

Abstract
The present population pharmacokinetic (PK) and pharmacodynamic (PD) study modeled the effects of covariates including drug adherence and the coadministration of protease inhibitors (PIs) on the pharmacokinetics of efavirenz (EFV) and the relationship between EFV exposure and virological failure in patients who failed initial PI treatment in Adult AIDS Clinical Trial Group (AACTG) study 398. We also report on the population PKs of the PIs nelfinavir (NFV) and indinavir (IDV). AACTG study 398 patients received EFV, amprenavir, adefovir dipivoxil, and abacavir and were randomized to take, in addition, one of the following: NFV, IDV, saquinavir (SQV), or placebo. The PK databases consisted of 531 EFV concentrations (139 patients), 219 NFV concentrations (75 patients), and 66 IDV concentrations (11 patients). Time to virological failure was ascertained for all patients in the PK databases. PK data were fit with a population PK model that assumed exclusive hepatic elimination (the well-stirred model). Notable findings with respect to EFV PK and PD are as follows. (i) The hepatic clearance of EFV is unaltered by NFV, IDV, or SQV coadministration. (ii) The hepatic clearance of EFV appears to be 28% higher in white non-Hispanics than in African Americans and Hispanics (P = 0.03). (iii) Higher adherence scores (as measured with the Medication Event Monitoring System) are associated with marginally increased levels of exposure to EFV. (iv) In patients with no prior experience with nonnucleoside reverse transcriptase inhibitors (NNRTIs), a given percent increase in the oral clearance (CL/F) of EFV is associated with a greater percent increase in the hazard of virological failure (P < 0.0003). Among NNRTI-experienced patients, however, hazard is relatively uncorrelated with EFV CL/F.
Efavirenz (EFV) is a nonnucleoside reverse transcriptase inhibitor (NNRTI) used in the treatment of patients with human immunodeficiency virus (HIV) infection (1). EFV is used in combination with either protease inhibitors (PIs) or nucleoside reverse transcriptase inhibitors (13, 28). Like the PIs, EFV is extensively metabolized by the cytochrome P450 enzymes, primarily CYP3A4 (1, 3). All currently available PIs are inhibitors of CYP3A4, and the inhibitory activities of these PIs range from weak (saquinavir [SQV]) to very potent (ritonavir) (8, 9, 21). Therefore, drug-drug interactions should be expected when EFV is coadministered with them. It has been reported that ritonavir produces a 21% increase in EFV concentrations (W. Fiske, I. H. Benedek, and J. L. Joseph, Abstr. 12th World AIDS Conf., abstr. 42269, 1998). However, very few data are available on the pharmacokinetic (PK) interactions between EFV and PIs such as indinavir (IDV), nelfinavir (NFV), and SQV at steady state in HIV-infected patients.
Adult AIDS Clinical Trial Group (AACTG) study 398 was a phase II trial which prescribed a background regimen of EFV, amprenavir (APV), abacavir, and adefovir dipivoxil for HIV-infected subjects with prior exposure to approved PIs and loss of virological suppression, as reflected by two consecutive plasma HIV RNA levels ≥1,000 copies/ml after at least 16 cumulative weeks of PI treatment. No prior exposure to EFV, APV, or adefovir dipivoxil was allowed. The study patients were selectively randomized to one of four treatment arms depending on their past PI exposure. In addition to the background drugs, the patients received IDV, NFV, SQV, or a placebo matched for one of the three PIs. Data on the PK of EFV, IDV, NFV, SQV, APV, and adefovir dipivoxil were collected. The viral responses of all patients were monitored. This paper presents data on the population PK of the first four antiviral drugs (the data on the PK of APV are the subject of a separate report [24], and the data on the PK of adefovir dipivoxil were not analyzed, as the drug is no longer being studied for the treatment of HIV infection) and the relationship between time to virological failure and EFV exposure.
MATERIALS AND METHODS
Study population and design.
The PK data are from a subset of the 481 HIV-positive patients participating in AACTG study 398 and nominally receiving the protocol-mandated drug regimens to which they were assigned. PK were evaluated at steady state. All patients were assigned to receive EFV (600 mg once a day [q.d.]), APV (1,200 mg twice a day [b.i.d.]), abacavir (300 mg b.i.d.), and adefovir dipivoxil (60 mg q.d.) with l-carnitine supplementation (500 mg q.d.). In addition, the patients received in four different treatment arms NFV (1,250 mg b.i.d.), IDV (1,200 mg b.i.d), SQV (soft-gel capsule formulation at 1,600 mg b.i.d.), or a placebo matched for one of the three PIs. Patients were selectively randomized to one of the four arms, with the selection depending on the PI used in the arm and the number and type of PIs to which the patient had previously been exposed. The institutional review boards of all participating institutions approved the study, and each subject gave written informed consent. For further details, see Hammer et al. (14).
Data. (i) PK data.
Three types of PK studies were performed during the trial. We designate these the 12-h PK study, the 6-h PK study, and the population PK study. Prior to each type of PK study, the patients were queried for the times of administration of their last three doses of each drug. Forty-six subjects participated in the 12-h PK study (of at least one drug) at approximately 2 weeks after the beginning of the study treatment, while 10 participated in the 6-h PK study at approximately 24 weeks. For the 12-h PK study, seven blood samples were obtained at time zero (predose sample) and at 1, 2, 4, 6, 8, and 12 h after supervised drug administration. For the 6-h PK study, samples were obtained only at the first five of these times. The protocol called for the latter design to be executed with equal numbers of individuals who had sustained a virological failure (as defined in the protocol) and who had not sustained a virological failure by 24 weeks. Execution problems prevented this plan from being realized. For the purposes of this analysis, subjects in the 6-h PK study were assumed to have been randomly chosen from the failure and nonfailure groups at 24 weeks, but not necessarily in proportion to the overall prevalence of patients who had sustained or not sustained a virological failure.
One hundred ninety-one patients participated in the population PK study for at least one drug during scheduled follow-up visits at weeks 16, 24, and/or 48, wherein a single blood sample was drawn. The protocol called for all patients to participate, but again, execution problems prevented this, and the subjects who were studied for each drug are regarded as a random sample of all subjects persisting in the study until the sampling times. Samples for the PK studies were drawn no sooner than 2 h after the stated times of dosing with the relevant drug(s). To ensure that not all samples were taken at the same time postdosing, for each patient at least one sample for population PK was scheduled to be obtained in the morning and another was scheduled to be obtained in the afternoon. Samples from all PK studies were assayed for EFV, NFV, IDV, and SQV concentrations. Plasma was separated by centrifugation and frozen at −70°C until it was analyzed.
Plasma EFV concentrations were measured by a method developed at Dupont and validated at the Laboratory for Antiviral Research at the State University of New York (Buffalo, N.Y.). By this method, EFV was extracted from 100 μl of plasma (anticoagulated with EDTA) by liquid-liquid extraction with ethylene dichloride, followed by centrifugation, aspiration of the aqueous layer, and evaporation under a nitrogen stream. Samples were reconstituted with the mobile phase and injected into a high-performance liquid chromatography (HPLC) system with UV detection at a wavelength of 250 nm. The calibration range is 100 to 10,000 ng/ml; the intra- and interassay variations were less than 12%.
The concentrations of NFV, IDV, and SQV were determined at the Clinical Pharmacology Laboratory at the Stanford University School of Medicine (Stanford, Calif.). The concentrations of NFV and SQV were determined by the same assay with an isocratic reverse-phase high-performance liquid chromatograph with UV detection. By this method, after solid-phase extraction, the final acetonitrile extracts of the cartridges were evaporated to dryness and the residues were reconstituted with fresh mobile phase. The prepared samples were analyzed on a Supercosil LC-DP column, which was eluted isocratically with a mobile phase containing phosphate buffer and acetonitrile. Ro-31-9564 (Roche Products, Ltd., Welwyn Garden City, United Kingdom) was used as an internal standard. NFV, SQV, and the internal standards were detected by determination of the UV absorbance at 240 nm. The linear dynamic ranges of the assays were 200 to 6,400 ng/ml for NFV and 87.5 to 5,600 ng/ml for SQV. The limits of quantification were 187.5 and 75 ng/ml for NFV and SQV, respectively. The inter- and intra-assay variations were within 15% for both compounds.
IDV and the internal standard (methyl IDV; Merck Research Laboratories) were extracted from 300 μl of each plasma sample by an ethyl ether extraction. The final ether layer was evaporated to dryness, and the residue was reconstituted with fresh mobile phase. The prepared samples were analyzed by reverse-phase HPLC with determination of the UV absorbance at 210 nm with a Microsorb MV C8 column. Elution was performed isocratically with a mobile phase containing phosphate buffer and acetonitrile. The range of IDV concentrations in plasma used to prepare the calibration curve was 78.1 to 5,000 ng/ml. The limit of quantification of IDV is 78.1 ng/ml. The inter- and intra-assay variations were within 10%.
Because of the protocol execution problems, we “cleaned” the data by removing outlier concentrations, e.g., those for patients with erroneous dosage histories, by checking the plausibility of each observed concentration against its prediction based on the most reliable data, the samples for the 12-h PK study, or the literature. In detail, a prior distribution for individual PK parameters for EFV was obtained by fitting a population PK model to the 12-h PK data (see also “PK model” below). Similar prior distributions were obtained for individual PK parameters for NFV, IDV, and SQV by reference to the literature (16, 29, 34) because the number of 12-h PK studies with these drugs was too small to reliably estimate a population model. The plausibility of each observed concentration was determined in terms of these prior predictive distributions, as follows. Concentrations with root mean square (population) weighted residuals lying outside the range of minus 2 to plus 5 prior standard deviations were flagged as implausible. Nineteen EFV, 25 NFV, 5 SQV, and no IDV concentrations were so flagged and were deleted. The remaining PK data consisted of 531 EFV concentrations (n = 139 patients), 219 NFV concentrations (n = 75 patients), 66 IDV concentrations (n = 11 patients), and 154 SQV concentrations (n = 51 patients). The total number of samples available for evaluation of the PK of NFV, IDV, and SQV were too few to permit investigation of the relationship between PK and covariates. The baseline characteristics tested as covariates in the EFV PK model (see below) are given in Table 1.
TABLE 1.
Baseline characteristics of AACTG study 398 patients for whom EFV PK data were available
| Characteristic | Value |
|---|---|
| No. (%) of patients | |
| NNRTI experienced | |
| No | 78 (56) |
| Yes | 61 (44) |
| Sex | |
| Male | 127 (91) |
| Female | 12 (9) |
| Race | |
| White, non-Hispanic | 95 (68) |
| Black, non-Hispanic | 22 (16) |
| Hispanic | 16 (11) |
| Asian, Pacific Islander | 4 (3) |
| American Indian, Alaska Native | 1 (1) |
| Other or unknown | 1 (1) |
| Age (yr)a | 40.2 (7.4) |
| Wt (kg)a | 77.5 (21.9) |
Data are given as means (standard deviations).
(ii) Adherence data.
Adherence to the medication regimen was assessed by two methods: by use of questionnaires (patient self-reporting and/or face-to-face interview) for all drugs and by use of electronic compliance monitoring caps (Medication Event Monitoring System [MEMS]; Aprex Corp., Union City, Calif.) for EFV (see the Appendix for details). Two subject-specific time-global adherence covariates were defined as the time-average of (i) the average across all individual questionnaire values for EFV and (ii) all EFV MEMS values. In addition, two subject- and time-specific adherence covariates were defined as (iii) adherence values from the questionnaire and (iv) values from MEMS; both of these were for the most recent 7 days prior to the PK study.
(iii) Virological outcome data.
Blood samples for determination of HIV RNA levels were collected in tubes with EDTA before study entry; at the time of entry into the study (day 0); at study weeks 2, 4, 8, 16, 24, 32, 40, and 48; at every 8 weeks thereafter; and at the time of confirmed virological failure. The plasma was separated by centrifugation and stored at −70°C until analysis. The Roche Amplicor Ultrasensitive assays for determination of HIV type 1 RNA levels in plasma were performed at Johns Hopkins University Laboratory (Baltimore, Md.). The lower limit of quantification was 200 copies/ml, and the upper limit of quantification was 750,000 copies/ml; samples with values >750,000 copies/ml were retested after they were diluted. The criteria defining virological failure are given in the Appendix.
PK model.
Separate population kinetic models were fit to the cleaned data for EFV, NFV, IDV, and SQV with the NONMEM (first-order method) computer program (4). The minimal PK model used, which served as the starting point for model building, is a one-compartment model with first-order absorption and exclusive hepatic elimination according to the classical well-stirred model (26, 33). The subject-specific parameters of this model for a generic drug (i.e., any one of EFV, IDV, NFV, or SQV) are the rate constant of absorption (Ka), volume of distribution (V), and clearance (CL), where CL is equal to Q CLint/(Q + CLint), in which Q is as defined below and CLint is intrinsic (hepatic) clearance. F is net bioavailability, which is equal to Fgut × Fhep, where Fhep, which is equal to Q/(Q + CLint), is the fraction of drug surviving a first passage through the liver and Fgut is the fraction of drug surviving the first passage across the gut wall. Assuming that drug is excluded from red blood cells, Q may be taken as hepatic plasma flow. To ensure identifiability, Q is usually fixed to a typical value; here, Q was equal to 50 liters/h. Fgut was arbitrarily fixed to unity. This left the parameters Ka, V, and CLint as the three unknown individual parameters.
To model intraindividual random effects (e.g., measurement errors), both additive and proportional components of error were used, as follows: y = f + f ɛ1 + ɛ2, where y is the observed concentration of drug, f is the model for its expectation, and the error ɛ = (ɛ1,ɛ2) is distributed N(0,Σ), where Σ is diagonal. The value of an element (e.g., Ka) of the parameter set for a given drug and specific individual i is given by Kai = Ka exp(ηi,Ka), where the vector ηi, which is equal to (ηi,Ka, ηi,CL, ηi,V)′, is multivariate normally distributed with a mean of zero (without loss of generality); the prime symbol indicates matrix transpose.
The typical (modal) population value for an element (e.g., Ka) of the individual parameter vector was modeled as a linear function of covariates X (such as age; categorical covariates are coded as indicator variables with values of 0 or 1): Ka = 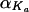 + X′
+ X′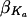 . Covariates evaluated for inclusion in the PK model for EFV were coadministered PIs (treatment arms), sex, race, age, weight, and the drug adherence measurements from the questionnaires and MEMS.
. Covariates evaluated for inclusion in the PK model for EFV were coadministered PIs (treatment arms), sex, race, age, weight, and the drug adherence measurements from the questionnaires and MEMS.
Model building.
The merit of a more complex model (one with more parameters) over a less complex submodel can be tested by a log-likelihood ratio test by use of Δobj, which is the difference in NONMEM objective functions (approximately minus two times the maximized log likelihood of the data) for the two models and which is referenced to its asymptotic chi-square distribution (with degrees of freedom equal to the difference in the number of free parameters) (7). However, because (i) the P values computed by the first-order method are known to be anticonservative (32) and (ii) the analyses herein are explanatory and not confirmatory (i.e., multiple significance tests are made, and they are data driven rather than hypothesis driven), P values should be regarded only as suggestive. For important inferences, a better approximation to the likelihood (the first-order conditional estimation method) was used and further statistical testing was done (see the next subsection). For the purposes of model building, we arbitrarily defined statistical significance as a nominal P value less than 0.01 in an attempt to preserve some conservatism.
Graphical displays for model building are based on residuals or weighted residuals. Between-subject residuals, useful for building the second-level model, are differences between the population average and subject-specific PK parameter estimates. The latter are available from NONMEM as maximum a posteriori (MAP) Bayes estimates. Observations thus predicted are called individual predictions (IPRED; in contrast to predictions made with estimates of population average parameters, which are called population predictions [PRED] and which are useful for judging the overall goodness of fit). Graphical displays for improving the within-subject first-level (PK) model are residuals or weighted residuals, obtained by using the individual predictions. A discussion of model building for hierarchical models can be found, for example, in the work by Pinheiro and Bates (25).
Inference.
To draw conclusions regarding the effects of covariates on EFV PK parameters when model building suggested that such effects might exist, we undertook more rigorous hypothesis testing. We used a two-sided t test based on subject-specific MAP Bayes estimates of parameters, contrasting those associated with one value of a dichotomous covariate (e.g., sex) with those associated with the other value, and a permutation test (5) by using Δobj as the test statistic but by referencing it to a simulation-based distribution rather than the chi-square distribution. See the Appendix for details of both tests.
A Cox proportional hazards model was used to test the potential relationship between PK of EFV and virological failure. Descriptive statistical analyses were performed with the software package Splus 5 for UNIX (Insightful Corporation, Seattle, Wash.).
RESULTS
We could not reliably fit a model to the SQV PK data (similar difficulties with the fitting of SQV PK data have been reported previously [29]), and therefore, we do not discuss them further.
Population PK parameters.
Parameter estimates for the final population PK models are given in Table 2. Figure 1 shows the goodness of fit of the final EFV PK model to the EFV data. This model is discussed further below.
TABLE 2.
EFV, IDV, and NFV PK parameter estimatesa
| Drug | CLint (liters/h) | CL/F (liters/h) | CVCL/F (%) | V/F (liters) | CVCL/F (%) | Ka (h−1) | CVKa (%) | tlag (h) |
|---|---|---|---|---|---|---|---|---|
| EFV | 9.0 (5.9) | 10.8 | 42 | 282 (7.2) | 7.9 | 1.39 (10) | 93 | |
| IDV | 123 (17) | 40 | 101 (24) | 88 | 1.5 (8.5) | 144 | 0.54 | |
| NFV | 41 (29) | 9 | 120 (2.8) | 2 | 0.4 (310) | 50 |
Data are given as means (percent standard errors, where available). CV, coefficient of interindividual variation; tlag, lag time.
FIG. 1.

Goodness-of-fit plots of the final PK model for EFV. Left panels, observed (OBS) concentrations (C) versus PRED and IPRED; heavy dashed lines, smoothing of concentrations versus PRED and IPRED. Right panels, population residuals (RES) and weighted individual residuals (IWRES) versus PRED and IPRED; heavy dashed lines, smoothing of RES (IWRES) versus PRED (IPRED). The fine dashed lines indicate lines of identity in the left panels and the ordinate value of 0 in the right panels.
EFV PK and covariates.
To assess factors associated with differences in the CLint of EFV between patients, covariates were included stepwise in the baseline PK model. The inclusion of effects for administration of a codrug (NFV, IDV, or SQV), age, or weight on the PK parameters CLint and Fgut did not improve the goodness of fit over that for the baseline model. The goodness of fit did improve with the inclusion of effects for race (and sex) on CLint. Since only 6 of a total of 139 patients were other than white non-Hispanic, African American, and Hispanic race (see Table 1), we did not use data for those patients to further investigate the apparent effect of race on CLint. The mean CLint of EFV was 29 and 27% higher for white non-Hispanics than for African Americans and Hispanics, respectively. African Americans and Hispanics are hereafter considered one group for the purposes of describing EFV PK, as the average PK parameters for the groups did not differ. By the use of only two groups for race, the mean EFV CLint appeared to be 28% higher for white non-Hispanics than African Americans and Hispanics (Fig. 2). To test this finding further, we applied both the permutation test and the t test. These tests indicate considerable support for the effect of race (P < 0.001 by the permutation test; P = 0.03 by the t test). The same tests provide only marginal support (if any) for an effect of sex (P = 0.27 by the permutation test; P = 0.01 by the t test).
FIG. 2.

MAP Bayes estimates of individual CL/F values obtained from the baseline PK model versus the covariate race (white non-Hispanic versus African American and Hispanic). Boxes, interquartile ranges (third quartile to first quartile); whiskers, 1.5 times the interquartile ranges; white horizontal lines, medians (50th percentile).
Imperfect adherence and misspecification of dosage are indistinguishable in the model. Hence, adherence was tested as a covariate that acts on Fgut. Only the time-specific MEMS measure demonstrated that such an effect was significant, and this was demonstrated only when MEMS was a covariate affecting the population PK data (those concentrations determined after an immediately preceding dose that was not observed). Specifically, a 16% increment in the time-specific MEMS value from the population average value of 0.86 to 1.0 (i.e., perfect drug adherence) was associated with only a marginal 4.3% increase in the apparent Fgut for the samples from the population PK study.
EFV exposure and virological failure.
Individual MAP Bayes estimates of EFV CL/F obtained from the baseline PK model correlated with time to virological failure. According to the Cox model, the hazard of virological failure was eightfold greater for subjects exposed to NNRTIs than for subjects unexposed to NNRTIs. Among the exposed subjects, the hazard of virological failure was relatively uncorrelated with CL/F. In patients not previously exposed to NNRTIs, however, the hazard of virological failure was 2.65-fold greater per 100% increase in CL/F (P < 0.0003). Figure 3 shows the relationship between EFV CL/F and the probability of virological failure at 1 year. Figure 3B shows that in individuals not previously exposed to NNRTIs, this probability increases from 0.45 to 0.7 with an increase in CL/F from the population average value to 150% of that value.
FIG. 3.

Probability of virological failure by 1 year versus scaled individual EFV CL/F among patients in AACTG study 398 with EFV PK data. (A) Patients previously exposed to NNRTIs; (B) patients not previously exposed to NNRTIs. CL/F is expressed as the percentage of the population average CL/F. Points are individual datum points: virological failure was given a value of 1, and virological nonfailure was given a value of 0. Data for only two individuals were censored before 300 days (plotted as nonfailures). The shaded areas in both plots delimit pointwise 90% confidence intervals obtained by bootstrapping (10) the data (500 replications). The solid central line in each plot is the mean of the bootstrapped smoothed data.
DISCUSSION
This study reports on the PK interactions between EFV and coadministered PIs, the effects of covariates on EFV PK, and the relationship between EFV PK and the hazard of virological failure in AACTG study 398. We first discuss our findings with respect to the population PK of NFV and IDV and subsequently discuss those with respect to EFV, the primary focus of this report.
The estimate of the mean CL/F of NFV herein (in the presence of coadministered EFV) is 41 liters/h, which is close to the value (41.9 to 45.1 liters/h) in the absence of EFV (or some other interacting drug) reported by Jackson et al. (16). Fiske et al. (W. D. Fiske, I. H. Benedek, S. J. White, et al., Abstr. 5th Conf. Retrovir. Opportunistic Infect., abstr. 349, 1998) reported an increase in the area under the concentration-time curve for NFV of 15 to 20% with EFV coadministration, perhaps due to inhibition by EFV of CYP2C19 (31), a major metabolizing enzyme for NFV (18; J. H. Lillibridge, C. A. Lee, Y. K. Pithavala, et al., Abstr. 12th Am. Assoc. Pharm. Sci. Annu. Meet. Exposition, abstr. 3035, 1998). This reported increase in the area under the concentration-time curve for NFV is small, and if it is real, it would easily be missed here, as we made no crossover comparison. On the other hand, induction of CYP3A4 by EFV might be expected to diminish the activity of NFV (23). Three of the five different metabolic pathways identified for NFV involve CYP3A4 (CYP2C19 and CYP2D6 are involved in the other two), which is also responsible for the metabolism of M8, which is a pharmacologically active metabolite of NFV (and which is formed via CYP2C19) (2; Lillibridge et al., Abstr. 12th Am. Assoc. Pharm. Sci. Annu. Meet. Exposition, abstr. 3035, 1998; T. M. Sandoval, H. M. Grettenberger, K. E. Zhang, et al., Abstr. 12th Am. Assoc. Pharm. Sci. Annu. Meet. Exposition, abstr. 1096, 1998).
Our estimate of IDV CL/F, 123 liters/h, is greater than previously reported values of 47 liters/h (34) and 55 liters/h (at a mean body weight of 70 kg) (17). This may be the result of induction of CYP3A4 by the drugs coadministered in this study, as IDV is primarily metabolized by this isozyme (6). EFV has been reported to increase the CL/F of IDV, although by only 35% (W. D. Fiske, D. Mayers, K. Wagner, et al., Abstr. 4th Conf. Retrovir. Opportunistic Infect., abstr. 535, 1997), a value still considerably less than the one that we found. Our higher value may be a consequence of dual induction by EFV and APV, as the latter, although generally regarded as an inhibitor of CYP3A, can also act as an inducer (1, 19, 23; U. S. Justesen, N. A. Klitgaard, K. Brosen, and C. Pedersen, Abstr, 9th Conf. Retrovir. Opportunistic Infect., abstr. 442, 2002). On the other hand, the IDV CL/F reported here is similar to the value (110 liters/h) reported by Letendre et al. (20) when IDV was administered without the concomitant administration of an NNRTI. However, those investigators used only one blood sample per patient.
Turning now to the population PK of EFV, the absence of PK differences between treatment arms indicates that coadministration of NFV, IDV, or SQV does not differentially affect the PK of EFV, at least to a degree discernible in our data. This result agrees with the results of a previous report (30), which described an investigation of the effect of NFV on EFV clearance. In that report the mean CL/F of EFV was 12.6 liters/h when the drug was given alone and 11.2 liters/h in the presence of NFV. In healthy volunteers, EFV concentrations are only marginally decreased by SQV and are not affected by IDV (DuPont Research Laboratories, data on file, 1998).
The estimated population average CL/F of EFV (10.8 liters/h) in the present study was slightly smaller than that previously reported in patients (30), although a recent report (23) has a value at steady state in healthy volunteers given EFV at 200 mg/day almost identical to that found in the present study.
Interestingly, EFV CLint appears to be 28% higher in white non-Hispanics than in African American and Hispanic patients in AACTG study 398. Such a difference between these groups was not observed in a prior study (A. S. Joshi, J. S. Barrett, M. Chai, W. D. Fiske, and T. M. Ludden, Abstr. Am. Assoc. Pharm. Sci. Annu. Meet. Exposition, abstr. 1316, 1998). Moreover, with all else being equal, higher levels of clearance should lead to a higher failure rate, but African Americans receiving EFV have recently been found to have a shorter, not a longer, median time to failure than Caucasians (422 versus 1,400 days) (S. Wegner, M. Vahey, M. Dolan, M. Wallace, N. Aronson, A. Barile, W. Emmons, S. Frazier, K. Stephan, M. Nau, S. Piscitelli, R. Harrigan, and B. Larder, Abstr. 9th Conf. Retrovir. Opportunistic Infect., abstr. 428, 2002).
As in any observational study, confounding is possible and may explain the apparent difference in clearance by race. One candidate as a confounder is drug adherence: it could explain the apparent effect of race if it differed between races. However, comparison of drug adherence as measured herein for white non-Hispanics versus that for African Americans and Hispanics did not show any differences compatible with such confounding. Also, the observation that the addition of an effect of race on CLint improved the PK model but that the addition of an effect of race on Fgut did not showed that adherence is not a confounder (adherence would be expected to appear as an effect on Fgut, not CLint). Lastly in this regard, differences in CLint between races from our data were not correlated with differences in baseline covariates (RNA, CD4 and CD8 cell, and albumin levels) between races in any mechanistically plausible way.
Possible explanations for the effect of race on hepatic clearance, if it is real, are differences in multidrug resistance transporter type 1 (MDR-1) genotypes and/or the activities of CYP450 enzymes such as CYP2D6 (12, 27). Indeed, Fellay et al. (12) reported a statistically significant relationship between the MDR-1 genotype (MDR-1 codes for P glycoproteins, which are differentially expressed between African Americans and Caucasians) and EFV PK. A relationship between the CYP2D6 genotype and the concentrations of EFV and NFV was also noted in the same study. As no genotype data were collected in AACTG study 398, no conclusions can be drawn regarding the influence of transporter and CYP450 isozymes on EFV PK in AACTG study 398 patients.
Other demographic covariate effects, including that of sex on EFV CLint, were not statistically significant. This is in line with data reported by DuPont Research Laboratories (Joshi et al., Abstr. Am. Assoc. Pharm. Sci. Annu. Meet. Exposition, abstr. 1316, 1998). However, since women represented only 9% of all patients in our study, the lack of a significant effect constitutes scant evidence against such an effect.
Interestingly, the inclusion of none of the adherence measures except the time-specific MEMS variable as covariates improved the model fit; the time-specific MEMS variable affected only the apparent bioavailability for the samples in the population PK study. Extrapolation to perfect drug adherence from 86% adherence (population average) predicts a maximal 4.3% increase in gut bioavailability (a surrogate for drug intake in the present study). The lack of a plausible relationship between any adherence measure and data from all PK studies (i.e., the 12- and 6-h PK studies, in addition to the population PK study), despite considerable variation in adherence, may be due to the fact that our data are dominated by EFV concentrations from the 12- and 6-h PK studies, both of which are rendered relatively insensitive to variation in adherence because ingestion of the prior dose was ensured.
The most interesting finding reported here is that a high EFV CL/F is associated with a reduced time to virological failure in NNRTI-naïve patients (among the NNRTI-experienced patients, virological failure is much more common and no relationship to EFV CL/F is observed). The fact that the antiviral effect of the NNRTI EFV is substantially compromised in patients with prior NNRTI experience was not unexpected (11, 15), nor was the presence of an exposure-response relationship (Fig. 3B) among those who were NNRTI naïve. Although Fiske et al. (W. D. Fiske et al., Abstr. 41st Intersci. Conf. Antimicrob. Agents Chemother., abstr 1-1727, 2001) found no discernible relationship between EFV PK and virologic response in 166 subjects, Marzolini et al. (22) observed that the probability of failure was inversely related to EFV concentrations in plasma drawn 8 to 20 h after administration of the daily EFV dose in their 130 patients on a steady-state regimen. A concentration-dependent response to EFV has also been observed by Joshi et al. (A. S. Joshi, J. S. Barrett, W. D. Fiske, et al., Abstr. 39th Intersci. Conf. Antimicrob. Agents Chemother., abstr. 1201, 1998).
Because of the study design, analysis method, and availability of PK-independent data on adherence in the present study, our results go beyond those described above and provide the first clear-cut evidence that variations in levels of EFV exposure resulting from true variability in PK may be responsible for variations in efficacy. Indeed, the striking finding in the present study, that there is no hint of flattening of the exposure-effect curve even at the lowest clearances (highest levels of EFV exposure), suggests that standard doses of EFV in patients such as those evaluated in AACTG study 398, a population restricted to patients who had failed PI treatment, yield levels of exposure below those associated with the maximal possible antiviral effect, even among those individuals with the highest levels of natural exposure.
Acknowledgments
This work was supported in part by NIH grants AI38858 and AI38855. L.L. is the recipient of support from the Canadian Institutes of Health Research and the Fonds de la Recherche en Santé du Québec.
We thank P. Vecchione, J. Giardini, K. Wood, J. F. Lu, T. F. Blaschke, and the AACTG 398 study team and participants.
APPENDIX
Adherence measures.Drug adherence is measured by two methods: by the use of electronic compliance monitoring caps and by the use of questionnaires. By both methods, two different adherence fractions (discussed further below) are computed and used as covariates: (i) the global time average of all adherence fraction values and (ii) the local time average of adherence fraction values for the week of the PK study.
(i) Adherence fraction from electronic compliance monitoring caps.Electronic compliance monitoring caps (MEMS; Aprex Corp.) record the time of each occasion that a medication vial is opened. At the time of initial dispensing of EFV, monitoring caps were assigned to each subject. After the subjects had taken all the capsules from a given medication bottle, the subjects were instructed to transfer the cap from that bottle to the next (full) one, from which they continued medicating themselves. The subjects were asked to bring their medication bottles and caps to the clinic at each study visit (weeks 2, 4, 8, 12, 16, 24, 32, 40, and 48 and every 8 weeks thereafter), where the cap data were downloaded to computer files and stored for later analysis. For our analyses, a daily MEMS adherence indicator variable for EFV was computed for each day on which the treatment record indicated that the patient was assigned to take the study-specified dose of EFV and on which the electronic compliance monitoring cap for EFV was in use. The indicator variable was set to 1 if the MEMS record indicated that the EFV medication bottle was opened at least once during the 24 h between 3 a.m. of the indicated day and 3 a.m. of the next day. The MEMS adherence value was otherwise set equal to 0 for all doses except those doses for which the individual stated that he or she had previously removed tablets for later ingestion. In that case, if weekly MEMS records indicated a pattern consistent with that claim, the MEMS adherence value for such a dose was fixed to 0.5. The global time average MEMS adherence fraction is the average of MEMS adherence values for all monitored doses; the local time average value for a given PK study is the average for all monitored doses in the preceding 7 days (time-specific MEMS).
(ii) Adherence fraction from adherence questionnaire.All subjects completed an adherence questionnaire (AACTG study 398 questionnaire QL0702) at study weeks 4, 8, 16, 24, 32, 40, and 48 and every 8 weeks thereafter. The questionnaire was completed by the study participant and/or by a face-to-face interview with study personnel. The patient was asked to specify the number of prescribed doses of each drug that he or she had failed to take on each of the preceding 4 days. An adherence fraction was calculated for EFV for each questionnaire as 1 minus the ratio of the total number of missed doses over the 4 days to the nominally prescribed number of doses over those days. The local and global time average questionnaire adherence fractions were calculated from these ratios. The global time average is the time-weighted average of all of the adherence fractions (one per questionnaire) available for an individual. The local time average for a given PK study is the single adherence fraction computed from the questionnaire administered closest in time to the study within 1 week, if available (time-specific adherence questionnaire).
Virological failure.Virological failure is defined (according to the protocol for AACTG study 398) as two consecutive plasma HIV RNA levels of ≥200 copies/ml after two consecutive levels below this limit, a confirmed rise in plasma HIV-1 RNA levels above the baseline level (the average of preentry and entry levels), a confirmed 1-log10 rise in the plasma HIV-1 RNA level above the nadir plasma HIV-1 RNA level (the lowest recorded level while receiving study treatment), a failure to achieve a >0.5-log10 decline in the plasma HIV-1 RNA level from the baseline level by week 8 of treatment, or confirmed HIV RNA levels ≥200 copies/ml at or after week 24.
Hypothesis tests. (i) t test.A t test is used to test the null hypothesis of equal means between groups with different covariate values (e.g., males and females) of subject-specific MAP Bayes estimates of parameters. For the purposes of this test, the MAP Bayes estimates are obtained with the baseline PK model, i.e., one that lacks all covariates. This induces a conservative bias because MAP Bayes estimates are shrinkage estimates; i.e., they are all biased toward the population mean common to both groups. Note, however, that the t-test assumption of independence is not strictly met, as all MAP Bayes estimates are correlated through the effect of the prior (population) model. It is not clear how this correlation will affect the validity of the test.
(ii) Permutation test.The permutation test uses the log-likelihood ratio statistic (Δobj) but does not make any assumptions about the reference distribution. For the sake of concreteness, the permutation test procedure given below is defined for a test for the effect of a dichotomous covariate (sex) on CLint.
First (step 1), define test statistic S; here, S is Δobj, the difference between the minimum objective function from the fit of a PK model with the covariate sex influencing CLint and the fit of a model without such an influence. Second (step 2), compute S0, the value of S with data from the original data set. Third (step 3), for m equal to 1 to M, create a new data set from the original data set by reassigning the value of the covariate sex for each individual [sex(i); i = 1,N] to the value of sex[r(i)], where r is a random permutation of the integers 1,N, and calculate and record the test statistic Sm = S for this data set. Finally (step 4), the P value for the permutation test is the fraction of Sm greater than S0. M (step 3) is taken to be large enough (e.g., 1,000) so that the standard error of the P value (step 4) is <10% of the P value itself.
REFERENCES
- 1.Adkins, J. C., and S. Noble. 1998. Efavirenz. Drugs 56:1055-1064. [DOI] [PubMed] [Google Scholar]
- 2.Baede-van Dijk, P. A., P. W. Hugen, C. P. Verweij-van Wissen, P. P. Koopmans, D. M. Burger, and Y. A. Hekster. 2001. Analysis of variation in plasma concentrations of nelfinavir and its active metabolite M8 in HIV-positive patients. AIDS 15:991-998. [DOI] [PubMed] [Google Scholar]
- 3.Barry, M., F. Mulcahy, C. Merry, S. Gibbons, and D. Back. 1999. Pharmacokinetics and potential interactions amongst antiretroviral agents used to treat patients with HIV infection. Clin. Pharmacokinet. 36:289-304. [DOI] [PubMed] [Google Scholar]
- 4.Beal, S., L. B. Sheiner, et al. 1998. NONMEM user's guides. University of California San Francisco, San Francisco, Calif.
- 5.Bradley, W. A. 1968. Distribution-free statistical tests. Prentice-Hall, Inc., Englewood Cliffs, N.J.
- 6.Chiba, M., M. Hensleigh, J. A. Nishime, S. K. Balani, and J. H. Lin. 1996. Role of cytochrome P450 3A4 in human metabolism of MK-639, a potent human immunodeficiency virus protease inhibitor. Drug Metab. Dispos. 24:307-314. [PubMed] [Google Scholar]
- 7.Cox, D. R., and D. V. Hinkley. 1974. Theoretical statistics. Chapman & Hall, Ltd., London, United Kingdom.
- 8.Decker, C. J., L. M. Laitinen, G. W. Bridson, S. A. Raybuck, R. D. Tung, and P. R. Chaturvedi. 1998. Metabolism of amprenavir in liver microsomes: role of CYP3A4 inhibition for drug interactions. J. Pharm. Sci. 87:803-807. [DOI] [PubMed] [Google Scholar]
- 9.Eagling, V. A., D. J. Back, and M. G. Barry. 1997. Differential inhibition of cytochrome P450 isoforms by the protease inhibitors, ritonavir, saquinavir and indinavir. Br. J. Clin. Pharmacol. 44:190-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Efron, B. 2000. The bootstrap and modern statistics. J. Am. Stat. Assoc. 95:1293-1296. [Google Scholar]
- 11.Falloon, J., M. Ait-Khaled, D. A. Thomas, C. L. Brosgart, J. J. Eron, Jr., J. Feinberg, T. P. Flanigan, S. M. Hammer, P. W. Kraus, R. Murphy, R. Torres, and H. Masur. 2002. HIV-1 genotype and phenotype correlate with virological response to abacavir, amprenavir and efavirenz in treatment-experienced patients. AIDS 16:387-396. [DOI] [PubMed] [Google Scholar]
- 12.Fellay, J., C. Marzolini, E. R. Meaden, D. J. Back, T. Buclin, J. P. Chave, L. A. Decosterd, H. Furrer, M. Opravil, G. Pantaleo, D. Retelska, L. Ruiz, A. H. Schinkel, P. Vernazza, C. B. Eap, and A. Telenti. 2002. Response to antiretroviral treatment in HIV-1-infected individuals with allelic variants of the multidrug resistance transporter 1: a pharmacogenetics study. Lancet 359:30-36. [DOI] [PubMed] [Google Scholar]
- 13.Haas, D. W., W. J. Fessel, R. A. Delapenha, H. Kessler, D. Seekins, M. Kaplan, N. M. Ruiz, L. M. Ploughman, D. F. Labriola, and D. J. Manion. 2001. Therapy with efavirenz plus indinavir in patients with extensive prior nucleoside reverse-transcriptase inhibitor experience: a randomized, double-blind, placebo-controlled trial. J. Infect. Dis. 183:392-400. [DOI] [PubMed] [Google Scholar]
- 14.Hammer, S. M., F. Vaida, K. K. Bennett, M. K. Holohan, L. Sheiner, J. J. Eron, L. J. Wheat, R. T. Mitsuyasu, R. M. Gulick, F. T. Valentine, J. A. Aberg, M. D. Rogers, C. N. Karol, A. J. Saah, R. H. Lewis, L. J. Bessen, C. Brosgart, V. DeGruttola, and J. W. Mellors. 2002. Dual vs single protease inhibitor therapy following antiretroviral treatment failure: a randomized trial. JAMA 288:169-180. [DOI] [PubMed] [Google Scholar]
- 15.Hirsch, M. S., F. Brun-Vezinet, R. T. D'Aquila, S. M. Hammer, V. A. Johnson, D. R. Kuritzkes, C. Loveday, J. W. Mellors, B. Clotet, B. Conway, L. M. Demeter, S. Vella, D. M. Jacobsen, and D. D. Richman. 2000. Antiretroviral drug resistance testing in adult HIV-1 infection: recommendations of an International AIDS Society-USA Panel. JAMA 283:2417-2426. [DOI] [PubMed] [Google Scholar]
- 16.Jackson, K. A., S. E. Rosenbaum, B. M. Kerr, Y. K. Pithavala, G. Yuen, and M. N. Dudley. 2000. A population pharmacokinetic analysis of nelfinavir mesylate in human immunodeficiency virus-infected patients enrolled in a phase III clinical trial. Antimicrob. Agents Chemother. 44:1832-1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kakuda, T. N., L. M. Page, P. L. Anderson, K. Henry, T. W. Schacker, F. S. Rhame, E. P. Acosta, R. C. Brundage, and C. V. Fletcher. 2001. Pharmacological basis for concentration-controlled therapy with zidovudine, lamivudine, and indinavir. Antimicrob. Agents Chemother. 45:236-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khaliq, Y., K. Gallicano, I. Seguin, K. Fyke, G. Carignan, D. Bulman, A. Badley, and D. W. Cameron. 2000. Single and multiple dose pharmacokinetics of nelfinavir and CYP2C19 activity in human immunodeficiency virus-infected patients with chronic liver disease. Br. J. Clin. Pharmacol. 50:108-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khanlou, H., E. Graham, M. Brill, and C. Farthing. 2002. Drug interaction between amprenavir and lopinavir/ritonavir in salvage therapy. AIDS 16:797-798. [DOI] [PubMed] [Google Scholar]
- 20.Letendre, S. L., E. V. Capparelli, R. J. Ellis, and J. A. McCutchan, and the HIV Neurobehavioral Research Center Group. 2000. Indinavir population pharmacokinetics in plasma and cerebrospinal fluid. Antimicrob. Agents Chemother. 44:2173-2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lillibridge, J. H., B. H. Liang, B. M. Kerr, S. Webber, B. Quart, B. V. Shetty, and C. A. Lee. 1998. Characterization of the selectivity and mechanism of human cytochrome P450 inhibition by the human immunodeficiency virus-protease inhibitor nelfinavir mesylate. Drug Metab. Dispos. 26:609-616. [PubMed] [Google Scholar]
- 22.Marzolini, C., A. Telenti, L. A. Decosterd, G. Greub, J. Biollaz, and T. Buclin. 2001. Efavirenz plasma levels can predict treatment failure and central nervous system side effects in HIV-1-infected patients. AIDS 15:71-75. [DOI] [PubMed] [Google Scholar]
- 23.Mouly, S., K. S. Lown, D. Kornhauser, J. L. Joseph, W. D. Fiske, I. H. Benedek, and P. B. Watkins. 2002. Hepatic but not intestinal CYP3A4 displays dose-dependent induction by efavirenz in humans. Clin. Pharmacol. Ther. 72:1-9. [DOI] [PubMed] [Google Scholar]
- 24.Pfister, M., L. Labbe, J. F. Lu, S. M. Hammer, J. Mellors, K. K. Bennett, S. Rosenkranz, and L. B. Sheiner. 2002. Effect of coadministration of nelfinavir, indinavir, and saquinavir on the pharmacokinetics of amprenavir. Clin. Pharmacol. Ther. 72:133-141. [DOI] [PubMed] [Google Scholar]
- 25.Pinheiro, J. C., and D. M. Bates. 2000. Statistics and computing. Mixed-effects models in S and S-plus. Springer, New York, N.Y.
- 26.Rowland, M., and T. N. Tozer. 1989. Clinical pharmacokinetics: concepts and applications, 2nd ed. Lea & Febiger, Philadelphia, Pa.
- 27.Schwartz, J. B. 2001. Race but not age affects erythromycin breath test results in older hypertensive men. J. Clin. Pharmacol. 41:324-329. [DOI] [PubMed] [Google Scholar]
- 28.Staszewski, S., J. Morales-Ramirez, K. T. Tashima, A. Rachlis, D. Skiest, J. Stanford, R. Stryker, P. Johnson, D. F. Labriola, D. Farina, D. J. Manion, N. M. Ruiz, et al. 1999. Efavirenz plus zidovudine and lamivudine, efavirenz plus indinavir, and indinavir plus zidovudine and lamivudine in the treatment of HIV-1 infection in adults. N. Engl. J. Med. 341:1865-1873. [DOI] [PubMed] [Google Scholar]
- 29.Vanhove, G. F., H. Kastrissios, J. M. Gries, D. Verotta, K. Park, A. C. Collier, K. Squires, L. B. Sheiner, and T. F. Blaschke. 1997. Pharmacokinetics of saquinavir, zidovudine, and zalcitabine in combination therapy. Antimicrob. Agents Chemother. 41:2428-2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Villani, P., M. B. Regazzi, F. Castelli, P. Viale, C. Torti, E. Seminari, and R. Maserati. 1999. Pharmacokinetics of efavirenz (EFV) alone and in combination therapy with nelfinavir (NFV) in HIV-1 infected patients. Br. J. Clin. Pharmacol. 48:712-715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.von Moltke, L. L., D. J. Greenblatt, B. W. Granda, G. M. Giancarlo, S. X. Duan, J. P. Daily, J. S. Harmatz, and R. I. Shader. 2001. Inhibition of human cytochrome P450 isoforms by nonnucleoside reverse transcriptase inhibitors. J. Clin. Pharmacol. 41:85-91. [DOI] [PubMed] [Google Scholar]
- 32.Wahlby, U., E. N. Jonsson, and M. O. Karlsson. 2001. Assessment of actual significance levels for covariate effects in NONMEM. J. Pharmacokinet. Pharmacodyn. 28:231-252. [DOI] [PubMed] [Google Scholar]
- 33.Wilkinson, G. R., and D. G. Shand. 1975. Commentary: a physiological approach to hepatic drug clearance. Clin. Pharmacol. Ther. 18:377-390. [DOI] [PubMed] [Google Scholar]
- 34.Zhou, X. J., D. V. Havlir, D. D. Richman, E. P. Acosta, M. Hirsch, A. C. Collier, P. Tebas, and J. P. Sommadossi. 2000. Plasma population pharmacokinetics and penetration into cerebrospinal fluid of indinavir in combination with zidovudine and lamivudine in HIV- 1-infected patients. AIDS 14:2869-2876. [DOI] [PubMed] [Google Scholar]