

Abstract
Zearalenones are produced by several Fusarium species and can cause reproductive problems in animals. Some aurofusarin mutants of Fusarium pseudograminearum produce elevated levels of zearalenone (ZON), one of the estrogenic mycotoxins comprising the zearalenones. An analysis of transcripts from polyketide synthase genes identified in the Fusarium graminearum database was carried out for these mutants. PKS4 was the only gene with an enoyl reductase domain that had a higher level of transcription in the aurofusarin mutants than in the wild type. An Agrobacterium tumefaciens-mediated transformation protocol was used to replace the central part of the PKS4 gene with a hygB resistance gene through double homologous recombination in an F. graminearum strain producing a high level of ZON. PCR and Southern analysis of transformants were used to identify isolates with single insertional replacements of PKS4. High-performance liquid chromatography analysis showed that the PKS4 replacement mutant did not produce ZON. Thus, PKS4 encodes an enzyme required for the production of ZON in F. graminearum. Barley root infection studies revealed no alteration in the pathogenicity of the PKS4 mutant compared to the pathogenicity of the wild type. The expression of PKS13, which is located in the same cluster as PKS4, decreased dramatically in the mutant, while transcription of PKS4 was unchanged. This differential expression may indicate that ZON or its derivatives do not regulate expression of PKS4 and that the PKS4-encoded protein or its product stimulates expression of PKS13. Furthermore, both the lack of aurofusarin and ZON influenced the expression of other polyketide synthases, demonstrating that one polyketide can influence the expression of others.
The genus Fusarium contains some of the most important toxigenic plant-pathogenic fungal species. These species are adapted to different ecological niches all over the world as saprophytes and as pathogens that have wide plant host ranges (36, 42). Fusarium graminearum (teleomorph, Gibberella zeae) is a devastating pathogen of cereals and causes Fusarium head blight (FHB), also known as scab, on wheat and barley (14, 42) and ear rot of maize (27). In the last decade FHB has reached epidemic proportions in the United States and has resulted in yield losses and price reductions due to inferior grain quality (30, 56). The economic damage caused by FHB includes reduced yields, discolored, shriveled “tombstone” kernels, mycotoxin contamination, and reduced seed quality (30).
Fusarium-infected grains often are contaminated with mycotoxins, such as trichothecenes, fumonisins, and zearalenones, which makes them unsuitable for use as food and feed (30). Directives from the European Union concerning Fusarium toxins in food set the maximum allowable level for zearalenone (ZON), one of the estrogenic mycotoxins comprising the zearalenones, at 100 ppb for unprocessed cereals other than corn beginning in July 2006 (GAIN report E35115; http://www.fas.usda.gov/gainfiles/200506/146130053.pdf). ZON is a major concern because it has estrogenic activity and can be a significant contaminant of maize, barley, wheat, and other cereals (15, 53). It causes hyperestrogenism, especially in pigs, and reproductive problems in experimental animals and livestock (8). ZON has several derivatives, such as α-zearalenol, that have greater estrogenic activity than ZON (47). ZON is produced by several Fusarium species, including F. graminearum, F. culmorum, F. cerealis (synonym, F. crookwellense), F. equiseti, F. semitectum (53), and F. pseudograminearum (5). There have also been reports of ZON production by other Fusarium species, but their accuracy has been questioned (53), as Fusarium taxonomy is complex and chemical analyses are not always clear-cut (29, 51).
ZON is produced by the acetate-polymalonate pathway and is a polyketide that is synthesized entirely from acetate-malonate units (9). Fungal polyketide synthases (PKSs) are large multidomain enzymes (type I PKSs) with an iterative function (4, 19, 20). A minimal PKS consists of the following domains: β-ketoacyl synthase, acyl transferase, and acyl carrier protein. ZON is a fully reduced macrolide, and a PKS involved in the biosynthesis of ZON probably also must have keto reductase, dehydratase, and enoyl reductase (ER) catabolic domains (9). Since the ER domain performs the last reduction step and is always found together with keto reductase and dehydratase domains, the most likely candidate for the biosynthesis of ZON is a PKS with an ER domain.
The main objectives of this study were to identify and characterize a PKS required for ZON production in Fusarium and to evaluate the importance of ZON for fungal virulence and development. Characterization of the genes and regulatory elements required for the biosynthesis of secondary metabolites may help workers identify factors that influence toxin production and lead to protocols for the detection of toxin-producing strains or to the development of strategies for reducing toxin contamination.
MATERIALS AND METHODS
Strains and culture conditions.
F. graminearum 1104-14 (24) was obtained from the Fusarium collection at the National Veterinary Institute in Norway, and F. pseudograminearum IBT 1544 was obtained from the IBT culture collection at the Technical University of Denmark. Two aurofusarin-deficient F. pseudograminearum mutants, PK2.1 and KHt62.1, were used (28). All strains were maintained as conidial suspensions (1 × 107 spores ml−1) in 20% glycerol at −80°C or in sterile soil.
For genomic DNA extraction, the fungi were cultured with shaking at 28°C in liquid complete medium (25). For RNA isolation, the F. pseudograminearum strains were cultivated on the medium described by Bell et al. (2) with maltose (20 g/liter) as the C source and urea (140 mg/liter) as the N source for 2 weeks at room temperature. For RNA isolation and high-performance liquid chromatography (HPLC) analysis of ZON production, F. graminearum strains were cultivated on polished parboiled rice at 25°C in the dark for up to 2 weeks; 20 g of rice (Uncle Ben's long-grain white rice) and 20 ml distilled H2O were mixed in a 500-ml Erlenmeyer flask, left at room temperature for 1 h, and then autoclaved at 121°C for 1 h. Each flask was inoculated with 1 × 106 spores. For real-time gene expression studies the fungal mycelia were harvested after 4, 7, 11, and 14 days of growth, and for each strain and incubation time three replicate flasks were used. Escherichia coli strains Top10 (Invitrogen, Carlsbad, Calif.) and JM109 (Promega, Madison, Wis.) were used for plasmid manipulations and propagation. E. coli was grown at 37°C in liquid Luria-Bertani (LB) medium or on LB agar with or without 50 ppm kanamycin (Sigma, St. Louis, MO) and 5-bromo-4-chloro-3-indolyl-β-d-galactopyranosie (X-Gal)/isopropyl-β-d-thiogalactopyranoside (IPTG) (Invitrogen) as appropriate (46). Agrobacterium tumefaciens LBA4404 carrying pAL4404, a disarmed version of the octopine-type Ti plasmid pTiAch5 (18), was purchased from Invitrogen.
DNA procedures.
Fungal genomic DNA was extracted essentially as described by Malz et al. (28). The fungal genomic DNA used for Southern analysis of ΔPKS4 mutants was extracted by the method of Rolland et al. (45). For PCR screening of mutants, DNA extraction was performed with microwave treatment of fungal samples (50). PCR was carried out with a GeneAmp PCR 9700 system (Applied Biosystems, Foster City, Calif.) with Dynazyme II (Finnzymes, Espoo, Finland) DNA polymerase. Primers were designed on the basis of the genome sequence determined by the Broad Institute (http://www.broad.mit.edu), the annotated MIPS F. graminearum genome database (31) (http://mips.gsf.de/genre/proj/fusarium/), the program SEARCHPKS (58), and the previously published PKS gene sequences of Kroken et al. (23). All primers used in this work are listed in Table 1.
TABLE 1.
PCR primersa
| Primer | Sequence (5′→3′) | Gene |
|---|---|---|
| Reverse transcriptase PCR primers | ||
| PKS1-1 | GCGCAGAGCTGAAGTATTTGTTGTCG | FG10548 |
| PKS1-2 | AAATGCGTCTTGTAGGTGGGTGGAGGTAAA | |
| PKS2-1 | GGCCTACGAACACCTGCTGACATACT | FG04694 |
| PKS2-2 | GCGCCTCGACGACCTCTGC | |
| PKS3-1 | TCGACGCATCCATCACCAACATC | FG12125 |
| PKS3-2 | AGCGGCGTATTCCCCAAGACTG | |
| PKS4-1 | AGATGGCCATGGTGCTTCGTGAT | FG12055 |
| PKS4-2 | GTGGGCTTCGCTAGACCGTGAGTT | |
| PKS5-1 | ACAGCCCGACATCCCACACTCAG | FG05794 |
| PKS5-2 | TGCCAATCTACGTTCCAAGGGTCAC | |
| PKS6-1 | CCCAGTGCCGGTGCTTGTGA | FG12109 |
| PKS6-2 | GCGCTGATAACCTTCGGCTACCTC | |
| PKS7-1 | ATCAGGGGCATCCAGCAAATCC | FG08795 |
| PKS7-2 | CCCAGCACAGGCCCAAGGTC | |
| PKS9-1 | GTCGATACCAATACGCCAATGCCTGATA | FG12121 |
| PKS9-2 | ATGCTGTGTTGCTGATGTTGTGGAAGAA | |
| PKS10-1 | CGCAGGCTATCGACGACCAAGTG | FG12100 |
| PKS10-2 | GCTGACGTGCTGCAAATGAATGTG | |
| PKS11-1 | AAGAGGATTTGCCGACCGATTTG | FG01790 |
| PKS11-2 | GCCTTGTATTTATGCATGTGCTCAGTGGG | |
| PKS12-1 | TGGTCCCAACGGTCAGCACATCA | FG12040 |
| PKS12-2 | GGAACGCCTTGGCCAATCTCATC | |
| PKS13-1 | AGCCGGGAAAGCCATACCATACAT | FG12014 |
| PKS13-2 | GCTTGCAATCTCGATCGCCATAAT | |
| PKS14-1 | AGGGCTCGATTGTTGCGGATTCT | FG03964 |
| PKS14-2 | AAAGCATGATGGTCGAGCCCTGTT | |
| PKS15-1 | CCGGCCTTACCCTCGCTTGAA | FG04588 |
| PKS15-2 | ATGATTGTTACGGGCTTGGGAGTGA | |
| FG03340-1 | TTCCTTGACACATTTGCCCGCTACC | FG03340 |
| FG03340-2 | ATGCATCACCCAATCGACCAAGAGT | |
| Primers for pAg1-H3-PKS4 construction | ||
| PKS4-1-PacI | ACCTTAATTAAGCCTTCTACCATCCAAACAGCAAACGAC | |
| PKS4-1-FseI | ACAGGCCGGCCGCAACATCTCCAGAAAGGGTGACATTC | |
| PKS4-2-ApaI | ACGGGGCCCTTTGTTGACGACGAGCGAGTATTGG | |
| PKS4-2-SacI | ATAGAGCTCTAAAACGAGGGTCTTGGAAGTATGTGG | |
| Primers for PCR verification of the PKS4 replacement | ||
| PKS4-Hyg-A1 | GAAGAGCGGAAAAGGTGAGCAGCAATAAGAAC | |
| PKS4-Hyg-A2 | GTGCTCAACGGCCTCAACCTACTACT | |
| PKS4-Hyg-B1 | AAGCGACATTTACCTTCATCTCTCTTGTGAT | |
| PKS4-Hyg-B2 | AACCATCGGCGCAGCTATTTACCC | |
| Primers for making a probe for Southern blot analysis of PKS4 mutants | ||
| PKS4-del-1 | TCGGGTTGCCGTTTACTTAC | |
| PKS4-del-2 | GTCGCTCCATGGATTAGGAA | |
| Primers for real-time expression studies | ||
| PKS4-PS.1 | GTGGGCTTCGCTAGACCGTGAGTT | FG12055 |
| PKS4-PS.2 | ATGCCCTGATGAAGAGTTTGAT | |
| PKS13-PS.1 | CCCCCAACTCGACGTCAAATCTAT | FG12014 |
| PKS13-PS.2 | TTCTTCCCGCCGACTTCAAAACA | |
| FGtubf | GGTCTCGACAGCAATGGTGTT | FG09530 |
| FGtubr | GCTTGTGTTTTTCGTGGCAGT | |
| EF1a-PS.1 | GGCTTTCACCGACTACCCTCCTCT | FG08811 |
| EF1a-PS.2 | ACTTCTCGACGGCCTTGATGACAC | |
| UBC-PS.1 | TCCCCTTACTCTGGCGGTGTC | FG10805 |
| UBC-PS.2 | TTGGGGTGGTAGATGCGTGTAGT |
Gene designations are based on the MIPS F. graminearum genome database. Restriction sites are underlined.
Generation of replacement mutants.
The A. tumefaciens vector pAg1-H3 (60) was used to create the PKS4 (FG12055) replacement vector. All gene designations are based on those used by Kroken et al. (23) and MIPS F. graminearum genome database (31). Genomic DNA of F. graminearum 1104-14, primers PKS4-1-PacI and PKS4-1-FseI, and primers PKS4-2-ApaI and PKS4-2-SacI were used in a PCR to produce the flanking regions PKS4-1 (2,092 bp) and PKS4-2 (1,808 bp). The PCR conditions were 35 cycles of denaturation at 94°C for 30 s (with a first-cycle hold for 5 min), annealing at 60°C for 1 min, and extension at 72°C for 2 min (with a last-cycle hold for 9 min). The fragments were cloned (TOPO TA cloning kit; Invitrogen), digested with the appropriate restriction enzymes (New England Biolabs, Ipswich, United Kingdom), gel purified (QIAquick gel extraction kit; QIAGEN), and ligated (T4 DNA ligase; Promega) into pAg1-H3 to create the replacement vector pAg1-H3-PKS4. This vector was used for homologous recombination and replacement of the central part of PKS4 with hygB.
The replacement vector pAgI-H3-PKS4 was introduced into A. tumefaciens LBA4404 by electroporation (Gene Pulser II; Bio-Rad, Hercules, Calif.) according to the manufacturer's instructions. A. tumefaciens-mediated transformation was carried out as described by Malz et al. (28). The number of transformed colonies was estimated, and 40 colonies were transferred to new DFM plates containing 150 ppm hygB. PKS4 replacement mutants were verified by PCR performed with primers PKS4-Hyg-A1 and PKS4-Hyg-A2 and primers PKS4-Hyg-B1 and PKS4-Hyg-B2 under the following conditions: 40 cycles of denaturation at 94°C for 30 s (with a first-cycle hold for 5 min), annealing at 60°C for 1 min, and extension at 72°C for 2 min (with a last-cycle hold for 9 min). The primers are located inside the hygB resistance gene and outside the replacement cassette in pAg1-H3-PKS4 but are within the flanking PKS4 sequence.
Southern blot analyses.
Genomic DNA (5 μg) used for Southern analysis of ΔPKS4 mutants was digested to completion with BglII, separated on a 0.8% agarose gel, and transferred to a GeneScreen Plus transfer membrane (NEN Life Science Products, Inc., Boston, Mass.) as described by Molenaar and Wilkins (34). The 1,684-bp hygB fragment used as a probe was obtained by digestion of plasmid pAg1-H3-PKS4 with BglII and AsiSI. The 794-bp fragment from the central region of PKS4, delPKS4 (located at positions 2635 to 3429 in GenBank accession number XM_382572) was obtained by performing PCR with primers PKS4-del-1 and PKS4-del-2 and with F. graminearum 1104-14 genomic DNA as the template. The 1,511-bp fragment from the PKS4-1 region was obtained by digestion of the same plasmid with BglII and XhoI. The probes were labeled by random priming with [α-32P]dCTP (Ready-To-Go DNA labeling beads; Amersham Biosciences, Uppsala, Sweden). Prehybridization, hybridization, and washing were performed by using methods described in Current Protocols in Molecular Biology (1); the prehybridization and hybridization solutions contained 5× SSC, 5× Denhardt solution, 1% (wt/vol) sodium dodecyl sulfate, and 100 mg ml−1 denatured salmon sperm DNA (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate), and the high-stringency wash solution used before autoradiography contained prewarmed (68°C) 0.1%× SSC and 0.1% sodium dodecyl sulfate.
Gene expression analysis.
RNA was purified from 50 mg fungal biomass by using a RNeasy plant mini kit (QIAGEN, Hilden, Germany) and was treated with RQ1 RNase-free DNase (Promega) or a Turbo DNA-free kit (Ambion, Austin, TX). For reverse transcriptase PCR total RNA was reverse transcribed with Superscript III (Invitrogen) and a random primer mixture of nine nucleotides. The first-strand cDNA product was then used in a PCR with the PKS primers and the following PCR conditions: 35 cycles of denaturation at 94°C for 30 s (with a first-cycle hold for 2 min), annealing at 57 to 59°C for 30 s, and extension at 72°C for 1 min (with a last-cycle hold for 11 min). The PCR products were compared by agarose gel electrophoresis.
Relative expression of the PKS4 and PKS13 (FG12014) genes was determined over time by real-time quantitative PCR using an Applied Biosystems 7900HT instrument (Applied Biosystems) with a standard 96-well block, SYBR Green PCR Master Mix, and SYBR Green reverse transcription-PCR reagents (Applied Biosystems). One-half of the inoculated rice was used for RNA extraction. The ZON content was determined by HPLC by using the second half of the culture. Housekeeping genes, including the genes encoding β-tubulin (FG09530), translation elongation factor 1α (FG08811), and ubiquitin conjugating enzyme (FG10805), were used as internal controls because the proteins belong to different protein families and were therefore unlikely to be coregulated. For the β-tubulin gene we used primers FGtubf and FGtubr (44). Specific primers for the other control genes and the target genes were designed by using Primer Select (DNAStar, Madison, WI). The specificity and efficiency of each primer pair were tested by constructing a standard curve over 5 logs, followed by a dissociation curve. All primer pairs had an efficiency of 1.91 to 1.93 and produced only a single amplicon. Target and internal control genes for each biological sample were examined on the same plate. A minimum of three PCRs were performed for each biological sample.
Housekeeping genes were stably expressed in this experiment, as determined by BestKeeper (43), and the BestKeeper Index was used to normalize target gene data. The comparative cycle threshold (CT) method (User Bulletin 2; ABI PRISM 7700 sequence detection system; Applied Biosystems) was used to calculate the level of gene expression relative to the calibrator.
Analysis of ZON production.
ZON production was determined by HPLC. Chemical analyses were carried out with ΔPKS4 mutants and the wild-type strain cultivated on polished parboiled rice. Samples were freeze-dried, and 0.2 g of each sample was transferred to a 14-ml screw-cap glass vial. HPLC analysis was carried out as described by Nielsen and Smedsgaard (37), but the procedure was modified by using a photo diode array detector (DAD), a fluorescence detector (FLD) with excitation at 230 nm and with an emission cutoff at 450 nm, and a Luna C18 II column (100 by 2 mm; inside diameter, 3 μm; Phenomenex, Torrance, Calif.). Samples (3 μl injected) were analyzed by using a water-CH3CN gradient that began at 15% CH3CN, increased linearly to 100% CH3CN over 20 min, and then remained the same for 5 min. Both solvents were acidified with 50 ppm trifluoroacetic acid. The DAD signal range was 200 to 600 nm, and the FLD was set to excitation at 230 nm with an emission cutoff at 450 nm. ZON was detected by comparison to data obtained from an analysis of a ZON standard (Sigma), using retention times recorded by the DAD and FLD supported by the characteristic UV-visible spectrum.
Pathogenicity tests.
Seeds of barley (Hordeum vulgare cv. Alexis) were sterilized in 5% (vol/vol) NaOCl and 0.1% (vol/vol) Tween 20 essentially as described by Miller et al. (32). For assessment of barley stem blight and root rot, the seeds were incubated for 1 h in an aqueous spore suspension containing 5 × 106 spores ml−1 of wild-type F. graminearum 1104-14 or ΔPKS4-T9. Control seeds were incubated in sterile distilled H2O. For each treatment, three replicates (replicates A, B, and C) were used. Each replicate contained three plants in a closed system. Inoculated and control seeds were germinated in prewetted sterile vermiculite and grown for 2 weeks at 15°C with a cycle consisting of 16 h of light and 8 h of darkness. The light intensity was 35,000 lx. A nutrient solution (38) was supplied after 2 days. Stems and leaves of the seedlings were scored for disease symptoms after 1 week and 2 weeks. Roots were examined for root rot after 2 weeks of growth.
Outcross experiments.
To test the genetic linkage of the ZON-deficient ΔPKS4-T9 mutant, we made crosses with the MAT1-1 deletion strain T39ΔM1-3 and with Z3639-M25, Z3643, and H-4 derivatives that had a green fluorescent protein sequence inserted into the MAT1-2 locus (26). The four heterothallic MAT deletion strains were used as females, and the ΔPKS4-T9 mutant and the 1104-14 wild-type strain were used as males in crosses on carrot agar, as described by Bowden and Leslie (6). Each outcross was performed on 10 carrot agar plates. Production of perithecia with emerging cirri containing viable ascopores was observed only with the ΔPKS4-T9 and 1104-14 controls.
RESULTS
Analysis of the PKS protein sequences with SEARCHPKS.
ZON is a reduced macrolide, and a PKS involved in the biosynthesis of ZON probably must have an ER domain (9). Analysis of the putative PKS protein sequences from the annotated MIPS F. graminearum genome database (31) with the program SEARCHPKS (58) revealed that 8 (PKS1 [FG10548], PKS2 [FG04694], PKS4 [FG12055], PKS5 [FG05794], PKS6 [FG12109], PKS7 [FG08795], PKS11 [FG01790], and FG03340) of the predicted 15 PKS protein sequences (23) had a potential ER domain. Primers for the PKSs were designed and tested with genomic DNA of F. graminearum 1104-14 and F. pseudograminearum IBT 1544. All of the primers gave a 400-bp amplification product, as expected (results not shown). These primers were used in reverse transcriptase PCR to identify a putative PKS involved in ZON biosynthesis.
PKS4 expression in the aurofusarin mutants and in the strain producing high levels of ZON.
Based on the HPLC analysis, ZON was produced by the aurofusarin-deficient mutants of F. pseudograminearum, PK2.1 and KHt62.1, but not by F. pseudograminearum wild-type strain IBT 1544 (28) under the culture conditions used. The PKS4 gene encoded the only PKS with an ER for which no transcript was detected by reverse transcriptase PCR in the wild type but which was detected in both mutants (Fig. 1A). For other PKSs (encoded by PKS3, PKS13, and PKS14) without an ER the gene expression patterns were similar to that of PKS4, with downregulation in the wild type compared to the aurofusarin mutants (Fig. 1B). The expression of the genes encoding most of the remaining PKSs without ER domains did not differ in the wild type and mutants (results not shown); the exception was PKS12, which is described elsewhere (28). PKS4 was expressed at a high level in F. graminearum wild-type strain 1104-14 after 7 and 11 days of growth on polished parboiled rice (Fig. 1C), which coincided with the accumulation of ZON.
FIG. 1.

(A) Reverse transcriptase PCR with the F. pseudograminearum wild type (WT) and aurofusarin mutants PK2.1 and KHt62.1 and with primers designed for PKSs with an ER domain. (B) Reverse transcriptase PCR with the F. pseudograminearum wild type and mutants on three PKSs without an ER domain. The F. pseudograminearum strains were cultivated for 2 weeks on a medium described by Bell et al. (2). (C) Reverse transcriptase PCR with PKS4-specific primers for F. graminearum strain 1104-14, which produces high levels of ZON, after 4, 7, and 11 days of growth on polished rice. All PCR products are about 400 bp long. The PKS designations are based on those of Kroken et al. (23).
A. tumefaciens-mediated replacement of the PKS4 gene.
Transformation of F. graminearum 1104-14 with pAg1-H3-PKS4 yielded approximately one hygB-resistant transformant per 1,000 fungal spores. Most of the transformants had the wild-type growth morphology (∼95%), but some exhibited restricted growth. Eight mutants with wild-type morphology were arbitrarily selected for further study. Homologous integration into the PKS4 gene was confirmed by PCR amplification with primers PKS4-Hyg-A1 and PKS4-Hyg-A2 and with primers PKS4-Hyg-B1 and PKS4-Hyg-B2. These primers were located inside the hygB gene and outside the replacement cassette, but they were within the flanking PKS4 sequence (Fig. 2) and yielded products only if homologous recombination occurred. The ΔPKS4 mutants T4, T5, T9, and T11 were double homologous recombinants that produced 2.4-kb and 2.6-kb amplification products with primers PKS4-Hyg-A1 and PKS4-Hyg-A2 and with primers PKS4-Hyg-B1 and PKS4-Hyg-B2 (Fig. 3). Mutants T7, T8, and T12 were single homologous recombinants; T7 and T8 produced amplification products with primers PKS4-Hyg-A1 and PKS4-Hyg-A2, and T12 produced an amplification product with primers PKS4-Hyg-B1 and PKS4-Hyg-B2. Mutant T6 produced no PCR product and was probably not a homologous recombinant.
FIG. 2.

Schematic diagram of the PKS4 (FG12055) gene in F. graminearum, showing putative catalytic domains and, at the bottom, the replacement cassette from pAg1-H3-PKS4 used to disrupt the gene. The small open boxes in PKS4 are putative introns. Primers PKS4-Hyg-A1 and PKS4-Hyg-A2 and primers PKS4-Hyg-B1 and PKS4-Hyg-B2 were used to verify replacement of PKS4 (Fig. 3). These primers are located inside the hygB gene and outside the replacement cassette in vector pAg1-H3-PKS4, but they are within the flanking PKS4 gene sequence and give a product only if homologous recombination occurs. For Southern blot analysis (Fig. 4), the following three probes were used: the 1,684-bp hygB probe from BglII/AsiSI, the 1,511-bp PKS4-1 probe from BglII/XhoI, and the 794-bp delPKS4 probe made with primers PKS4-del-1 and PKS4-del-2 from the deleted sequence of PKS4. KS, β-ketoacyl synthase; AT, acyl transferase; ACP, acyl carrier protein; DH, dehydratase; KR, keto reductase; ER, enoyl reductase.
FIG. 3.

PCR products from genomic DNA of wild-type strain F. graminearum 1104-14 (WT) and ΔPKS4 mutants amplified with primers PKS4-Hyg-A1 and PKS4-Hyg-A2 (lanes A) and with primers PKS4-Hyg-B1 and PKS4-Hyg-B2 (lanes B). The 2.4-kb and 2.6-kb bands resulted from homologous recombination and replacement of the central part of PKS4 with hygB. For the locations of the primers, see Fig. 2. ΔPKS4 mutants T4, T5, T9, and T11 are double homologous recombinants, while T7, T8, and T12 are single homologous recombinants.
Southern analysis confirmed the PCR results. In seven mutants, the hygB probe hybridized to a 4,100-bp fragment (Fig. 4A), as expected if the entire replacement cassette was inserted into the fungal genome. Additional hybridizing fragments from T7, T11, and T12 probably resulted from partial insertions of the hygB gene and the replacement cassette. Hybridization with the delPKS4 probe (Fig. 4B) showed that the central region of the PKS4 gene was deleted in mutants T4, T5, T9, and T11 but was present in the other four transformants. Hybridization with the PKS4-1 probe (Fig. 4C) resulted in identification of multiple hybridizing bands in mutants T7 and T12 but only a single hybridizing fragment in the other mutants. Based on the results of the Southern blot and PCR analyses, T4, T5, and T9 were mutants in which the central region of the PKS4 gene was replaced by a single copy of the hygB gene through double homologous recombination. In mutants T7, T8, and T12 the hygB gene was inserted into the PKS4 gene by a single homologous recombination event. In T7 and T8 the insertion was in PKS4-1, and in T12 it was in the PKS4-2 region. In mutants T7 and T12 a second nonhomologous recombination event also occurred. In mutant T11 the replacement cassette also was inserted into the PKS4 gene by a double homologous recombination event; in addition, this mutant had a second insert in which part of the cassette (hygB was present, but PKS4-1 was not present) was inserted by nonhomologous recombination.
FIG. 4.

Southern blot analysis of F. graminearum PH1, wild type (WT), and ΔPKS4 mutants. Genomic DNA was digested with BglII. (A) Analysis with probe made from a BglII/AsiSI digest of the hygB sequence from pAg1-H3-PKS4. (B) Analysis with probe made from a PCR product obtained with primers PKS4-del-1 and PKS4-del-2 from the deleted sequence of PKS4. (C) Analysis with probe made from a BglII/XhoI digest of the PKS4-1 sequence from pAg1-H3-PKS4. For the locations of probes, see Fig. 2.
Functional analysis of ΔPKS4 mutants.
The wild-type strain produced ZON, but neither ZON nor any of its derivatives was detected in the ΔPKS4 mutants tested (Fig. 5). There were no significant quantitative differences in auro- and rubrofusarin production between the wild-type strain and the mutants. In a pathogenicity test, ΔPKS4-T9 caused the same severe root rot symptoms in barley roots that the wild-type strain caused (Table 2). Treated and nontreated barley seeds germinated within 2 days. Seedlings coated with conidia of F. graminearum 1104-14 or ΔPKS4-T9 all had the typical bleaching and impaired growth of stems and leaves. After 2 weeks, the roots showed typical signs of root rot. The control seedlings were healthy. The mycelial morphology on potato dextrose agar, YES, and carrot agar was similar to that of the wild type.
FIG. 5.

HPLC traces for the wild type (WT) and ZON mutant ΔPKS4-T9 after 11 days of growth on rice.
TABLE 2.
Analysis of pathogenicity of the wild type and ΔPKS4-T9 mutant for 14-day-old barley seedlingsa
| Replicate | Bleachingb | Impaired growthc | Visible hyphaed | Rot rote |
|---|---|---|---|---|
| Control | ||||
| A | − | − | − | − |
| B | − | − | − | − |
| C | − | − | − | − |
| Strain 1104-14 | ||||
| A | +++ | +++ | + | +++ |
| B | ++ | +++ | + | +++ |
| C | ++ | ++ | + | ++ |
| Mutant ΔPKS4-T9 | ||||
| A | +++ | +++ | + | ++ |
| B | +++ | +++ | + | +++ |
| C | ++ | ++ | + | +++ |
Three replicates (replicates A, B, and C) were used, and each replicate contained three plants in a closed system.
−, no bleaching; ++, green/yellow bleaching; +++, heavy yellow bleaching.
−, growth not impaired; ++, growth impaired; +++, growth heavily impaired.
−, not detected; +, detected.
−, no browning; ++, browning; +++, heavy browning.
Real-time gene expression analysis of PKS4 and PKS13.
In wild-type strain F. graminearum 1104-14, both PKS4 and PKS13 were expressed at low levels after 4 days of growth (Fig. 6). The relative gene expression then increased before it returned to the low level after 14 days of incubation. After 7 and 11 days of growth the level of expression of PKS13 was twice the level of expression of PKS4. In the gene replacement mutant ΔPKS4-T9 the expression of PKS13 was ∼1% that in the wild type after 11 days. The levels of PKS4 expression were similar in the wild type and ΔPKS4-T9 after 4 and 7 days of incubation. However, in ΔPKS4-T9 the level of expression of PKS4 was lower after 11 days and slightly higher after 14 days than the level of expression in the wild-type strain. The quantitative real-time PCR assays were highly reproducible, with standard deviations of <0.5 in replicate PCRs with the same RNA sample. More variation was observed in the results for replicates with RNA samples from different cultures, and the standard deviations of ΔCT ranged from 0.35 to 5.16. High levels of ZON were produced under the culture conditions used. After 11 days the level in the culture inoculated with strain 1104-14 was 58 ppm.
FIG. 6.

Relative expression of PKS4 and PKS13 in the wild-type strain (WT) and the ΔPKS4-T9 mutant cultured for 4, 7, 11, and 14 days on rice. Expression data were normalized to the BestKeeper Index based on the internal control genes encoding β-tubulin (FG09530), translation elongation factor 1α (FG08811), and ubiquitin conjugating enzyme (FG10805) and were calibrated with the level of expression of PKS4 in ΔPKS4-T9 cultured for 4 days under ZON-producing conditions. The results are the averages for three replicate cultures and a minimum of three independent amplifications.
PKS expression in the ZON mutant.
To determine if the lack of ZON altered the expression of other PKSs, we performed a reverse transcriptase PCR analysis of the wild type and ΔPKS4-T9 after 7 days of growth on rice. In ΔPKS4-T9 expression of PKS5, PKS11, and PKS12 increased and expression of PKS10 decreased (Fig. 7). For the eight other PKSs there was no clear difference between the wild type and ΔPKS4-T9 under these conditions.
FIG. 7.
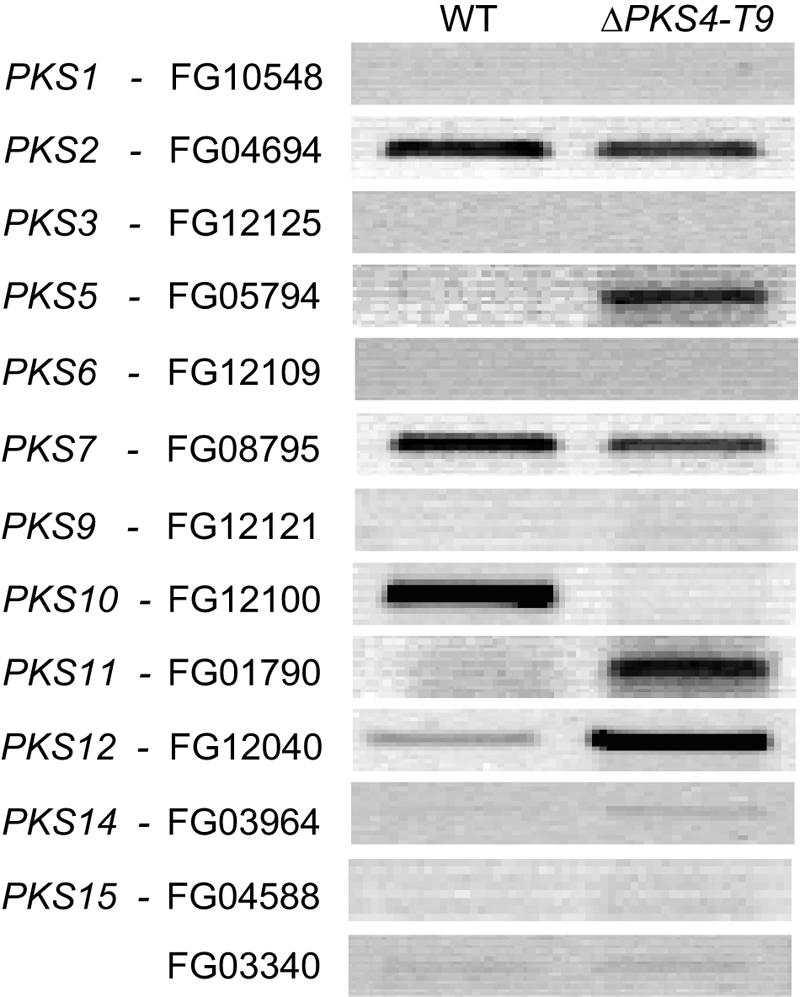
Reverse transcriptase PCR for F. graminearum wild type (WT) and ZON mutant ΔPKS4-T9 with primers designed for all PKS genes except PKS4 and PKS13. The strains were cultivated for 7 days on rice at 25°C in the dark.
DISCUSSION
ZON is a nonsteroid estrogen that is a contaminant of cereal grains used for human consumption or animal feed. Knowledge of ZON biosynthesis is required to understand how and why this mycotoxin is produced and whether its production influences the production of other secondary metabolites or fungal development. Urry et al. (52) determined the chemical structure of ZON, and other workers (16, 33) described its biosynthesis by head-to-tail incorporation of acetate units via the acetate-malonate coenzyme A (polyketide) pathway. Fifteen PKS genes have been identified in F. graminearum (23), and recent studies have identified the products of five of these genes (12, 21, 22, 28, 49); however, the products and functions of the rest of these genes have not been determined. These studies provide important information concerning the genetic background for biosynthesis of the different PKSs and should aid in determining their functions and regulation.
Eight of the 15 annotated PKS protein sequences of F. graminearum have an ER domain, which is predicted to be required for the biosynthesis of ZON. Reverse transcriptase PCR of F. pseudograminearum aurofusarin mutants with increased ZON levels (28) showed that there was specific upregulation of only one gene encoding a PKS with an ER domain, the PKS4 gene. The genes for secondary metabolites often are clustered (3, 48), and PKS4 is adjacent to PKS13, a gene encoding a nonreducing polyketide synthase. Interestingly, PKS13 and PKS4 have similar expression patterns, and the expression of both genes was reduced in the wild-type strain that produced no detectable ZON under the culture conditions used. PKS4 also was expressed at a high level in a strain of F. graminearum known to produce high levels of ZON. Replacement of the central part of PKS4 with hygB turned off ZON production. The genes encoding several PKSs of F. graminearum have orthologues in other fungi that do not produce ZON. A BLAST search with the whole protein or the nucleotide sequence of PKS4 showed that PKS4 is one of only four genes encoding PKSs of F. graminearum with an ER domain that have no known orthologues in other species. This finding, combined with the transcription data, gene knockout data, and reports of other workers (12, 22), strengthens the hypothesis that PKS4 is involved in ZON production.
The real-time transcription pattern of PKS4 and PKS13 in the wild type, in which there is a rapid increase followed by a sudden reduction in gene expression, is in agreement with previous Northern analysis results described by Kim et al. (22). The different time frames for the expression pattern observed in the two experiments might be explained by the different growth media used, SG liquid medium and parboiled rice. The primers used for analysis of PKS4 gene expression were located at the 3′ end of the coding region. However, in mutant ΔPKS4-T9, the hygB cassette was inserted in the central part of PKS4 and in the direction opposite the direction of transcription, as determined with the unmodified PKS4 promoter; consequently, the transcription of PKS4 should not be influenced by our gene replacement mutation. PCR tests with the different mutants showed that the promoter and terminator region of PKS4 are not changed. The basically unaltered PKS4 transcription level observed in ΔPKS4-T9 indicates that PKS4 probably is not regulated by ZON or a derivative of ZON. This conclusion is consistent with the results of Kim et al. (22), who showed that the putative transcriptional regulator of ZON biosynthesis, ZEB2, was not expressed at the same level in the wild type and the isoamyl alcohol oxidase mutant zeb1 (the isoamyl alcohol oxidase catalyzes the oxidation step for the conversion of β-zearalenol to ZON).
Our results also show that inactivation of PKS4 dramatically decreases the expression of PKS13, the second PKS gene in the cluster, and confirm the results reported by Kim et al. (22). This suggests that the PKS4-encoded protein may work independently and not as part of a multiprotein gene complex together with the PKS13-encoded protein, as observed for a fatty acid synthase and a PKS in the biosynthesis of aflatoxin B1 (55). The PKS4-encoded protein or its product may stimulate the expression of PKS13 but not the expression of PKS4. A potential biosynthetic pathway for ZON is as follows: the PKS4-encoded protein synthesizes and reduces the first 10 carbon additions, and this moiety is then released and used as a starter unit (a trigger for gene expression) for the nonreducing PKS13-encoded protein, which completes the carbon addition, similar to the pathway suggested by Gaffoor and Trail (13).
Several PKSs are differentially expressed in the ZON mutant and in the aurofusarin mutants compared to the expression in the wild-type strains. In the aurofusarin mutants, PKS3, PKS4, PKS13, and PKS14 had a different expression pattern than the F. pseudograminearum wild type. Gaffoor et al. (12) grouped PKS4, PKS13, and PKS14 due to the similarity of their expression patterns. The gene expression in two different Fusarium species and under two different culture conditions grouped these three PKS genes together. ZON mutant ΔPKS4-T9 expressed higher levels of PKS5, PKS11, and PKS12 and lower levels of PKS10 under conditions under which ZON was produced by the wild type. Thus, in the aurofusarin mutants there was increased expression of ZON genes and the ZON mutant of PKS12, an aurofusarin-related gene (21, 28). Malz et al. (28) found that a lack of aurofusarin alters ZON production, and we found that expression of ZON biosynthetic genes also is affected in the aurofusarin mutants. In ΔPKS4-T9 and the wild-type strain, there were no significant quantitative differences in aurofusarin production, so the potential influence on gene expression could have been a coincidence. PKS5 was upregulated in our ZON mutant cultivated on rice, but it was not detected under any conditions in a wild-type strain tested by Gaffoor et al. (12).
There is evidence that plant-pathogenic fungi are enriched in genes for secondary metabolism, such as the genes encoding PKSs and nonribosomal peptide synthetases (NRPS), whereas fungi that are primarily saprophytes appear to have fewer copies of such genes (59). Secondary metabolites may play a far more significant role in plant-pathogen interactions than previously anticipated. An NRPS (NPS15) gene is also found in the ZON gene cluster, and its expression seems to be affected by mutations in the ZON gene cluster (22). The chemical structure of ZON reveals no obvious evidence that an NRPS is required, so these results might be coincidental. Deletion of NPS15 did not affect ZON production (22). Trichothecenes participate as virulence factors in F. graminearum (17) and are both phytotoxic and mycotoxic. Studies of barley roots inoculated with F. graminearum 1104-14, which produces high levels of ZON, and with ΔPKS4-T9 revealed no differences in pathogenicity. Similar results were reported by Gaffoor et al. (12) for wheat head infections and by Kim et al. (22) for barley head infections. In addition, for aurofusarin mutants that produced higher levels of ZON than the wild-type strains produced there was not a proportional increase in pathogenicity (28). In conclusion, ZON does not appear to be important for infection of wheat and barley.
During revision of the manuscript, both the PKS4 and PKS13 genes were reported to be required for ZON production, and the linkage of phenotype to genotype was established with genetic crosses (12, 13, 22). No sexual progeny were observed in our outcross experiments. The Norwegian strain F. graminearum 1104-14 (24) did not cross with MAT deletion strains from the United States or Korea. F. graminearum has been divided into nine lineages (40, 41, 54), but our strains have not been placed in any of these lineages at this time. F. graminearum strains from Norway seem to be less aggressive than foreign isolates, and they produce relatively few macroconidia on potato dextrose agar (Oleif Elen, personal communication). Genetic diversity has the potential to cause fertility barriers. Alternative explanations are that there are problems with male fertility or that there are particular strain-by-strain interactions, as reported by Bowden and Leslie (6). ΔPKS4-T9 and its wild-type parent produced similar numbers of perithecia when they were cultured on carrot agar. These results are consistent with the conclusion that ZON is not required for or involved in regulation of production of perithecia in F. graminearum (12, 22, 57).
Agrobacterium-mediated transformation in F. graminearum was very efficient, and the frequency of replacement of the PKS4 gene was high. Of eight transformants examined by PCR and Southern analysis, four resulted from double homologous recombination events and three resulted from single homologous recombination events. We used a 2-kb region on either side of the hygB cassette, which is shorter than the size recommended by Zhang et al. (60) for gene replacement using the pAg1-H3 vector. The transformation frequency of F. graminearum which we obtained in this study was much higher than that obtained by Malz et al. (28), using the pPK2 vector (7) or the pKHt vector (35). Thus, A. tumefaciens-mediated transformation in F. graminearum can result in a high frequency of homologous integration events and represents an attractive alternative to protoplast-based fungal transformation protocols. The existence of several efficient tools for knocking out genes in F. graminearum should allow generation of mutants in which the effects of various secondary metabolites on fungal and plant biology can be evaluated.
This study helped identify and characterize genes encoding enzymes that catalyze critical steps in the synthesis of toxins in an economically important fungus. The connection of PKS4 to ZON may be important for developing strategies to combat the fungus (e.g., chemicals from a genetically modified plant or a single-site fungicide that specifically inactivates PKS4 or other genes in the ZON biosynthetic pathway). More mutants and detailed gene expression studies are needed to elucidate the ZON biosynthetic pathway. There have been reports of collective detection of trichothecene-producing Fusarium species with a PCR-based assay that detects the tri5 gene, the gene encoding trichodiene synthase (10, 11, 39). A similar assay for all ZON-producing Fusarium spp. could be based on PKS4 or other genes in the cluster and might function as a preliminary screening test for grain samples for possible ZON contamination. Analyses of fungal strains that differ in toxin production are needed to generate complementary data on the regulation of these compounds and to provide a more complete picture of the importance of these compounds for fungal biology and pathogenicity.
Acknowledgments
We especially thank Kirsten Henriksen, M. Jafar Razzaghian, Karen Petersen, Sussanne Iversen, and Ann Siri Borg Hentze for valuable technical assistance. We thank Barbara Kosiak for providing F. graminearum 1104-14 and John Leslie, Gillian Turgeon, and Yin-Won Lee for providing the MAT mutant strains. We thank J. S. Tkacz for providing the pAg1-H3 vector.
Grants from the Research Council of Norway (grant 147003/130), the Danish Research Foundation, the Danish Ministry of Food Science, and the Centre for Advanced Food Studies (LMC) funded this work.
REFERENCES
- 1.Ausubel, F. M., R. E. Kingston, D. D. Moore, J. G. Seidman, and K. Struhl. 1987. Current protocols in molecular biology. John Wiley & Sons Inc., Hoboken, N.J.
- 2.Bell, A. A., M. H. Wheeler, J. G. Liu, and R. D. Stipanovic. 2003. United States Department of Agriculture Agricultural Research Service studies on polyketide toxins of Fusarium oxysporum f. sp. vasinfectum: potential targets for disease control. Pest. Manag. Sci. 59:736-747. [DOI] [PubMed] [Google Scholar]
- 3.Bhatnagar, D., K. C. Ehrlich, and T. E. Cleveland. 2003. Molecular genetic analysis and regulation of aflatoxin biosynthesis. Appl. Microbiol. Biotechnol. 61:83-93. [DOI] [PubMed] [Google Scholar]
- 4.Bingle, L. E. H., T. J. Simpson, and C. M. Lazarus. 1999. Ketosynthase domain probes identify two subclasses of fungal polyketide synthase genes. Fungal Genet. Biol. 26:209-223. [DOI] [PubMed] [Google Scholar]
- 5.Blaney, B. J., and R. L. Dodman. 2002. Production of zearalenone, deoxynivalenol, nivalenol, and acetylated derivatives by Australian isolates of Fusarium graminearum and F. pseudograminearum in relation to source and culturing conditions. Aust. J. Agric. Res. 53:1317-1326. [Google Scholar]
- 6.Bowden, R. L., and J. F. Leslie. 1999. Sexual recombination in Gibberella zeae. Phytopathology 89:182-188. [DOI] [PubMed] [Google Scholar]
- 7.Covert, S. F., P. Kapoor, M. H. Lee, A. Briley, and C. J. Nairn. 2001. Agrobacterium tumefaciens-mediated transformation of Fusarium circinatum. Mycol. Res. 105:259-264. [Google Scholar]
- 8.Desjardins, A. E., and R. H. Proctor. 2001. Biochemistry and genetics of Fusarium toxins, p. 50-69. In B. A. Summerell, J. F. Leslie, D. Backhouse, W. L. Bryden, and L. W. Burgess (ed.), Fusarium. Paul E. Nelson Memorial Symposium. American Phytopathological Society Press, St. Paul, Minn.
- 9.Dewick, P. N. 2001. The actetate pathway: fatty acids and polyketides, p. 35-117. In P. N. Dewick (ed.), Medicinal natural products. A biosynthetic approach, 2nd ed. John Wiley & Sons Ltd, West Sussex, England.
- 10.Doohan, F. M., D. W. Parry, and P. Nicholson. 1999. Fusarium ear blight of wheat: the use of quantitative PCR and visual disease assessment in studies of disease control. Plant Pathol. 48:209-217. [Google Scholar]
- 11.Edwards, S. G., S. R. Pirgozliev, M. C. Hare, and P. Jenkinson. 2001. Quantification of trichothecene-producing Fusarium species in harvested grain by competitive PCR to determine efficacies of fungicides against fusarium head blight of winter wheat. Appl. Environ. Microbiol. 67:1575-1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaffoor, I., D. W. Brown, R. Plattner, R. H. Proctor, W. H. Qi, and F. Trail. 2005. Functional analysis of the polyketide synthase genes in the filamentous fungus Gibberella zeae (anamorph Fusarium graminearum). Eukaryot. Cell 4:1926-1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaffoor, I., and F. Trail. 2006. Characterization of two polyketide synthase genes involved in zearalenone biosynthesis in Gibberella zeae. Appl. Environ. Microbiol. 72:1793-1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goswami, R. S., and H. C. Kistler. 2004. Heading for disaster: Fusarium graminearum on cereal crops. Mol. Plant Pathol. 5:515-525. [DOI] [PubMed] [Google Scholar]
- 15.Hagler, W. M., Jr., N. R. Towers, C. J. Mirocha, R. M. Eppley, and W. L. Bryden. 2001. Zearalenone: mycotoxin or mycoestrogen?, p. 321-331. In B. A. Summerell, J. F. Leslie, D. Backhouse, W. L. Bryden, and L. W. Burgess (ed.), Fusarium. Paul E. Nelson Memorial Symposium. American Phytopathological Society Press, St. Paul, Minn.
- 16.Hagler, W. M., and C. J. Mirocha. 1980. Biosynthesis of [14C]zearalenone from [1-14C]acetate by Fusarium roseum gibbosum. Appl. Environ. Microbiol. 39:668-670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harris, L. J., A. E. Desjardins, R. D. Plattner, P. Nicholson, G. Butler, J. C. Young, G. Weston, R. H. Proctor, and T. M. Hohn. 1999. Possible role of trichothecene mycotoxins in virulence of Fusarium graminearum on maize. Plant Dis. 83:954-960. [DOI] [PubMed] [Google Scholar]
- 18.Hoekema, A., P. R. Hirsch, P. J. J. Hooykaas, and R. A. Schilperoort. 1983. A binary plant vector strategy based on separation of Vir-region and T-region of the Agrobacterium tumefaciens Ti-plasmid. Nature 303:179-180. [Google Scholar]
- 19.Hopwood, D. A. 1997. Genetic contributions to understanding polyketide synthases. Chem. Rev. 97:2465-2497. [DOI] [PubMed] [Google Scholar]
- 20.Kennedy, J., K. Auclair, S. G. Kendrew, C. Park, J. C. Vederas, and C. R. Hutchinson. 1999. Modulation of polyketide synthase activity by accessory proteins during lovastatin biosynthesis. Science 284:1368-1372. [DOI] [PubMed] [Google Scholar]
- 21.Kim, J. E., K. H. Han, J. Jin, H. Kim, J. C. Kim, S. H. Yun, and Y. W. Lee. 2005. Putative polyketide synthase and laccase genes for biosynthesis of aurofusarin in Gibberella zeae. Appl. Environ. Microbiol. 71:1701-1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim, Y. T., Y. R. Lee, J. M. Jin, K. H. Han, H. Kim, J. C. Kim, T. Lee, S. H. Yun, and Y. W. Lee. 2005. Two different polyketide synthase genes are required for synthesis of zearalenone in Gibberella zeae. Mol. Microbiol. 58:1102-1113. [DOI] [PubMed] [Google Scholar]
- 23.Kroken, S., N. L. Glass, J. W. Taylor, O. C. Yoder, and B. G. Turgeon. 2003. Phylogenomic analysis of type I polyketide synthase genes in pathogenic and saprobic ascomycetes. Proc. Natl. Acad. Sci. USA 100:15670-15675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Langseth, W., A. Bernhoft, T. Rundberget, B. Kosiak, and M. Gareis. 1998. Mycotoxin production and cytotoxicity of Fusarium strains isolated from Norwegian cereals. Mycopathologia 144:103-113. [DOI] [PubMed] [Google Scholar]
- 25.Leach, J., B. R. Lang, and O. C. Yoder. 1982. Methods for selection of mutants and in vitro culture of Cochliobolus heterostrophus. J. Gen. Microbiol. 128:1719-1729. [Google Scholar]
- 26.Lee, J., T. Lee, Y. W. Lee, S. H. Yun, and B. G. Turgeon. 2003. Shifting fungal reproductive mode by manipulation of mating type genes: obligatory heterothallism of Gibberella zeae. Mol. Microbiol. 50:145-152. [DOI] [PubMed] [Google Scholar]
- 27.Logrieco, A., G. Mule, A. Moretti, and A. Bottalico. 2002. Toxigenic Fusarium species and mycotoxins associated with maize ear rot in Europe. Eur. J. Plant Pathol. 108:597-609. [Google Scholar]
- 28.Malz, S., M. N. Grell, C. Thrane, F. J. Maier, P. Rosager, A. Felk, K. S. Albertsen, S. Salomon, L. Bohn, W. Schäfer, and H. Giese. 2005. Identification of a gene cluster responsible for the biosynthesis of aurofusarin in the Fusarium graminearum species complex. Fungal Genet. Biol. 42:420-433. [DOI] [PubMed] [Google Scholar]
- 29.Marasas, W. F. O., P. E. Nelson, and T. A. Tousson. 1984. Toxigenic Fusarium species. Identity and mycotoxicology. The Pennsylvania State University Press, University Park.
- 30.McMullen, M., R. Jones, and D. Gallenberg. 1997. Scab of wheat and barley: a re-emerging disease of devastating impact. Plant Dis. 81:1340-1348. [DOI] [PubMed] [Google Scholar]
- 31.Mewes, H. W., C. Amid, R. Arnold, D. Frishman, U. Guldener, G. Mannhaupt, M. Munsterkotter, P. Pagel, N. Strack, V. Stumpflen, J. Warfsmann, and A. Ruepp. 2004. MIPS: analysis and annotation of proteins from whole genomes. Nucleic Acids Res. 32:D41-D44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller, H. J., G. Henken, and J. A. Vanveen. 1989. Variation and composition of bacterial populations in the rhizospheres of maize, wheat, and grass cultivars. Can. J. Microbiol. 35:656-660. [Google Scholar]
- 33.Mirocha, C. J., and S. V. Pathre. 1979. Mycotoxins—their biosynthesis in fungi: zearalenone biosynthesis. J. Food Prot. 42:821-824. [DOI] [PubMed] [Google Scholar]
- 34.Molenaar, A. J., and R. J. Wilkins. 1991. A simple and convenient way of blotting nucleic acids. BioTechniques 10:334-335. [PubMed] [Google Scholar]
- 35.Mullins, E. D., X. Chen, P. Romaine, R. Raina, D. M. Geiser, and S. Kang. 2001. Agrobacterium-mediated transformation of Fusarium oxysporum: an efficient tool for insertional mutagenesis and gene transfer. Phytopathology 91:173-180. [DOI] [PubMed] [Google Scholar]
- 36.Nelson, P. E., T. A. Tousson, and R. J. Cook. 1981. Fusarium diseases: biology and taxonomy. The Pennsylvania State University Press, University Park.
- 37.Nielsen, K. F., and J. Smedsgaard. 2003. Fungal metabolite screening: database of 474 mycotoxins and fungal metabolites for dereplication by standardized liquid chromatography-UV-mass spectrometry methodology. J. Chromatogr. A 1002:111-136. [DOI] [PubMed] [Google Scholar]
- 38.Nielsen, N. E. 1984. Crop production in re-circulating nutrient solution according to the principle of regeneration, p. 421-446. In Proceedings of 6th International Congress of Soilless Culture. International Society of Soilless Culture, Lunteren, The Netherlands.
- 39.Niessen, M. L., and R. F. Vogel. 1998. Group specific PCR-detection of potential trichothecene-producing Fusarium species in pure cultures and cereal samples. Syst. Appl. Microbiol. 21:618-631. [DOI] [PubMed] [Google Scholar]
- 40.O'Donnell, K., H. C. Kistler, B. K. Tacke, and H. H. Casper. 2000. Gene genealogies reveal global phylogeographic structure and reproductive isolation among lineages of Fusarium graminearum, the fungus causing wheat scab. Proc. Natl. Acad. Sci. USA 97:7905-7910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O'Donnell, K., T. J. Ward, D. M. Geiser, H. C. Kistler, and T. Aoki. 2004. Genealogical concordance between the mating type locus and seven other nuclear genes supports formal recognition of nine phylogenetically distinct species within the Fusarium graminearum clade. Fungal Genet. Biol. 41:600-623. [DOI] [PubMed] [Google Scholar]
- 42.Parry, D. W., P. Jenkinson, and L. Mcleod. 1995. Fusarium ear blight (scab) in small-grain cereals—a review. Plant Pathol. 44:207-238. [Google Scholar]
- 43.Pfaffl, M. W., A. Tichopad, C. Prgomet, and T. P. Neuvians. 2004. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper-Excel-based tool using pair-wise correlations. Biotechnol. Lett. 26:509-515. [DOI] [PubMed] [Google Scholar]
- 44.Reischer, G. H., M. Lemmens, A. Farnleitner, A. Adler, and R. L. Mach. 2004. Quantification of Fusarium graminearum in infected wheat by species specific real-time PCR applying a TaqMan Probe. J. Microbiol. Methods 59:141-146. [DOI] [PubMed] [Google Scholar]
- 45.Rolland, S., C. Jobic, M. Fevre, and C. Bruel. 2003. Agrobacterium-mediated transformation of Botrytis cinerea, simple purification of monokaryotic transformants and rapid conidia-based identification of the transfer-DNA host genomic DNA flanking sequences. Curr. Genet. 44:164-171. [DOI] [PubMed] [Google Scholar]
- 46.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 47.Shier, W. T., A. C. Shier, W. Xie, and C. J. Mirocha. 2001. Structure-activity relationships for human estrogenic activity in zearalenone mycotoxins. Toxicon 39:1435-1438. [DOI] [PubMed] [Google Scholar]
- 48.Sidhu, G. S. 2002. Mycotoxin genetics and gene clusters. Eur. J. Plant Pathol. 108:705-711. [Google Scholar]
- 49.Song, Z. S., R. J. Cox, C. M. Lazarus, and T. J. Simpson. 2004. Fusarin C biosynthesis in Fusarium moniliforme and Fusarium venenatum. Chembiochemistry 5:1196-1203. [DOI] [PubMed] [Google Scholar]
- 50.Tendulkar, S. R., A. Gupta, and B. B. Chattoo. 2003. A simple protocol for isolation of fungal DNA. Biotechnol. Lett. 25:1941-1944. [DOI] [PubMed] [Google Scholar]
- 51.Thrane, U. 2001. Development in the taxonomy of Fusarium species based on secondary metabolites, p. 29-49. In B. A. Summerell, J. F. Leslie, D. Backhouse, W. L. Bryden, and L. W. Burgess (ed.), Fusarium. Paul E. Nelson Memorial Symposium. American Phytopathological Society Press, St. Paul, Minn.
- 52.Urry, W. H., H. L. Wehrmeis, E. B. Hodge, and P. H. Hidy. 1966. Structure of zearalenone. Tetrahedron Lett. 27:3109-3114. [Google Scholar]
- 53.Vidnes, A., B. Paulsen, and C. Bergsten. 2003. Report from SCOOP task 3.2.10 “Collection of occurrence data of Fusarium toxins in food and assessment of dietary intake by the population of EU member states”—Subtask II: zearalenone. [Online.] http://europa.eu.int/comm/food/fs/scoop/task3210.pdf. [DOI] [PubMed]
- 54.Ward, T. J., J. P. Bielawski, H. C. Kistler, E. Sullivan, and K. O'Donnell. 2002. Ancestral polymorphism and adaptive evolution in the trichothecene mycotoxin gene cluster of phytopathogenic Fusarium. Proc. Natl. Acad. Sci. USA 99:9278-9283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Watanabe, C. M. H., and C. A. Townsend. 2002. Initial characterization of a type I fatty acid synthase and polyketide synthase multienzyme complex NorS in the biosynthesis of aflatoxin B1. Chem. Biol. 9:981-988. [DOI] [PubMed] [Google Scholar]
- 56.Windels, C. E. 2000. Economic and social impacts of Fusarium head blight: changing farms and rural communities in the northern Great Plains. Phytopathology 90:17-21. [DOI] [PubMed] [Google Scholar]
- 57.Windels, C. E., C. J. Mirocha, H. K. Abbas, and W. P. Xie. 1989. Perithecium production in Fusarium graminearum populations and lack of correlation with zearalenone production. Mycologia 81:272-277. [Google Scholar]
- 58.Yadav, G., R. S. Gokhale, and D. Mohanty. 2003. SEARCHPKS: a program for detection and analysis of polyketide synthase domains. Nucleic Acids Res. 31:3654-3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yoder, O. C., and B. G. Turgeon. 2001. Fungal genomics and pathogenicity. Curr. Opin. Plant Biol. 4:315-321. [DOI] [PubMed] [Google Scholar]
- 60.Zhang, A., P. Lu, A. M. Dahl-Roshak, P. S. Paress, S. Kennedy, J. S. Tkacz, and Z. An. 2003. Efficient disruption of a polyketide synthase gene (pks1) required for melanin synthesis through Agrobacterium-mediated transformation of Glarea lozoyeasis. Mol. Genet. Genomics 268:645-655. [DOI] [PubMed] [Google Scholar]