

Abstract
The complex imprinted Gnas locus encodes several gene products including Gsα, the ubiquitously expressed G protein α-subunit required for receptor-stimulated cAMP generation, and the neuroendocrine-specific Gsα isoform XLαs. XLαs is only expressed from the paternal allele, while Gsα is biallelically expressed in most tissues. XLαs knockout mice (Gnasxlm+/p−) have poor suckling and perinatal lethality, implicating XLαs as critical for postnatal feeding. We have now examined the metabolic phenotype of adult Gnasxlm+/p− mice. Gnasxlm+/p− mice had reduced fat mass and lipid accumulation in adipose tissue, with increased food intake and metabolic rates. Gene expression profiling was consistent with increased lipid metabolism in adipose tissue. These changes likely result from increased sympathetic nervous system activity rather than adipose cell-autonomous effects, as we found that XLαs is not normally expressed in adult adipose tissue and Gnasxlm+/p− mice had increased urinary norepinephrine levels but not increased metabolic responsiveness to a β3-adrenergic agonist. Gnasxlm+/p− mice were hypolipidemic and had increased glucose tolerance and insulin sensitivity. The similar metabolic profile observed in some prior paternal Gnas knockout models results from XLαs deficiency (or deficiency of the related alternative truncated protein XLN1). XLαs (or XLN1) is a negative regulator of sympathetic nervous system activity in mice.
The incidence of both obesity and diabetes has markedly increased over the past two decades. A basic understanding of the regulation of energy and glucose metabolism is required for the identification of potential new therapeutic targets. One gene shown to be important for metabolic regulation is GNAS on chromosome 20q13 in humans (Gnas on chromosome 2 in mice) (1,2). GNAS/Gnas encodes several protein products, including the ubiquitously expressed G protein α-subunit Gsα1 that couples receptors to cAMP generation, the alternative Gsα isoform XLαs, and the 55 kDa neuroendrocrine-specific secretory protein (NESP55). Heterozygous mutations resulting in partial Gsα deficiency lead to obesity in mice (3) and in patients with Albright hereditary osteodystrophy (1,2).
GNAS/Gnas is a complex imprinted gene whose major gene products are generated by alternative promoters and first exons that splice onto a common exon (exon 2) (1,2) (Fig. 1). NESP55 and XLαs are expressed from oppositely imprinted Nesp and Gnasxl promoters: NESP55 is only expressed from the maternal allele while XLαs is only expressed from the paternal allele. In contrast, the Gsα promoter is imprinted in a tissue-specific manner, with Gsα being biallelically expressed in most tissues but primarily expressed from the maternal allele in a small number of tissues.
Figure 1. Expression patterns of encoded proteins from the GNAS/Gnas locus.
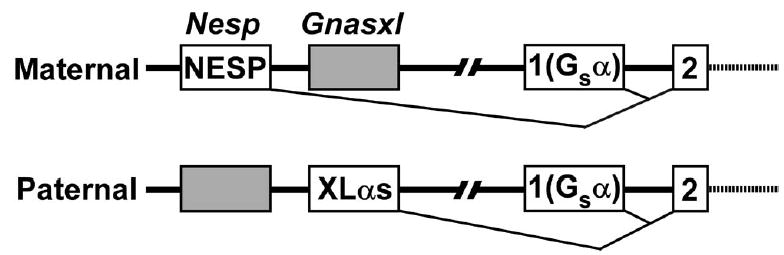
The maternal (above) and paternal (below) alleles of Gnas are shown with active (white) and inactive (gray) promoters and first exons encoding NESP55 (Nesp), XLαs (Gnasxl), and Gsα (exon 1), respectively, splicing to the common exon 2 (not drawn to scale). Exons downstream of exon 2 and promoters and exons for noncoding transcripts are not shown.
XLαs is structurally identical to Gsα except for a long amino terminal extension encoded within its specific first exon (4,5). In contrast to Gsα, which is ubiquitously expressed at high levels in virtually all tissues, XLαs has a more restricted tissue distribution with expression primarily limited to neuroendocrine tissues (4,6). XLαs has also been shown to be expressed in adipose tissue in four day old mice (7). In vitro biochemical studies have shown that XLαs is capable of stimulating adenylyl cyclase (8,9) and mediating receptor-stimulated cAMP production (9), although its role in vivo is less clear. The XLαs promoter also generates a neural-specific truncated form of XLαs (XLN1) produced by splicing of exon 3 onto an alternative terminal exon (6). Whether the resultant truncated protein has any biological function or whether it can act as a dominant negative inhibitor of Gsα or XLαs signaling is unknown. NESP55 does not appear to be an important metabolic regulator in either humans (10) or mice (11).
We originally generated a Gnas knockout in which exon 2, an exon common to all transcripts, was disrupted (12). Heterozygotes with mutation of the paternal allele (E2m+/p−), leading to complete XLαs deficiency and partial Gsα deficiency, failed to suckle and most died soon after birth with hypoglycemia (S. Yu, L.S.W., unpublished observations). Those that survived had markedly reduced adiposity, increased metabolic rate and activity levels, and greater than normal glucose tolerance and insulin sensitivity (13–15). E2m+/p− mice had increased urinary norepinephrine excretion, suggesting that these metabolic effects may be the consequence of increased sympathetic nervous system activity (14). In contrast, E1m+/p− mice with paternal deletion of Gsα exon 1 leading only to partial Gsα deficiency had normal suckling and survival and developed obesity, glucose intolerance, and insulin resistance (3). Comparison of E2m+/p− and E1m+/p− mice indirectly implicates XLαs as an important regulator of postnatal feeding as well as energy and glucose metabolism in older animals and suggests that XLαs and Gsα may have opposite effects on metabolic regulation in the whole animal.
We have recently generated mice with deficiency of only XLαs (Gnasxlm+/p−) and showed that these mice have poor suckling and perinatal survival (7), similar to that observed in E2m+/p− mice (12) and other mouse models with disruption or loss of the paternal Gnas allele (16–18), implicating XLαs as critical for postnatal feeding adaptation in mice. We have now examined the metabolic phenotype of adult Gnasxlm+/p− mice, and show that these mice have markedly reduced adiposity, increased metabolic rate with increased lipid metabolism in adipose tissue, and increased glucose tolerance and insulin sensitivity. We provide evidence that these metabolic changes are due to increased sympathetic nervous system activity rather than adipocyte cell-autonomous effects. XLαs (or possibly XLN1) appears to be an important negative regulator of sympathetic nervous system activity in mice. We also show that XLαs expression in adipose tissue is developmentally regulated.
EXPERIMENTAL PROCEDURES
Animals
Gnasxlm+/p− mice (7) were repeatedly mated with CD1 wild-type mice (Charles River, Wilmington, MA) for >5 generations and were maintained on a standard pellet diet (NIH-07, 5% fat by weight) and 12 h:12 h light/dark cycle. Except when noted, all experiments were performed on 12–14 week old male mutant mice and wild type littermates. Experiments were approved by the NIDDK Animal Care and Use Committee.
Body Composition, Food Intake, Metabolic Rate, and Activity Measurements
Body composition was measured using the Minispec mq10 NMR analyzer (Bruker Optics Inc., Woodlands, TX) which was calibrated with corn oil for fat mass and rat muscle for lean mass. Food intake, metabolic rates (oxygen consumption rate by indirect calorimetry) and activity levels were determined as previously described (14). The effect of the β3-specific adrenergic agonist CL316243 (19) on metabolic rate was measured as follows. At 9 A.M., mice were placed into calorimetry chambers which were prewarmed to 30°C. At 1 P.M., CL316243 at the indicated dose (from a 1 mg/ml stock in saline) or a saline vehicle control was injected ip. and, after a 1 h delay, data were collected for 3 h. Food and water were available at all times.
Microscopic Analysis
For standard histology, samples were fixed overnight in 4% paraformaldehyde, embedded in paraffin, cut and stained with hematoxylin and eosin. For mitochondrial staining, 10 hm cryostat sections were stained for succinate dehydrogenase (SDH) by incubation in 130 mM Tris-HCl buffer (pH 7.4) containing 0.2 mM nitroblue tetrazolium and 60 mM sodium succinate (Sigma, St. Louis, MO) at 37°C for 30 min. The slides were then rinsed with deionized water, dehydrated, and washed with xylene before coverslip mounting.
Hyperinsulinemic-Euglycemic Clamp Studies
Hyperinsulinemic-euglycemic clamps studies and in vivo glucose flux analysis were performed as previous described (15). Insulin levels on samples during the clamp studies were measured by RIA kit (Linco Research, St. Charles, MO).
Blood, Tissue, and Urine Chemistries
Blood was obtained by retroorbital bleed. Serum glucose, cholesterol, and triglyceride levels were measured with an auto-analyzer by the NIH Clinical Chemistry Laboratory. Serum insulin, leptin, glucagon, and adiponectin were measured by Linco Research assay services. For C-peptide/insulin ratios, both were measured by RIA (Linco). Serum resistin was measured using an ELISA kit (Linco). Serum corticosterone was measured by Ani-Lytics, Inc., Gaithersburg, MD. Serum free fatty acids (FFA) were measured using a kit from Roche Diagnostics, Branchburg, NJ. Triglyceride and glycogen content of hindlimb muscles were measured as previously described (15). Urine was collected by bladder puncture and catecholamines were measured by HPLC (20), and corrected by creatinine concentration within the same samples, which were measured by Ani-Lytics.
Triglyceride Clearance Test
Clearance of triglycerides (12.5 μl of peanut oil/g body weight delivered by gavage) from the circulation was measured in mice after a 4 h fast. Blood was taken before gavage and hourly for 6 h after gavage and plasma triglycerides were measured (Kit#TR22421, Thermo DMA, Waltham, MA).
Glucose and Insulin Tolerance Tests
Glucose and insulin tolerance tests were performed in overnight fasted mice after ip. injection of glucose (2 mg/g) or insulin (Humulin, 0.50 mIU/g). Blood glucose levels in tail vein bleeds were measured using a Glucometer Elite (Bayer, Elkhart, IN) immediately before and at indicated times after injection.
Portal Vein Insulin Injection and Immunoblot Analysis
Overnight-fasted mice were anesthetized with avertin (0.25 mg/g body weight ip.). Insulin (100 hl of 15 hg/ml, Sigma) was injected via the portal vein. At 2 or 4 min after injection, interscapular BAT, epididymal WAT, and hind leg muscles were dissected and immediately frozen. Tissues were then homogenized and protein concentrations of protein extracts were measured as previously described (15). Immunoprecipitations were performed on tissue extracts (1 mg protein) using an anti-Akt1 Ab (Upstate Biotechnology, Lake Placid, NY) as per manufacturer’s instructions, followed by immunoblot analysis using an anti-Akt1 phospho-Ser473 Ab (Upstate). Ab binding was determined by ECL (ECL kit, Amersham, Arlington Heights, IL). The same blots were stripped and rehybridized with the anti-Akt1 Ab.
Quantitative Reverse Transcription-PCR
RNA was extracted from frozen tissues using TRIzol (Invitrogen, Carlsbad, CA) or Mini RNeasy Kit (Qiagen, Germantown, MD) and treated with DNase (DNA-free, Ambion, Austin, TX) to remove DNA contamination. Reverse transcription was performed using the SuperScript First-Strand Synthesis system (Invitrogen). Gene expression levels were measured by quantitative RT-PCR using a real time PCR machine (MxP3000, Stratagene, La Jolla, CA). PCR reactions (20 μl total volume) included cDNA, 100 nM primers and 10 hl of SYBR Green MasterMix (Applied Biosystems, Foster City, CA). To get relative quantification, standard curves were simultaneously generated with serial dilutions of cDNA and results were normalized to β-actin mRNA levels in each sample, which were determined simultaneously by the same method. Specificity of each RT-PCR product was indicated by its dissociation curve and the presence of a single band of expected size on acrylamide gel electrophoresis. Sequences for gene-specific primers are listed on Supplementary Table 2.
Semiquantitative RT-PCR
XLαs mRNA expression was examined in BAT and WAT by semi-quantitative RT-PCR. After first-strand synthesis, duplex PCR was performed (32 cycles, 60°C annealing temperature) using a pair of XLαs-specific primers (1 μM each) and β-actin-specific primers (100 nM each) (primer sequences listed on Supplemental Table 2). Products were separated on agarose gels and gels were stained with ethidium bromide.
Statistical Analysis
Data are expressed as mean ± S.E.M.. Statistical significance between groups was determined using unpaired Student t test (two-tailed) with differences considered significant at p < 0.05.
RESULTS
Gnasxlm+/p− Mice Have Reduced Survival and Adiposity
In this study we examined the survival and adult metabolic phenotype of Gnasxlm+/p− mice which were placed in a CD1 genetic background in order to compare these mice to other Gnas knockout models which we have studied on the same genetic background. In most other inbred genetic backgrounds E2m+/p− mice have no survival beyond several days (S. Yu, L.S.W., unpublished observations). In the CD1 background most Gnasxlm+/p− mice failed to suckle and died soon after birth, as was previously described in both these mice (7) and E2m+/p− mice (12). By 3 weeks of age, Gnasxlm+/p− mice had a survival rate that was ~27% of that expected by Mendelian inheritance (8 of 59 total offspring) and the effect appeared to be similar in males and females. This survival rate is similar to the survival rate that we previously reported for E2m+/p− mice in the CD1 background (23%) (12).
Growth curves (Fig. 2A) demonstrate that both male and female Gnasxlm+/p− mice grew very slowly through the first 30 days relative to their wild-type littermates, and the resulting weight differences were maintained during adulthood. Adult Gnasxlm+/p− mice weighed ~45% less than wild-type littermates, and had a small but significant reduction in nasoanal body length (Fig. 2B). Body composition analysis by NMR showed that Gnasxlm+/p− mice had markedly reduced fat mass and increased lean mass relative to total body weight (Fig. 2C). Consistent with this, the relative weights of retroperitoneal and epidydimal white adipose tissue (WAT) pads and interscapular brown adipose tissue (BAT) pads were greatly reduced in Gnasxlm+/p− mice, while the relative weights of most other organs were unchanged (Fig. 2D). The relative weight of testes and lungs were slightly, although significantly, increased in Gnasxlm+/p− mice as compared to controls. Histology of both BAT and WAT from Gnasxlm+/p− mice showed reduced lipid accumulation and size of adipocytes (Fig. 2E). BAT from Gnasxlm+/p− mice had markedly increased SDH staining compared to controls (Fig. 2F), indicating that the mutant mice had greater mitochondrial content in BAT. Overall, our findings show that, similar to E2m+/p− mice (12,14), Gnasxlm+/p− mice have reduced preweaning survival, poor growth, and maintain a very lean phenotype in adulthood.
Figure 2. Body and organ weights and body composition.
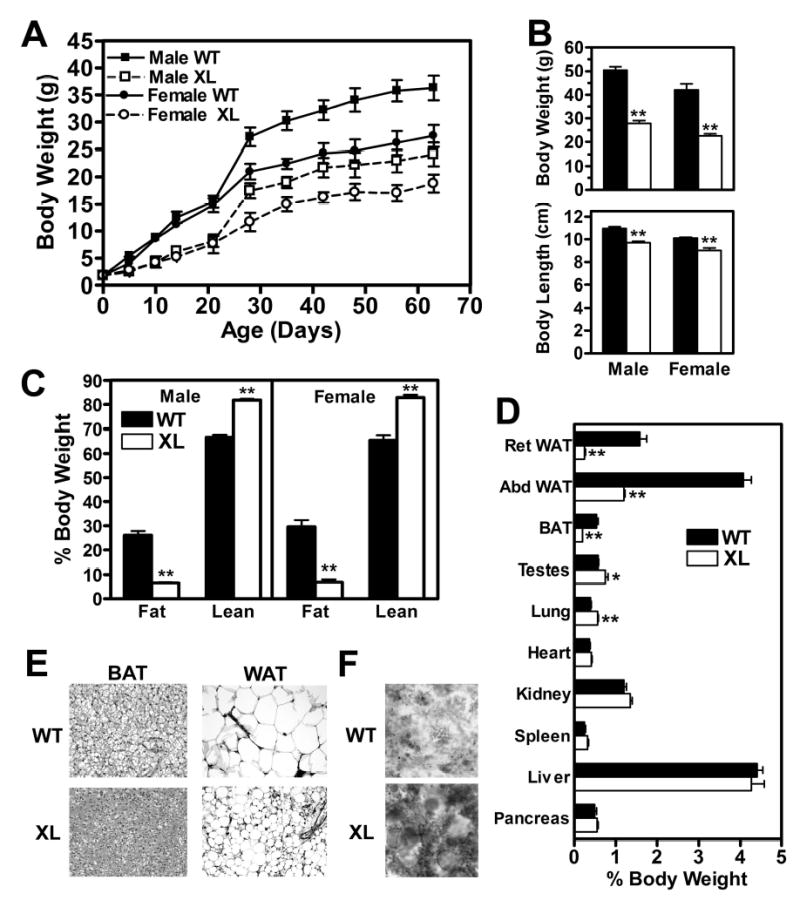
A, Growth curves of male (▪,□) and female (•,○) wild-type (WT; ▪,•, solid lines) and Gnasxlm+/p− (XL; x,z, dashed lines) mice (n = 14-17/group). B, Body weights (top panel) and nasoanal body length (lower panel) of wild-type (closed bars in all panels) and Gnasxlm+/p− mice (open bars in all panels) (n = 5–7/group). C, Fat and lean mass expressed as percent body weight (n = 5–8/group). In B and C males are on the left and females on the right. D, Organ weights in males expressed as percent body weight (n = 4-5/group). BAT, interscapular BAT; Ret and Epi WAT, retroperitoneal and epidydimal WAT. E, BAT (left) and epididymal WAT (right) from wild-type (WT, top) and Gnasxlm+/p− mice (XL, bottom). F, SDH (mitochondrial) staining of BAT from wild-type (top) and mutant (bottom) mice. *p < 0.05 or **p < 0.01 vs. wild-type.
Hypermetabolism in Gnasxlm+/p− Mice is Associated with Increased Norepinephrine Levels
The markedly reduced adiposity of Gnasxlm+/p− mice indicates that they may have either reduced food intake or increased energy expenditure rates relative to controls. Both male and female Gnasxlm+/p− mice had significantly increased food intake when corrected for body mass (Fig. 3A), indicating that reduced adiposity is not due to hypophagia. Hyperphagia in Gnasxlm+/p− mice may be a physiologic response to low circulating leptin levels (Table 1), which is an expected consequence of their severely lean phenotype. Both resting and total energy expenditure (O2 consumption) rates were very significantly increased at both ambient temperature (24°C) and thermoneutral temperature (30°C) (Fig. 3B). There were no differences in the respiratory exchange ratio (ratio of CO2 produced to O2 consumed) (Fig. 3C) or activity levels (Fig. 3D) between wild-type and mutant mice. The metabolic changes are similar to those previously observed in E2m+/p− mice, although we found E2m+/p− mice to be hyperactive (14) while Gnasxlm+/p− mice were not. Sex and age differences between the mice examined in this and the prior E2m+/p− mouse study may account for these discrepant results.
Figure 3. Studies of energy balance, activity, β3-adrenergic responsiveness, and urine catecholamine excretion.
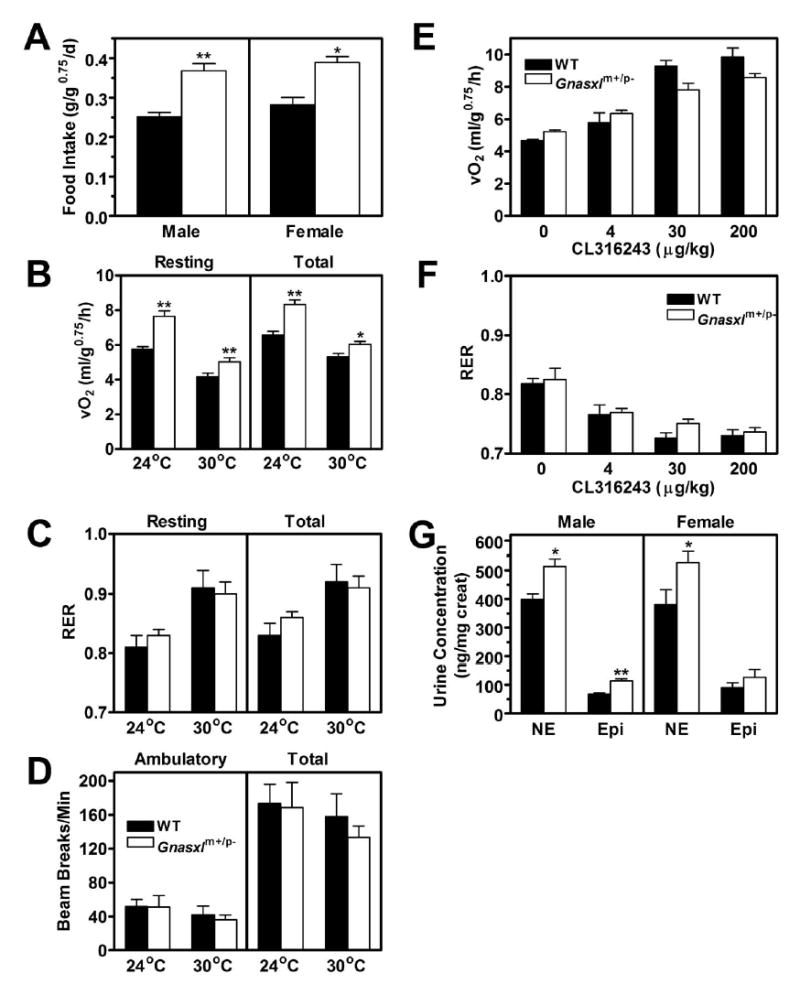
A, Food intake in male (left) and female (right) wild-type (closed bars) and Gnasxlm+/p− (open bars) mice (n = 5-11/group). B, Resting and total metabolic rate, C, respiratory exchange ratio (RER), and D, ambulatory and total activity levels measured over 24 h at indicated temperatures (n = 6/group). E, Resting metabolic rate and F, RER at thermoneutral temperature (30°C) after indicated doses of CL316243 administered to male mice (n = 3/group). G, Urine norepinephrine (NE) and epinephrine (Epi) levels expressed as ng/mg creatinine in male and female mice (n = 7-9/group). *p < 0.05 or **p < 0.01 vs. wild-type.
Table 1.
Serum chemistries
| Wild-Type | Gnasxlm+/p− | |
|---|---|---|
| Glucose (mg/dl) | 180 ± 8 | 152 ± 6** |
| Cholesterol (mg/dl) | 132 ± 7 | 82 ± 9** |
| Triglycerides (mg/dl) | 104 ± 16 | 45 ± 18** |
| Insulin (ng/ml) | 4.14 ± 0.90 | 0.73 ± 0.11** |
| Glucagon (pM) | 101 ± 22 | 145 ± 96 |
| Leptin (pM) | 692 ± 79 | 39 ± 19** |
| Adiponectin (μg/ml) | 17.0 ± 1.1 | 11.8 ± 0.8** |
| Resistin (ng/ml) | 8.2 ± 1.2 | 10.1 ± 1.6 |
| Corticosterone (ng/ml) | 169 ± 58 | 203 ± 48 |
p<0.01 vs. wild-type littermates
We next examined whether the hypermetabolic state could be explained by increased metabolic responsiveness to sympathetic stimulation by measuring the resting metabolic rate after administration of various doses of the β3-adrenergic agonist CL316243 at thermoneutral temperature (30°C). Resting metabolic rate at thermoneutrality reflects the metabolic rate in the absence of adaptive thermogenesis with minimal endogenous stimulation of adipose tissue by the sympathetic nervous system (21,22). As β3 adrenergic receptors are specifically expressed in adipose tissue and CL316246 has been shown to specifically activate adipose tissue (23), increases in metabolic rate after CL316246 reflect the metabolic responsiveness of adipose tissue to catecholamine stimulation by β3-adrenergic receptors, a major receptor stimulated by sympathetic nerves in adipose tissue. Our results show that adipose tissue in Gnasxlm+/p− mice was not more sensitive to β3-adrenergic stimulation (Fig. 3E). In fact Gnasxlm+/p− mice tended to have an increased baseline activity (p = 0.079) and a somewhat reduced response to higher doses of the β3 agonist, suggesting that they may be subtly resistant to β3-adrenergic stimulation. There were no significant differences in the respiratory exchange ratio in response to the β3 agonist (Fig. 3F). These results are similar to what was previously observed before and after maximal β3 agonist stimulation in E2m+/p− mice (14).
We also assessed the ability of the β3 agonist to acutely raise serum FFA levels, which is a measure of the acute lipolytic response in adipose tissue. In the fed state Gnasxlm+/p− mice tended to have elevated serum FFA levels compared to controls (p = 0.098) even though they had a reduced adipose mass (Fig. 4A). The increase in FFA levels measured 20 min after injection of CL316243 (100 μg/kg) was significantly lower in Gnasxlm+/p− mice than in controls. The ratio of unstimulated/stimulated FFA levels was significantly higher in Gnasxlm+/p− mice (0.33 ± 0.05 in Gnasxlm+/p− vs. 0.21 ± 0.02 in control mice). These results are similar to the slightly reduced metabolic responsiveness to CL316243 observed in the mutant mice. In the fasted state (when insulin levels are low) Gnasxlm+/p− mice had 2-fold higher serum FFA levels than controls (Fig. 4B), indicative of a very high lipolytic rate in mutant mice. After administration of insulin (0.75 mIU/g body weight), FFA levels fell to similar levels in Gnasxlm+/p− and control mice, indicating that the ability of insulin to suppress lipolysis is maintained in Gnasxlm+/p− mice.
Figure 4. Studies of lipid metabolism.
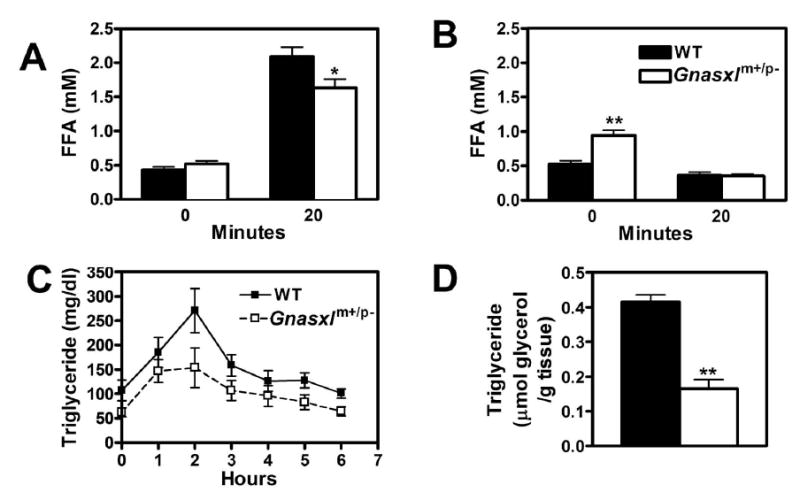
A, Serum FFAs measured in non-fasted wild-type (closed bars) and Gnasxlm+/p− (open bars) mice before (time 0) and 20 min after ip. administration of CL316243 (100 μg/kg body weight; n = 6/group). B, Mice were fasted for 9 h, and serum FFAs were measured before or 20 min after ip. insulin administration (0.75 mIU/g body weight; n = 5–6/group). C, Serum triglyceride levels measured in wild-type (▪, solid lines) and Gnasxlm+/p− (□, dashed lines) mice before and hourly after lipid (peanut oil) administration by oral gavage (n = 6/group). The areas under the curve are 972 + 135 for wild-type and 648 + 114 for Gnasxlm+/p− mice (p < 0.05). D, Triglyceride content of hindlimb muscles from non-fasting wild-type and mutant mice (n = 7/group). Studies in panels A-C were performed in 6 mo old female mice. *p < 0.05 or **p < 0.01 vs. wild-type.
To assess overall sympathetic nerve activity we measured urinary norepinephrine concentrations normalized to creatinine to correct for differences in urinary concentration and in lean mass. Similar to E2m+/p− mice (14), both male and female Gnasxlm+/p− mice had significantly increased urine NE levels (Fig. 3G). Urine epinephrine levels, which primarily reflect adrenomedullary activity, were also increased, although the difference was only significant in males. There was no significant difference in urine creatinine concentration between the two groups (data not shown). Overall, our results suggest that increased metabolic rate in adult Gnasxlm+/p− mice is primarily the result of increased sympathetic nervous system activity (and possibly adrenomedullary secretion), rather than an inherent difference in sensitivity to metabolic stimulation.
Gnasxlm+/p− Mice have Improved Glucose Tolerance and Insulin Sensitivity
Random serum glucose, insulin, cholesterol, and triglyceride levels were all significantly lower in Gnasxlm+/p− as compared to control mice (Table 1). Similar to E2m+/p− mice (15), Gnasxlm+/p− mice had an increased clearance rate of an oral triglyceride load and very low muscle triglyceride levels (Figs. 4C, D), consistent with an increased rate of overall lipid metabolism. Glucose and insulin tolerance tests showed that Gnasxlm+/p− mice had increased glucose tolerance and insulin sensitivity compared to controls (Fig. 5A, B). Improved insulin sensitivity is not explained by altered serum levels of adipocytokines, as circulating levels of leptin and the insulin-sensitizing factor adiponectin were reduced relative to controls, while resistin levels were unaffected in Gnasxlm+/p− mice (Table 1). Consistent with the severely lean phenotype, serum leptin levels were very low in Gnasxlm+/p− mice (Table 1). In contrast to young Gnasxlm+/p− pups (7) and similar to adult E2m+/p−mice (15), serum glucagon levels were unaffected in adult Gnasxlm+/p− mice (Table 1). Corticosterone levels were also unaffected in Gnasxlm+/p− mice (Table 1). Low tissue triglyceride levels are generally associated with increased insulin sensitivity and may contribute to the increased insulin sensitivity observed in Gnasxlm+/p− mice.
Figure 5. Studies of glucose metabolism and insulin sensitivity.
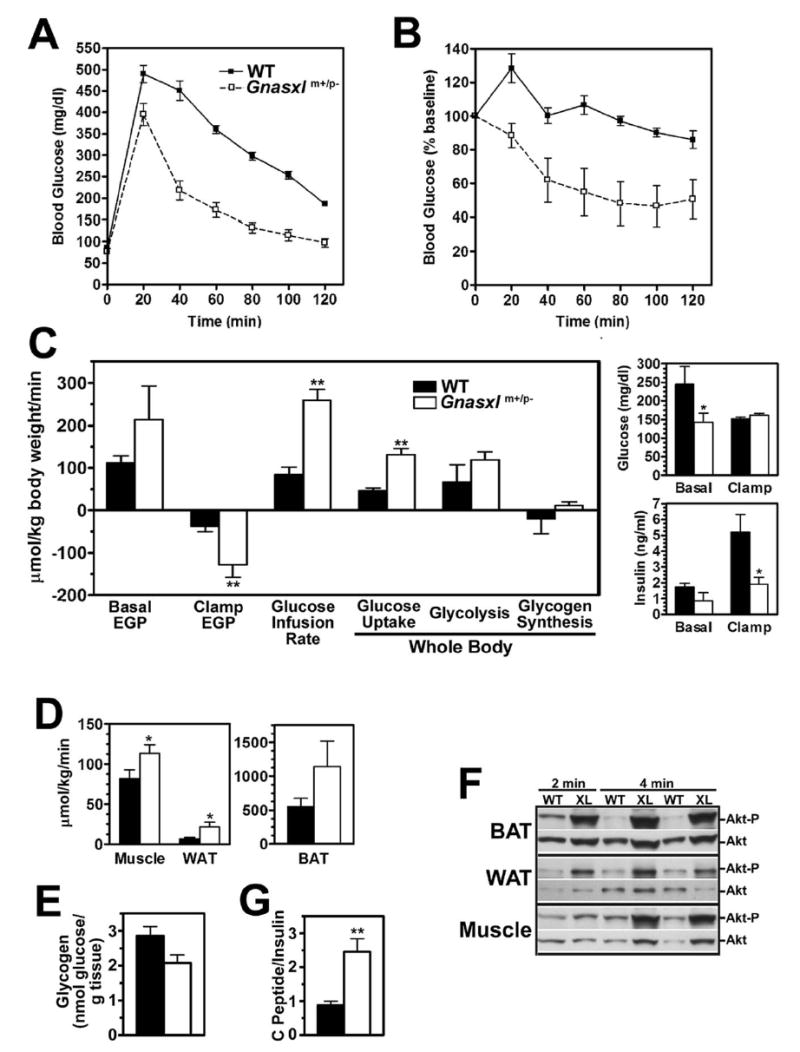
Blood glucose in Gnasxlm+/p− mice (□, dashed lines) and wild-type littermates (▪, solid lines) measured at indicated time points before and after A, glucose or B, insulin administration (n = 4-5 per group; results for insulin tolerance test expressed as % of baseline). Genotype had a significant effect by two-factor ANOVA for both studies. C, Basal (pre-clamp) and clamp endogenous glucose production rate, glucose infusion rate, and rates of whole body glucose uptake, glycolysis, and glycogen synthesis in wild-type (closed bars) and Gnasxlm+/p− mice (open bars) during hyperinsulinemic-euglycemic clamp studies (n = 7/group). Basal and clamp glucose (top panel) and insulin levels (bottom panel) are shown to the right. D, Glucose uptake rates in gastrocnemius muscle, WAT, and BAT, respectively, during the clamp study. E, Glycogen content of hindlimb muscles obtained from non-fasting wild-type (open bar) and mutant (closed bar) mice (n = 5–7/group). F, Results of immunoblotting using anti-Akt1 phospho-Ser473 (top of each panel) followed by anti-Akt Ab (bottom of each panel) in BAT, WAT, and muscle tissue extracts derived from wild-type (WT) and Gnasxlm+/p− (XL) mice which were sacrificed at indicated times after insulin administration. G, Serum C-peptide/insulin ratios from non-fasted 8 mo old male mice (n = 5-6/group). *p < 0.05 or **p < 0.01 vs. wild-type.
Euglycemic-hyperinsulinemic clamp studies were performed to further characterize insulin action in specific tissues. Prior to insulin infusion, both basal glucose and insulin were lower in Gnasxlm+/p− mice (Fig. 5C). During the clamp glucose levels were maintained at similar levels in Gnasxlm+/p− mice and wild-type controls (Fig. 5C). Although receiving similar infusion rates corrected for body size, Gnasxlm+/p− mice had significantly lower insulin levels during the clamp (Fig. 5C), suggesting that they may have increased insulin clearance. The serum C-peptide/insulin ratio measured in non-fasting mice was three-fold higher in Gnasxlm+/p− than in control mice (Fig. 5G), which is also consistent with these mice having an increased insulin clearance rate. Despite the lower insulin levels attained, Gnasxlm+/p− mice had significantly higher rates of glucose infusion and whole body glucose uptake (Fig. 5C), consistent with increased insulin sensitivity. Increased whole body glucose utilization is most accounted for by increased whole body glycolysis, although this difference did not reach statistical significance. There were no differences in whole body glycogen synthesis.
Glucose uptake was significantly increased in muscle and WAT and increased 2-fold in BAT (Fig. 5D). Consistent with these findings, phosphorylation of the downstream kinase Akt1 in response to acute insulin administration was greater in WAT, BAT, and skeletal muscle of mutant mice (Fig. 5F). Gene expression studies showed the insulin receptor (IR), insulin receptor substrate 1 (IRS1), and Akt2 to be overexpressed in WAT and the insulin-sensitive glucose transporter Glut4 to be overexpressed in skeletal muscle of Gnasxlm+/p− mice (Fig. 6). Despite the increased muscle glucose uptake observed during the clamp study, Gnasxlm+/p− mice tended to have reduced muscle glycogen levels in the fed state (Fig. 5E, p = 0.058). Potential causes of reduced muscle glycogen levels include hypoinsulinemia (Table 1, Fig. 5C), increased glucose utilization via glycolysis, and/or increased glycogenolysis.
Figure 6. Gene expression studies.
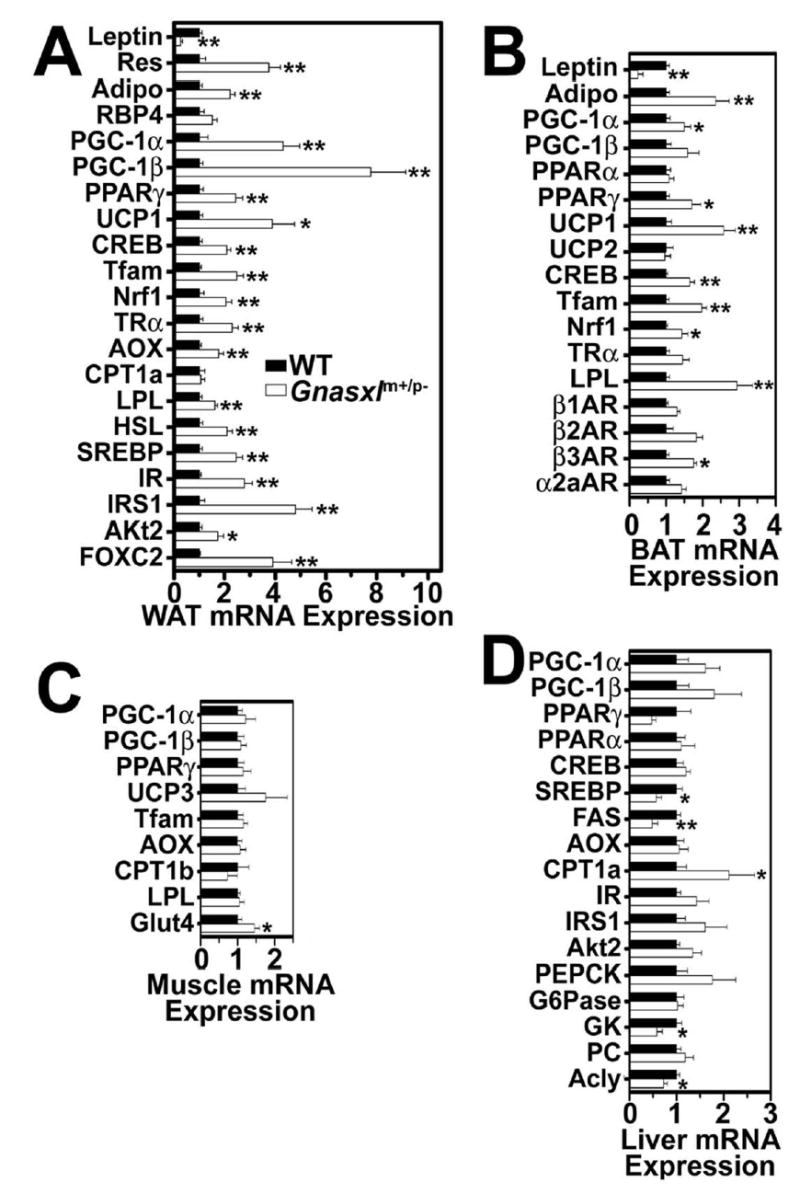
mRNA levels of indicated genes (corrected for β-actin mRNA expression) in A, WAT, B, BAT, C, muscle, and D, liver, respectively, from non-fasted wild-type (closed bars, means normalized to 1) and Gnasxlm+/p− mice (open bars) (n = 5/group). Res, resistin; Adipo, adiponectin; CPT, carnitine palmitoyltransferase; G6Pase, glucose-6-phosphatase; PC, pyruvate carboxylase; β1-, β2-, β3-, α2a-AR, β1-, β2- , β3-, α2a-adrenergic receptor. Other abbreviations are defined in the text. *p < 0.05 or **p < 0.01 vs. wild-type.
Basal (pre-clamp) endogenous glucose production rate, which primarily reflects glucose output from the liver, tended to be higher in Gnasxlm+/p− mice prior to the clamp, although this difference was not significant (Fig. 5C). Suppression of endogenous glucose production rate during the clamp was much greater in Gnasxlm+/p− mice, despite the lower insulin levels attained in these mice. Comparison of Gnasxlm+/p− mice during the clamp with wild-type mice prior to clamp, where insulin levels were similar in the two groups, showed that Gnasxlm+/p− mice had much lower endogenous glucose production at similar circulating insulin levels (Fig. 5C). Overall our results show that, similar to E2m+/p−mice (13,15), Gnasxlm+/p− mice have increased glucose tolerance and insulin sensitivity in muscle, adipose tissue, and liver.
Gene Expression Studies in Tissues Involved in Energy and Glucose Metabolism
We examined gene expression patterns in WAT, BAT, liver, and muscle from non-fasted mice by real-time quantitative RT-PCR (Fig. 6). Consistent with their reduced adiposity, Gnasxlm+/p− mice had reduced leptin and increased adiponectin expression in both BAT and WAT. Resistin was also overexpressed in WAT of Gnasxlm+/p− mice. These changes in leptin, adiponectin, and resistin expression mimic those observed in E2m+/p− mice (14,15). The discrepancies between gene expression in adipose tissue and serum levels of resistin and adiponectin are most likely due to the fact that Gnasxlm+/p− have markedly reduced adipose tissue mass, and similar discrepancies between gene expression and serum protein levels have been previously observed in other knockout mouse models (15,24). The adipocytokine RBP4, which has been reported to reduce insulin sensitivity (25), tended to be expressed at higher levels in WAT of Gnasxlm+/p− mice, although the difference was not significant.
Peroxisome proliferator activated receptor (PPAR) γ-coactivator 1α (PGC1α) is a cAMP-inducible gene that activates genes required for thermogenesis, mitochondrial function, and lipid oxidation (26). This gene, as well as the downstream thermogenic uncoupling protein 1 (UCP1) gene and the mitochondriagenic genes nuclear respiratory factor 1 (Nrf1) and mitochondrial transcription factor A (Tfam) were all significantly upregulated in WAT and BAT of Gnasxlm+/p− mice, consistent with increased sympathetically-mediated cAMP formation and mitochondrial biogenesis in adipose tissue (see Fig. 2F). While UCP1 was shown to be induced in both BAT and WAT, WAT had <1000 fold lower UCP1 mRNAs levels than BAT. The lipid oxidation gene acyl CoA oxidase (AOX) was also significantly upregulated in WAT. Lipoprotein lipase (LPL) and hormone-sensitive lipase (HSL), which catalyze triglyceride uptake and hydrolysis, respectively, as well as other adipogenic and signaling genes (PGC1β, PPARγ, sterol regulatory element binding protein 1c [SREBP1c], cAMP-response element binding protein [CREB], and thyroid hormone receptor α [TRα]) were also upregulated in WAT of Gnasxlm+/p− mice. Several of these genes (PPARγ, CREB, LPL) were upregulated in BAT of Gnasxlm+/p− mice as well (Fig. 5B). Increased LPL expression is consistent with the increased triglyceride clearance observed in Gnasxlm+/p− mice (Fig. 4C). Gnasxlm+/p− mice also have 4-fold higher expression of the forkhead transcription factor FOXC2 in WAT. Adipose-specific FOXC2 overexpression leads to increased β-adrenergic/Gsα/cAMP signaling and a metabolic phenotype similar to that present in Gnasxlm+/p− mice (27,28). Expression of the β2- and β3-adrenergic receptor genes were increased (only significant for β3 receptors), while there were no changes in β1-or α2a-adrenergic receptors. Overall these changes in gene expression are consistent with increased cAMP-driven metabolic activation and energy dissipation (lipolysis and lipid oxidation) in adipose tissue of adult Gnasxlm+/p−mice. In contrast, there were no significant changes in genes involved in energy metabolism and thermogenesis in skeletal muscle (Fig. 6C), suggesting that the increase metabolic rate of Gnasxlm+/p− is primarily due to increased energy dissipation in adipose tissue.
The major changes in liver (Fig. 6D) include reduced expression of genes involved in lipogenesis, (PPARγ, sterol regulatory element binding protein 1c [SREBP1c], ATP citrate lyase [Acly], and fatty acid synthase [FAS]) and increased expression of the lipid oxidation gene for carnitine palmitoyltransferase 1a (CPT1a). These changes, as well as the increased lipid oxidation in adipose tissue, probably lead to the low serum and tissue triglyceride levels and increased triglyceride clearance in Gnasxlm+/p−and E2m+/p− mice (14,15). The glucose metabolizing gene glucokinase (GK) was downregulated while the gluconeogenic gene for phosphoenolpyruvate carboxykinase (PEPCK) tended to be somewhat upregulated, although the difference for PEPCK was not statistically significant. Insulin stimulates lipogenesis and glucose metabolism and inhibits gluconeogenesis, while catecholamines lead to opposite effects on hepatic glucose metabolism (29). The gene expression changes in livers of Gnasxlm+/p− mice most likely result from these mice having very low insulin levels and increased catecholamine levels, and are consistent with the tendency for these mice to have higher basal endogenous glucose production rates which were observed during the clamp study.
XLαs Expression in Adipose Tissue is Lost During Postnatal Development
To determine if XLαs deficiency in adipose tissue could contribute to the adult Gnasxlm+/p− metabolic phenotype, we examined XLαs mRNA expression in adipose tissue of wild-type mice at various ages by duplex RT-PCR. As we showed previously (7), XLαs mRNA was expressed in both BAT and WAT during the first week postpartum (Fig. 7). However XLαs expression in both BAT and WAT decreased over the first 2 weeks after birth leading to barely detectable expression at postpartum days 15 and 20 and total absence in adult mice. Integrity and amount of RNA was confirmed by successful coamplication of β-actin mRNA in all samples (Figure 7, A and B, lower major band). XLαs expression is developmentally regulated in adipose tissue, being present only during the early postnatal period. It is therefore unlikely that genetic loss of XLαs should lead to cell-autonomous changes in adipocytes in adult mice.
Figure 7. XLαs gene expression in adipose tissue.
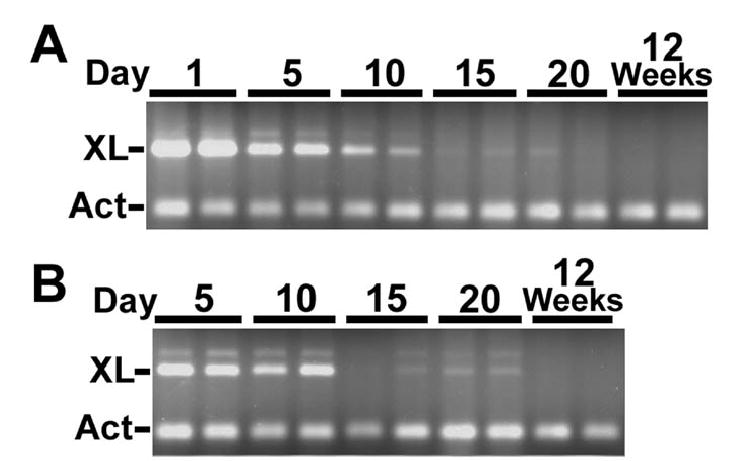
Duplex PCR with XLαs- (XL) and β-actin-specific (Act) primers, respectively, on A, BAT and B, WAT RNA samples from normal mice at postpartum ages indicated above each lane. The faint band present in some lanes is likely to be from amplication of transcripts including a small alternatively spliced exon downstream of XLαs exon 1 (6,48).
DISCUSSION
In this study we show that XLαs deficiency leads to reduced adiposity and increased metabolic rate and insulin sensitivity. These changes, as well as reduced survival, poor suckling, and reduced body weight of Gnasxlm+/p− pups (7), are also observed in E2m+/p− mice (12). These results confirm that the E2m+/p− phenotype results from XLαs deficiency, which has a dominant effect over the partial Gsα deficiency in these mice. This explains why E1m+/p− mice, which have loss of Gsα expression from the paternal allele but no disruption of XLαs expression, lack the Gnasxlm+/p− or E2m+/p− phenotype, and in fact develop obesity and insulin resistance (3).
Fat stores reflect the balance between energy intake and energy expenditure. Reduced adiposity of Gnasxlm+/p− mice results from increased energy expenditure. Both Gnasxlm+/p− and E2m+/p− mice (14,15) have increased energy expenditure associated with increased triglyceride clearance, consistent with an increase in lipid oxidation. In Gnasxlm+/p− mice the most striking changes in morphology and gene expression are in WAT, with a gene profile pattern consistent with increased metabolic activity. Multiple genes involved in lipolysis (hormone-sensitive lipase), and fatty acid oxidation and thermogenesis (PGC-1, UCP-1, AOX, TRα, Nrf1, and Tfam), most of which are known to be stimulated by β-adrenergic/Gsα/cAMP signaling pathways (26,30), are upregulated in WAT, and to a lesser extent in BAT, of Gnasxlm+/p− mice. Consistent with increased expression of genes involved in oxidative metabolism and mitochondrial biogenesis, we showed that BAT from Gnasxlm+/p− mice has increased mitochondrial content. In contrast, genes regulating energy metabolism were unaffected in skeletal muscle or liver, except for an increase in CPT1a in the latter. It therefore appears that Gnasxlm+/p− mice are lean primarily as a result of increased lipid mobilization and oxidation in adipose tissue. A similar metabolic profile was observed in mice with adipose-specific overexpression of the forkhead transcription factor FOXC2 (27,28) and mice in which the C/EBPα gene was replaced with the C/EBPβ gene (30). Both of these models have increased β-adrenergic/Gsα/cAMP signaling in adipocytes, and increased Gsα expression in cultured white adipocytes was shown to promote mitochondrial biogenesis and lipid oxidation (30). Interestingly FOXC2 is overexpressed in WAT of Gnasxlm+/p− mice, suggesting that this gene may have a role in the induction of lipid metabolism in adipose tissue by sympathetic nervous system stimulation.
The sympathetic nervous system stimulates lipolysis in WAT (31,32) and lipolysis, lipid oxidation, and thermogenesis in BAT (26) via β-adrenergic/Gsα/cAMP signaling pathways. The metabolic and molecular changes in adipose tissue of Gnasxlm+/p− mice could result from a cell-autonomous effect leading to increased Gsα/cAMP signaling in adipocytes or from increased sympathetic nervous system activity. Our results for adult mutants are most consistent with altered sympathetic nerve activity rather than adipocyte cell-autonomous differences as the primary mechanism for the changes observed in Gnasxlm+/p− mice. Similar to E2m+/p− mice (14), Gnasxlm+/p− mice had increased urine norepinephrine levels, indicative of increased sympathetic nervous system activity, and increased urine epinephrine levels in males, indicative of increased adrenal medullary activity. Adult Gnasxlm+/p− mice had increased energy expenditure rates and serum FFA levels in the basal state, and reduced responses in both of these parameters after administration of an adipose-specific β3-adrenergic agonist. These results suggest that these mice are not intrinsically more sensitive to catecholamine stimulation (at least not through β3 adrenergic pathways) but rather have increased sympathetic nervous system activity. We cannot rule out the possibility that increased β-adrenergic receptor gene expression, which we documented in BAT from Gnasxlm+/p− mice, may lead to increased sensitivity of adipose tissue to catecholaminergic stimulation and increased metabolic activation. However, this is not likely to be the primary defect, as we show that XLαs is not expressed in adipose tissue of adult wild-type mice, and therefore genetic loss of XLαs should produce no intrinsic changes to adipocytes. We can not rule out the possibility that XLαs deficiency in adipocytes during the perinatal period leads to altered adipose function later in development.
Hyperleptinemia, which results from increased leptin expression in adipose tissue, acts in the central nervous system to inhibit food intake and stimulate sympathetic nervous system activity, leading to increased energy expenditure. Sympathetic stimulation of β-adrenergic/Gsα/cAMP signaling in adipocytes inhibits leptin expression (33). Hypoleptinemia likely contributes to the hyperphagia in Gnasxlm+/p− mice. However, the increased sympathetic nervous system activity in Gnasxlm+/p− mice is opposite to what is expected in the setting of hypoleptinemia, suggesting that these mice have a primary defect in sympathetic nervous system regulation. Chronically increased sympathetic stimulation of adipose tissue contributes to reduced leptin expression and increased adiponectin and resistin expression, similar to what we have observed in E2m+/p− mice (14,15,34,35).
XLαs is expressed within the distribution of the sympathetic trunk in the central nervous system (7,36), suggesting that it could play a primary role as a negative regulator of sympathetic nervous system activity. Gsα deficiency may lead to opposite metabolic effects by acting at sites that are distinct from those expressing XLαs. For example, Gsα presumably mediates the effects of melanocortins to stimulate sympathetic nervous system activity within the hypothalamus (31,32,37-39). Another possibility is that the alternative truncated Gnasxl product XLN1 is a dominant-negative inhibitor of Gsα signaling. XLαs has also been shown to be expressed in the adrenal gland (6) and therefore XLαs deficiency could perhaps additionally lead to dysregulation of catecholamine secretion from the adrenal medulla.
Similar to E2m+/p− mice (13), Gnasxlm+/p− mice have increased glucose tolerance and low insulin levels. While XLαs has been shown to be expressed in pancreatic islets (6), hypoinsulinemia is not likely due to impaired insulin secretion from β-cells, as insulin secretion is intact in E2m+/p− mice (15). Rather, like E2m+/p− mice (13,15), Gnasxlm+/p− mice have increased insulin sensitivity in BAT, WAT, muscle, and liver. The low insulin levels attained in Gnasxlm+/p− during the clamp study and as well as their elevated serum C-peptide/insulin ratios suggest that increased insulin clearance is a likely contributing factor to their hypoinsulinemia. The insulin receptor gene is overexpressed in WAT and to a lesser extent in liver of Gnasxlm+/p− mice, which may result in increased insulin clearance through receptor binding and internalization.
Increased insulin sensitivity of Gnasxlm+/p− mice can not be explained by changes in circulating levels of the adipocytokines leptin, adiponectin, or resistin. More likely, increased insulin sensitivity is secondary to reduced tissue triglyceride content resulting from chronically increased lipid oxidation and energy expenditure, which we have now documented in Gnasxlm+/p− mice and had documented previously in E2m+/p− mice (15). Reduced expression of hepatic lipogenic genes, most likely secondary to chronic hypoinsulinemia, also contributes to hypolipidemia in Gnasxlm+/p− mice. Sympathetic nerve stimulation also stimulates glucose uptake in BAT and muscle by insulin-independent mechanisms (40-42). Hypoinsulinemia and elevated catecholamine levels (29) may lead to increased basal glucose production, as well as the upregulation of PEPCK and downregulation of GK in livers of Gnasxlm+/p− mice.
We have now shown that XLαs is only expressed in adipose tissue during the early postnatal period, which suggests that this Gsα isoform may play a specific role during development. The parental conflict hypothesis of genomic imprinting predicts that paternally expressed imprinted genes promote the delivery of resources to the offspring during the gestational and preweaning period, and loss of these genes leads to reduced fetal and early postnatal growth (43,44). Another paternally expressed imprinted gene Igf2, which encodes a fetal growth factor, is also only expressed at high levels during gestation and early development (45,46). The parental conflict theory predicts that the evolutionary drive for expression of these imprinted genes may be lost after weaning. Small paternal 20q13 deletions including the GNAS locus have been associated with a global dysmorphic and developmental syndrome including pre- and post-natal growth retardation, abnormal adipose tissue distribution, and feeding difficulties (47), suggesting that XLαs deficiency may lead to similar defects in humans. However, the role of XLαs in humans remains unclear, as Albright hereditary osteodystrophy patients with paternal GNAS mutations predicted to disrupt XLαs expression do not develop a specific syndrome similar to 20q13 deletion or reminiscent of that observed in Gnasxlm+/p− mice.
Footnotes
This work was supported by the NIDDK Intramural Research Program, U.S. Department of Health and Human Services. We wish to thank G. Eisenhofer and K. Pacak for advice and assistance in measurements of urine catecholamines and R. Vinitsky for technical assistance.
The abbreviations used are: BAT, WAT, brown and white adipose tissue; Gsα, stimulatory G protein α subunit; XLαs, extra-large Gsα isoform; XLN1, alternative transcript from Gnasxl promoter; NESP55, 55 kDa neuroendocrine-specific protein; FFA, free fatty acids; SDH, succinate dehydrogenase; UCP, uncoupling protein; AOX, acyl-CoA oxidase; LPL, lipoprotein lipase; HSL, hormone-sensitive lipase; CREB, cAMP response element binding protein; FAS, fatty acid synthase; Acly, ATP citrate lyase; PEPCK, phosphoenolpyruvate carboxykinase; PPAR, peroxisomal proliferator activated receptor; PGC, PPARγ coactivator; CPT, carnitine palmitoyltransferase; PEPCK, phosphoenolpyruvate carboxykinase; GK, glucokinase; G6Pase, glucose-6-phosphatase; SREBP, sterol regulated element binding protein; Tfam, mitochondrial transcription factor A; Nrf1, nuclear respiratory factor 1; PC, pyruvate carboxylase; Glut4, glucose transporter 4; IR, insulin receptor; IRS1, insulin receptor substrate 1, CREB, cAMP response element binding protein; TRα, thyroid hormone receptor α, RBP4, retinol binding protein 4.
References
- 1.Weinstein LS, Liu J, Sakamoto A, Xie T, Chen M. Endocrinology. 2004;145:5459–5464. doi: 10.1210/en.2004-0865. [DOI] [PubMed] [Google Scholar]
- 2.Weinstein LS, Yu S, Warner DR, Liu J. Endocr Rev. 2001;22:675–705. doi: 10.1210/edrv.22.5.0439. [DOI] [PubMed] [Google Scholar]
- 3.Chen M, Gavrilova O, Liu J, Xie T, Deng C, Nguyen AT, Nackers LM, Lorenzo J, Shen L, Weinstein LS. Proc Natl Acad Sci USA. 2005;102:7386–7391. doi: 10.1073/pnas.0408268102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kehlenbach RH, Matthey J, Huttner WB. Nature. 1994;372:804–809. doi: 10.1038/372804a0. [DOI] [PubMed] [Google Scholar]
- 5.Li T, Vu TH, Zeng ZL, Nguyen BT, Hayward BE, Bonthron DT, Hu JF, Hoffman AR. Genomics. 2000;69:295–304. doi: 10.1006/geno.2000.6337. [DOI] [PubMed] [Google Scholar]
- 6.Pasolli HA, Klemke M, Kehlenbach RH, Wang Y, Huttner WB. J Biol Chem. 2000;275:33622–33632. doi: 10.1074/jbc.M001335200. [DOI] [PubMed] [Google Scholar]
- 7.Plagge A, Gordon E, Dean W, Boiani R, Cinti S, Peters J, Kelsey G. Nat Genet. 2004;36:818–826. doi: 10.1038/ng1397. [DOI] [PubMed] [Google Scholar]
- 8.Klemke M, Pasolli HA, Kehlenbach RH, Offermanns S, Schultz G, Huttner WB. J Biol Chem. 2000;275:33633–33640. doi: 10.1074/jbc.M006594200. [DOI] [PubMed] [Google Scholar]
- 9.Bastepe M, Gunes Y, Perez-Villamil B, Hunzelman J, Weinstein LS, Jüppner H. Mol Endocrinol. 2002;16:1912–1919. doi: 10.1210/me.2002-0054. [DOI] [PubMed] [Google Scholar]
- 10.Liu J, Litman D, Rosenberg MJ, Yu S, Biesecker LG, Weinstein LS. J Clin Invest. 2000;106:1167–1174. doi: 10.1172/JCI10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Plagge A, Isles AR, Gordon E, Humby T, Dean W, Gritsch S, Fischer-Colbrie R, Wilkinson LS, Kelsey G. Mol Cell Biol. 2005;25:3019–3026. doi: 10.1128/MCB.25.8.3019-3026.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu S, Yu D, Lee E, Eckhaus M, Lee R, Corria Z, Accili D, Westphal H, Weinstein LS. Proc Natl Acad Sci USA. 1998;95:8715–8720. doi: 10.1073/pnas.95.15.8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu S, Castle A, Chen M, Lee R, Takeda K, Weinstein LS. J Biol Chem. 2001;276:19994–19998. doi: 10.1074/jbc.M010313200. [DOI] [PubMed] [Google Scholar]
- 14.Yu S, Gavrilova O, Chen H, Lee R, Liu J, Pacak K, Parlow AF, Quon MJ, Reitman ML, Weinstein LS. J Clin Invest. 2000;105:615–623. doi: 10.1172/JCI8437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen M, Haluzik M, Wolf NJ, Lorenzo J, Dietz KR, Reitman ML, Weinstein LS. Endocrinology. 2004;145:4094–4102. doi: 10.1210/en.2004-0038. [DOI] [PubMed] [Google Scholar]
- 16.Skinner JA, Cattanach BM, Peters J. Genomics. 2002;80 doi: 10.1006/geno.2002.6842. [DOI] [PubMed] [Google Scholar]
- 17.Cattanach BM, Peters J, Ball S, Rasberry C. Hum Mol Genet. 2000;9:2263–2273. doi: 10.1093/oxfordjournals.hmg.a018917. [DOI] [PubMed] [Google Scholar]
- 18.Cattanach BM, Kirk M. Nature. 1985;315:496–498. doi: 10.1038/315496a0. [DOI] [PubMed] [Google Scholar]
- 19.Bloom JD, Dutia MD, Johnson BD, Wissner A, Burns MG, Largis EE, Dolan JA, Claus TH. J Med Chem. 1992;35:3081–3084. doi: 10.1021/jm00094a025. [DOI] [PubMed] [Google Scholar]
- 20.Eisenhofer G, Goldstein DS, Stull R, Keiser HR, Sunderland T, Murphy DL, Kopin IJ. Clin Chem. 1986;32:2030–2033. [PubMed] [Google Scholar]
- 21.Pennycuik PR. Aust J Exp Biol Med Sci. 1967;45:331–346. doi: 10.1038/icb.1967.33. [DOI] [PubMed] [Google Scholar]
- 22.Zaror-Behrens G, Himms-Hagen J. Am J Physiol. 1983;244:E361–366. doi: 10.1152/ajpendo.1983.244.4.E361. [DOI] [PubMed] [Google Scholar]
- 23.Gavrilova O, Marscus-Samuels B, Reitman ML. Diabetes. 2000;49:1910–1916. doi: 10.2337/diabetes.49.11.1910. [DOI] [PubMed] [Google Scholar]
- 24.Chen HC, Jensen DR, Myers HM, Eckel RH, Farese RV., Jr J Clin Invest. 2003;111:1715–1722. doi: 10.1172/JCI15859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang Q, Graham TE, Mody N, Preitner F, Peroni OD, Zabolotny JM, Kotani K, Quadro L, Kahn BB. Nature. 2005;436:356–362. doi: 10.1038/nature03711. [DOI] [PubMed] [Google Scholar]
- 26.Puigserver P, Spiegelman BM. Endocr Rev. 2003;24:78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- 27.Cederberg A, Gronning LM, Ahren B, Tasken K, Carlsson P, Enerback S. Cell. 2001;106:563–573. doi: 10.1016/s0092-8674(01)00474-3. [DOI] [PubMed] [Google Scholar]
- 28.Kim JK, Kim HJ, Park SY, Cederberg A, Westergren R, Nilsson D, Higashimori T, Cho YR, Liu ZX, Dong J, Cline GW, Enerback S, Shulman GI. Diabetes. 2005;54:1657–1663. doi: 10.2337/diabetes.54.6.1657. [DOI] [PubMed] [Google Scholar]
- 29.Chu CA, Sindelar DK, Neal DW, Allen EJ, Donahue EP, Cherrington AD. J Clin Invest. 1997;99:1044–1056. doi: 10.1172/JCI119232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chiu C-H, Lin W-D, Huang S-Y, Lee Y-H. Genes Dev. 2004;18:1970–1975. doi: 10.1101/gad.1213104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bartness TJ, Kay Song C, Shi H, Bowers RR, Foster MT. Proc Nutr Soc. 2005;64:53–64. doi: 10.1079/pns2004409. [DOI] [PubMed] [Google Scholar]
- 32.Song CK, Jackson RM, Harris RB, Richard D, Bartness TJ. Am J Physiol. 2005;289:R1467–R1476. doi: 10.1152/ajpregu.00348.2005. [DOI] [PubMed] [Google Scholar]
- 33.Rayner DV. Proc Nutr Soc. 2001;60:357–364. doi: 10.1079/pns2001101. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Y, Matheny M, Zolotukhin S, Tumer N, Scarpace PJ. Biochim Biophys Acta. 2002;1584:115–122. doi: 10.1016/s1388-1981(02)00298-6. [DOI] [PubMed] [Google Scholar]
- 35.Martinez JA, Margareto J, Marti A, Milagro FI, Larrarte E, Moreno Aliaga MJ. J Physiol Biochem. 2001;57:287–288. doi: 10.1007/BF03179821. [DOI] [PubMed] [Google Scholar]
- 36.Pasolli HA, Huttner WB. Neurosci Lett. 2001;301:119–122. doi: 10.1016/s0304-3940(01)01620-2. [DOI] [PubMed] [Google Scholar]
- 37.Matsumura K, Tsuchihashi T, Abe I, Iida M. Brain Res. 2002;948:145–148. doi: 10.1016/s0006-8993(02)03045-7. [DOI] [PubMed] [Google Scholar]
- 38.Ellacott KL, Cone RD. Recent Prog Horm Res. 2004;59:395–408. doi: 10.1210/rp.59.1.395. [DOI] [PubMed] [Google Scholar]
- 39.Yasuda T, Masaki T, Kakuma T, Yoshimatsu H. Exp Biol Med. 2004;229:235–239. doi: 10.1177/153537020422900303. [DOI] [PubMed] [Google Scholar]
- 40.Liu X, Perusse F, Bukowiecki LJ. Am J Physiol. 1994;266:R914–R920. doi: 10.1152/ajpregu.1994.266.3.R914. [DOI] [PubMed] [Google Scholar]
- 41.Shimizu Y, Kielar D, Minokoshi Y, Shimazu T. Biochem J. 1996;314:485–490. doi: 10.1042/bj3140485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haque MS, Minokoshi Y, Hamai M, Iwai M, Horiuchi M, Shimazu T. Diabetes. 1990;48:1706–1712. doi: 10.2337/diabetes.48.9.1706. [DOI] [PubMed] [Google Scholar]
- 43.Moore T, Haig D. Trends Genet. 1991;7:45–49. doi: 10.1016/0168-9525(91)90230-N. [DOI] [PubMed] [Google Scholar]
- 44.Haig D. Annu Rev Genet. 2004;38:553–585. doi: 10.1146/annurev.genet.37.110801.142741. [DOI] [PubMed] [Google Scholar]
- 45.Lee JE, Pintar J, Efstratiadis A. Development. 1990;110:151–159. doi: 10.1242/dev.110.1.151. [DOI] [PubMed] [Google Scholar]
- 46.Singh JS, Rall LB, Styne DM. Biol Neonate. 1991;60:7–18. doi: 10.1159/000243383. [DOI] [PubMed] [Google Scholar]
- 47.Genevieve D, Sanlaville D, Faivre L, Kottler ML, Jambou M, Gosset P, Boustani-Samara D, Pinto G, Ozilou C, Abeguile G, Munnich A, Romana S, Raoul O, Cormier-Daire V, Vekemans M. Eur J Hum Genet. 2005;13:1033–1039. doi: 10.1038/sj.ejhg.5201448. [DOI] [PubMed] [Google Scholar]
- 48.Abramowitz J, Grenet D, Birnbaumer M, Torres HN, Birnbaumer L. Proc Natl Acad Sci USA. 2004;101:8366–8371. doi: 10.1073/pnas.0308758101. [DOI] [PMC free article] [PubMed] [Google Scholar]