
Horse isolates of R. equi typically harbor an 85- to 90-kb virulence plasmid, of which an example has been fully sequenced (11, 32). This plasmid encodes virulence-associated protein A or VapA, a 17.4-kDa surface lipoprotein presumed to be involved in pathogenesis but whose role in the infectious process remains unknown (10, 14). VapA is encoded by the vapA gene, which is present in a plasmidic, 27.5-kb pathogenicity island together with six other vapA homologues (32). In nonhorse R. equi isolates, including human isolates, the VapA protein/vapA gene is much less frequently found than a variant protein/allele designated VapB/vapB. The VapB antigen is structurally and immunologically closely related to VapA but is larger (18.2 to 20 kDa as detected by sodium dodecyl sulfate-polyacrylamide gel electrophoresis immunoblotting) and is encoded by plasmids of various sizes (79 to 100 kb), not yet characterized genetically. The vapB plasmids are not found in equine isolates, suggesting that vapB+ strains are not pathogenic for the horse (22, 31, 34). Except for soil isolates from horse breeding farms, in which vapA-type plasmids are common, environmental isolates of R. equi do not usually carry plasmids, or if they do, these are smaller in size and most often vapA and vapB negative (31).
Laboratory diagnosis of rhodoccocal infections currently relies on classical bacteriological methods involving the isolation of the organism from clinical samples or postmortem material (23). However, these culture-based procedures are lengthy and sometimes lack adequate sensitivity due either to prior antibiotic treatments or, in the case of respiratory specimens (typically bronchoalveolar lavage [BAL] aspirate or sputum), to the presence of multiple bacterial contaminants (28). An added problem is the difficulties posed by the identification due to the micro- and macroscopic morphological variability exhibited by these bacteria and the lack of accuracy of biochemical tests for R. equi species determination (8, 15, 29, 36). There is therefore considerable interest in developing new, simpler tests for the rapid and reliable detection and identification of R. equi for use in both veterinary and medical clinical microbiology laboratories.
Several molecular methods for R. equi have been described based on amplification of DNA sequences by conventional PCR (1, 3, 12, 15, 22, 28, 30). Although comparatively faster than culture-based methods, conventional PCR, however, provides only qualitative results and requires post-PCR procedures. The “open” post-PCR processing of massive amounts of amplicon increases the risk of false-positive results due to sample cross-contamination. This risk is minimized in the real-time PCR technique as the reaction and fluorescent probe-based amplicon detection are brought about simultaneously in a closed tube. Real-time monitoring of the amplification curve via fluorescence emission permits also a much more sensitive detection of positive reactions and at the same time, importantly, an accurate quantification of the target DNA present in the sample (35). However, to date only one quantitative real-time PCR (Q-PCR) assay has been reported for R. equi (13). This assay targets the vapA gene and therefore detects only strains carrying vapA+ plasmids, which as mentioned above are rarely found in human and most other nonhorse R. equi isolates, thus limiting its applicability to the field of equine medicine. Moreover, the quantification accuracy of this assay can be compromised by strain-to-strain differences in plasmid copy number or plasmid DNA extraction efficiency.
We recently identified the R. equi cholesterol oxidase gene choE and demonstrated that this chromosomal locus is universally conserved in these bacteria (21) and is a suitable target for their specific and sensitive detection by conventional PCR (15). Here we report the design and development of a novel dual-reaction Q-PCR method that allows both the species-specific quantification of R. equi and determination of its “horse-associated” subtype via detection of choE and vapA sequences, respectively. The method includes an internal amplification control (IAC) for monitoring the occurrence of false-negative results due to PCR failure or inhibition.
MATERIALS AND METHODS
Bacterial strains, culture media, and growth conditions.
A total of 255 bacterial strains were used in this study: 197 were Rhodococcus spp. (178 R. equi and 19 non-R. equi isolates) and 58 belonged to different actinomycete genera, including cholesterol oxidase-producing species. The R. equi strains included horse (n = 81), human (n = 35), pig (n = 30), bovine (n = 8), soil (n = 13), and ancillary (n = 11, from sheep, goat, dog, cat, pheasant, primate, iguana, and unknown origin) isolates from 14 countries (Argentina, Australia, Brazil, Canada, China, Dominican Republic, Germany, France, Hungary, Ireland, Japan, Slovenia, Spain, and the United Kingdom). All were confirmed as R. equi by analysis of colony morphology, API Coryne biochemical profiling, synergistic hemolysis (CAMP-like) test with Listeria ivanovii (21), and our previously described conventional choE-PCR test (15). R. equi isolate 103S from J. Prescott (University of Guelph, Canada), deposited as PAM 1126 in our collection, was used as the reference strain. This strain was originally isolated from a case of foal pneumonia and is currently being used for the determination of the complete genome sequence of R. equi by the International R. equi Genome Consortium (www.sanger.ac.uk/Projects/R_equi). A detailed list of R. equi isolates is available in the supplemental material as Table S1. The non-R. equi isolates are listed in Table S2 in the supplemental material. Bacteria were maintained at −80°C in a medium containing 2% tryptone, 4% skimmed milk, and 16% glycerol. Rhodococcus spp. were grown at 30°C in brain heart infusion (BHI) and non-Rhodococcus isolates at 37°C in YME medium (0.4% yeast extract, 1% malt extract, 0.4% glucose), supplemented with 1.5% agar for plate cultures. All media were purchased from Oxoid (Hampshire, United Kingdom), except BHI (from Difco-BD, Detroit, MI).
DNA isolation and quantification.
Bacterial genomic DNA was isolated from overnight cultures on solid medium using a cetyltrimethylammonium bromide (CTAB)-based protocol. Bacterial colonies from half a petri dish were collected with a loop, suspended in 1 ml phosphate-buffered saline, pelleted at 4,000 × g for 10 min, and incubated for 1 h at 37°C after resuspension in 567 μl Tris-EDTA buffer and 3 μl 100-mg/ml (30,000 units) lysozyme (Sigma) solution. Subsequently, 30 μl 10% sodium dodecyl sulfate and 3 μl 20-mg/ml (1.8 units) proteinase K (Sigma) was added and the mixture was incubated again for 1 h at 37°C. Then, 170 μl 5 M NaCl, 80 μl CTAB-NaCl solution (10% CTAB in 0.7 M NaCl), and 5 μl 100-mg/ml (50 Kunitz units) RNase A (Sigma) were added followed by a 30-min incubation at 65°C. After cooling to room temperature, the mixture was extracted with phenol-chloroform and chloroform-isoamyl alcohol followed by DNA precipitation with isopropanol and washing with 70% ethanol (27). DNA was resuspended in 100 μl 10 mM Tris-HCl, pH 8.0, and its amount and quality were determined spectrophotometrically by calculating the ratio of optical density at 260 nm to that at 280 nm and visually by agarose gel electrophoresis.
Oligonucleotides.
The oligonucleotide primers and TaqMan probes used in this study were designed using Primer Express 2.0 software (Applied Biosystems, Foster City, CA) and purchased from Metabion AG (Martinsried, Germany). They are listed in Table 1. The IAC probe was labeled with HEX (6-carboxy-2′,4,4′,5′,7,7′-hexachlorofluorescein) and the choE probe with 6-carboxyfluorescein (FAM).
TABLE 1.
Oligonucleotides used in this study
| Target | Oligonucleotide name | Application | Sequence | Tma (°C) | G-C (%) | Reference(s) |
|---|---|---|---|---|---|---|
| choE | reqF | Q-PCR forward primer | 5′-CGA CAA GCG CTC GAT GTG-3′ | 59 | 61 | This study |
| reqR | Q-PCR reverse primer | 5′-TGC CGA AGC CCA TGA AGT-3′ | 59 | 56 | This study | |
| reqP | TaqMan probe | 5′-FAM-TGG CCG ACA AGA CCG ATC AGC C-TAMRAb-3′ | 69 | 64 | This study | |
| COX-F | PCR forward primer | 5′-GTC AAC AAC ATC GAC CAG GCG-3′ | 62.3 | 57.1 | 15 | |
| COX-R | PCR reverse primer | 5′-CGA GCC GTC CAC GAC GTA CAG-3′ | 64.7 | 66.7 | 15 | |
| vapA | RvapA114F | Q-PCR forward primer | 5′-CAG CAG TGC GAT TCT CAA TAG TG-3′ | 59 | 48 | This study |
| RvapA188R | Q-PCR reverse primer | 5′-GAA GTC GTC GAG CTG TCA TAG CT-3′ | 59 | 52 | This study | |
| RvapA140P | TaqMan probe | 5′-FAM-CAG AAC CGA CAA TGC CAC TGC CTG-TAMRA-3′ | 69 | 58 | This study | |
| IP1 | PCR forward primer | 5′-AC TCT TCA CAA GAC GGT-3′ | 46 | 50 | 22, 30 | |
| IP2 | PCR reverse primer | 5′-TAG GCG TTG TGC CAG CTA-3′ | 55.1 | 55.6 | 22, 30 | |
| vapB | H1 | PCR forward primer | 5′-TGA TGA AGG CTC TTC ATA A-3′ | 47.6 | 36.8 | 22 |
| H2 | PCR reverse primer | 5′-TTA TGC AAC CTC CCA GTT G-3′ | 53.2 | 47.4 | 22 | |
| IAC | IACP | TaqMan probe | 5′-HEX-CGC CTG CAA GTC CTA AGA CGC CA-TAMRA-3′ | 68 | 61 | 24 |
| hly | riacF | Forward primer IAC construction | 5′-CGA CAA GCG CTC GAT GTG CAT GGC ACC ACC-3′ | 81 | 63 | This study |
| riacR | Reverse primer IAC construction | 5′-CGA CAA GCG CTC GAT GTG ATC CGC GTG TTT-3′ | 78 | 57 | This study |
Theoretical melting temperature.
TAMRA, 6-carboxytetramethylrhodamine.
IAC construction.
The IAC consisted of a 100-bp chimerical DNA containing a portion of the listeriolysin (hly) gene from Listeria monocytogenes (GenBank accession no. M24199), which we previously validated as a Q-PCR probe target (24), flanked by the R. equi-specific choE gene sequences targeted by reqF and reqR primers (Table 1). This chimeric DNA molecule was generated by two rounds of PCR as previously described (26). The first PCR used 1 ng L. monocytogenes DNA template and primers riacF and riacR (Table 1), which contained the corresponding hly target sequences plus a 5′ tail with the reqF or reqR primer sequence. The second PCR used the purified first-round PCR product (diluted 1:1,000) as a template and the reqF and reqR primers. PCR conditions were as previously described (26). The IAC PCR product was purified using the QIAquick gel extraction kit (QIAGEN, Hilden, Germany), quantified, and diluted to the appropriate concentration in 10 mM Tris-HCl, pH 8.0, in the presence of 500 ng/ml of acetylated bovine serum albumin as a blocking agent to minimize binding of the negatively charged IAC DNA to the plastic microtubes. With the exception of its target sequence in the L. monocytogenes hly gene (nucleotide positions 114 to 177), the IAC did not display significant similarity to any DNA sequence deposited in public databases, as determined by BLAST-N searches (National Center for Biotechnology Information, Bethesda, Md.; http://www.ncbi.nlm.nih.gov). The IAC amplicon, 100 bp in size, was longer than the 68-bp choE-specific amplicon, facilitating the differentiation of the two PCR products by gel electrophoresis.
Q-PCR.
The assays were performed essentially as described previously (24) in 20-μl reaction volumes containing 1× PCR buffer II; 6 mM MgCl2; 200 μM dATP, dCTP, and dGTP; 400 μM dUTP; 300 nM specific primers; 150 nM probe (for the duplex choE-IAC system, 100 nM of IAC probe was added); 1 unit of AmpliTaq Gold DNA polymerase (Applied Biosystems-Roche Molecular Systems Inc., Branchburg, N.J.); 0.2 units of AmpErase uracil N-glycosylase; and 5 μl of the target DNA solution. Reactions were run on an iCycler IQ platform (Bio-Rad Laboratories Inc., Hercules, CA) with the following program: 2 min at 50°C, 10 min at 95°C, and 50 cycles of 15 s at 95°C and 1 min at 60°C. Q-PCR results were analyzed using the Optical System Software v3.0a (Bio-Rad Laboratories Inc., Hercules, CA). Quantification was obtained by interpolation in a standard regression curve of cycle threshold (CT) values generated from samples of known DNA concentrations. One molecule of R. equi DNA, or genome equivalent, corresponds to approximately 5.5 fg of DNA considering a genome size of 5.2 Mb as determined for PAM 1126, according to the following equation: DNA amount in fg = bp × 660 Da/bp × 1.6 × 10−27 kg/Da × 1 × 10−18 fg/kg (25). Q-PCRs with CT values of >50 were considered negative. The 95% confidence interval was calculated for every serial dilution according to a binomial distribution (with the statistical software SPSS 12.0S for Windows v8.0 [SPSS Inc., Chicago, Ill.]) (25, 26). Unless otherwise stated, all reactions were performed in triplicate.
Quantitative detection of R. equi in BAL fluid.
An overnight culture in BHI of R. equi PAM 1126 was centrifuged for 3 min at 3,000 × g, the bacterial pellet was resuspended in sterile phosphate-buffered saline, and the suspension was serially 10-fold diluted in BAL fluid obtained from a healthy adult horse with the use of 0.9% NaCl intravenous infusion solution (Baxter Healthcare Corp.) as vehicle. The concentration of R. equi in the BAL fluid dilutions was determined by standard plate counting. DNA was extracted from R. equi-contaminated BAL dilutions as follows: 1-ml samples were transferred to clean 1.5-ml microtubes and centrifuged for 5 min at 10,000 × g at 4°C; the pellets were resuspended in 100 μl Instagene Matrix suspension (Bio-Rad Laboratories, Hercules, CA) by vortexing, and the suspensions were incubated at 56°C for 20 min; after 10 s of vigorous vortexing, these were incubated at 100°C for 8 min and then placed on ice and centrifuged for 5 min at 14,000 × g at 4°C; finally, 50 μl of the supernatants was transferred to a fresh microtube and stored at −20°C until use. Each PCR used 5 μl of DNA preparation.
RESULTS
Design and optimization of choE- and vapA-specific Q-PCR assays.
To specifically identify R. equi, we used the conserved choE gene (21) as a target. A previously developed conventional PCR assay based on detection of choE sequences was 100% specific and sensitive for R. equi taking as a positive result the expected 959-bp amplicon (15). Using this PCR assay, smaller products are occasionally observed with other rhodococcal species (reference 12 and our unpublished observations). To minimize the risk of unspecific reactions, we identified a new choE target region suitable for Q-PCR primers and probe design by careful analysis of all cholesterol oxidase gene sequences deposited in public databases using the CLUSTALW multiple-alignment tool (European Bioinformatics Institute, EMBL; www.ebi.ac.uk). The new choE primers, reqF and reqR (Table 1), amplify a 100% specific, conserved 68-bp DNA fragment corresponding to positions 938 to 1005 of the coding sequence deposited in GenBank under accession no. AJ242746 (21).
Gene regions suitable for vapA-specific Q-PCR oligonucleotides were selected by visual inspection of CLUSTALW multiple alignments of all known sequences of the vap multigene family. These include, in addition to vapA, six other vap genes (vapC to -H, identified in vapA+ virulence plasmids from strains ATCC 33701 and 103) (32) and vapB identified in plasmids from “nonequine” R. equi isolates (22). Primer pair RvapA114F-RvapA188R amplifies a vapA-specific, conserved 75-bp DNA fragment corresponding to positions 114 to 188 of the gene sequence deposited in GenBank with accession no. NC002576 (32).
The BLAST-N tool v.2.2.12 was used (with default settings and low-complexity filter off) to confirm in silico that none of the selected oligonucleotides recognized any registered DNA sequence other than the target sequence. Primers, TaqMan probes, and MgCl2 concentrations were optimized for Q-PCR assays by using 1 ng of template DNA from R. equi strain PAM 1126. The minimum primer and probe concentrations that gave the lowest CT value and the highest fluorescence intensity were retained as the standard optimal conditions (see Materials and Methods).
Optimization of duplex choE-IAC Q-PCR assay.
The optimal IAC probe concentration (i.e., the minimum concentration not resulting in an increase of CT) (26), 100 nM, was experimentally determined by performing Q-PCRs in the presence of 1,000 IAC molecules, no R. equi DNA, 150 nM FAM-labeled choE probe, and increasing amounts (from 25 to 250 nM) of HEX-labeled IAC probe. Since an excess of IAC may inhibit the target-specific reaction, Q-PCRs were also carried out in the presence of various IAC amounts (10,000, 1,000, 100, and 10 molecules per reaction) and a fixed amount (30 genome equivalents) of R. equi PAM 1126 DNA. The maximum IAC amount with no inhibitory effect on the choE-specific FAM signal was 100 copies.
Specificity and sensitivity of the choE-IAC Q-PCR.
The capacity of the choE Q-PCR assay to discriminate between target and nontarget bacteria was assessed using 1 ng of genomic DNA from 178 R. equi strains from a variety of sources (including clinical isolates from different animal species and environmental isolates), 19 non-R. equi rhodococcal species strains, and 58 strains from 18 different non-Rhodococcus actinomycete genera. The choE Q-PCR assay was 100% sensitive and 100% specific as all 178 R. equi strains tested gave a positive choE signal whereas none of the 77 nontarget bacteria did (detailed results in Table S1 in the supplemental material). Rhodococcus fascians, reported by others (12) to give an unspecific (smaller) amplicon by conventional PCR using our previously described primers (15), did not give any significant signal in the choE Q-PCR assay. All the reactions generated a positive IAC (HEX) signal, ruling out that the absence of choE (FAM) signal observed in non-R. equi isolates was due to failure of the PCR. All the nonactinomycete bacteria that we have tested to date, including a variety of common gram negatives and gram positives, have yielded negative results in the choE Q-PCR assay (not shown).
Specificity and sensitivity of the vapA Q-PCR.
As with the choE PCR, none of the 77 nontarget bacteria (non-Rhodococcus strains and non-R. equi rhodococci) gave a positive amplification signal with the vapA Q-PCR. However, this assay detected the target sequence in only 48% of R. equi isolates (85 out of 178), as expected from the varied composition of the strain panel tested, which contained only a proportion of horse-derived bacteria (see above and Table S1 in the supplemental material). The distribution of vapA+ isolates per animal species is shown in Fig. 1.
FIG. 1.

Distribution of the vapA allele according to isolate origin. The number of isolates within each category is indicated above the bars; within the bars are the percentages of vapA+ (gray section) and vapA-negative (empty section) isolates. “Other” includes sheep, goat, dog, cat, pheasant, primate, iguana, and unknown origin. Most soil isolates are from equine-related environments, explaining the relatively high percentage of vapA+ isolates (31).
The discrimination capacity of the vapA Q-PCR assay was assessed on a selection of 60 R. equi strains using as reference method a previously described dual-reaction conventional PCR system that differentiates vapA+ isolates from vapB-negative or vapA- and vapB-negative isolates using two pairs of primers (22). Prior to applying this method, we confirmed its 100% efficacy on a representative sample of R. equi strains with known vapA/B genotypes (17, 18). As shown in Table 2, there was a perfect concordance between the results obtained by the two techniques. As all the strains tested positive with the choE-IAC system, these results indicated that our vapA Q-PCR assay is 100% specific and 100% sensitive.
TABLE 2.
Specificity and sensitivity of vapA Q-PCRa
| Strain | Origin | Conventional PCR resultb
|
Q-PCR result for vapA | Concordancec | |
|---|---|---|---|---|---|
| vapA | vapB | ||||
| PAM 1126 | Horse | + | − | + | + |
| PAM 1286 | Iguana | − | − | − | + |
| PAM 1335 | Horse | + | − | + | + |
| PAM 1340 | Horse | + | − | + | + |
| PAM 1346 | Horse | + | − | + | + |
| PAM 1348 | Soil | − | − | − | + |
| PAM 1350 | Soil | + | − | + | + |
| PAM 1351 | Soil | + | − | + | + |
| PAM 1358 | Horse | + | − | + | + |
| PAM 1365 | Horse | + | − | + | + |
| PAM 1367 | Horse | + | − | + | + |
| PAM 1371 | Horse | + | − | + | + |
| PAM 1374 | Horse | + | − | + | + |
| PAM 1376 | Human | − | + | − | + |
| PAM 1387 | Unknown | − | − | − | + |
| PAM 1404 | Horse | + | − | + | + |
| PAM 1406 | Human | − | + | − | + |
| PAM 1408 | Horse | + | − | + | + |
| PAM 1410 | Horse | + | − | + | + |
| PAM 1413 | Human | − | + | − | + |
| PAM 1414 | Human | − | + | − | + |
| PAM 1415 | Human | − | − | − | + |
| PAM 1416 | Horse | + | − | + | + |
| PAM 1418 | Horse | + | − | + | + |
| PAM 1422 | Horse | + | − | + | + |
| PAM 1424 | Horse | + | − | + | + |
| PAM 1425 | Horse | + | − | + | + |
| PAM 1427 | Horse | + | − | + | + |
| PAM 1430 | Horse | + | − | + | + |
| PAM 1431 | Horse | + | − | + | + |
| PAM 1436 | Soil | + | − | + | + |
| PAM 1437 | Horse | + | − | + | + |
| PAM 1441 | Soil | − | − | − | + |
| PAM 1447 | Pig | − | + | − | + |
| PAM 1448 | Human | − | + | − | + |
| PAM 1453 | Horse | + | − | + | + |
| PAM 1463 | Human | − | − | − | + |
| PAM 1467 | Pig | − | + | − | + |
| PAM 1468 | Pig | − | − | − | + |
| PAM 1469 | Pig | − | + | − | + |
| PAM 1473 | Pig | − | + | − | + |
| PAM 1474 | Pig | − | + | − | + |
| PAM 1475 | Pig | − | + | − | + |
| PAM 1479 | Pig | − | + | − | + |
| PAM 1480 | Pig | − | + | − | + |
| PAM 1483 | Pig | − | − | − | + |
| PAM 1485 | Pig | − | − | − | + |
| PAM 1487 | Pig | − | − | − | + |
| PAM 1488 | Pig | − | − | − | + |
| PAM 1493 | Pig | − | + | − | + |
| PAM 1495 | Pig | − | + | − | + |
| PAM 1499 | Pig | − | − | − | + |
| PAM 1500 | Pig | − | + | − | + |
| PAM 1504 | Pig | − | − | − | + |
| PAM 1518 | Pig | − | − | − | + |
| PAM 1533 | Pig | − | − | − | + |
| PAM 1547 | Pig | − | − | − | + |
| PAM 1549 | Pig | − | − | − | + |
| PAM 1550 | Pig | − | − | − | + |
| PAM 1563 | Bovine | − | − | − | + |
Note that vapA+ and vapB+ scores are mutually exclusive, indicating that vapB is most likely an allelic variant of vapA. +, positive; −, negative.
Concordance between results of vapA conventional PCR and our vapA Q-PCR.
Detection and quantification limits of the choE-IAC and vapA Q-PCR assays.
The detection and quantification limits of the developed PCR assays were determined by using R. equi PAM 1126 genomic DNA. Amplification reactions were performed with a range of DNA concentrations equivalent to approximately 1 × 106, 1 × 105, 1 × 104, 1 × 103, 1 × 102, 10, and 1 target molecule. Figure S1 in the supplemental material illustrates typical amplification profiles and the regression curves obtained with each Q-PCR assay; Table 3 shows the mean CT values for a total of nine replicates in three independent experiments. The two Q-PCR assays yielded similar results in terms of absolute detection values. Positive amplification in all nine replicates of each DNA dilution was achieved when 10 or more target molecules were present, and as few as one target molecule could be detected with 67 to 78% probability for choE- and vapA-based Q-PCR assays, respectively (Table 3). The slopes of the linear regression curves calculated over a 6-log range were similar to the theoretical optimum of −3.32 (26) (choE, −3.337; choE-IAC duplex reaction, −3.335; vapA, −3.379) and showed that the amplifications were very efficient (E = 0.994 ± 0.001, 0.995 ± 0.002, and 0.977 ± 0.002 for choE-, choE-IAC-, and vapA-based Q-PCR assays, respectively). Moreover, R2 values were above the optimal 0.995 (0.998 for choE and choE-IAC reactions, 0.999 for vapA reaction), indicating that the Q-PCRs that we developed are appropriately linear. The confidence intervals based on the standard deviations of CT values did not overlap each other down to 10 target molecules, indicating that reliable quantification was possible above this limit.
TABLE 3.
Determination of the detection and quantification limits of R. equi choE, choE-IAC, and vapA Q-PCR assaysa
| Approx no. of genome eq/reaction | Confidence interval limitb
|
Signal ratio for assayc:
|
CT for assayd:
|
|||||
|---|---|---|---|---|---|---|---|---|
| Lower | Upper | choE | choE-IAC | vapA | choE | choE-IAC | vapA | |
| 1 × 106 | 997,600 | 1,003,300 | 9/9 | 9/9 | 9/9 | 19.83 ± 0.02 | 19.90 ± 0.04 | 18.80 ± 0.02 |
| 1 × 105 | 99,643 | 100,358 | 9/9 | 9/9 | 9/9 | 23.32 ± 0.05 | 23.40 ± 0.06 | 22.25 ± 0.06 |
| 1 × 104 | 9,887 | 10,113 | 9/9 | 9/9 | 9/9 | 26.05 ± 0.02 | 26.01 ± 0.03 | 25.60 ± 0.03 |
| 1 × 103 | 964 | 1,036 | 9/9 | 9/9 | 9/9 | 29.50 ± 0.02 | 29.60 ± 0.04 | 29.00 ± 0.03 |
| 1 × 102 | 89 | 111 | 9/9 | 9/9 | 9/9 | 32.88 ± 0.03 | 32.95 ± 0.05 | 32.20 ± 0.04 |
| 1 × 101 | 7 | 14 | 9/9 | 9/9 | 9/9 | 36.77 ± 0.15 | 36.80 ± 0.14 | 35.80 ± 0.12 |
| 1 | 0 | 2 | 6/9 | 6/9 | 7/9 | 38.38 ± 0.41 | 38.70 ± 0.34 | 38.20 ± 0.29 |
Nontemplate controls were negative in the three Q-PCR assays (CT values of >50 in all the replicates).
Calculated for the expected number of template molecules at each dilution at 95% confidence level.
Number of positive results out of nine reactions.
Cycle number at which fluorescence intensity equals a fixed threshold. Mean values ± standard errors of the means were calculated with a prefixed threshold of 200. Differences in CT values were statistically significant with P < 0.05.
Quantitative detection of R. equi in BAL fluid.
We assessed the applicability of the choE-IAC Q-PCR for the quantitative detection of R. equi bacteria in artificially contaminated BAL fluid. Taking into consideration that 100 μl of DNA was obtained from the processing of 1 ml of BAL fluid, and that 5 μl of DNA preparation was used for the PCR, our choE-IAC assay consistently detected down to approximately 100 R. equi cells/ml or 5 genomic units per reaction (Table 4). Quantitative amplification parameters were optimal, with linearity (R2) above 0.99 down to 1 × 103 R. equi CFU/ml and overall PCR efficiency of 0.94. Relative accuracy values (24, 27) ranged between 88.95% and 113.53%, indicating a high degree of correspondence between the quantitative results obtained by the reference method (number of R. equi CFU/ml as determined by plate counting) and the results obtained by the choE Q-PCR method (Table 4).
TABLE 4.
Quantitative detection of R. equi bacteria in BAL fluida
| Approx R. equi CFU/ml | Approx no. of R. equi genome eq/reactionb | Signal ratioc | CTd | Relative accuracye |
|---|---|---|---|---|
| 1 × 107 | 5 × 105 | 9/9 | 20.67 ± 0.10 | 100.05 |
| 1 × 106 | 5 × 104 | 9/9 | 24.10 ± 0.10 | 103.59 |
| 1 × 105 | 5 × 103 | 9/9 | 27.82 ± 0.04 | 88.95 |
| 1 × 104 | 5 × 102 | 9/9 | 30.93 ± 0.13 | 113.53 |
| 1 × 103 | 5 × 101 | 9/9 | 34.68 ± 0.20 | 95.57 |
| 1 × 102 | 5 × 100 | 9/9 | 37.90 ± 0.21 | NAf |
| 1 × 101 | 5 × 10−1 | 0/9 | >50 | NA |
| 1 | 5 × 10−2 | 0/9 | >50 | NA |
| 0g | 0 | 0/9 | >50 | NA |
Results from three independent experiments with three replicates each. Overall efficiency E is 0.94, and linearity R2 is 0.99.
Estimated number of R. equi genome equivalents in each PCR run assuming 100% DNA extraction efficiency (each reaction mixture contained 5 μl of a DNA preparation of 100 μl extracted from 1 ml BAL fluid).
Number of positive results out of nine reactions.
As defined in Table 3, footnote d.
Degree of correspondence between the quantitative results obtained by standard plate counting (R. equi CFU/ml) and the results obtained by the choE Q-PCR method (27, 29).
NA, not applicable.
Noncontaminated BAL fluid.
DISCUSSION
We describe here a Q-PCR method that permits the sensitive and specific, accurate quantitative detection of the pathogenic actinomycete R. equi. This is achieved by targeting sequences from the chromosomal choE gene, previously identified in our laboratory and shown to provide a useful marker for the molecular detection and identification of R. equi (15, 21). A previous Q-PCR assay for R. equi, recently reported by others (14), targets the plasmidic gene vapA and thus detects only R. equi bacteria carrying this allelic variant. vapA+ strains are associated with infections in the horse and hence are predominantly found in equine-associated specimens. However, R. equi can be isolated from a variety of other animal species in which, with few exceptions, vapA+ strains are rarely found (17, 19, 22, 34) (Fig. 1). Indeed, our data show that only a small proportion (20%) of human clinical isolates are vapA+, consistent with previously reported figures on the prevalence of VapA+/vapA+ R. equi bacteria in human specimens (12, 22, 34). The vapA+ plasmid genotype appears to be particularly rare among bovine and pig isolates, as shown by our data (0% positives; Fig. 1) and other studies (6, 17). Overall, more than 50% of the isolates tested in this study were vapA negative (Fig. 1), clearly showing that a vapA-only-based detection assay misses a very significant proportion of common R. equi strains. Importantly, 15% of the horse clinical isolates included in our study were vapA negative (Fig. 1), questioning the value of vapA as a sensitive molecular diagnostic marker for R. equi even if its application is restricted to equine specimens.
Besides being universally highly conserved in R. equi, the choE gene offers also the advantage that it is present on the chromosome in monocopy (21), thus permitting an accurate quantification of the genomic units present in a sample. In contrast, the detection (and quantification in terms of genome equivalents) of a plasmidic gene, as is vapA, relies on the efficiency of plasmid DNA extraction and, critically, also on the number of copies of the plasmid carried by each individual strain. From the CT values obtained for choE (control monocopy gene) and vapA we have estimated that 39% of the isolates contained two or more plasmid copies per genome (Table 5), indicating that Q-PCR data based on vapA would overestimate on a significant number of occasions the R. equi bacterial load present in the sample by at least a factor of two.
TABLE 5.
Numbers of copies of the vapA gene in R. equi isolatesa
| Avg no. of copies of vapA for value type | % of isolates | |
|---|---|---|
| Exptl continuous range | Theoretical discrete | |
| 0.57b-1.49 | 1 | 61.2 |
| 1.50-2.49 | 2 | 25.9 |
| 2.50-3.49 | 3 | 5.9 |
| 3.50-4.49 | 4 | 0 |
| 4.50-5.49 | 5 | 1.2 |
| 5.50-6.49 | 6 | 1.2 |
| 6.50-7.49 | 7 | 2.4 |
| 7.50-8.49 | 8 | 0 |
| 8.50-9.49 | 9 | 1.2 |
| Total | 100 | |
Estimated from Q-PCR results for choE and vapA in vapA+ isolates (at least three replicates per reaction per isolate) using 1 ng of DNA and the following formula: number of vapA copies = 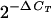 , where ΔCT = CTvapA − CTchoE, assuming 100% plasmid DNA extraction efficiency relative to chromosomal DNA. This calculation is possible because the PCR efficiencies for both targets, 0.977 and 0.995, were close to the optimal value E = 1, meaning that duplication of each amplicon occurs in each cycle across a wide linear range. The coefficient of variance was 7% and 11% for choE and vapA Q-PCR systems, respectively.
, where ΔCT = CTvapA − CTchoE, assuming 100% plasmid DNA extraction efficiency relative to chromosomal DNA. This calculation is possible because the PCR efficiencies for both targets, 0.977 and 0.995, were close to the optimal value E = 1, meaning that duplication of each amplicon occurs in each cycle across a wide linear range. The coefficient of variance was 7% and 11% for choE and vapA Q-PCR systems, respectively.
Minimum value obtained (see Table S1 in the supplemental material).
Although vapA is clearly unsuitable as a target for the species-specific quantitative detection of R. equi by Q-PCR, it is indisputable that this gene has diagnostic value as a predictor of horse pathogenicity (31). Moreover, horse isolates are quantitatively the most significant component of R. equi epidemiology. Indeed, this fully justifies the incorporation of vapA detection capabilities in any diagnostic method targeting R. equi. Bearing this in mind, we designed our Q-PCR method for R. equi as a dual-reaction system with independent assays for choE and vapA, the former as an entry-level, primary test aiming at the quantitative detection of R. equi species, and the latter as a complementary, optional test for vapA+ genotype determination. This modular design provides full flexibility as the vapA Q-PCR assay will not always be needed, in particular in medical (human) microbiology laboratories, due to its specific relevance for the horse. It also helps avoid possible problems of loss of analytical performance, as is sometimes seen in multiplex PCR assays (7, 16). This is important as the primary choE Q-PCR test includes an IAC, thus being already de facto a duplex-format assay.
A major limitation to the application of Q-PCR-based tests in diagnostic laboratories is the relatively common occurrence of false-negative results due to the presence of PCR inhibitors in the sample. To tackle this problem, we included an IAC in our choE Q-PCR assay. An IAC consists of a nontarget DNA fragment that is coamplified with the target sequence, preferably with the same primers used for the test reaction (4, 26). To achieve this, we constructed the IAC by fusing the forward and reverse choE target sequences to both ends of an unrelated DNA fragment to which a second fluorescent probe (the IAC probe) hybridized. The use of two differently labeled fluorescent probes in the same reaction permitted the simultaneous detection/quantification of the target DNA and assessment of PCR efficiency. The inclusion of an IAC did not have any significant impact on the performance of the choE Q-PCR assay (Table 3).
A critical aspect in the design of molecular diagnostic methods for microbial pathogens is achieving low detection and quantification limits. This goal is of particular interest in the case of R. equi. Indeed, rhodococcal foal pneumonia initially follows an insidious course (9, 23), and it has been suggested that accurate detection (and quantification) of low levels of R. equi in respiratory specimens during the “silent” phase of infection, before the development of gross lesions in the lungs and the manifestation of clinical symptoms, could be of diagnostic value (13, 20). On purified DNA, our choE Q-PCR assay could detect approximately one target genome equivalent in at least 67% of the replicates and 10 genome equivalents in all cases. Accurate quantification, with excellent linearity (R2 = 0.998) and efficiency (E = 0.994 without IAC and 0.995 with IAC), was possible down to ≈10 R. equi genome equivalents per reaction. A similar performance was achieved when the technique was applied to the enumeration of bacterial cells in BAL fluid, a specimen commonly used in the clinical diagnosis of R. equi pneumonia (9, 28). Here, due to the DNA extraction protocol used, which involved processing of 1-ml BAL fluid samples, and the inclusion per reaction of a fraction of DNA extract (5 out of 100 μl), the practical detection limit for R. equi bacteria was 100 CFU/ml (or 5 genome equivalents per reaction). It should be possible to lower this detection limit by refining the sample processing so that DNA is extracted from larger specimen amounts and recovered in smaller volumes. Moreover, accurate determination of horse-pathogenic (i.e., vapA+) R. equi numbers in environmental samples or in fecal or nasal specimens could also provide a predictive tool to assess the risk of R. equi clinical infection in stud farms where the organism is endemic. The optimal performance parameters of our vapA Q-PCR (R2 = 0.999, E = 0.977) indicate that this assay is suitable for this purpose.
In conclusion, the dual-reaction Q-PCR method that we have developed for the rapid (results in less than 2 h), species-specific quantitative monitoring and determination of the vapA+ (equine) subtype of R. equi provides a diagnostic tool potentially very useful in both medical and veterinary diagnostic laboratories.
Supplementary Material
Acknowledgments
We thank all those who kindly provided us with the R. equi strains used in this study (J. Agüero, Spain; T. Buckley, Ireland; R. Callejo, Argentina; A. Caterino-de-Araujo, Brazil; T. Chakraborty, Germany; V. García, Dominican Republic; J. M. García-Arenzana, Spain; A. Enríquez, Spain; A. Kodjo, France; J. L. Hernández, Spain; P. Martin-Rabadán, Spain; A. Martín-Sánchez, Spain; A. Morton, Australia; M. Pate, Slovenia; S. Petry, France; J. F. Prescott, Canada; S. Ricketts, United Kingdom; F. Quigley, Ireland; I. Simarro, Spain; S. Takai, Japan). Thanks are also due to D. Leadon for stimulating and constant cooperation, C. Helps for help and advice on the iCycler machine, and A.-L. Cohen for help in the identification and curation of R. equi isolates.
This work was supported by a Leverhulme grant from the Institute of Advanced Studies, University of Bristol. D.R.-L. is a fellow of the European Union's Marie-Curie mobility program.
Footnotes
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Arriaga, J. M., N. D. Cohen, J. N. Derr, M. K. Chaffin, and R. J. Martens. 2002. Detection of Rhodococcus equi by polymerase chain reaction using species-specific nonproprietary primers. J. Vet. Diagn. Investig. 14:347-353. [DOI] [PubMed] [Google Scholar]
- 2.Arya, B., S. Hussian, and S. Hariharan. 2004. Rhodococcus equi pneumonia in a renal transplant patient: a case report and review of literature. Clin. Transplant. 18:748-752. [DOI] [PubMed] [Google Scholar]
- 3.Bell, K. S., J. C. Philp, N. Christofi, and D. W. Aw. 1996. Identification of Rhodococcus equi using the polymerase chain reaction. Lett. Appl. Microbiol. 23:72-74. [DOI] [PubMed] [Google Scholar]
- 4.Cone, R. W., A. C. Hobson, and M. L. Huang. 1992. Coamplified positive control detects inhibition of polymerase chain reactions. J. Clin. Microbiol. 30:3185-3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davis, W. P., B. A. Steficek, G. L. Watson, B. Yamini, H. Madarame, S. Takai, and J. A. Render. 1999. Disseminated Rhodococcus equi infection in two goats. Vet. Pathol. 36:336-339. [DOI] [PubMed] [Google Scholar]
- 6.Flynn, O., F. Quigley, E. Costello, D. O'Grady, A. Gogarty, J. McGuirk, and S. Takai. 2001. Virulence-associated protein characterisation of Rhodococcus equi isolated from bovine lymph nodes. Vet. Microbiol. 78:221-228. [DOI] [PubMed] [Google Scholar]
- 7.Foy, C. A., and H. C. Parkes. 2001. Emerging homogeneous DNA-based technologies in the clinical laboratory. Clin. Chem. 47:990-1000. [PubMed] [Google Scholar]
- 8.Funke, G., F. N. Renaud, J. Freney, and P. Riegel. 1997. Multicenter evaluation of the updated and extended API (RAPID) Coryne database 2.0. J. Clin. Microbiol. 35:3122-3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giguère, S., and J. F. Prescott. 1997. Clinical manifestations, diagnosis, treatment, and prevention of Rhodococcus equi infections in foals. Vet. Microbiol. 56:313-334. [DOI] [PubMed] [Google Scholar]
- 10.Giguère, S., M. K. Hondalus, J. A. Yager, P. Darrah, D. M. Mosser, and J. F. Prescott. 1999. Role of the 85-kilobase plasmid and plasmid-encoded virulence-associated protein A in intracellular survival and virulence of Rhodococcus equi. Infect. Immun. 67:3548-3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haites, R. E., G. Muscatello, A. P. Begg, and G. F. Browning. 1997. Prevalence of the virulence-associated gene of Rhodococcus equi in isolates from infected foals. J. Clin. Microbiol. 35:1642-1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Halbert, N. D., R. A. Reitzel, R. J. Martens, and N. D. Cohen. 2005. Evaluation of a multiplex polymerase chain reaction assay for simultaneous detection of Rhodococcus equi and the vapA gene. Am. J. Vet. Res. 66:1380-1385. [DOI] [PubMed] [Google Scholar]
- 13.Harrington, J. R., M. C. Golding, R. J. Martens, N. D. Halbert, and N. D. Cohen. 2005. Evaluation of a real-time quantitative polymerase chain reaction assay for detection and quantitation of virulent Rhodococcus equi. Am. J. Vet. Res. 66:755-761. [DOI] [PubMed] [Google Scholar]
- 14.Jain, S., B. R. Bloom, and M. K. Hondalus. 2003. Deletion of vapA encoding virulence associated protein A attenuates the intracellular actinomycete Rhodococcus equi. Mol. Microbiol. 50:115-128. [DOI] [PubMed] [Google Scholar]
- 15.Ladrón, N., M. Fernández, J. Aguero, B. González Zorn, J. A. Vázquez-Boland, and J. Navas. 2003. Rapid identification of Rhodococcus equi by a PCR assay targeting the choE gene. J. Clin. Microbiol. 41:3241-3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mackay, I. M. 2004. Real-time PCR in the microbiology laboratory. Clin. Microbiol. Infect. 10:190-212. [DOI] [PubMed] [Google Scholar]
- 17.Makrai, L., S. Takai, M. Tamura, A. Tsukamoto, R. Sekimoto, Y. Sasaki, T. Kakuda, S. Tsubaki, J. Varga, L. Fodor, N. Solymosi, and A. Major. 2002. Characterization of virulence plasmid types in Rhodococcus equi isolates from foals, pigs, humans and soil in Hungary. Vet. Microbiol. 88:3777-3784. [DOI] [PubMed] [Google Scholar]
- 18.Makrai, L., S. Takayama, B. Denes, I. Hajtos, Y. Sasaki, T. Kakuda, S. Tsubaki, A. Major, L. Fodor, J. Varga, and S. Takai. 2005. Characterization of virulence plasmids and serotyping of Rhodococcus equi isolates from submaxillary lymph nodes of pigs in Hungary. J. Clin. Microbiol. 43:1246-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morton, A. C., A. P. Begg, G. A. Anderson, S. Takai, C. Lämmler, and G. F. Browning. 2001. Epidemiology of Rhodococcus equi strains on thoroughbred horse farms. Appl. Environ. Microbiol. 67:2167-2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muscatello, G., and G. F. Browning. 2004. Identification and differentiation of avirulent and virulent Rhodococcus equi using selective media and colony blotting DNA hybridization to determine their concentrations in the environment. Vet. Microbiol. 100:121-127. [DOI] [PubMed] [Google Scholar]
- 21.Navas, J., B. Gonzalez-Zorn, N. Ladrón, P. Garrido, and J. A. Vazquez-Boland. 2001. Identification and mutagenesis by allelic exchange of choE, encoding a cholesterol oxidase from the intracellular pathogen Rhodococcus equi. J. Bacteriol. 183:4796-4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oldfield, C., H. Bonella, L. Renwick, H. I. Dodson, G. Alderson, and M. Goodfellow. 2004. Rapid determination of vapA/vapB genotype in Rhodococcus equi using a differential polymerase chain reaction method. Antonie Leeuwenhoek 85:317-326. [DOI] [PubMed] [Google Scholar]
- 23.Prescott, J. F. 1991. Rhodococcus equi: an animal and human pathogen. Clin. Microbiol. Rev. 4:20-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodríguez-Lázaro, D., M. Hernández, M. Scortti, T. Esteve, J. A. Vázquez-Boland, and M. Pla. 2004. Quantitative detection of Listeria monocytogenes and Listeria innocua by real-time PCR: assessment of hly, iap, and lin02483 targets and AmpliFluor technology. Appl. Environ. Microbiol. 70:1366-1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodríguez-Lázaro, D., M. D'Agostino, A. Herrewegh, M. Pla, N. Cook, and J. Ikonomopoulos. 2005. Real-time PCR-based methods for quantitative detection of Mycobacterium avium subsp. paratuberculosis in water and milk. Int. J. Food Microbiol. 101:93-104. [DOI] [PubMed] [Google Scholar]
- 26.Rodríguez-Lázaro, D., M. Pla, M. Scortti, H. J. Monzó, and J. A. Vázquez-Boland. 2005. A novel real-time PCR for Listeria monocytogenes that monitors analytical performance via an internal amplification control. Appl. Environ. Microbiol. 71:9008-9012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sambrook, J., and R. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 28.Sellon, D. C., T. E. Besser, S. L. Vivrette, and R. S. McConnico. 2001. Comparison of nucleic acid amplification, serology, and microbiologic culture for diagnosis of Rhodococcus equi pneumonia in foals. J. Clin. Microbiol. 39:1289-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Soto, A., J. Zapardiel, and F. Soriano. 1994. Evaluation of API Coryne system for identifying coryneform bacteria. J. Clin. Pathol. 47:756-759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takai, S., T. Ikeda, Y. Sasaki, Y. Watanabe, T. Ozawa, S. Tsubaki, and T. Sekizaki. 1995. Identification of virulent Rhodococcus equi by amplification of gene coding for 15- to 17-kilodalton antigens. J. Clin. Microbiol. 33:1624-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takai, S. 1997. Epidemiology of Rhodococcus equi infections: a review. Vet. Microbiol. 56:167-176. [DOI] [PubMed] [Google Scholar]
- 32.Takai, S., S. A. Hines, T. Sekizaki, V. M. Nicholson, D. A. Alperin, M. Osaki, D. Takamatsu, M. Nakamura, K. Suzuki, N. Ogino, T. Kakuda, H. Dan, and J. F. Prescott. 2000. DNA sequence and comparison of virulence plasmids from Rhodococcus equi ATCC 33701 and 103. Infect. Immun. 68:6840-6847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takai, S., R. J. Martens., A. Julian., M. G. Riberio., M. Rodrigues de Farias., Y. Sasaki., K. Inuzuka., T. Kakuda., S. Tsubaki, and J. F. Prescott. 2003. Virulence of Rhodococcus equi isolated from cats and dogs. J. Clin. Microbiol. 41:4468-4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takai, S., P. Tharavichitkul, P. Takarn, B. Khantawa, M. Tamura, A. Tsukamoto, S. Takayama, N. Yamatoda, A. Kimura, Y. Sasaki, T. Kakuda, S. Tsubaki, N. Maneekarn, T. Sirisanthana, and T. Kirikae. 2003. Molecular epidemiology of Rhodococcus equi of intermediate virulence isolated from patients with and without acquired immune deficiency syndrome in Chiang Mai, Thailand. J. Infect. Dis. 188:1717-1723. [DOI] [PubMed] [Google Scholar]
- 35.Walker, N. 2002. A technique whose time has come. Science 296:557-559. [DOI] [PubMed] [Google Scholar]
- 36.Weinstock, D. M., and A. E. Brown. 2002. Rhodococcus equi: an emerging pathogen. Clin. Infect. Dis. 34:1379-1385. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.