

Summary
Growing evidence supports the view that enzymatic activity results from a subtle interplay between chemical kinetics and molecular motions. A systematic analysis is performed here to delineate the type and level of coupling between catalysis and conformational mechanics. The dynamics of a set of 98 enzymes representative of different EC classes are analyzed with the Gaussian network model (GNM) and compared with experimental data. In more than 70% of the examined enzymes, the global hinge centers predicted by the GNM are found to be colocalized with the catalytic sites experimentally identified. Low translational mobility (<7%) is observed for the catalytic residues, consistent with the fine-tuned design of enzymes to achieve precise mechanochemical activities. Ligand binding sites, while closely neighboring catalytic sites, enjoy a moderate flexibility to accommodate the ligand binding. These findings could serve as additionalcriteria for assessing drug binding residues and could lessen the computational burden of substrate docking searches.
Introduction
Understanding the relationship between protein structure and biochemical function is of utmost importance for effective design or inhibition of proteins. Despite the rapidly increasing number of known structures and the advances in techniques for probing activity, relatively few studies have systematically investigated the connection between catalytic function and conformational dynamics. While several groups have examined the molecular dynamics of individual enzymes, only recently has conformational dynamics been recognized as a mechanism supporting catalytic activity (Benkovic and Hammes-Schiffer, 2003; Daniel et al., 2002; Diaz et al., 2003; Luo and Bruice, 2004; Ringe and Petsko, 2004; Tousignant and Pelletier, 2004; Agarwal et al., 2004; Eisenmesser et al., 2002; Clark, 2004; Kohen et al., 1999; Wolf-watz et al., 2004).
Thornton and collaborators recently created a data set (CATRES) in which structural and physicochemical data on 615 catalytic residues have been compiled (Bartlett and Thornton, 2002). The catalytic residues in the data set were defined according to well-defined criteria and experimental data reported for 176 nonhomologous enzymes. Properties compiled in CATRES include amino acid type, secondary structure, solvent accessibility, flexibility, conservation, and quaternary structure and function. In particular, the low temperature factors of catalytic residues, along with their preferred coiled conformations, are discussed. Using a neural network algorithm and spatial clustering, Thornton and coworkers could predict the catalytic site with an accuracy rate of 69% for a number of test enzymes (Gutteridge et al., 2003). More recently, a new resource, the Catalytic Site Atlas (CSA) (http://www.ebi.ac.uk/thornton-srv/databases/CSA/), was made available by the same group (Porter et al., 2004). The CSA contains both the hand-curated CATRES entries and homologous entries generated by multiple sequence alignments and covers about 27% of the enzyme structures deposited in the Protein Data Bank (PDB) (Berman et al., 2000).
Recently, Ma and coworkers have brought attention to the possibility of accurately describing protein dynamics in the absence of amino acid sequence and atomic coordinates (Ming et al., 2002a, 2002b). The main point is to take rigorous account of the protein architecture, described by the interresidue contact topology, through using an elastic network (EN) formalism (Bahar et al., 1997; Atilgan et al., 2001). This and other studies based on EN models lend support to the view that proteins possess mechanical characteristics uniquely defined by their particular architecture, regardless of their chemical properties. It also raises other questions. To what extent are these structure-induced mechanical properties functional? Is there any coupling between conformational mechanics and chemical activity? Can we identify potentially functional residues by merely examining the enzyme dynamics?
We present here the results from a set of 98 nonredundant, nonhomologous enzymes, 24 of which are inhibitor bound enzymes extracted from the PDB (Set 1; Table 1), and 74 of which are monomeric enzymes taken from CATRES (Set 2). Set 1 provides information on 104 catalytic residues and 159 ligand binding residues. Set 2 provides information on 253 catalytic residues.
Table 1.
Correlation between Functional Sites from Experiments and Computations
| Experimental Datac | Theoretical Resultsd,e | |||||
|---|---|---|---|---|---|---|
| PDBa | Protein Name | Sizeb | Catalytic Res. | Ligand Binding Res. | Key Mechanical Res. | Odds Ratio |
| 10GS | Human glutathione S-transferase P1-1 | 2 × 209 | 7, 8, 13, 38, 44, 51, 52, 64, 65, 98 | A, B: 8, 10, 13, 38, 51, 52, 64, 65, 98, 108 | A, B: 47–50 | 6.4 |
| 1A16 | Aminopeptidase P | 440 | 260, 271, 354, 361, 383, 406 | 350, 354, 361, 404 | 168–181 | 4.2 |
| 1A30 | HIV-1 protease | 2 × 99 | A25, A30, B25 | A27, A29, A48–A50, B23, B81, B84 | A, B: 25–28, 47–54 | 4.2 |
| 1A3B | Human α-thrombin heavy chain | 245 + 14 | 57, 195 | 57, 60A, 60D, 189, 194,195, 215, 219 | 95–102, 121–123, 132–138, 158–176, 198–208, 212–220, 221–228 | 2.1 |
| 1A42 | Human carbonic anhydrase II | 260 | 64, 92, 94, 96, 119 | 64, 92, 106, 131, 198, 199, 200, 202 | 44–53, 142–148, 186–191, 210–215, 243–245 | 2.3 |
| 1A47 | CGTase | 683 | 101, 141, 228, 230, 258, 328, 329 | 197, 371 | 131–148, 247–262, 496–510 | 3.1 |
| 1A5I | Plasminogen activator | 244 | 57, 102, 156, 194, 195 | 57 | 90–105, 120–123, 135–141, 155–161, 183–192, 194–209 | 2.8 |
| 1A5V | Asv integrase | 54–199 | 64, 121, 157 | 62, 119, 154, 155, 158 | 62–67, 76–82, 153–158 | 5.4 |
| 1AEC | Actinidin | 218 | 25 | 19 ,24, 26, 66, 68, 69, 162 | 7–19, 113–115 | 5.6 |
| 1AL8 | Glycolate oxidase | 359 | 24, 108, 129, 257 | 24, 108, 129, 161, 254, 257 | 80–106, 150–161, 225–258 | 2.4 |
| 1ARZ | E. coli dihydrodipicolinate reductase | 4 × 273 | B–D: 159, 160, 163 | B–D: 12, 13, 16, 17, 34, 39, 81, 84, 88, 102, 104, 127, 129, 163, 169, 170 | A, B: 134–195, 197–239; C, D: 147–164, 189–216 | .61 |
| 1B3N | β-ketoacyl carrier protein synthase | 412 | 163, 398–401 | 107, 108, 111, 163, 193, 198, 202, 303, 340, 342, 398–401 | 41–56, 145–219 | 3.6 |
| 1B6A | Methionine aminopeptidase 2 | 110–478 | 231 | 219, 328, 331, 339, 340, 376, 444, 447 | 163–271, 363–381, 445–462 | 2.4 |
| 1BGQ | N-terminal domain of yeast Hsp90 | 214 | 40, 44, 79, 80, 84, 92, 93, 98, 123, 124, 171, 173 | 34, 44, 79, 83, 124, 171 | 27–42, 82–93, 127–141, 149–165 | 1.4 |
| 1BH6 | Subtilisin DY | 275 | 32, 64, 221 | 64, 99–101, 125– 127, 155, 221 | 20–26, 122–126, 204–207, 214–217 | 2.15 |
| 1BVV | Endo-1,4-xylanase | 185 | 69, 78, 172 | 9, 80, 112, 116, 166 | 59–109, 128–140, 162–177 | 2.2 |
| 1BLC | β-lactamase | 31–290 | 70 | 69, 70, 234 | 65–72, 206–215 | 6.2 |
| 1BR6 | Ricin | 268 | 80, 81, 121,123, 177, 180 | 78, 80, 81, 121, 180 | 14–33, 45–52, 168–180 | 3.3 |
| 1BIO | Complement factor D | 16–243 | 57, 102, 195 | 195, 189, 214, 218 | 122–124, 136–153, 155–160 | 6.9 |
| 1BK9 | Phospholipase A2 | 134 | 48, 52, 99 | 5, 9, 30, 45, 48, 49 | 3–22, 43–54, 100–111 | 5.0 |
| 1BXO | Penicillopepsin | 323 | 33, 213 | 75, 216 | 146–180 | 5.3 |
| 1CP3 | Apopain | 35 + 227 | 121, 122, 161–165 | 64, 161, 163, 205, 207, 209, 214 | 169–195, 261–274 | 1.2 |
| 1CQQ | Human rhinovirus 3C protease | 180 | 40, 71, 145, 147 | 142, 143, 144, 145, 147, 161, 165, 170 | 61–63, 70–72, 86–89 | 2.9 |
| 1CR6 | Murine soluble epoxide hydrolase | 2 × 544 | A, B: 333, 334, 465, 495, 523 | A, B: 333, 334, 465, 523 | A, B: 225–241 | - |
References: 10GS: (Oakley et al., 1997); 1A16: (Wilce et al., 1998); 1A30: (Louis et al., 1998); 1A3B: (Zdanov et al., 1993); 1A42: (Stams et al., 1998); 1A47: (Wind et al., 1998); 1A5I: (Renatus et al., 1997); 1A5V: (Lubkowski et al., 1998); 1AEC: (Varughese et al., 1992); 1AL8: (Stenberg and Lindqvist, 1997); 1ARZ: (Scapin et al., 1997); 1B3N: (Moche et al., 1999); 1B6A: (Liu et al., 1998); 1BGQ: (Roe et al., 1999); 1BH6: (Eschenburg et al., 1998); 1BVV: (Sidhu et al., 1999); 1BLC: (Chen and Herzberg, 1992); 1BR6: (Yan et al., 1997); 1BIO: (Jing et al., 1998); 1BK9: (Zhao et al., 1998); 1BXO: (Khan et al., 1998); 1CP3: (Mittl et al., 1997); 1CQQ: (Matthews et al., 1999); 1CR6: (Argiriadi et al., 1999).
1A3B has two subunits of 14 and 245 residues. 1A5V, 1B6A, and 1BLC PDB coordinates refer to the indicated ranges.
Underlined residues have mobility scores < 0.10 and exhibit minima of types I–III in mode 1.
Hinge residues with mobility scores < 0.05, at crossover between positive and negative displacements in mode 1.
Odds ratio = p/p0 (probability of finding a catalytic residue among key mechanical sites, relative to that in all residues).
Figure 1 illustrates the distribution of mean square (ms) fluctuations, as exhibited by the experimental temperature factors, for all residues (Figure 1A), catalytic residues (Figure 1B), and ligand binding residues (Figure 1C) in this set, as well as the distribution of these enzymes among the six EC classes (Figure 1D). The B factors scale with ms fluctuations as Bi = 8π2 <(ΔRi)2 >/3, where 1 ≤ i ≤ N refers to the residue position along the sequence. We already note upon comparison of Figures 1A and 1B that catalytic residues tend to have smaller fluctuations compared to the behavior of average residues. The origin of this behavior will be clarified below by examining the involvement of active sites in the collective motions predicted by an EN model, the Gaussian network model (GNM) (Bahar et al., 1997).
Figure 1.
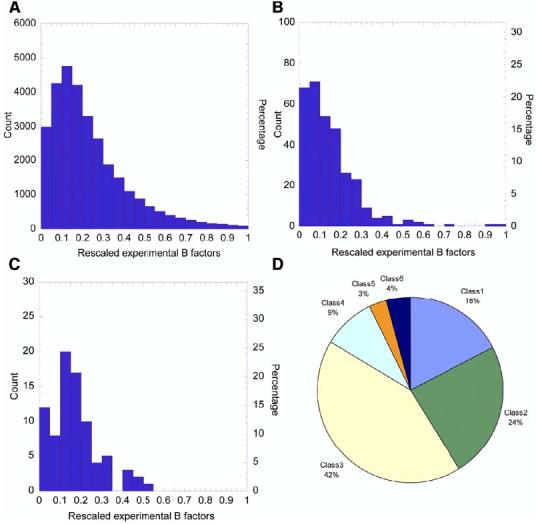
Distribution of Residue Fluctuations and Enzyme Classes in the Examined Data Set (A–C) Distribution of temperature (“B”) factors for (A) all (30,419) residues in the examined data set of 93 monomeric enzymes (Table S1 in the Supplemental Data), (B) the 324 catalytic residues of these enzymes, and (C) the 82 ligand binding residues of the subset of 19 monomeric enzymes in Set 1. The abscissa refers to the B factors, divided into 20 intervals of equal size, and the ordinate indicates the number of counts in each interval. The original Gaussian-like distribution of all residues is skewed toward low B factors in the case of catalytic residues and shows the same tendency, but to a weaker extent, in the case of ligand binding residues. The mean values are <B> = 0.24 for “all” residues, 0.14 for catalytic residues, and 0.17 for inhibitor binding residues. (D) shows the distribution of enzyme classes (E.C.) in the observed sets of proteins.
Hinge-bending flexibility has been pointed out in several studies to be an important mechanism that underlies functional changes in protein conformations (Bahar et al., 1998; Banks et al., 1979; Frauenfelder and MacMahon, 1998; Falcon and Matthews, 1999; Hirano and Hirano, 2002; Levitt et al., 1985; Pang et al., 2003; Xiang et al., 2001; Zhang et al., 2003; Ma and Karplus, 1998; McCammon et al., 1976; Cregut et al., 1998; Sinha et al., 2001). Hinge motions may be instrumental in facilitating ligand binding (Bahar et al., 1998; Towler et al., 2004), mediating allosteric effects (Xu et al., 2003), or fine tuning function (Gutteridge et al., 2003). We have shown for HIV-1 reverse transcriptase (RT), for example, that the hinge residues in the p66 palm subdomain form a stable anchoring region about which the thumb and fingers enjoy rotational mobility (Bahar et al., 1999). The mechanical role of the p66 palm goes hand-in-hand with its biochemical function, inasmuch as RT catalyzes nucleotide addition in the p66 palm, and, not surprisingly, RT inhibitors bind the palm to interfere with the global motions (Temiz and Bahar, 2002).
We focus here on the low-frequency motions, also called global motions (as opposed to local motions subject to high-frequency modes), and ask if or how the global dynamics and enzymatic function are correlated. The dominant role of the slow modes in effectuating the functional motions has been suggested in early normal mode analyses (NMAs) (see, for example, Karplus and McCammon, 1983; Go et al., 1983) and has been confirmed in many studies (e.g., Tama et al., 2000; Kitao and Go, 1999). The present study demonstrates that catalytic residues tend to be positioned near key mechanical sites (hinges, anchoring points) that are uniquely defined for particular architectures. The probability of finding a catalytic residue among the key mechanical sites is found to be 3–4 times higher than from a random search. The observed predisposition of enzymes to couple their chemical and mechanical properties holds promise for its assisting in the identification and design of functional sites or inhibitors.
Results and Discussion
Catalytic Residues Coincide or Communicate with Global Hinge Regions
Figure 2A displays the fluctuation profiles in the global modes of motion for a few enzymes selected from Set 1. Fluctuation profiles for the complete set of 98 enzymes can be accessed at http://ignm.ccbb.pitt.edu/. The abscissa represents the residue index, and the curve displays the distribution of residue fluctuations (squared) in the slowest modes predicted by the GNM. Peaks indicate the most mobile regions, and minima are those regions anchored in space, some of which act as global hinge residues (shown by the arrows). Global hinge residues are at the interfaces between domains, or clusters of residues, which move in opposite directions within the global modes. We will refer to minima in the slow modes as key mechanical sites.
Figure 2.
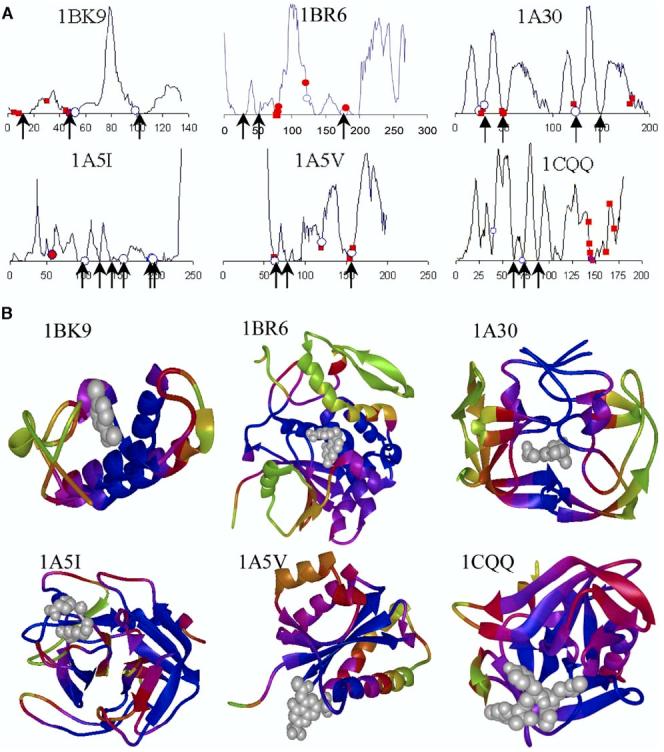
Mobilities in the Global Modes for Example Proteins (A) Fluctuation profiles in the global mode (k = 1) and position of catalytic and inhibitor binding residues illustrated for six enzymes from Set 1. Residues involved in catalytic function are marked with an open circle, inhibitors binding sites are marked with a closed square, and residues serving both catalytic and inhibitor binding functions are marked with a closed circle. Arrows indicate the hinge sites. (B) Color-coded ribbon diagrams showing the localization of inhibitors (gray ball-and-stick) near the most constrained (blue) regions. See Table 1 for the list of chemically (from experiments) and mechanically (from computations) key residues for all ligand/inhibitor bound enzymes in our data set.
Figure 2B suggests that most of the active residues tend to occupy minima in the fluctuation profiles. Notably, the catalytic residues preferably coincide with, or sequentially neighbor, key mechanical sites regardless of the enzyme function or size. Figure 2B displays the color-coded ribbon structures corresponding to the proteins in Figure 2A. The dark blue regions (minima in the slow modes) point to the residues subject to the strongest constraints in the global modes. Although these residues are not contiguous along the sequence, they usually cluster in space so as to consolidate the anchor/hinge region that coordinates the global movements. Examination of their structural properties and context shows that they are not necessarily coiled regions or domain linkages, but may occasionally occur in secondary structural motifs, such as kinks in helices.
Quantitative Assessment of Mobilities in the Global Modes
In order to make a quantitative assessment of the dynamics of active residues or ligand binding in relation to key mechanical residues, we assigned a mobility score, Mik, to each residue i in the kth mode. Mik is the square fluctuation normalized with respect to the most mobile residue in the kth mode of the particular enzyme. The highest peak in the slow mode profile of each enzyme (Figure 2A) is thus assigned a mobility score of unity, and the lowest is assigned a score of zero. Additionally, we examined the relative mobilities along the sequence, because a given residue may appear rigid, due to the constraints imposed on its bonded neighbors. Residues that precisely lie at local minima are referred to as key mechanical residues of type I, and their first and second neighbors are described as types II and III, respectively.
The global mobility scores corresponding to the more collective mode among the two slowest modes were computed for all catalytic and inhibitor binding residues of our data set, which led to the distributions shown in Figure 3. The scores for the individual residues can be accessed in the iGNM database at http://ignm.ccbb.pitt.edu/ (Yang et al., 2005a, 2005b). The distributions shown in Figures 3A and 3B may be compared to the respective distributions in Figures 1B and 1C. This comparison reveals that the skewed distribution of ms fluctuations indicated by the B factors is further pronounced when attention is confined to the mobilities in the most collective modes. 78% of the catalytic residues show mobility scores below 0.10 in the slow modes (66% below 0.05), as can be seen from the inset in Figure 3A. In contrast, the same interval (<0.10) in the B factors' distribution (i.e., all modes) contains 43% of catalytic residues and 25% of all residues. The insets in Figure 3 display the cumulative percentage of catalytic residues (Figure 3A) and inhibitor binding residues (Figure 3B) in different mobility ranges, compared to those observed for all residues in the same mobility ranges. We also compare the predictions from global modes, versus all modes, which clearly demonstrates the high propensity of catalytic residues to have low mobilities in the global mode profiles. The low-frequency mode shapes thus provide a means of discriminating potentially active sites.
Figure 3.
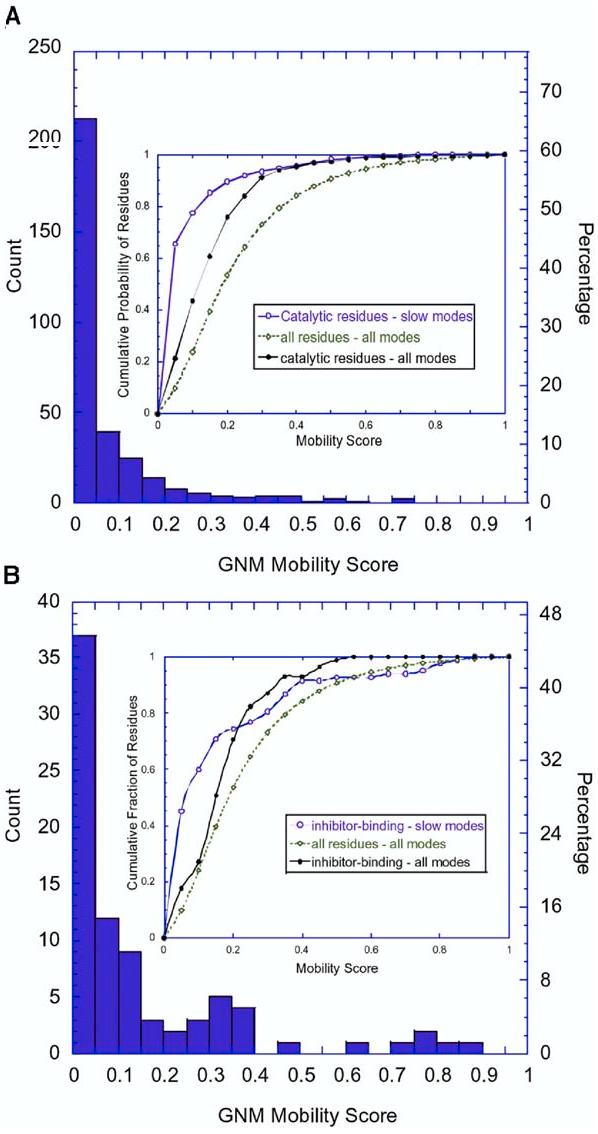
Distribution of Mobilities Predicted by the GNM (A and B) The results are shown for (A) 325 catalytic residues and (B) 81 ligand binding residues in the examined set of 98 enzymes. The abscissa refers to the mobility scores of the residues found in the most collective GNM mode. The inset displays the cumulative fraction of (A) catalytic residues and (B) inhibitor binding residues in different ranges of mobility based on the slow modes, compared to the cumulative fraction of catalytic residues and all residues in all modes (see the labels).
The results found by averaging the scores over all proteins are presented in Table 2. <M1>cat and <M2>cat refer to the average behavior of catalytic residues in the slowest modes, modes 1 and 2, and <M1>lig and <M2>lig are their counterparts for ligand binding residues. The averages were calculated by first evaluating an average over catalytic residues for each protein, and then by averaging over all proteins, thereby removing biases that arise from the different numbers of functional sites per protein. These results show that the catalytic residues possess highly suppressed mobilities in the first two slowest (dominant) modes. The low mobilities indicate their participation in (or close proximity to) the key mechanical sites of the molecules. These residues are for the most part fixed/anchored in space, while the other regions undergo motions about them. The low mobility of the catalytic residues was already apparent in their B factors, although this effect was less pronounced due to the superimposition of all modes in the B factors. Extraction of the global modes shows that the reduced mobilities are essentially caused by their constrained global dynamics, rather than by local packing effects.
Table 2.
Mobility Scores for Catalytic and Ligand Binding Residues
| Active Sites | Ligand Binding Sites | |||||
|---|---|---|---|---|---|---|
| Enzymes | <M1>cat | <M2>cat | <MB>cat | <M1>lig | <M2>lig | <MB>lig |
| All (Set 1) | ||||||
| Average over proteins | 6.72 | 5.94 | 12.55 | 12.15 | 9.72 | 16.72 |
| Standard deviation | 8.60 | 9.91 | 7.44 | 13.37 | 10.31 | 7.24 |
| All (Set 2) | ||||||
| Average over proteins | 9.06 | 6.75 | 13.77 | |||
| Standard deviation | 9.40 | 8.46 | 9.09 | |||
| Monomers (Set 1) | ||||||
| Average over proteins | 4.55 | 3.47 | 11.49 | 11.06 | 8.89 | 15.25 |
| Standard deviation | 5.22 | 4.67 | 7.55 | 12.82 | 10.42 | 6.94 |
| Multimers (Set 1) | ||||||
| 10GS (dimer) | 21.30 | 41.08 | 24.45 | 27.31 | 23.05 | 25.62 |
| 1A30 (dimer) | 10.13 | 5.13 | 11.56 | 5.24 | 6.94 | 24.38 |
| 1CP3 (dimer) | 35.90 | 2.73 | 21.51 | 39.33 | 3.71 | 22.21 |
| 1CR6 (dimer) | 12.98 | 18.11 | 20.90 | 12.92 | 17.60 | 18.00 |
| 1ARZ (tetramer) | 0.10 | 0.13 | 12.13 | 2.81 | 5.46 | 27.15 |
The mobility score 0 < Mik < 1 for a given residue, i, is the ratio of its square fluctuation to that of the residue exhibiting the highest fluctuation in the kth mode. <Mk>cat and <Mk>lig are averages taken over catalytic residues (columns 4–6) or ligand binding residues (columns 7–9) for modes k = 1 and 2. <MB>cat is the equivalent ratio based on temperature factors. The results for the reduced set (19 out of 24) of monomeric enzymes are shown separately. The last five rows display the results for selected multimeric enzymes.
Table 1 compares the chemically active (catalytic and ligand binding) residues identified in previous experimental studies (columns 4 and 5) and the key mechanical residues (column 6) predicted by present computations. The underlined residues in columns 4 and 5 are those, among the experimentally reported chemically active residues, that are indicated by the GNM to be key mechanical sites of types I–III and which have mobility scores < 0.10. The close correspondence between chemical activity and the mechanical role is evident by the large fraction of underlined residues. Column 6 lists the hinge sites distinguished by mobility scores below 0.05 in the slowest mode. These residues are proposed here to be the most critical residues, from a mechanical point of view, that could serve as targets for interfering with the dynamics of the enzyme.
Ligand Binding Residues Enjoy Higher Mobility Despite Their Close Proximity to Catalytic Sites
The ligand binding sites exhibit relatively higher flexibility (<M1-2>lig ≈ 0.11 and <MB>lig ≈ 0.17; see Table 2) and larger variations compared to the catalytic sites, although they are also somewhat constrained in the global motions. We note that some of the ligand binding residues also act as catalytic residues. Exclusion of these residues (46 out of 159) leads to an increase in mobility scores (<M1>lig = 0.148, <M2>lig = 0.104, and <MB>lig = 0.19) accompanied with larger variations. This suggests that ligand binding residues occupy proximal positions with respect to the catalytic residues but enjoy higher fluctuation. The latter may indeed be required for efficient recognition of substrate and optimization of intermolecular interactions. The close proximity of inhibitors to catalytic sites and their moderate mobility suggest that they block the catalytic function by interacting with the fluctuating residues, for instance, in the entrance of a catalytic pocket. Our findings support the observation that regions of high and low structural stabilities participate in binding sites (Freire, 1999).
Dimerization Induces New Cooperative Modes that Engage the Catalytic Site
Among the 98 enzymes presently examined, 4 (10GS, 1A30, 1CP3, and 1CR6) are dimers, and 1 (1ARZ) is a tetramer. It is of interest to assess how multimerization affects the mobilities of active sites. A pure monomer set was generated by removing the multimers from the Set 1. It is shown in Table 2 that the <M1>cat and <M2>cat values, as well as their standard deviations, decrease in this case, which is consistent with the higher mobilities of chemically active residues in the multimers (Table 2). Multimerization usually provides a means of achieving structural and dynamic properties that would otherwise be inaccessible to the monomers. It is of interest to see if the new structures and structure-induced modes of motion (especially low-frequency global motions) impart stability and/or mechanical properties that affect catalytic residues. It can be anticipated that a high mobility/disorder at the catalytic site might be detrimental from the point of view of the precise regulation and communication ability of the active site.
Figure 4 displays the color-coded ribbon diagrams obtained for the first (left column) and second (right column) slowest modes of these multimers. Catalytic residues are shown in red ball-and-stick representation, and inhibitor binding residues are shown in color-coded (according to mobilities) ball-and-stick representation.
Figure 4.
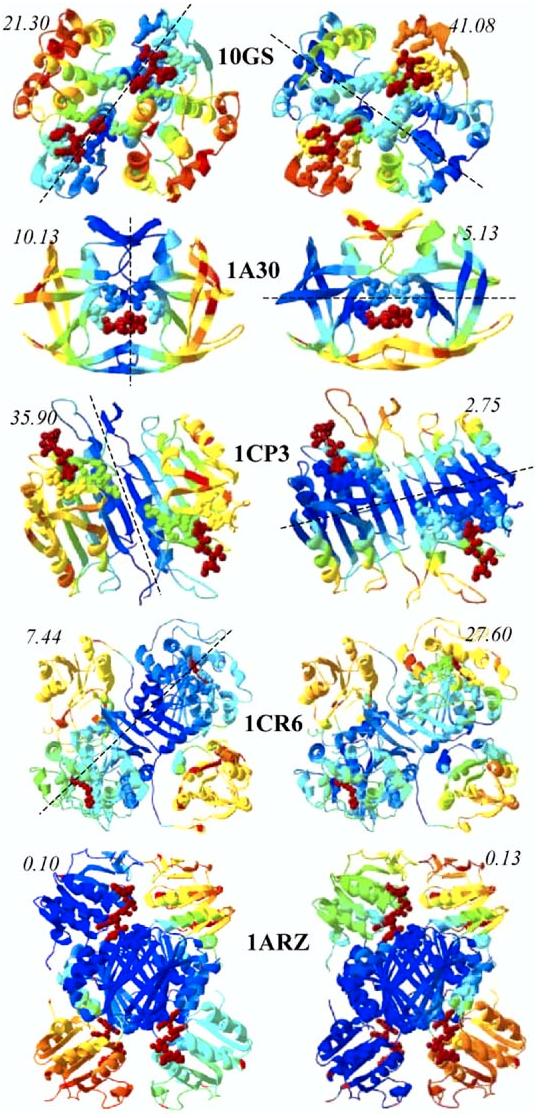
Global Mode Shapes of Five Multimeric Enzymes Included in Our Data Set The left and right ribbon diagrams refer to modes 1 and 2, respectively. Catalytic residues are shown by red ball representation. The dashed line indicates the loci of global hinge regions. The mobility scores corresponding to catalytic residues are indicated in each case.
The dimers 10GS, 1A30, and 1CP3 exhibit symmetric motions with respect to extended hinge regions that span the entire structures. The hinge region lies at the interface between the monomers in mode 1 in all three cases, whereas it runs through the cores of the monomers in mode 2, as indicated by the dashed lines. Interestingly, the motions of the monomers in the second mode of the dimers almost exactly reproduce those in their first mode, which were computed for the monomers taken alone (data not shown). These data are consistent with the decrease in the wavelength of the second slowest vibrational mode by 1/2 compared to the first, whereas one expects the higher frequency modes to be even more localized. Hence, mode 1 is the mode that appears to be exclusively induced in the dimeric form. In 10GS, this mode ensures the localization of the catalytic sites in a mechanically important region. The mobility score of the catalytic residues in this mode is 0.21, which is decreased by a factor of two compared to the mobility in mode 2 (0.41), and this coupling to a global hinge region may be anticipated to be functional. A similar effect can be conjectured in 1CR6, where dimerization secures the colocalization of the catalytic site with a global hinge region. Alternatively, in 1A30 and 1CP3 mode 2 already confines the catalytic site in a mechanically key region, as evident from the low mobility scores. Dimerization seems to be a structural, rather than dynamic requirement in these cases. This process permits the catalytic site to be sequestered from solvent in HIV-1 protease (1A30). Finally, the catalytic residues are positioned at the interface between the core domain and the two different peripheral domains in the two slowest modes of 1ARZ, suggesting the activation of different domains in different modes.
Catalytic Residues Occupy or Neighbor Key Mechanical Sites
The correspondence between the loci of the catalytic residues and key mechanical sites, revealed upon comparison of the position of the residues that control the chemical activity (from experiments) and global dynamics (from computations), is a feature of fundamental functional importance that deserves further examination. The ligand binding residues are found to exhibit a broad range of mobilities. The catalytic residues, on the other hand, are severely constrained. The averages over the 93 monomeric enzymes are <M1>cat = 0.085 and <M2>cat = 0.066 in the first and second slowest modes, respectively, as opposed to <M1>all = 0.236 and <M2>all = 0.154 for all residues. From another perspective, 228, out of 325, catalytic residues included in our data set serve as key mechanical sites of type I, II, and III (with respective proportions of 107:82:39) when considering the weighted average of the two slowest modes. Therefore, more than 70% of the examined catalytic residues communicate with key mechanical sites, if not directly engaging in a mechanical role, and their ms fluctuations are, on the average, 2–3 times smaller than those of “average” residues. This reveals a simple but important feature in the design of enzymes: catalytic activity takes place at cooperatively constrained regions distinguished by suppressed fluctuations in the collective dynamics. And, a corollary is to select inhibitor target sites from amongst the residues lying at the minima of the global mode shapes.
One could hypothesize based on this observation that catalytic residues are immobilized in order to protect the delicate arrangement of functional groups. It is important to note, however, that in many cases the low mobility is not a consequence of their being rigidly embedded in a given (functional) domain, but of lying near the crossover regions between substructures subject to oppositely correlated motions, as illustrated in the Experimental Procedures for an example enzyme. The catalytic residues are therefore localized in/near key mechanical sites that control the global motions. These sites have limited, if any, translational mobility while enjoying rotational flexibility. The communication of chemically active sites with mechanically active sites supports the idea of a functional coupling between catalysis and global conformational dynamics.
From another perspective, our analysis shows that 86% (80/93) of the enzymes have at least one key mechanical residue of type II and III and that 94% (87/93) have at least one key mechanical residue of type I, II, and III in their active site. Examination of the catalytic residues that do not exhibit restricted fluctuations in the global modes (<30% of all catalytic residues) shows that their mobilities could be associated with functional requirements in the particular enzymes. For example, Tyr75 in 1BXO is located at the tip of a “flap”—a β hairpin loop that forms the catalytic pocket against which inhibitors pack. The large fluctuations of Tyr75 are needed to accommodate the docking of big inhibitors such as PPi3 or PPi4. Another example in His64 is 1A42, which acts as a proton shuttle between zinc bound solvent and bulk solvent, and which switches between “in” and “out” conformations depending on the pH. The rotational motions of His64 ensure the occurrence of catalytic reaction at suitable pH.
Enzymes Are Predisposed to Couple Their Chemical and Mechanical Activities
Several studies have demonstrated that the global mode shapes are insensitive to structural details but are uniquely defined by the overall three-dimensional structure (see, for example, Kitao and Go, 1999). The global mode profiles may indeed be viewed as the signatures of particular architectures. Consistent with this observation, the inclusion or exclusion of a few contacts are usually inconsequential, because the observed dynamics are a collective property of ∼103 interresidue contacts (for a protein of n = 300 residues and an average coordination number of 7 per residue). The restricted motions at the catalytic sites are not due to the presence of substrates at those sites but are intrinsic mechanical properties of the enzymes themselves irrespective of bound molecules. We also note that active sites are frequently in clefts, which may be functional in excluding water molecules and/or maximizing the contact surface at the ligand binding site.
Figure 5 shows the global modes obtained for liganded and unliganded forms of a protein (β-lactamase in Figure 5A) and for two different ligand bound forms of another protein (plasminogen, Figure 5B). The close similarity of the two curves in each panel illustrates the robustness of the global modes. We note, however, that inhibitor binding may alter the mobility of a few key sites, while leaving the overall profile almost unchanged, and the maintenance of the fluctuation profile may indeed be critical for inducing or propagating functional motions. The similarity in the global dynamics of liganded and unliganded forms suggests that particular regions of proteins are already predisposed to serve as catalytic centers prior to substrate or inhibitor binding, and substrate binding essentially stabilizes the conformations, or induces the motions, that are intrinsically favored or accessible by the enzyme under native state conditions.
Figure 5.
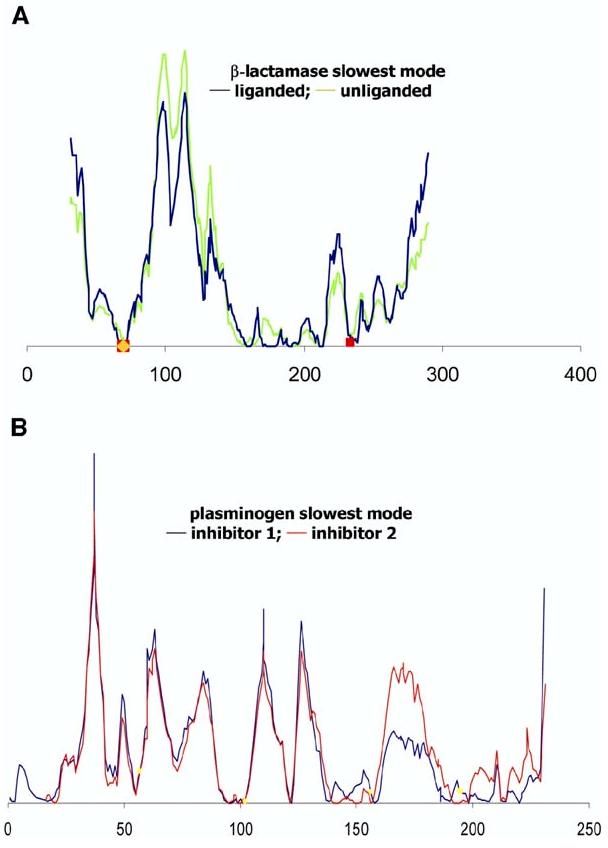
Comparison of the Dynamics of Liganded and Unliganded Forms of Two Enzymes (A and B) Results are presented for (A) β-lactamase in liganded (1BLC, black) and unliganded (1BK9, green) forms and (B) plasminogen activator bound to different ligands. The black curve refers to the complex with inhibitor Glu-Gly-Arg chloromethyl ketone (1A5I), and the red curve refers to the complex with the inhibitor 2-(2-hydroxy-5-methoxy-phenyl)-1H-benzoimidazole-5-carboxamidine (1GI9). The critical mechanical sites (minima) are preserved in both forms, illustrating that the global dynamics retain the same qualitative features in the liganded and unliganded forms, while the amplitudes of motions may be affected.
Participation in Key Mechanical Sites: A Criterion for Identifying Functional Sites
The present analysis suggests that the low mobilities in the global modes can be adopted as a new criterion for discriminating catalytic sites. The utility of this criterion may be assessed by a simple probabilistic analysis. Let us first sort all mechanically key residues of types I and II whose mobility score found from the weighted average of modes 1 and 2 is Mi,1–2 < 0.05. Let us consider the odds ratio p/p0 of detecting a catalytic site among these key mechanical residues (p), compared to a random search over all residues (p0). p0 is simply the fraction of active residues in the examined enzyme. The ratio p/p0 was computed for all enzymes in Set 1. The results are listed in the last column of Table 1. p/p0 is found to be 3.4 on average, with a standard deviation of 1.8, which means the odds of having a catalytic residue among the key mechanical sites is 3–4 times higher than the fraction of catalytic residues in the sequence. Our recent study supports the utility of considering global mode profiles in addition to physicochemical features for predicting catalytic sites (Chen and Bahar, 2004), an issue that will be further pursued in a future study.
Experimental Procedures
Sample Proteins
Our data set consists of two sets of enzymes. First, all ligand-protein complexes available in the PDB were downloaded. Structures having higher than 90% sequence identity were removed; the remaining >100 structures were reduced to 24 (Set 1, Table 1) after requiring (i) the availability of explicit experimental data on inhibitor binding and catalytic residues, (ii) the size of the inhibitor to be small-to-moderate (up to 35 heavy atoms), and (iii) all atomic coordinates to be deposited, except those at the truncated domains that do not interfere with the catalytic site. Set 2 consists of 74 nonhomologous, monomeric proteins extracted from CATRES (Bartlett and Thornton, 2002). Three of these have a substrate composed of less than ten residues (PDB identifiers: 2PHK, 8PCH, and 8TLN). The complete data set of enzymes is given in the Supplemental Data available with this article online, Table S1.
Definition of Catalytic Residues
According to the definition introduced by Bartlett et al., 2002, a given residue is catalytic if (i) it is directly involved in a catalytic function, (ii) it affects the residues or water molecules directly involved in catalysis, (iii) it can stabilize a transient intermediate, or (iv) it interacts with a substrate or cofactor that facilitates the local chemical reaction. These criteria were adopted for defining the catalytic residues in Set 2. Those in Set 1 were identified from either (i) experimental data that explicitly indicate the involvement in catalytic function or (ii) from the label “SITE” in the PDB entry. We note that not all PDB files of enzymes include these labels, hence the need to examine the literature. This definition was confirmed to point to the same “active” amino acids when applied to Set 1, except for the inclusion of 1–2 additional residues in a few cases.
The inhibitor binding sites listed in Table 1 are those reported in previous experimental studies which act to bind the inhibitor (ligand). They may, or may not, overlap with an active site.
Gaussian Network Model. General Formulation and Mode Decomposition
The GNM is based on two assumptions (Bahar et al., 1997; Haliloglu et al., 1997): (i) The structure is viewed as an EN, the nodes of which are the individual residues usually represented by their α carbons, (ii) a uniform spring constant,γ, is adopted for all residue pairs located within a cutoff distance, rc, inspired by the atomic NMA of Tirion (1996).γ is of the order of 1 kcal/(mol Å2), and rc is typically 7.0 ± 0.5 Å (Bahar et al., 1997; Kundu et al., 2002). The topology of the network is defined by the NxN Kirchhoff matrix Γ, the off-diagonal elements of which are −1 if nodes i and j are connected and zero otherwise, and the columns sum up to zero. The crosscorrelations between the fluctuations ΔRi and ΔRi of residues i and j are found from (Bahar et al., 1997):
| (1) |
using the statistical mechanical theory of polymer networks (Flory, 1976). Here, kB is the Boltzmann constant, T is the absolute temperature, and [Γ −1]ij is the ijth element of the inverse of Γ.
The equilibrium dynamics of the folded protein are described by a set of N-1 normal modes (Haliloglu et al., 1997). The kth eigenvector (uk) of Γ gives the profile of residue displacements along the mode k, and the kth eigenvalue,λk, scales with its frequency. The contribution of mode k to the ms fluctuations <(ΔRi)2> = (3kBT/γ) Γ −1]ii of residue i is
| (2) |
where [uk]i is the ith diagonal element of uk. We note that the eigenvectors are normalized; the quantity as a function of i thus represents the probability distribution of residue mobilities in the kth mode. The degree of collectivity of mode k (Tama and Sane-jouand, 2001) is computed from
| (3) |
The slowest modes are usually the most collective ones, except for a few cases in which they refer to a large motion of an exposed loop or a chain terminus. We examine the position of catalytic and inhibitor binding residues relative to the global hinge regions associated with the slowest modes. The global hinges are identified from the crossover between the positive and negative elements of the eigenvectors u1 and u2.
Illustration of GNM Analysis and Comparison with Experiments
Figure 6 illustrates the computations for an example protein, endo-1,4-β-xylanase (1BVV) (Oakley et al., 1997) from Set 1. The eigenvectors u1 and u2 are plotted in Figure 6A. The global hinge centers are located at the crossover between the positive and negative displacements. Four groups of global hinge residues are shared by the two modes: T67–G70, V81–S84, R89–P90, and Y166–M169, indicated by the arrows. Figure 6B shows the ribbon diagrams colored from red (most mobile) to blue (most rigid) according to the square fluctuations of residues in the slowest two modes, and Figure 6C displays the structural regions exhibiting opposite direction fluctuations in these modes, colored red (positive) and blue (negative) in the two modes. The hinge residues lie at the interface between these anticorrelated regions. The catalytic and/or inhibitor binding residues reported in the literature are labeled in Figure 6A.
Figure 6.
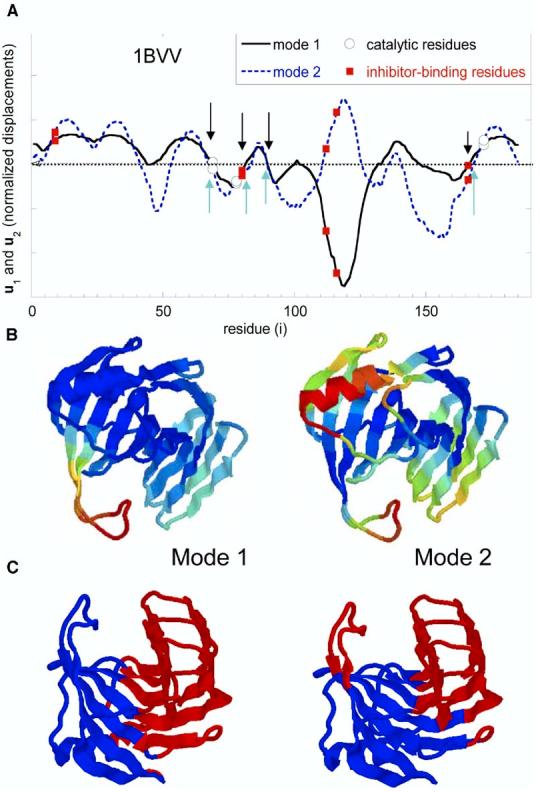
Distribution of Displacements (A) Distribution of displacements along the slowest modes, modes 1 (solid) and 2 (dotted), as a function of residue index, computed for endo-1,4-β-xylanase. Catalytic and inhibitor binding residues are indicated by open circles and closed squares, respectively. The crossover regions between negative and positive motions are the predicted global hinge sites, G41–W42, Y53–N54, T67–G70, V81–S84, R89–P90, T110–T111, F125–T126, Q133–P137, S140–N41, T147–N148, Y166–M169 (by combining both modes), four of which that are indicated by arrows are common to modes 1 and 2. (B) Ribbon diagrams colored blue-green-yellow-orange-red in the order of increasing mobility of residues along modes 1 and 2. The (normalized) mobilities, , are directly found from the values in (A) , squared. (C) The regions subject to opposite direction movements in modes 1 and 2, deduced from (A). Regions colored red and blue correspond to “+” and “−” displacements, respectively, along the first (left) or second (right) mode axes.
Note that mode 1 essentially sets in motion an extended loop at the entrance of the catalytic pocket permitting the opening/closing of the catalytic site, while mode 2 engages a larger portion of the structure. The respective collectivities of modes 1 and 2 calculated from equation 3 are Ω1 = 0.246 and Ω2= 0.576.
Of interest is the comparison of experimentally known residues with the hinge sites predicted by the GNM. Three catalytic residues have been reported for 1BVV (Oakley et al., 1997), one of which (Y69) coincide with a global hinge residue, and the other two (E78 and E172) of which are positioned close to the hinge centers V81–S84 and Y166–M169, supporting the view of a coupling/communication between catalytic and mechanically important sites.
The inhibitor binding residues, on the other hand, show a more varied behavior: one (Y166) coincides with a global hinge residue, two (Y80 and R112) are first sequential neighbors to hinge residues V81 and T111 (mode 2 only), and a third (P116) is rather exposed, distinguished by a high flexibility. The latter may indeed be instrumental for efficient recognition of substrates. These results suggest that ligand binding may involve a network of residues that include both highly flexible “recognition” sites as well as constrained residues establishing the communication with the mechanically and chemically active sites.
Supplementary Material
Acknowledgments
We thank Dr. Gail Bartlett for her generous offer of the catalytic residue information in CATRES. Partial support from National Science Foundation grant 0225636 and National Institutes of Health grant 1R01LM007994-01A1 is gratefully acknowledged.
Footnotes
Supplemental Data
Supplemental Data including the names, EC numbers, and PDB codes of the 98 proteins examined in this study are available at http://www.structure.org/cgi/content/full/13/6/893/DC1/.
References
- Agarwal P, Geist A, Gorin A. Protein dynamics and enzymatic catalysis: investigating the peptidyl-prolyl cis-trans isomerization activity of cyclophilin A. Biochemistry. 2004;43:10605–10618. doi: 10.1021/bi0495228. [DOI] [PubMed] [Google Scholar]
- Argiriadi MA, Morisseau C, Hammock BD, Christianson DW. Detoxification of environmental mutagens and carcinogens: structure-based mechanism and evolution of liver epoxide hydrolase. Proc. Natl. Acad. Sci. USA. 1999;96:10637–10642. doi: 10.1073/pnas.96.19.10637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atilgan AR, Durell SR, Jernigan RL, Demirel MC, Keskin O, Bahar I. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 2001;80:505–515. doi: 10.1016/S0006-3495(01)76033-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahar I, Atilgan AR, Erman B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold. Des. 1997;2:173–181. doi: 10.1016/S1359-0278(97)00024-2. [DOI] [PubMed] [Google Scholar]
- Bahar I, Atilgan AR, Demirel MC, Erman B. Vibrational dynamics of folded proteins: significance of slow and fast motions in relation to function and stability. Phys. Rev. Lett. 1998;80:2733–2736. [Google Scholar]
- Bahar I, Erman B, Jernigan RL, Atilgan AR, Covell DG. Collective motions in HIV-1 reverse transcriptase: examination of flexibility and enzyme function. J. Mol. Biol. 1999;285:1023–1037. doi: 10.1006/jmbi.1998.2371. [DOI] [PubMed] [Google Scholar]
- Banks RD, Blake CCF, Evans PR, Haser R, Rice DW, Hardy GW, Merret M, Phillips AW. Sequence, structure and activity of phosphoglycerate kinase: a possible hinge-bending enzyme. Nature. 1979;279:773–777. doi: 10.1038/279773a0. [DOI] [PubMed] [Google Scholar]
- Bartlett G, Thornton JM. Analysis of catalytic residues in enzyme active sites. J. Mol. Biol. 2002;324:105–121. doi: 10.1016/s0022-2836(02)01036-7. [DOI] [PubMed] [Google Scholar]
- Bartlett G, Porter C, Borkakoti N, Thornton JM. Analysis of catalytic residues in enzyme active sites. J. Mol. Biol. 2002;324:105–121. doi: 10.1016/s0022-2836(02)01036-7. [DOI] [PubMed] [Google Scholar]
- Benkovic SJ, Hammes-Schiffer S. A perspective on enzyme catalysis. Science. 2003;301:1196–1202. doi: 10.1126/science.1085515. [DOI] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Herzberg O. Inhibition of beta-lactamase by clavulanate. Trapped intermediates in cryocrystallographic studies. J. Mol. Biol. 1992;224:1103–1113. doi: 10.1016/0022-2836(92)90472-v. [DOI] [PubMed] [Google Scholar]
- Chen SC, Bahar I. Mining frequent patterns in protein structures: a study of protease families. Bioinformatics. 2004;20:i77–i85. doi: 10.1093/bioinformatics/bth912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark DS. Characteristics of nearly dry enzymes in organic solvents: implications for biocatalysis in the absence of water. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2004;359:1299–1307. doi: 10.1098/rstb.2004.1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregut D, Drin G, Liautard JP, Chiche L. Hinge-bending motions in annexins: molecular dynamics and essential dynamics of apo-annexin V and of calcium bound annexin V and I. Protein Eng. 1998;11:891–900. doi: 10.1093/protein/11.10.891. [DOI] [PubMed] [Google Scholar]
- Daniel RM, Dunn RV, Finney JL, Smith JC. The role of dynamics in enzyme activity. Annu. Rev. Biophys. Biomol. Struct. 2002;32:69–92. doi: 10.1146/annurev.biophys.32.110601.142445. [DOI] [PubMed] [Google Scholar]
- Diaz N, Sordo TL, Merz KM, Jr., Suarez D. Insights into the acylation mechanism of class A beta-lactamases from molecular dynamics simulations of the TEM-1 enzyme complexed with benzylpenicillin. J. Am. Chem. Soc. 2003;125:672–684. doi: 10.1021/ja027704o. [DOI] [PubMed] [Google Scholar]
- Eisenmesser EZ, Bosco DA, Akke M, Kern D. Enzyme dynamics during catalysis. Science. 2002;295:1480–1481. doi: 10.1126/science.1066176. [DOI] [PubMed] [Google Scholar]
- Eschenburg S, Genov N, Peters K, Fittkau S, Stoeva S, Wilson KS, Betzel C. Crystal structure of subtilisin DY, a random mutant of subtilis in Carlsberg. Eur. J. Biochem. 1998;257:309–318. doi: 10.1046/j.1432-1327.1998.2570309.x. [DOI] [PubMed] [Google Scholar]
- Falcon CM, Matthews KS. Glycine insertion in the hinge region of lactose repressor protein alters DNA binding. J. Biol. Chem. 1999;274:30849–30857. doi: 10.1074/jbc.274.43.30849. [DOI] [PubMed] [Google Scholar]
- Flory PJ. Statistical thermodynamics of random networks. Proc. R. Soc. Lond. A. 1976;351:351–380. [Google Scholar]
- Frauenfelder H, MacMahon B. Dynamics and functions of proteins: the search of general concepts. Proc. Natl. Acad. Sci. USA. 1998;95:4795–4797. doi: 10.1073/pnas.95.9.4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freire E. The propagation of binding interactions to remote sites in proteins: analysis of the binding of the monoclonal antibody D1.3 to lysozyme. Proc. Natl. Acad. Sci. USA. 1999;96:10118–10122. doi: 10.1073/pnas.96.18.10118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go N, Noguti T, Nishikawa T. Dynamics of a small globular protein in terms of low-frequency vibrational modes. Proc. Natl. Acad. Sci. USA. 1983;80:3696–3700. doi: 10.1073/pnas.80.12.3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutteridge A, Thornton JM, Bartlett G. Using a neural network and spatial clustering to predict the location of active sites in enzymes. Biochemistry. 2003;37:11940–11948. doi: 10.1016/s0022-2836(03)00515-1. [DOI] [PubMed] [Google Scholar]
- Haliloglu T, Bahar I, Erman B. Gaussian dynamics of folded proteins. Phys. Rev. Lett. 1997;79:3090–3093. [Google Scholar]
- Hirano M, Hirano T. Hinge-mediated dimerization of SMC protein is essential for its dynamic interaction with DNA. EMBO J. 2002;21:5733–5744. doi: 10.1093/emboj/cdf575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing H, Babu YS, Moore D, Kilpatrick JM, Liu XY, Volanakis JE, Narayana SV. Structures of native and complexed complement factor D: implications of the atypical His57 conformation and self-inhibitory loop in the regulation of specific serine pro-tease activity. J. Mol. Biol. 1998;282:1061–1081. doi: 10.1006/jmbi.1998.2089. [DOI] [PubMed] [Google Scholar]
- Karplus M, McCammon J. Dynamics of proteins: elements and functions. Annu. Rev. Biochem. 1983;53:263–300. doi: 10.1146/annurev.bi.52.070183.001403. [DOI] [PubMed] [Google Scholar]
- Khan AR, Parrish JC, Fraser ME, Smith WW, Bartlett PA, James MN. Lowering the entropic barrier for binding conformationally flexible inhibitors to enzymes. Biochemistry. 1998;37:16839–16845. doi: 10.1021/bi9821364. [DOI] [PubMed] [Google Scholar]
- Kitao A, Go N. Investigating protein dynamics in collective coordinate space. Curr. Opin. Struct. Biol. 1999;9:164–169. doi: 10.1016/S0959-440X(99)80023-2. [DOI] [PubMed] [Google Scholar]
- Kohen A, Cannio R, Bartolucci S, Klinman JP. Enzyme dynamics and hydrogen tunnelling in a thermophilic alcohol dehydrogenase. Nature. 1999;399:417–418. doi: 10.1038/20981. [DOI] [PubMed] [Google Scholar]
- Kundu S, Melton JS, Sorensen DC, Phillips GN., Jr. Dynamics of proteins in crystals: comparison of experiment with simple models. Biophys. J. 2002;83:723–732. doi: 10.1016/S0006-3495(02)75203-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt M, Sander C, Stern PS. Protein normal-mode dynamics: trypsin inhibitor, crambin, ribonuclease and lysozyme. J. Mol. Biol. 1985;181:423–447. doi: 10.1016/0022-2836(85)90230-x. [DOI] [PubMed] [Google Scholar]
- Liu S, Widom J, Kemp CW, Crews CM, Clardy J. Structure of human methionine aminopeptidase-2 complexed with fumagillin. Science. 1998;282:1324–1327. doi: 10.1126/science.282.5392.1324. [DOI] [PubMed] [Google Scholar]
- Louis JM, Dyda F, Nashed NT, Kimmel AR, Davies DR. Hydrophilic peptides derived from the transframe region of Gag-Pol inhibit the HIV-1 protease. Biochemistry. 1998;37:2105–2110. doi: 10.1021/bi972059x. [DOI] [PubMed] [Google Scholar]
- Lubkowski J, Yang F, Alexandratos J, Wlodawer A, Zhao H, Neamati N, Pommier Y, Merkel G, Skalka AM. Structure of the catalytic domain of avian sarcoma virus integrase with a bound HIV-1 integrase-targeted inhibitor. Proc. Natl. Acad. Sci. USA. 1998;95:4831–4836. doi: 10.1073/pnas.95.9.4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Bruice TC. Anticorrelated motions as a driving force in enzyme catalysis: the dehydrogenase reaction. Proc. Natl. Acad. Sci. USA. 2004;101:13152–13156. doi: 10.1073/pnas.0405502101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Karplus M. The allosteric mechanism of the chaperonin GroEL: a dynamic analysis. Proc. Natl. Acad. Sci. USA. 1998;95:8502–8507. doi: 10.1073/pnas.95.15.8502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews D, Dragovich PS, Webber SE, Fuhrman SA, Patick AK, Zalman LS, Hendrickson TF, Love RA, Prins TJ, Marakovits JT, et al. Structure-assisted design of mechanism-based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc. Natl. Acad. Sci. USA. 1999;96:11000–11007. doi: 10.1073/pnas.96.20.11000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCammon JA, Gelin BR, Karplus M, Wolynes PG. Hinge-bending mode in lysozyme. Nature. 1976;262:325–326. doi: 10.1038/262325a0. [DOI] [PubMed] [Google Scholar]
- Ming D, Kong Y, Wakil SJ, Brink J, Ma J. Domain movements in human fatty acid synthase by quantized elastic deformational model. Proc. Natl. Acad. Sci. USA. 2002a;99:7895–7899. doi: 10.1073/pnas.112222299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming D, Kong YF, Lambert MA, Huang Z, Ma JP. How to describe protein motion without amino acid sequence and atomic coordinates. Proc. Natl. Acad. Sci. USA. 2002b;99:8620–8625. doi: 10.1073/pnas.082148899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittl PR, Di Marco S, Krebs JF, Bai X, Karanewsky DS, Priestle JP, Tomaselli KJ, Grutter MG. Structure of recombinant human CPP32 in complex with the tetrapeptide acetyl-Asp-Val-Ala-Asp fluoromethyl ketone. J. Biol. Chem. 1997;272:6539–6547. doi: 10.1074/jbc.272.10.6539. [DOI] [PubMed] [Google Scholar]
- Moche M, Schneider G, Edwards P, Dehesh K, Lindqvist Y. Structure of the complex between the antibiotic cerulenin and its target, beta-ketoacyl-acyl carrier protein synthase. J. Biol. Chem. 1999;274:6031–6034. doi: 10.1074/jbc.274.10.6031. [DOI] [PubMed] [Google Scholar]
- Oakley AJ, Bello ML, Battistoni A, Ricci G, Rossjohn J, Villar HO, Parker MW. The structures of human glutathione transferase P1–1 in complex with glutathione and various inhibitors at high resolution. J. Mol. Biol. 1997;274:84–100. doi: 10.1006/jmbi.1997.1364. [DOI] [PubMed] [Google Scholar]
- Pang A, Arinaminpathy Y, Sansom M, Biggin P. Interdomain dynamics and ligand binding: molecular dynamics simulations of glutamine binding protein. FEBS Lett. 2003;550:168–174. doi: 10.1016/s0014-5793(03)00866-4. [DOI] [PubMed] [Google Scholar]
- Porter CT, Bartlett GJ, Thornton JM. The catalytic site atlas: a resource of catalytic sites and residues identified in enzymes using structural data. Nucleic Acids Res. 2004;32:D129–D133. doi: 10.1093/nar/gkh028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renatus M, Stubbs MT, Huber R, Bringmann P, Donner P, Schleuning WD, Bode W. Catalytic domain structure of vampire bat plasminogen activator: a molecular paradigm for proteolysis without activation cleavage. Biochemistry. 1997;36:13483–13493. doi: 10.1021/bi971129x. [DOI] [PubMed] [Google Scholar]
- Ringe D, Petsko GA. The ‘glass transition’ in protein dynamics: what it is, why it occurs, and how to exploit it. Biophys. Chem. 2004;105:667–680. doi: 10.1016/s0301-4622(03)00096-6. [DOI] [PubMed] [Google Scholar]
- Roe SM, Prodromou C, O'Brien R, Ladbury JE, Piper PW, Pearl LH. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J. Med. Chem. 1999;42:260–266. doi: 10.1021/jm980403y. [DOI] [PubMed] [Google Scholar]
- Scapin G, Reddy SG, Zheng R, Blanchard JS. Three-dimensional structure of Escherichia coli dihydrodipicolinate reductase in complex with NADH and the inhibitor 2,6-pyridinedicarboxylate. Biochemistry. 1997;36:15081–15088. doi: 10.1021/bi9719915. [DOI] [PubMed] [Google Scholar]
- Sidhu G, Withers SG, Nguyen NT, McIntosh LP, Ziser L, Brayer GD. Sugar ring distortion in the glycosyl-enzyme intermediate of a family G/11 xylanase. Biochemistry. 1999;38:5346–5354. doi: 10.1021/bi982946f. [DOI] [PubMed] [Google Scholar]
- Sinha N, Kumar S, Nussinov R. Inter-domain interactions in hinge-bending transitions. Structure. 2001;9:1165–1181. doi: 10.1016/s0969-2126(01)00687-6. [DOI] [PubMed] [Google Scholar]
- Stams T, Chen Y, Boriack-Sjodin PA, Hurt JD, Liao J, May JA, Dean T, Laipis P, Silverman DN, Christianson DW. Structures of murine carbonic anhydrase IV and human carbonic anhydrase II complexed with brinzolamide: molecular basis of isozyme-drug discrimination. Protein Sci. 1998;7:556–563. doi: 10.1002/pro.5560070303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenberg K, Lindqvist Y. Three-dimensional structures of glycolate oxidase with bound active-site inhibitors. Protein Sci. 1997;6:1009–1015. doi: 10.1002/pro.5560060506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tama F, Sanejouand YH. Conformational change of proteins arising from normal mode calculations. Protein Eng. 2001;14:1–6. doi: 10.1093/protein/14.1.1. [DOI] [PubMed] [Google Scholar]
- Tama F, Gadea FX, Marques O, Sanejouand YH. Building-block approach for determining low-frequency normal modes of macromolecules. Proteins. 2000;41:1–7. doi: 10.1002/1097-0134(20001001)41:1<1::aid-prot10>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Temiz NA, Bahar I. Inhibitor binding alters the directions of domain motions in HIV-1 reverse transcriptase. Proteins. 2002;49:61–70. doi: 10.1002/prot.10183. [DOI] [PubMed] [Google Scholar]
- Tirion MM. Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys. Rev. Lett. 1996;77:1905–1908. doi: 10.1103/PhysRevLett.77.1905. [DOI] [PubMed] [Google Scholar]
- Tousignant A, Pelletier JN. Protein motions promote catalysis. Chem. Biol. 2004;11:1037–1042. doi: 10.1016/j.chembiol.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Towler P, Staker B, Prasad SG, Menon S, Tang J, Parsons T, Ryan D, Fisher M, Williams D, Dales NA, et al. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J. Biol. Chem. 2004;279:17996–18007. doi: 10.1074/jbc.M311191200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varughese KI, Su Y, Cromwell D, Hasnain S, Xuong NH. Crystal structure of an actinidin-E-64 complex. Biochemistry. 1992;31:5172–5176. doi: 10.1021/bi00137a012. [DOI] [PubMed] [Google Scholar]
- Wilce MC, Bond CS, Dixon NE, Freeman HC, Guss JM, Lilley PE, Wilce JA. Structure and mechanism of a proline-specific aminopeptidase from Escherichia coli. Proc. Natl. Acad. Sci. USA. 1998;95:3472–3477. doi: 10.1073/pnas.95.7.3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wind RD, Uitdehaag JC, Buitelaar RM, Dijkstra BW, Dijkhuizen L. Engineering of cyclodextrin product specificity and pH optima of the thermostable cyclodextrin glycosyltransferase from Thermoanaerobacterium thermosulfurigenes EM1. J. Biol. Chem. 1998;273:5771–5779. doi: 10.1074/jbc.273.10.5771. [DOI] [PubMed] [Google Scholar]
- Wolf-watz M, Thai V, Henzler-Wiilman K, Hadjipavlou G, Eisenmesser EZ, Kern D. Linkage between dynamics and catalysis in a thermophilic-mesophilic enzyme pair. Nat. Struct. Mol. Biol. 2004;11:945–949. doi: 10.1038/nsmb821. [DOI] [PubMed] [Google Scholar]
- Xiang J, Sun J, Sampson NS. The importance of hinge sequence for loop function and catalytic activity in the reaction catalyzed by triosephosphate isomerase. J. Mol. Biol. 2001;307:1103–1112. doi: 10.1006/jmbi.2001.4536. [DOI] [PubMed] [Google Scholar]
- Xu C, Tobi D, Bahar I. Allosteric changes in protein structure computed by a simple mechanical model: hemoglobin T <-> R2 transition. J. Mol. Biol. 2003;333:153–168. doi: 10.1016/j.jmb.2003.08.027. [DOI] [PubMed] [Google Scholar]
- Yan X, Hollis T, Svinth M, Day P, Monzingo AF, Milne GW, Robertus JD. Structure-based identification of a ricin inhibitor. J. Mol. Biol. 1997;266:1043–1049. doi: 10.1006/jmbi.1996.0865. [DOI] [PubMed] [Google Scholar]
- Yang LW, Liu X, Jursa CJ, Holliman M, Rader AJ, Karimi H, Bahar I. iGNM: a protein functional motion database containing essential dynamics for 23149 PDB structures. http://ignm.ccbb.pitt.edu/ 2005a doi: 10.1093/bioinformatics/bti469. [DOI] [PMC free article] [PubMed]
- Yang LW, Liu X, Jursa CJ, Holliman M, Rader AJ, Karimi H, Bahar I. iGNM: a database of protein functional motions based on a Gaussian Network Model. Bioinformatics. 2005b;266 doi: 10.1093/bioinformatics/bti469. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdanov A, Wu S, DiMaio J, Konishi Y, Li Y, Wu X, Edwards BF, Martin PD, Cygler M. Crystal structure of the complex of human alpha-thrombin and nonhydrolyzable bifunctional inhibitors, hirutonin-2 and hirutonin-6. Proteins. 1993;17:252–265. doi: 10.1002/prot.340170304. [DOI] [PubMed] [Google Scholar]
- Zhang ZY, Shi YY, Liu HY. Molecular dynamics simulations of peptides and proteins with amplified collective motions. Biophys. J. 2003;84:3583–3593. doi: 10.1016/S0006-3495(03)75090-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Tang L, Wang X, Zhou Y, Lin Z. Structure of a snake venom phospholipase A2 modified by p-bromo-phenacyl-bromide. Toxicon. 1998;36:875–886. doi: 10.1016/s0041-0101(97)00169-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.