

Abstract
A fixed artesunate-mefloquine combination, comprising three daily doses of 8 mg of mefloquine/kg of body weight and 4 mg of artesunate/kg, has been developed recently. This study was designed to construct a population pharmacokinetic model describing this new dosage regimen of mefloquine given as loose tablets together with artesunate. In two randomized trials in Thailand which evaluated the efficacy, safety, and tolerability of this new regimen, the members of a subgroup of 50 patients were randomized to have capillary blood sampling before treatment and at five randomly assigned time points during the 63-day follow-up period. Mefloquine levels in capillary whole blood were assayed by liquid chromatography with UV detection. A pharmacokinetic model for mefloquine was constructed using mixed-effects modeling. A one-compartment model with first-order absorption and elimination was selected to describe the kinetic properties of mefloquine. For capillary whole-blood mefloquine, the area under the concentration curve (AUC) was 40% higher than previous estimates for patients given the equivalent conventional-dose regimen (mefloquine given as 15 mg/kg and then 10 mg/kg on the second and third days of treatment). The half-life (t1/2) of the carboxylic acid metabolite was estimated as 26 days, and the metabolite was eliminated more slowly than the parent drug (population t1/2 estimate, 10.5 days). Splitting the 25 mg/kg dose of mefloquine into three doses of 8 mg/kg each resulted in improved oral bioavailability compared to the conventional split-dose regimen results. This new regimen is well tolerated and results in an equivalent therapeutic response.
Mefloquine, a fluorinated 4-quinoline methanol compound, was developed by the Walter Reed Army Institute of Research over 35 years ago. Mefloquine has two asymmetric carbon atoms and is used as an oral treatment containing a racemic mixture of equal proportions. The pharmacokinetic properties are stereospecific. Mefloquine is moderately well absorbed orally and extensively distributed and is >98% bound to plasma proteins. The terminal elimination half-life is approximately 3 weeks for healthy subjects and 2 weeks for subjects with malaria (6). The main metabolite identified in man is 2-8-bis-trifluoromethyl-4-quinoline carboxylic acid (MMQ), which is inactive against Plasmodium falciparum.
Mefloquine was introduced first as a single-dose therapy for falciparum malaria in Thailand in 1984, but initial high cure rates were not sustained (11). In a bid to halt the loss of antimalarial monotherapies to resistance in rapid succession, the strategy of artemisinin-based combination therapy with mefloquine was developed (19) and was adopted in Thailand in 1994.
In cases of malaria, absorption of mefloquine is dose limited and is reduced in the acute phase of illness. Splitting the dose and delaying administration after the first dose of an artemisinin derivative increase mefloquine absorption (12, 14). This is attributed partly to the rapid clinical and parasitological responses to artesunate. Smaller studies with artemisinin and other derivatives have given inconsistent results with respect to the pharmacokinetic-pharmacodynamic interaction with mefloquine in acute malaria (5, 9, 15). To improve oral bioavailability and tolerability, the mefloquine dose of 25 mg/kg of body weight was split into a 15 mg/kg dose followed by a 10 mg/kg dose. This is the dose regimen currently recommended in conjunction with artesunate.
A new fixed combination of mefloquine and artesunate has been developed (Drugs for Neglected Diseases Initiative; http://www.dndi.org). Coformulation of the drugs reduces the pill burden and, more importantly, eliminates the possibility of patients taking only one component of the combination or of providers selling only one drug to reduce costs. As the tolerability of mefloquine is dose related, dividing the total dosage into three rather than two doses might be preferable provided this approach does not impact efficacy adversely.
The aim of this study was to construct a population pharmacokinetic model for mefloquine given once daily in an 8 mg base/kg dose with artesunate and to estimate the key pharmacokinetic parameters. Patients were participants in two community-based clinical trials on the northwestern border of Thailand that compared mefloquine and artesunate with dihydroartemisinin-piperaquine for the treatment of uncomplicated falciparum malaria. The safety and efficacy results of those trials have been reported in full elsewhere (1, 2).
Study site.
The study took place in the clinics of the Shoklo Malaria Research Unit in Tak province along the Thailand-Burma border, an area of unstable low and seasonal malaria transmission where P. falciparum is highly drug resistant (8).
Ethical review.
The protocol was approved by the Ethical Committee of the Faculty of Tropical Medicine, Mahidol University, Bangkok, Thailand, and the Oxford Tropical Research Ethics Committee (OXTREC), Oxford University, United Kingdom.
MATERIALS AND METHODS
Patients aged 1 to 65 years with symptomatic uncomplicated falciparum infection who were either of the Karen ethnic group or Burmese and weighed at least 5 kg were recruited from four clinics. Exclusion criteria included known mefloquine treatment in the previous 2 months, severe malaria, pregnancy, and lactation. The comparator drug in these randomized trials was dihydroartemisinin-piperaquine given in three different dosing regimens. Members of a subgroup of patients were subjected to blood sampling for mefloquine levels. The study was explained to patients in their own languages, and written consent (or a thumbprint in the case of patients unable to read or write) was obtained.
Randomization and test blinding.
After the initial computer-generated randomization (STATA 7 software) to allocate patients to the different treatment arms, 65 subjects in the mefloquine-artesunate-treated group were allocated randomly to blood sampling groups for mefloquine drug levels. The treatment and sampling allocations were both concealed in sealed envelopes.
Mefloquine-artesunate dosing regimen.
Mefloquine 250 mg tablets (Mequin Atlantic Laboratories Corp. Ltd.) were administered at a dose of 8 mg/kg of body weight/day (rounded to the nearest quarter-tablet volume) with artesunate 50 mg tablets (Guilin Factory no.1, Guangdong, China) at a dose of 4 mg/kg/day for 3 days.
Sample times.
Each patient in the pharmacokinetic study had a capillary blood sample taken pretreatment and was randomized into a group to have five or six samples taken in the following time windows: 3, 7 to 14, 21, and 28 to 63 days (17). Samples (100 to 120 μl) were taken into heparinized capillary tubes. The whole blood was then transferred to a polypropylene tube and frozen immediately at −20°C. Samples were transferred in batches to the main laboratory, where they were stored at −80°C. The concentrations of mefloquine and carboxymefloquine were determined using solid-phase extraction (SPE) combined with liquid chromatography (LC) in a slight modification of a method reported in a published assay (4). A 100-μl volume of internal standard (5 μg/ml) in hydrochloric acid (0.01 mol/liter) was added to an equivalent volume of whole blood in an Eppendorf microtube. The samples were then precipitated with 25 μl zinc sulfate and 275 μl acetonitrile, mixed for 10 s, and left undisturbed for 10 min. The precipitated samples were then centrifuged at 15,000 × g and the supernatants transferred to 5 ml polypropylene tubes already containing 1,150 μl phosphate buffer (pH 3) (0.1 mol/liter). The samples were then loaded onto preconditioned C18-SD SPE columns (3 M Empore). The SPE columns were washed and dried before being eluted into 5 ml polypropylene tubes with 400 μl methanol. The SPE eluates were evaporated to dryness at 65°C under a gentle stream of air. The samples were then reconstituted in 100 μl methanol-hydrochloric acid at 0.01 mol/liter (50:50 [vol/vol]), and 50 μl was injected into the LC system. The LC system used was a LaChrom Elite system consisting of an L2130 LC pump, an L2200 injector, an L2300 column oven set at 25°C, and an L2400 UV detector (Hitachi). The detector was set at 222 nm. Data acquisition was performed using LaChrom Elite software (VWR International). The compounds were analyzed using an Alltima C18 column (Alltech) (5 μm; 150 by 4.6 mm) and a mobile phase containing methanol-phosphate buffer (pH 2.5; 0.1 mol/liter) (60/40 [vol/vol]) at a flow rate of 1.0 ml/min. The lower limit of quantification of the assay was 70 ng/ml. The coefficients of variation (CV) for mefloquine during the analysis (n = 28) were 4.5%, 3.5%, and 3.0% at 300 ng/ml, 800 ng/ml, and 2500 ng/ml, respectively. The CV for carboxymefloquine during the analysis (n = 28) were 8.0%, 6.2%, and 6.8% at 300 ng/ml, 800 ng/ml, and 2500 ng/ml, respectively.
Recurrence of malaria infection during follow-up.
For patients who had a reappearance of P. falciparum during the study follow up period, recrudescence was distinguished from reinfection by PCR genotyping as described previously (3).
Pharmacokinetic modeling and statistical methods.
Nonlinear mixed-effects modeling was used to fit population models to the capillary whole-blood concentration profiles of mefloquine and its metabolite. One- and two-compartment models were considered. In each case a one-compartment model with first-order absorption and first-order elimination was selected as the kinetic model. Intersubject variability values in the pharmacokinetic parameters were modeled with log-normal error models, for example, (CL/Fi) = (CL/F)exp(ηiCL/F), where apparent clearance (CL/Fi) is the pharmacokinetic parameter for the individual (represented by “i”), CL/F is the population mean, and ηiCL/F is the random effect with a mean of 0 and variance σCL/F, the intersubject variability for the parameter. The intrasubject variability was modeled with normal error models. The magnitude of the intersubject variability is expressed as the CV approximated by the square root of the variance estimate, while the intrasubject variability is expressed as the standard deviation of the residual error. The intersubject variability is expressed as an asymmetric 90% prediction interval, for example, for the parameter CL/F equal to exp[log(CL/F) ± 1.645 σCL/F].
The variability in pharmacokinetic parameters was investigated by examining the following covariates: weight, level of parasitemia and temperature at enrollment, duration of fever before enrollment in hours, presence or absence of vomiting in the 24 h before enrollment, presence or absence of gametocytes, type of infection (falciparum monoinfection or mixed infection), and hematocrit. Continuous covariates were centered on their median values so that the population estimates would represent those of an average patient. A forward variable-selection procedure was employed to determine the model that best fitted the data. The likelihood ratio test and Akaike information criterion (AIC) were used to compare models with different covariates. A P value of 0.01 was taken as the cutoff for statistical significance. The effect of each covariate on the variance of the random effects was also examined. Paired individual predictions of elimination-rate constants for mefloquine and for the metabolite were compared using the nonparametric rank sign test. Normally distributed data were quantitated by means and 95% confidence intervals and compared using Student's t test or a chi-square test. Non-normally distributed data were quantitated by median and range and compared using the Mann-Whitney U test. Statistical programs used were the NLME procedure (7) of the Splus program (SPLUS 6 for Windows; Mathsoft, Inc.) for pharmacokinetic calculations, SPSS 11.0 for Windows (SPSS Inc.), EpiInfo (version 1.0, 2000; Centers for Disease Control and Prevention), and STATA/SE (version 8, Stata Corp. LP).
RESULTS
Between July 2002 and April 2004, 1,029 patients were recruited into two studies; of these patients, 343 received artesunate and mefloquine. Sixty-five patients were randomized to the pharmacokinetic study. It was possible to measure 312 mefloquine concentrations for 61 patients, at a median of six time points (range, two to seven time points). Eleven patients were excluded from the analysis: 8 had detectable levels of mefloquine pre treatment, 2 had missing pretreatment samples, and 1 vomited the drug. Therefore, 50 patients were included in the final pharmacokinetic analysis. The baseline characteristics of these patients and of the larger mefloquine-artesunate-treated population are shown in Table 1. There were more female patients and more patients with a higher baseline level of parasitemia among the patients who had provided samples for pharmacokinetic analysis than in the rest of the population (P = 0.01; Mann-Whitney U Test). The median total dose of mefloquine received by patients in the pharmacokinetic study was 24 mg/kg (range, 22 to 32 mg/kg).
TABLE 1.
Baseline characteristics of the patients
| Parameter | Patients in final pharmacokinetic analysis | Patients not in pharmacokinetic analysis |
|---|---|---|
| n | 50 | 293 |
| No. (%) of males | 25 (50) | 200 (68.3) |
| Median (range) age (yr) | 19 (2-55) | 20 (1-63) |
| Median (range) weight (kg) | 44.5 (10-63) | 45 (7-66) |
| Geometric mean (range) of parasitemia/μl | 20,417 (363-173,780) | 10,240 (100-229,087) |
| No. (%) of mixed infections | 6 (12) | 29 (9.8) |
Pharmacokinetic modeling.
The measured mefloquine concentrations are shown in Fig. 1. A one-compartment model with first-order absorption and first-order elimination gave a good fit to the data assessed by examination of residuals and was therefore selected. The fundamental parameters used to characterize the one-compartment model were first-order absorption rate constant (ka), apparent clearance (CL/F) and apparent volume of distribution (V/F). The sampling schedule meant it was not possible to estimate the absorption rate constant (ka) for mefloquine, and this was set from previously determined data (14) to 7 per day, while apparent clearance and volume of distribution were modeled as random effects. A model based on the assumption that the within-group error variance increased with fitted values gave the best fit to the data. The predicted population pharmacokinetic profile is shown (Fig. 2).
FIG. 1.

Scatter plot of capillary whole-blood mefloquine concentrations (in nanograms per milliliter).
FIG. 2.

Predicted population pharmacokinetic profile for mefloquine administered at 8 mg/kg/day with artesunate for 3 days.
In the analysis of covariates, admission temperature correlated negatively with volume of distribution (P = 0.007), while increasing body weight was associated with a reduction in clearance (P = 0.002). The pharmacokinetic parameters for the base model are shown in Table 2, and the parameters for the model with covariates are shown in Table 3.
TABLE 2.
Population pharmacokinetic parameters for the base model of mefloquine
| Parameterb | Mefloquinea administered with artesunate in three doses (8 mg/kg/day)
|
|
|---|---|---|
| Estimated value (SE) | 90% Prediction interval | |
| CL/F (liters kg−1day−1) | 0.755 (0.041) | 0.436-1.306 |
| V/F (liters kg−1) | 10.172 (0.777) | 4.395-23.541 |
| ke (day−1) | 0.074 | |
| Elimination t1/2 (days) | 9.4 | |
| AUC0→∞ (ng/ml · day) | 31,788 | |
| σCL (CV) (%) | 33 | |
| σV (CV) (%) | 51 | |
| σɛ (ng/ml) | 179 | |
Data represent results for 50 patients and 201 concentrations (AIC = 2,908).
CL/F, apparent clearance; V/F, apparent volume of distribution; ke, elimination rate constant; t1/2, elimination half-life; AUC0→∞, area under the whole-blood concentration-time curve; σ2CL, unexplained between-subject variance around the population average CL/F; σ2V, unexplained between-subject variance around the population average V/F; σ2ɛ, variance of the residual error.
TABLE 3.
Population pharmacokinetic model with covariates for mefloquinea
| Parameterb | Estimated value (SE) | 90% Prediction interval |
|---|---|---|
| Group | ||
| CL/F (liters/kg/day) | 0.722 (0.034) | 0.463-1.126 |
| Effect of weight on CL/F | −0.011 (0.003) | |
| V/F (liters kg−1) | 10.966 (0.863) | 4.987-24.113 |
| Effect of temp on V/F | −1.739 (0.594) | |
| ke (day−1) | 0.066 | |
| Elimination t1/2 (day) | 10.5 | |
| AUC0→∞ (ng/ml · day) | 33,241 | |
| σ2CL (CV) (%) | 27 | |
| σ2V (CV) (%) | 48 | |
| σ2ɛ (ng/ml) | 179 | |
| Each patientc | ||
| AUC0→∞ (ng/ml · day) | 31,395 | 20,523-49,239 |
| Cmax (ng/ml) | 2,202 | 915-3,730 |
| Tmax (day) | 2.4 | 2.3-2.6 |
Data represent results for 50 patients and 201 concentrations (AIC = 2,892).
All parameter estimates are for the average person (admission temperature, 37°C; weight, 45 kg). CL/F, apparent clearance; V/F, apparent volume of distribution; ke, elimination rate constant; t1/2, elimination half-life; AUC0→∞, area under the whole-blood concentration-time curve; σ2CL, unexplained between-subject variance around the population average CL/F; σ2V, unexplained between-subject variance around the population average V/F; σ2ɛ, variance of the residual error.
Values in columns 2 and 3 were calculated using the actual dose given and the actual time of dosing and represent median and 90% range values, respectively.
Figure 3 shows a scatter graph of measured concentrations of the metabolite. The model selected to characterize metabolite profiles was also a one-compartment model with three parameters: 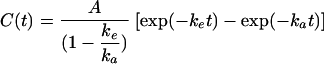 , where ke is the elimination-rate constant and ka is the absorption rate constant. Among the covariates, none of the variables had an effect on metabolite absorption and only increasing weight was associated independently with a lower elimination-rate constant (P = 0.002; likelihood ratio test). The metabolite was eliminated significantly more slowly than mefloquine (z = 5.58 [P < 0.001; sign test]) (Table 4). The metabolite area under the concentration time curve from 0 h to infinity ([AUC0→∞]) was significantly higher than the mefloquine AUC0→∞ (z = 6.09 [P < 0.001; sign test]) by a factor of 1.7 (median; 90% range, 0.69 to 3.7).
, where ke is the elimination-rate constant and ka is the absorption rate constant. Among the covariates, none of the variables had an effect on metabolite absorption and only increasing weight was associated independently with a lower elimination-rate constant (P = 0.002; likelihood ratio test). The metabolite was eliminated significantly more slowly than mefloquine (z = 5.58 [P < 0.001; sign test]) (Table 4). The metabolite area under the concentration time curve from 0 h to infinity ([AUC0→∞]) was significantly higher than the mefloquine AUC0→∞ (z = 6.09 [P < 0.001; sign test]) by a factor of 1.7 (median; 90% range, 0.69 to 3.7).
FIG. 3.

Scatter plot of capillary whole-blood concentrations of the carboxylic acid metabolite (in nanograms per milliliter).
TABLE 4.
Population parameters for the base model of mefloquine's carboxylic acid metabolitea
| Parameterb | MMQ
|
|
|---|---|---|
| Estimated value (SE) | 90% Prediction interval | |
| ka (day−1) | 0.304 (0.030) | 0.275-0.335 |
| Ac | 642.1 (42.5) | 398.5-1,034.6 |
| ke (day−1) | 0.032 (0.004) | 0.013-0.079 |
| Elimination t1/2 (days) | 21.6 | |
| AUC0→∞ (ng/ml · day) | 61,047 | |
| σka (CV) (%) | 6 | |
| σA (CV) (%) | 29 | |
| σke (CV) (%) | 55 | |
| σɛ (ng/ml) | 223.4 | |
Data represent results for 50 patients and 201 concentrations (AIC = 2921).
ka, absorption rate constant; ke, elimination rate constant; t1/2, elimination half-life; AUC0→∞, area under the whole-blood concentration-time curve; 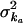 , unexplained between-subject variance around population average ka;
, unexplained between-subject variance around population average ka; 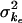 , unexplained between subject variance around population average ke; σ2A, unexplained between-subject variance around the population average A; σ2ɛ, variance of residual error.
, unexplained between subject variance around population average ke; σ2A, unexplained between-subject variance around the population average A; σ2ɛ, variance of residual error.
Results represent calculations based on the model 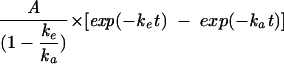 .
.
Treatment failures.
Three patients with PCR-confirmed treatment failure took part in the pharmacokinetic study. The predicted AUCs (in nanograms per milliliter day) for these three patients were 21,648, 27,312, and 27,317, respectively (90% range for all patients shown in Table 3), values which fall into the lower 36% range of all AUCs. The predicted values for the maximum concentrations of drug in serum were 396, 1,027, and 3,706 ng/ml; the first two values were among the lowest 8% of the predicted values for this population.
DISCUSSION
Mefloquine was absorbed well in this new split-dose regimen given with artesunate. The population estimate of the AUC0→∞ for mefloquine given as a dose of 8 mg/kg of body weight per day for 3 days was nearly 40% higher than a previous population estimate of AUC0→∞ of 24,343 ng/ml · day in the same population treated with the conventional equivalent dose regimen of mefloquine given as 15 mg/kg and then 10 mg/kg on the second and third days of treatment (14). The terminal elimination half-life was estimated at 10.5 days, within the expected range for patients with malaria. Estimates of apparent volume of distribution and clearance were lower than those described previously for the conventional regimen (estimated values, 1.027 liters/kg/day [standard error, 0.083; 90% prediction interval, 0.600 to 1.759] and 13.99 liters/kg/day [standard error, 0.70; 90% prediction interval, 9.81 to 19.95], respectively), although prediction intervals were similar. As demographic details and levels of disease severity were similar, this is most likely explained by a higher fraction of the drug being absorbed. Analysis of covariates identified relationships between body weight and clearance and between admission temperature and volume of distribution which were statistically significant. A linear relationship between body weight and mefloquine clearance has been described previously (13). The carboxy-metabolite AUC0→∞ was nearly double that for mefloquine itself, and elimination was slower. In previous studies of healthy volunteers the AUC0→∞ of the metabolite was three to five times larger than that of the parent drug, reaching higher plasma concentrations than mefloquine after approximately 2 weeks and then declining at a similar rate.
Mefloquine elimination is usually best quantified by biexponential or multiexponential disposition kinetics. It was not possible to fit a two-compartment model to these data, partly because there were insufficient samples taken early in the elimination phase. This is a disadvantage of the sampling schedule, which was tailored to minimize additional visits, with patients attending the clinic daily for 3 days after enrollment and then at weekly intervals to monitor treatment efficacy. Despite this, the long duration of follow up after drug administration means that the one-compartment model should give satisfactory estimates of the main kinetic parameters. Direct comparison of these results with those from previous studies should also take into account that a different formulation of the drug was used. Previous studies have shown that the different products are not bioequivalent (10, 18). Tolerability of this three-dose mefloquine regimen was good. Administration of mefloquine on the first day of treatment has been associated with a higher frequency of vomiting than administration of the first dose after a delay of 24 h (16). The rate of early vomiting was very low in this study. This may be the result of dividing the total dosage into three separately administered doses and giving a lower dose than that used in the conventional regimen each day, since intolerance to mefloquine is usually dose related. Efficacy was extremely good, with a cure rate above 95% after 9 weeks of follow up. These results are encouraging for the new fixed-dose combination, which has been tested recently in phase 3 trials. Mefloquine is already widely available as monotherapy in Southeast Asia but is not used in many African countries; this comparative lack of availability in African countries might delay the emergence of mefloquine resistance in those countries when the drug is deployed only as part of a fixed combination.
Acknowledgments
We thank all the patients who took part in the study and the staff of the Shoklo Malaria Research Unit, especially the laboratory staff for processing the samples and Al Brockman for PCR genotyping analysis. Thanks to Julie Simpson for helpful comments on the manuscript.
The study was partially supported by the INCO-DEV Programme (“Confirming the International Role of Community Research for Development”), European Commission, Proposal No. ICA4-2001-10193. The Shoklo Malaria Research Unit is part of the Wellcome Trust-Mahidol University, Oxford Tropical Medicine Research Programme sponsored by the Wellcome Trust of Great Britain.
REFERENCES
- 1.Ashley, E. A., S. Krudsood, L. Phaiphun, S. Srivilairit, R. McGready, W. Leowattana, R. Hutagalung, P. Wilairatana, A. Brockman, S. Looareesuwan, F. Nosten, and N. J. White. 2004. Dose optimisation randomized controlled studies of dihydroartemisinin-piperaquine for the treatment of uncomplicated multi-drug resistant falciparum malaria in Thailand. J. Infect. Dis. 190:1773-1824. [DOI] [PubMed] [Google Scholar]
- 2.Ashley, E. A., R. McGready, R. Hutagalung, L. Phaiphun, T. Slight, S. Proux, K. L. Thwai, M. Barends, S. Looareesuwan, N. J. White, and F. Nosten. 2005. A randomized controlled study of a simple once daily regimen of dihydroartemisinin-piperaquine for the treatment of uncomplicated multi-drug resistant falciparum malaria. Clin. Infect. Dis. 41:426-432. [DOI] [PubMed] [Google Scholar]
- 3.Brockman, A., R. E. Paul, T. J. Anderson, I. Hackford, L. Phaiphun, S. Looareesuwan, F. Nosten, and K. P. Day. 1999. Application of genetic markers to the identification of recrudescent Plasmodium falciparum infections on the northwestern border of Thailand. Am. J. Trop. Med. Hyg. 60:14-21. [DOI] [PubMed] [Google Scholar]
- 4.Green, M. D., Y. Bergqvist, D. L. Mount, S. Corbett, and M. J. D'Souza. 1999. Improved validated assay for the determination of mefloquine and its carboxy metabolite in plasma, serum and whole blood using solid-phase extraction and high-performance liquid chromatography. J. Chromatogr. B 727:159-165. [DOI] [PubMed] [Google Scholar]
- 5.Hung, L. Q., P. J. de Vries, T. Q. Binh, P. T. Giao, N. V. Nam, R. Holman, and P. A. Kager. 2004. Artesunate with mefloquine at various intervals for non-severe Plasmodium falciparum malaria. Am. J. Trop. Med. Hyg. 71:160-166. [PubMed] [Google Scholar]
- 6.Karbwang, J., and N. J. White. 1990. Clinical pharmacokinetics of mefloquine. Clin. Pharmacokinet. 19:264-279. [DOI] [PubMed] [Google Scholar]
- 7.Lindstrom, M. J., and D. M. Bates. 1990. Nonlinear mixed effects models for repeated measures data. Biometrics 46:673-687. [PubMed] [Google Scholar]
- 8.Luxemburger, C., K. L. Thwai, N. J. White, H. K. Webster, D. E. Kyle, L. Maelankirri, T. Chongsuphajaisiddhi, and F. Nosten. 1996. The epidemiology of malaria in a Karen population on the western border of Thailand. Trans. R. Soc. Trop. Med. Hyg. 90:105-111. [DOI] [PubMed] [Google Scholar]
- 9.Na-Bangchang, K., J. Karbwang, P. Molunto, V. Banmairuroi, and A. Thanavibul. 1995. Pharmacokinetics of mefloquine, when given alone and in combination with artemether, in patients with uncomplicated falciparum malaria. Fundam. Clin. Pharmacol. 9:576-582. [DOI] [PubMed] [Google Scholar]
- 10.Na-Bangchang, K., J. Karbwang, J., P. A. Palacios, R. Ubalee, S. Saengtertsilapachai, and W. H. Wernsdorfer. 2000. Pharmacokinetics and bioequivalence evaluation of three commercial tablet formulations of mefloquine when given in combination with dihydroartemisinin in patients with acute uncomplicated falciparum malaria. Eur. J. Clin. Pharmacol. 55:743-748. [DOI] [PubMed] [Google Scholar]
- 11.Nosten, F., F. ter Kuile, T. Chongsuphajaisiddhi, C. Luxemburger, H. K. Webster, M. Edstein, L. Phaipun, K. L. Thew, and N. J. White. 1991. Mefloquine-resistant falciparum malaria on the Thai-Burmese border. Lancet 337:1140-1143. [DOI] [PubMed] [Google Scholar]
- 12.Price, R., J. A. Simpson, P. Teja-Isavatharm, M. M. Than, C. Luxemburger, D. G. Heppner, T. Chongsuphajaisiddhi, F. Nosten, and N. J. White. 1999. Pharmacokinetics of mefloquine combined with artesunate in children with acute falciparum malaria. Antimicrob. Agents Chemother 43:341-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simpson, J. A., L. Aarons, R. Price, and N. J. White. 2002. The influence of body weight on the pharmacokinetics of mefloquine. Br. J. Clin. Pharmacol. 53:337-338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simpson, J. A., R. Price, F. ter Kuile, P. Teja-Isavatharm, F. Nosten, T. Chongsuphajaisiddhi, S. Looareesuwan, L. Aarons, and N. J. White. 1999. Population pharmacokinetics of mefloquine in patients with acute falciparum malaria. Clin. Pharmacol. Ther. 66:472-484. [DOI] [PubMed] [Google Scholar]
- 15.Svensson, U. S., H. Alin, M. O. Karlsson, Y. Bergqvist, and M. Ashton. 2002. Population pharmacokinetic and pharmacodynamic modelling of artemisinin and mefloquine enantiomers in patients with falciparum malaria. Eur. J. Clin. Pharmacol. 58:339-351. [DOI] [PubMed] [Google Scholar]
- 16.ter Kuile, F. O., F. Nosten, C. Luxemburger, D. Kyle, P. Teja-Isavatharm, L. Phaipun, R. Price, T. Chongsuphajaisiddhi, and N. J. White. 1995. Mefloquine treatment of acute falciparum malaria: a prospective study of non-serious adverse effects in 3673 patients. Bull. W. H. O. 73:631-642. [PMC free article] [PubMed] [Google Scholar]
- 17.ter Kuile, F. O., P. Teja-Isavatharm, M. D. Edstein, D. Keeratithakul, G. Dolan, F. Nosten, L. Phaipun, H. K. Webster, and N. J. White. 1994. Comparison of capillary whole blood, venous whole blood, and plasma concentrations of mefloquine, halofantrine, and desbutyl-halofantrine measured by high-performance liquid chromatography. Am. J. Trop. Med. Hyg. 51:778-784. [DOI] [PubMed] [Google Scholar]
- 18.Weidekamm, E., G. Rusing, H. Caplain, F. Sorgel, and C. Crevoisier. 1998. Lack of bioequivalence of a generic mefloquine tablet with the standard product. Eur. J. Clin. Pharmacol. 54:615-619. [DOI] [PubMed] [Google Scholar]
- 19.White, N. J. 1999. Antimalarial drug resistance and combination chemotherapy. Philos. Trans. R. Soc. Lond. B Biol. Sci. 354:739-749. [DOI] [PMC free article] [PubMed] [Google Scholar]