

Abstract
Elicitor-induced activation of the potato pathogenesis-related gene PR-10a requires a 30-bp promoter sequence termed the ERE (elicitor response element) that is bound by the nuclear factor PBF-2 (PR-10a binding factor 2). In this study, PBF-2 has been purified to near homogeneity from elicited tubers through a combination of anion-exchange and DNA affinity chromatography. Evidence demonstrates that inactive PBF-2 is stored in the nuclei of fresh tubers and becomes available for binding to the ERE upon elicitation. A protein with an apparent molecular mass of 24 kD (p24) is a DNA binding component of PBF-2. A cDNA encoding p24 has been cloned and encodes a novel protein with a potential transcriptional activation domain that could also act as a single-stranded DNA binding domain. Both PBF-2 and the cDNA-encoded protein bind with high affinity to the single-stranded form of the ERE in a sequence-specific manner. The inverted repeat sequence of the ERE, TGACAnnnnTGTCA, is critical for binding of this factor in vitro and for PR-10a expression in vivo, supporting the role of PBF-2 as a transcriptional regulator.
INTRODUCTION
Plants defend themselves against fungal pathogens by a variety of mechanisms, including preexisting physical barriers and inducible defenses (Lamb et al., 1989). Attack by an avirulent strain of pathogen results in a rapid localized necrosis at the site of infection (termed the hypersensitive response), which contributes to pathogen limitation (Keen, 1992). With a few exceptions, deployment of the inducible defenses requires massive gene induction (Lamb et al., 1989). Despite the importance of transcriptional activation during the plant defense response, very little is known about the players involved and the exact mechanisms that lead to defense gene induction.
PR (pathogenesis-related) genes are among the best characterized genes induced by pathogens. Heterogeneous in structure and function, PR genes are subdivided into 11 groups (Van Loon et al., 1994). Although the function of certain PR proteins is unknown, some display in vitro antifungal properties (Schlumbaum et al., 1986; Vigers et al., 1991; Ponstein et al., 1994; Niderman et al., 1995). Genes of the PR-10 group are present in numerous dicots (Somssich et al., 1988; Breiteneder et al., 1989; Matton and Brisson, 1989; Walter et al., 1990) and monocots (Warner et al., 1992; Moons et al., 1997). Evidence is accumulating that some PR-10 proteins might possess ribonuclease activity (Moiseyev et al., 1994; Bufe et al., 1996; Swoboda et al., 1996). More recently, structural and sequential homology between the PR-10 proteins and a group of latex proteins has been described (Osmark et al., 1998). Genes of the PR-10 group encode small, primarily acidic intracellular proteins with molecular masses ranging from 15 to 18 kD and have been shown to be transcriptionally regulated (Linthorst, 1991).
In only two cases have cis elements and their trans-acting factors been characterized in the promoters of PR-10 genes. These studies revealed that the processes of transcriptional activation by elicitation differ among the various PR-10 genes, even in the same species (Korfhage et al., 1994; Rushton et al., 1996; Euglem et al., 1999). In potato, a 30-bp elicitor response element (ERE) responsible for induction by the elicitor arachidonic acid is recognized by two nuclear factors, PBF-1 and PBF-2 (for PR-10a binding factors 1 and 2), the binding of which to the ERE correlates with the accumulation of PR-10a mRNA, suggesting that they are involved in the elicitor-dependent activation of this gene (Matton et al., 1993; Després et al., 1995). The binding of these factors to the ERE and the expression of PR-10a are both regulated by phosphorylation (Després et al., 1995). Furthermore, PR-10a gene expression and PBF-2 DNA binding activity are controlled by a functional homolog of protein kinase C (Subramaniam et al., 1997). A better understanding of the mechanisms that control expression of PR-10a and binding of PBF-2 to the ERE requires that we determine how this factor interacts with the DNA element to regulate gene expression.
This study demonstrates that PBF-2 binds with high affinity to the coding strand (CS) and the noncoding strand (NCS) of the ERE. PBF-2 was purified to near homogeneity through a combination of anion-exchange chromatography and DNA affinity chromatography. A 24-kD protein (p24) is a DNA binding component of PBF-2, as indicated by UV cross-linking to the ERE and interference of PBF-2 binding by p24 antibodies. Interestingly, after purification, PBF-2 was also recovered from fresh potato tubers in quantities comparable with that from elicited tubers. This suggests that PBF-2 is sequestered in an inactive state in the nuclei of fresh potato tubers and is activated upon elicitation. Mutational analyses demonstrated that PBF-2 is a single-stranded DNA (ssDNA) binding factor that binds with sequence specificity to the inverted repeat (IR) sequence TGACAnnnnTGTCA. In vivo, the TATA-proximal 3′ half of the ERE is critical for PR-10a expression. This region contains the core PBF-2 binding site, supporting the role of PBF-2 as a transcriptional regulator. A cDNA for p24 was cloned and shown to encode a novel protein with a feature found in many transcriptional activators, including a ssDNA binding factor. Recombinant p24 protein also binds with high affinity to ssDNA and displays the same sequence specificity as purified PBF-2.
RESULTS
PBF-2 Is a ssDNA Binding Factor
Crude extracts of elicited potato tubers were previously found to contain two factors, PBF-1 and PBF-2, that bind the –130 to –105 ERE region of the PR-10a promoter (Després et al., 1995). An analysis of the PBF binding site revealed that these factors bound with high affinity to ssDNA. A crude nuclear extract from 18-hr elicited tubers was incubated with labeled double-stranded and single-stranded CS and NCS of the ERE. As presented in Figure 1, both PBF-1 and PBF-2 bound with a greater affinity to ssDNA (lanes 3 and 4) than to double-stranded DNA (dsDNA) (lane 2). PBF-2 was capable of binding with high affinity to both the CS and NCS (lanes 3 and 4), whereas PBF-1 bound strictly to the NCS (lane 3). Attempts to isolate cDNA clones by screening expression libraries with oligonucleotide probes were unsuccessful. Therefore, a purification procedure to isolate PBF-1 and PBF-2 from elicited potato tuber discs was devised to further characterize these factors.
Figure 1.

PBF-1 and PBF-2 Bind to Single- and Double-Stranded Forms of the ERE.
EMSA was conducted in which crude nuclear extract from 18-hr elicited tubers was incubated with double-stranded ERE (lane 2, DS) or with the coding strand (lane 3, CS) or with the noncoding strand (lane 4, NCS) of the ERE. No extract was added in lane 1 (−E). PBF-1 and PBF-2 shifts are indicated by arrows.
Purification of PBF-2
Isolated nuclei were lysed in the buffer used for electrophoretic mobility shift assays (EMSA buffer: 20 mM Hepes-KOH, pH 7.9, 200 mM NaCl [except where stated], 1.5 mM MgCl2, and 0.2 mM EDTA) and loaded directly onto an anion-exchange resin (Q Sepharose Fast Flow). Proteins in the flowthrough effluent were collected and precipitated with polyethylene glycol (PEG). Since both PBF-1 and PBF-2 bound the NCS of the ERE (Figure 1), this strand was coupled to paramagnetic beads and used for DNA affinity chromatography.
As indicated in Figure 2A, the crude nuclear extracts contained both PBF-1 and PBF-2 (lane 1). However, the flowthrough fraction of the Q Sepharose chromatography contained only PBF-2 (lane 2). Attempts to recover PBF-1 from other Q Sepharose fractions were unsuccessful. Further purification of PBF-2 was obtained by two rounds of DNA affinity chromatography (lanes 5 and 8). Neither the flowthrough effluent nor the wash fractions contained PBF-2, which suggests high-affinity binding of PBF-2 to the DNA affinity columns (lanes 3, 4, 6, and 7). Figure 2B shows the silver stain of proteins recovered in the DNA affinity chromatography steps (lanes 2 and 3) in comparison with the total proteins in crude nuclear extract (lane 1). The first round of chromatography purified three main polypeptides with apparent molecular masses of 105, 48, and 24 kD (lane 2). The second round of DNA affinity chromatography further purified PBF-2 to a single 24-kD protein (lane 3).
Figure 2.

PBF-2 Contains a 24-kD DNA Binding Protein.
(A) EMSA performed with probe of the NCS of the ERE. Crude nuclear extract from 18-hr elicited tubers (lane 1), the Q Sepharose flowthrough fraction (lane 2), and the flowthrough, wash, and eluant fractions of the first (lanes 3 to 5, respectively) and second (lanes 6 to 8, respectively) DNA affinity chromatography steps. A 50-μg aliquot of crude extract (lane 1) and 1/100 of the total volume of each step of purification (lanes 2 to 8) were examined by EMSA. The intensity of the retarded bands in lanes 2 to 8 reflects the total binding activity of PBF-2. Arrows at right indicate PBF-1 and PBF-2 shifts.
(B) After migration on a 12% SDS–polyacrylamide gel, 100 μg of protein from crude extract (lane 1) and 1/4 of the elution volume from the first (lane 2) and second (lane 3) DNA affinity chromatography (Aff.) steps were silver-stained. In lane 4, DNA affinity-purified PBF-2 was UV cross-linked to radiolabeled NCS, digested with DNase I, electrophoresed on an SDS–polyacrylamide gel, and visualized by autoradiography (C-L). Apparent molecular masses of the three prominent bands after the first DNA affinity chromatography procedure are indicated at right.
To determine whether p24 interacts directly with the DNA, purified PBF-2 was UV cross-linked to radiolabeled NCS, and the DNA probe was subsequently digested. SDS-PAGE of cross-linked proteins and autoradiography revealed a single band with an apparent molecular mass of 24 kD, confirming that p24 interacts with DNA (C-L; lane 4).
Table 1 summarizes the purification of PBF-2 from 18-hr elicited tubers as measured by the amount of labeled NCS shifted by PBF-2 in EMSA. Q Sepharose anion-exchange chromatography resulted in a 2.6-fold increase in purification. PEG precipitation served as a concentration step and retained 95% of PBF-2 activity. After DNA affinity chromatography, the amount of protein eluted was nonquantifiable; therefore, specific activity is not presented. The yield of PBF-2 activity increased during purification to a maximum of 1210% after the first affinity step. This suggested that PBF-2 may be present in tubers but is unavailable for binding before elicitation—an observation that prompted us to investigate the possible presence of PBF-2 in fresh tubers.
Table 1.
Purification of PBF-2 from Elicited Potato Tuber Discs
| Fraction | Total Protein (mg) | Total Activitya (pmol of DNA) | Specific Activity (pmol/mg) | Purification (fold) | Yield (%) |
|---|---|---|---|---|---|
| Crude nuclear | 17.4 | 2.2 | 1.3 × 10−1 | 1.0 | 100 |
| Q Seph.b | 10.4 | 3.5 | 3.4 × 10−1 | 2.6 | 160 |
| PEGc | 10.0 | 3.4 | 3.4 × 10−1 | 2.6 | 150 |
| Aff. 1d | —e | 26.7 | — | — | 1210 |
| Aff. 2f | — | 7.0 | — | — | 320 |
Total activity determined by measuring labeled probe bound by PBF-2 in EMSA and calculating total picomoles of DNA bound in each fraction.
Q Seph., Q Sepharose Fast Flow anion exchange chromatography.
PEG, protein precipitated by 25% PEG 8000.
Aff. 1, first round of DNA affinity chromatography.
Nonquantifiable.
Aff. 2, second round of DNA affinity chromatography.
PBF-2 Is Stored Inactive in the Nuclei of Fresh Potato Tubers
Table 2 indicates that a negligible amount of PBF-2 binding activity was detected in crude extracts of fresh potato tubers. However, after subjecting the extract to Q Sepharose chromatography, PBF-2 binding activity was recovered in quantities comparable with that of elicited tubers. This confirmed that the increase in PBF-2 binding after elicitation was mainly the result of either the removal of an inhibitor or a modification of PBF-2 rather than de novo synthesis. The first round of DNA affinity chromatography also resulted in an increase in the yield of PBF-2, similar to that observed with elicited tubers. This increase in yield may be attributed to a distinct DNA-dependent mechanism. The total yield of PBF-2 after two rounds of DNA affinity chromatography was identical to that obtained from elicited tubers.
Table 2.
Purification of PBF-2 from Fresh Potato Tubers
| Fraction | Total Protein (mg) | Total Activitya (pmol of DNA) | Specific Activity (pmol/mg) | Purification (fold) | Yield (%) |
|---|---|---|---|---|---|
| Crude nuclear | 19.6 | 0.1 | 5.1 × 10−3 | 1.0 | 100 |
| Q Seph.b | 11.6 | 2.2 | 1.9 × 10−1 | 37.3 | 2,200 |
| PEGc | 11.1 | 2.1 | 1.9 × 10−1 | 37.3 | 2,100 |
| Aff. 1d | —e | 26.1 | — | — | 26,100 |
| Aff. 2f | — | 7.1 | — | — | 7,100 |
Total activity determined by measuring labeled probe bound by PBF-2 in EMSA and calculating total picomoles of DNA bound in each fraction.
Q Seph., Q Sepharose Fast Flow anion exchange chromatography.
PEG, protein precipitated by 25% PEG 8000.
Aff. 1, first round of DNA affinity chromatography.
Nonquantifiable.
Aff. 2, second round of DNA affinity chromatography.
p24 Is a Novel Protein with a Feature of Many Transcriptional Activators
Purified p24 was excised and digested with trypsin in-gel. After capillary electrophoresis, two peptides were selected and sequenced by Edman degradation. The partial amino acid sequence of the two peptides (SPEFSPLDSGAFK and VEPLPDG) showed no significant similarity to proteins of known function according to the BLAST search program (Altschul et al., 1997). However, the large peptide sequence was found to be encoded by a partial expressed sequence tag (EST) from tomato carpel tissue extracted 5 days preanthesis to 5 days postanthesis (AI488224.1). This EST showed homology to an Arabidopsis bacterial artificial chromosome (BAC) clone (AC002521) from chromosome II that also encoded the other peptide sequence of p24. This information was used to design polymerase chain reaction (PCR) primers. The sequence of a fragment amplified from potato genomic DNA encoded the large peptide and aligned with the tomato EST and the Arabidopsis BAC clone (data not shown). The primers also amplified a single fragment from a potato cDNA library constructed from tubers elicited with arachidonic acid for 72 hr. Using a PCR-based cDNA pooling approach (Israel, 1993), we isolated a cDNA clone for p24. The frequency of this cDNA clone in the 72-hr elicited cDNA library was found to be ∼1/210,000, suggesting that the p24 gene is not highly expressed after long periods of elicitation.
The cDNA clone revealed a single open reading frame encoding a protein of 274 amino acids. As indicated in Figure 3, both peptides from the purified p24 were present in the encoded protein. This protein has a predicted molecular mass of 30.3 kD and a pI of 9.16. ESTs encoding proteins with strong similarity to p24 can be found in the GenBank database from evolutionarily distant plants such as loblolly pine, rice, maize, Arabidopsis, and tomato.
Figure 3.

Sequence of the p24 Protein.
The protein sequence represents the single open reading frame found in the p24 cDNA. Sequences of the two peptides obtained from the purified p24 protein are underlined. The polyglutamine stretch is shaded. The GenBank accession number for the sequence is AF233342.
Strikingly, a stretch of 11 glutamine residues interrupted only by one proline residue can be found in the N-terminal half of the protein. Such glutamine stretches are part of the proline/glutamine class of transcriptional activation domains. Polyglutamine stretches have been shown to activate transcription in human HeLa cells (Gerber et al., 1994) and in plant cells (Schwechheimer et al., 1998).
p24 Is a DNA Binding Component of PBF-2
To confirm that p24 is a DNA binding component of PBF-2, antibodies were raised against the large peptide (SPEFSPLDSGAFK). As demonstrated by the protein gel blot in Figure 4A, the p24 antibodies detected a protein with an apparent molecular mass of 24 kD in the fraction eluted from the first round of DNA affinity chromatography (lane 2, +PBF-2). However, the antibody did not cross-react with any proteins in a purified potato tuber extract that did not produce a PBF-2 shift (lane 1, −PBF-2).
Figure 4.

Anti-p24 Antibodies Interfere with PBF-2 Binding.
(A) Protein gel blot of a 400 mM NaCl anion-exchange chromatography fraction that does not produce a PBF-2 shift (−PBF-2; lane 1) and of an extract from 250 g of 9-hr elicited tubers after the first round of DNA affinity chromatography that does produce a PBF-2 shift (+PBF-2; lane 2), as detected with anti-p24 peptide antibodies. Equal amounts of protein were loaded in each lane. Mobility of molecular mass standards is indicated at left in kilodaltons.
(B) EMSA of protein eluted from the first round of DNA affinity chromatography in the presence of p24 antibodies (+p24 Ab; lane 2) and of preimmune serum (+PI; lane 1) containing an equivalent amount of protein. A preincubation was conducted overnight at 4°C followed by a 15-min incubation with radioactive NCS probe. The PBF-2 shift is indicated (arrowhead).
Furthermore, if p24 is a component of PBF-2, then incubation of p24 antibody with an extract of the first round of DNA affinity chromatography should either produce a supershift or interfere with PBF-2 binding in EMSA. As shown in Figure 4B, a decrease in PBF-2 binding was observed (lane 2, + PBF-2 Ab) compared with a reaction mixture containing an equal amount of preimmune serum (lane 1, + PI). These results support our earlier observation that a 24-kD protein from the purified PBF-2 factor can be cross-linked to the ERE (Figure 2B, lane 4) and confirm that p24 is a DNA binding component of PBF-2.
PBF-2 and Recombinant p24 Protein Bind to ssDNA with Sequence Specificity
A comparison of the NCS and CS of the ERE reveals a common 14-base IR sequence (TGACAnnnnTGTCA) present in the same orientation on both strands. As demonstrated in Figure 1, PBF-2 bound both the CS and NCS of the ERE with high affinity. Therefore, the common IR sequence may represent the core binding site of PBF-2. Mutational analyses within the IR were undertaken to test this hypothesis. Figure 5A represents mutations of the NCS tested for their effect on PBF-2 binding. As demonstrated in Figure 5B, mutant oligonucleotides containing only IR1 (TGACA; m3) or IR2 (TGTCA; m4) were not bound by PBF-2. Similarly, mutations affecting simultaneously both IR1 and IR2 led to a strong reduction (m1) or an abolishment (m2) of PBF-2 binding. These results suggest that both IR1 and IR2 contribute to the interaction of PBF-2 with the ERE. The mutant m5, in which the majority (75%) of nucleotides outside the IR sequence are altered, resulted in a net increase in PBF-2 binding. This increase may have been the result of stabilization of the PBF-2 complex afforded by the presence of surrounding G/C-rich sequences. The interaction of PBF-2 with m5 suggests that no specific sequences of the ERE outside the IR sequence are required for PBF-2 binding.
Figure 5.

PBF-2 Binds Specifically to the IR Sequence of the ERE.
(A) Oligonucleotides are shown in the 5′ to 3′ orientation. The IR sequences of the CS and NCS are underlined, and IR1 and IR2 are indicated. m1 to m5 represent mutants of the NCS. The common sequence between wild-type NCS and mutated NCS oligonucleotides is indicated by dashed lines; mutant nucleotides are represented by lowercase boldface letters.
(B) EMSA of proteins eluted from the first round of DNA affinity chromatography, performed with the oligonucleotides presented in (A). The PBF-2 shift is indicated (arrowhead).
(C) EMSA of purified recombinant p24 protein (Rp24) with the oligonucleotides presented in (A). The Rp24 shift is indicated (arrowhead).
To demonstrate that the protein encoded by the p24 clone possesses characteristics similar to those of PBF-2, we expressed the p24 cDNA as a histidine tag fusion protein and tested whether it could bind the single-stranded ERE. However, repeated attempts to express the full-length p24 coding region in Escherichia coli failed to yield enough protein for EMSA, suggesting that the cDNA or the encoded protein was toxic to these cells. A truncated version of the protein (Rp24) containing a deletion of the 67 N-terminal amino acids showed much less toxicity to E. coli. As demonstrated in Figure 5C, Rp24 bound to the single-stranded ERE in vitro, confirming that the cDNA clone encodes a DNA binding protein. Furthermore, the recombinant protein bound to mutant ERE sequences with the same specificity as PBF-2 purified from tubers, providing further evidence that the cloned cDNA encodes p24.
PBF-2 Preferentially Binds the Single-Stranded Form of the IR Sequence
IR sequences may promote the self-annealing of two identical DNA strands or the annealing of IR on the same strand to form a hairpin structure. To examine this problem, the IR of the NCS was sequentially mutated two nucleotides at a time as presented in Figure 6A. Most of these mutants would disrupt the IR sequence and destabilize a double-stranded structure. DNA with a hairpin structure is known to have greater electrophoretic mobility than does linear DNA. Furthermore, the migration pattern of hairpin structure formed by short ssDNA on nondenaturing gels is dependent on the length of the stem (Baumann et al., 1989). As demonstrated in Figure 6B, the NCS mutants displayed electrophoretic mobilities characteristic of hairpin formation. Mutations resulting in a stem of three nucleotides (m6, m7, m11, and m12) displayed the slowest mobilities, whereas those mutants that decreased the stem by only one nucleotide (m8 and m10) migrated at a rate that was intermediate between the full-length five-nucleotide stem of the wild type and the three-nucleotide stem.
Figure 6.

PBF-2 Preferentially Binds the Single-Stranded Form of the IR Sequence.
(A) The sequences of wild-type (NCS) and mutated NCS oligonucleotides (m6 to m12) are presented in the 5′ to 3′ orientation with the IR sequence underlined and IR1 and IR2 indicated. The common sequence between wild-type and mutated oligonucleotides is indicated by dashed lines; mutant nucleotides are represented by lowercase boldface letters.
(B) Differential mobility of the NCS oligonucleotides presented in (A) under EMSA conditions. 20,000 counts min−1 (cpm) of each oligonucleotide probe was loaded, and the gel was exposed for 1 hr. WT, wild type.
(C) EMSA of extract from the first round of DNA affinity chromatography after incubation with radiolabeled oligonucleotides presented in (A) as probes. PBF-2 shifts are indicated.
Because the NCS displayed the characteristics of a hairpin structure, it is possible that PBF-2 interacts with dsDNA in the hairpin stem. In such a case, mutations that destabilize the hairpin structure should lead to a reduction in PBF-2 binding. Results presented in Figure 6C show that mutations that destabilized the hairpin structure (m6, m7, m11, and m12) increased PBF-2 binding over that of the wild type. Mutants with a stem only one residue short of wild type (m8 and m10) and the mutation not affecting the stem (m9) had PBF-2 binding capacities similar to the wild type. These results confirm that PBF-2 preferentially binds the single-stranded form of its core binding site.
The 3′ IR Sequence of the ERE Is Critical for PR-10a Activation
Earlier studies demonstrated that the ERE is sufficient and necessary for the activation of PR-10a. The studies also showed a strong correlation between activation of PR-10a and binding of PBF-2 to the ERE (Després et al.,1995). Those findings are supported in this study by the observation that PBF-2 is present in fresh tubers but is unavailable for binding to DNA. Only upon elicitation is PBF-2 able to bind to the ERE and promote PR-10a expression.
Because the IR sequence of the ERE is required for PBF-2 binding, we were interested in testing the role of the IR on the expression of PR-10a. Mutations in this promoter region, presented in Figure 7A, were constructed and the expression of PR-10a was monitored by way of β-glucuronidase (GUS) activity in transient expression assays in potato protoplasts. Earlier results had shown that the expression levels of GUS in electroporated protoplasts matched closely those obtained after elicitor treatment of transgenic potato tubers carrying deletions in the PR-10a promoter (Matton et al., 1993; Després et al., 1995). As indicated in Figure 7B, mutations m13 to m17 of the 5′ half of the ERE resulted in either the same as wild type or increased gene expression. Only the two mutations, m18 and m19 of the 3′ half of the IR sequence (TGTCA), decreased GUS activity to 20 and 40% of wild type, respectively. Therefore, the 3′ IR of the ERE, but not the 5′ IR, is critical for PR-10a activation in vivo.
Figure 7.

The 3′ IR Sequence of the ERE Is Required for PR-10a Expression.
(A) Mutations within the ERE sequence (m13 to m19) were constructed and fused to the GUS reporter gene. Wild-type double-stranded ERE is presented with the IR sequence in boldface. The common sequence between wild-type and mutated oligonucleotides is indicated by dashed lines, and mutant nucleotides are represented by lowercase boldface letters for the CS of the ERE.
(B) The ERE mutants presented in (A) were fused to a GUS reporter gene and electroporated into potato leaf protoplasts. The histogram represents the GUS activity of each ERE mutant relative to the wild-type pLP9 construct (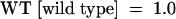 ). Transfection efficiencies were corrected by coelectroporating a luciferase reporter gene. Results are for two batches of protoplasts prepared at different times, each plasmid having been electroporated three times with each batch of protoplasts. Error bars indicate the standard deviation of the mean.
). Transfection efficiencies were corrected by coelectroporating a luciferase reporter gene. Results are for two batches of protoplasts prepared at different times, each plasmid having been electroporated three times with each batch of protoplasts. Error bars indicate the standard deviation of the mean.
The DNA binding results demonstrated that mutations in both IR1 and IR2 are required to decrease PBF-2 binding (Figures 5 and 6). However, the in vivo results indicated that only the 3′ IR of the ERE is sufficient for PR-10a expression. This apparent contradiction is reconciled by the model outlined in Figure 8. Figure 8A shows the structure of the ERE that may be present in vivo. In this model, PBF-2 interacts with IR1 on the NCS and IR2 on the CS. Similarly, as indicated in Figure 8B, PBF-2 could interact in vitro with both IR1 and IR2 of the NCS or the CS on bent DNA. This model suggests that PBF-2 would have easier access to its binding site if the DNA does not form a stable hairpin, which is corroborated by the evidence presented in Figure 6. Furthermore, according to the model, m18 and m19, which reduced expression in vivo (Figure 7), are analogous to m1 and m2. As predicted, m1 and m2 interfered with PBF-2 and Rp24 binding, respectively, in vitro (Figure 5).
Figure 8.

Proposed Model of PBF-2 Binding in Vivo and in Vitro.
(A) In vivo, PBF-2 binds a melted ERE recognizing IR1 of the NCS and IR2 of the CS. The nucleotides of IR1 and IR2 are indicated in boldface. dsERE, double-stranded ERE.
(B) In vitro, PBF-2 binds to both IR1 and IR2 of ssDNA with its core binding site oriented the way it is found in vivo. The nucleotides of IR1 and IR2 in both CS and NCS are indicated in boldface.
DISCUSSION
In this study, PBF-2 was purified to near homogeneity and shown to contain a protein with an apparent molecular mass of 24 kD. p24 can be cross-linked to DNA, and antibodies raised against a p24 peptide interfere with PBF-2 binding in EMSA, indicating that p24 is a DNA binding component of PBF-2. The sequence of a cDNA clone encoding p24 indicates that it is a novel protein with a polyglutamine stretch, found in many transcriptional activators. Furthermore, both PBF-2 and Rp24 bind preferentially to ssDNA with the same sequence specificity.
PBF-2 was also recovered from nuclear extracts of fresh potato tubers in amounts comparable with those recovered from elicited tubers, but anion-exchange chromatography was required to uncover its binding activity. Two proteins with apparent molecular masses of 48 and 105 kD copurified with p24 up to the first round of DNA affinity chromatography, suggesting that p24 may form part of a multiprotein complex. Support for this comes from a gel filtration chromatography procedure, in which PBF-2 eluted at a molecular mass of ∼100 kD (A. Joyeux, unpublished results). The loss of the 48- and 105-kD proteins after the second round of DNA affinity chromatography may have resulted from the high salt concentration (1 M) used to elute the first DNA affinity column, which could irreversibly dissociate the proteins from p24.
The Polyglutamine Tract of p24 Is Found in Many Transcriptional Activators
The cDNA clone encoding p24 was isolated from a cDNA library constructed from elicited potato tubers. The predicted molecular mass of the encoded protein differs from the apparent molecular mass of p24 purified from tubers, suggesting that the protein may be processed. The fact that we observed strong specific DNA binding activity with a truncated form of the recombinant protein suggests that tuber-purified p24 may indeed be derived from a precursor protein encoded by the p24 cDNA.
The most striking feature of the p24 protein sequence is the homopolymeric stretch of 11 glutamine residues. Such stretches are found in many transcription factors and are known to activate transcription in human HeLa cells (Gerber et al., 1994). Glutamine-rich activation domains are also known to activate transcription preferentially from proximal promoter positions in concert with an enhancer element (Seipel et al., 1992). Similarly, PBF-2 acts from a proximal element (−135 to –105) of the PR-10a promoter, and distal enhancer elements (−155 to –135 and –670 to –441) are required for high expression of PR-10a (Després et al., 1995). Recently, a fusion protein containing the GAL4 DNA binding domain, the VP16 acidic activation domain, and a stretch of 51 glutamine residues activated transcription 14-fold more than did the GAL4/VP16 fusion in plant cells (Schwechheimer et al., 1998). Studies in animal cells have shown that fusion proteins with a tract of 10 glutamines displayed the most transcription, whereas proteins with >26 glutamine residues showed progressively less transcription (Gerber et al., 1994). These observations strongly suggest that the glutamine stretch found in p24 has the potential to activate transcription.
More recently, the glutamine-rich domain of the Drosophila melanogaster GAGA factor (GAF) has been shown to bind ssDNA. Furthermore, the glutamine-rich domain of GAF promotes the stabilization of melted dsDNA (Wilkins and Lis, 1999). Similarly, the p24 glutamine stretch may help stabilize ssDNA in the ERE.
Single-Stranded ERE Mimics the Double-Stranded PBF-2 Binding Site
In vitro results indicate that the core PBF-2 binding site is the IR sequence TGACAnnnnTGTCA. In vivo (i.e., dsDNA), the 3′ half of the ERE is required for optimal gene expression (Figure 7). This region contains the PBF-2 core binding site with IR1 (TGACA) on the NCS and IR2 (TGTCA) on the CS. Because PBF-2 binds with greater affinity to ssDNA, it is proposed to bind melted ERE in vivo, as presented in Figure 8A. In this model, PBF-2 contacts its core binding site on both the CS and NCS of the ERE. In vitro, however, PBF-2 would bind a bent form of the ssDNA, which would mimic the dsDNA binding site, as presented in Figure 8B.
The symmetry of the IR sequence in the ERE makes it a possible site for protein dimerization (Lamb and McKnight, 1991). However, PBF-2 binding displays the properties of monomeric binding. In vitro, removal of either half of the IR abolished PBF-2 binding and did not result in an intermediate shift corresponding to a monomer. Furthermore, cross-linking of PBF-2 to the NCS of the ERE followed by SDS-PAGE without digestion of the DNA probe resulted in a band at ∼30 kD (p24/DNA complex) rather than the 48 kD predicted for dimer formation (data not shown). Finally, in vivo, only the 3′ half of the ERE is critical for PR-10a expression, rather than both halves.
Elicitor-Induced Activation of PBF-2
As indicated in Table 2, PBF-2 is present in the nuclei of fresh tubers in an inactive form. Activation of PBF-2 may resemble the model proposed for the salicylic acid–induced transcription from the as-1 element of the cauliflower mosaic virus (Jupin and Chua, 1996). Binding of the cellular factor SARP (salicylic acid response protein) to the as-1 element correlates with the activation of transcription after salicylic acid treatment. SARP, in turn, is proposed to be sequestered by the inhibitory protein SAI (salicylic acid inhibitor) and released by treatment with salicylic acid. Disruption of protein–protein interactions in extracts of untreated leaves leads to a strong increase in SARP binding activity. Furthermore, the increase in SARP DNA binding activity observed after treatment with salicylic acid does not involve de novo synthesis of protein. Interestingly, the DNA binding activity was sensitive to phosphatase treatment, similar to that observed with PBF-2 (Després et al., 1995), which suggests a role for phosphorylation in salicylic acid–induced gene activation. Phosphorylation events also play a critical role in activating PR-10a and establishing the defense response in potato (Subramaniam et al., 1997). p24 may also be kept inactive in the form of a precursor in fresh tubers. Unfortunately, this hypothesis could not be tested because anti-p24 antibodies did not detect the protein in crude nuclear extracts from either fresh or elicited tubers.
The presence of PBF-2 activity in the nuclei of fresh potato tubers to the extent recovered from elicited tubers makes PBF-2 activation a potentially rapid response to pathogen infection. In addition to activating PR-10a, PBF-2 could activate the transcription of other factors involved in the defense response, such as the WRKY proteins. The factors WRKY1 and WRKY3 bind the promoters of two parsley PR-10a homologs and are transcriptionally activated by elicitation (Rushton et al., 1996). Interestingly, the promoters of both these proteins contain GTCA motifs, which have been shown to be important for WRKY1 promoter activity in vivo (Euglem et al., 1999). A possible defense pathway induced by elicitation may involve the phosphorylation-dependent activation of proteins such as PBF-2 followed by the transcriptional activation of other factors involved in the defense response.
Transcriptional Activation of PR-10a by PBF-2
Sequence-specific ssDNA binding proteins have been shown to participate in the regulation of transcription. Among the best characterized are the FUSE binding protein, the heterogeneous ribonucleoprotein K, and the cellular nucleic acid binding protein, all of which bind the promoter of the c-myc protooncogene and stimulate its expression (Takimoto et al., 1993; Duncan et al., 1994; Michelotti et al., 1995). Other examples of ssDNA binding proteins that regulate transcription include single-strand D-box binding factor (ssDBF), which represses estrogen-induced transcription of the chicken apo VLDL II gene (Smidt et al., 1995); CBF-A, which represses SMα-A transcription in C2 myotubule cells (Kamada and Miwa, 1992); and MEF-1/Pur α and MyEF-2, which enhance and repress transcription of the myelin basic protein, respectively, in mouse cells (Haas et al., 1993, 1995; Muralidharan et al., 1997). In both prokaryotes and eukaryotes, DNA in non-B conformations, such as melted DNA, Z-DNA, and cruciform structures, forms as a consequence of transcriptionally induced supercoiling in vitro and in vivo (Liu and Wang, 1987; Giaver and Wang, 1988; Horwitz and Loeb, 1988; Dröge and Nordheim, 1991; Dayne et al., 1992; Dai et al., 1997). Torsional stress that accumulates in upstream sequences as a result of transcription can be relieved by unwinding, generating single strands. This process is facilitated by the presence of A/T-rich sequences (Kowalski et al., 1988). Proteins such as PBF-2 with a greater affinity for ssDNA than for dsDNA may stabilize melted forms of helically unstable sites, thereby relieving transcriptionally produced superhelical torsion, similar to the model proposed for the c-myc promoter (Duncan et al., 1994; Bazar et al., 1995; Michelotti et al., 1996).
Accumulation of small amounts of PR-10a mRNA is induced by wounding and increases greatly after elicitation (Matton and Brisson, 1989). The transcriptional activity induced by wounding could result in upstream negative supercoiling and possible localized melting of the ERE, 80% of which is A or T residues. When elicited, activated PBF-2 could recognize, bind, and stabilize the melted ERE, thereby amplifying the PR-10a transcriptional activity. The overall destabilization of DNA would increase the potential for the formation of site-specific loops or bends (Kahn et al., 1994) that could act as hinges, allowing for interaction of distal cis-regulatory elements with the downstream promoter. Analogous to the c-myc system, multiple ssDNA binding factors could bind non-B-DNA of the PR-10a promoter to regulate transcription. Preliminary results suggest that SEBF, a nuclear factor shown to bind a repressor region of the PR-10a promoter (Després et al., 1995), is also a sequence-specific ssDNA binding factor (B. Boyle and N. Brisson, unpublished results).
Concluding Remarks
PBF-2 represents a novel DNA binding factor that potentially could exploit non-B-DNA to activate gene expression. It may function in concert with potato homologs of other transcription factors implicated in the defense response that recognize dsDNA motifs such as the WRKY proteins (Rushton et al., 1996; Euglem et al., 1999), TGA6 (Xiang et al., 1997; Zhang et al., 1999), AHBP-1b (Kawata et al., 1992; Zhang et al., 1999), and other leucine zipper proteins (Izawa et al., 1993; Foster et al., 1994). The TGAC sequence found in the 5′ IR sequence of PR-10a is a W-box type element known to bind the WRKY proteins (Rushton et al., 1996). The increased in vivo activity observed when this sequence was mutated (Figure 7) suggests that a repressor site may have been mutated. In parsley, WRKY2 mRNA decreases in response to elicitor, suggesting that this factor may have a negative regulatory role (Rushton et al., 1996). A potato WRKY2 homolog could potentially bind the TGAC sequence of the ERE and act to inhibit PR-10a transcription. The inhibitor might stabilize dsDNA, which would be less favorable for PBF-2 binding. The regulation of PR-10a at the ERE is likely to involve a combination of activation and derepression events, as proposed for the Arabidopsis PR-1 gene (Lebel et al., 1998).
METHODS
Plant Material
Potato tubers (Solanum tuberosum cv Kennebec) were obtained from the Québec Ministry of Agriculture “Les Buissons” Research Station (Pointe-aux-Outardes, Québec). Tubers were stored in the dark at 4°C and brought to room temperature 24 hr before use. Leaf mesophyll protoplasts were isolated from 7- to 8-week-old potato plants (cv Kennebec) grown in growth chambers as described by Magnien et al. (1980).
Preparation of Crude Nuclear Extracts
Potatoes for fresh extracts were washed in water, surface-sterilized in 70% ethanol for 5 min, rinsed with distilled water, dried, and weighed. Nuclear protein extracts from elicited potato tubers were prepared as described previously (Marineau et al., 1987; Després et al., 1995), with slight modifications. One kilogram of fresh tubers or elicited tuber discs was homogenized in a blender at maximum speed for 1 min in 1 liter of ice-cold buffer containing 1 M hexylene glycol, 20 mM 2-mercaptoethanol, 10 mM Pipes-KOH, pH 6.0, 10 mM MgCl2, 0.5 mM spermidine, and 0.15 M spermine. After the first centrifugation, the nuclear pellets were rinsed with 50 mL of EMSA buffer (20 mM Hepes-KOH, pH 7.9, 200 mM NaCl, 1.5 mM MgCl2, and 0.2 mM EDTA) and resuspended in 16 mL of the same buffer. The nuclei were lysed by sonication, incubated on ice for 30 min, and centrifuged for 60 min at 9000g at 4°C. The supernatant was recovered and used as crude extract.
Purification of PBF-2
Q Sepharose chromatography was performed as a batch procedure by adding crude nuclear extract from 1 kg of potato tubers to 15 mL of Q Sepharose Fast Flow resin (Amersham Pharmacia Biotech, Baie d'Urfé, Québec) preequilibrated at 200 mM NaCl with the EMSA buffer. After incubation for 15 min on ice, the slurry was passed through glass wool to separate the resin from the flowthrough fraction containing PBF-2. One volume of 50% polyethylene glycol (PEG) solution was added to the flowthrough fraction, and proteins were precipitated overnight at −20°C. The next day, the mixture was thawed and centrifuged at 4°C for 60 min at 9000g. The pellet was resuspended in 4 mL of EMSA buffer and centrifuged at 4°C for 15 min at 11,000g. Aliquots of the supernatant were stored at −80°C with 10% glycerol, and the remaining solution was used immediately for DNA affinity chromatography.
The DNA sequence 5′-CTAGACCATTTTTGACATTTGTGTCATTTTATCTAG corresponding to the noncoding strand (NCS) of the elicitor response element (ERE) was chemically synthesized with biotin at the 3′ position (Keystone Labs, Camarillo, CA). This biotinylated oligonucleotide was coupled to streptavidin-coated paramagnetic beads (Sigma; 1 μg of streptavidin per 1 μL of beads) by incubating in EMSA buffer containing 2 M NaCl for 15 min. Coupling was monitored by adding a trace of biotinylated oligonucleotide labeled with phosphorus-32. The DNA-coupled affinity beads were equilibrated by three washes with 200 mM NaCl EMSA buffer. The extract from the equivalent of 250 g of starting material (1 mL of PEG-precipitated Q Sepharose fraction) was incubated with 500 μL of affinity beads for 15 min at room temperature. For the first round of DNA affinity chromatography, EDTA was added directly to the binding reaction to a final concentration of 100 mM to inhibit nucleases. The beads were washed twice with 500 μL of 200 mM NaCl EMSA buffer. The bound proteins were then eluted twice with 100 μL of EMSA buffer containing 1 M NaCl. The eluent was immediately equilibrated to 200 mM NaCl by adding 800 μL of EMSA buffer devoid of NaCl. The second round of DNA affinity chromatography was identical to the first except that EDTA was omitted from the 15-min incubation period. Aliquots were stored at −80°C with 10% glycerol added. Proteins in each fraction were separated by 12% SDS-PAGE and visualized by silver staining (Oakley et al., 1980).
For peptide sequencing, p24 was purified from 10 kg of elicitor-treated potato tuber discs. Sequencing was performed by the Harvard Microchemistry Facility (Cambridge, MA).
Cloning of p24
A partial tomato expressed sequence tag (EST) sequence (AI488224.1) coding for the p24 large peptide (SPEFSPLDSGAFK) and an Arabidopsis bacterial artificial chromosome (BAC) clone (AC002521) coding for both peptides were aligned and polymerase chain reaction (PCR) primers were designed to flank the large peptide. The primers derived from the tomato EST sequence had sequences of 5′-ATATACAAAGGGAAGGCAGCT and 5′-GATAGA-TCCAATTTCAGTCAC. These primers were first used to amplify potato genomic DNA. A single DNA fragment of ∼550 bp was amplified, cloned, partially sequenced, and shown to share 99% identity over 109 bp with the tomato sequence and to code for the large peptide (data not shown).
A modified version of the method described by Israel (1993) was used to screen a potato cDNA library made in lambda ZAP (Stratagene, La Jolla, CA) from mRNAs isolated from potato tubers elicited with arachidonic acid for 72 hr (Matton and Brisson, 1989). Approximately 960,000 plaque-forming units (pfu) were used to infect Escherichia coli XL-1 Blue (Stratagene), and portions were placed in a 96-well microtiter plate at 10,000 pfu per well in 100 μL. The phages were amplified for 8 hr at 37°C. A portion from each well was mixed with an equal volume of water, and 3 μL from each sample was added to a PCR reaction containing 2.5 mM MgCl2, 50 mM KCl, 10 mM Tris-HCl, pH 9.0, 0.01% gelatin, 0.1% Triton X-100, 200 μM deoxyribonucleotide triphosphates, and 0.4 units of Taq polymerase (Roche Molecular Biochemicals, Laval, Québec) with 10 pmol each of the primers described above. The DNA was amplified in a standard PCR reaction, with an annealing temperature of 53°C, in a Whatman/Biometra T-gradient PCR apparatus. Approximately 336,000 pfu from a positive well was transferred to another 96-well plate at 4000 pfu per well, amplified, and analyzed by PCR as described above. This process was repeated a total of four times, with ∼3000 and 250 pfu per well in the third and fourth screens, respectively. Isolation of the p24 cDNA clone was done by hybridization of plaques from a positive well with the genomic DNA fragment described above. A positive clone was excised by using the ExAssist system (Stratagene), according to the manufacturer's instructions. The clone was sequenced on both strands by using Thermosequenase (Amersham Pharmacia Biotech) in a cycle-sequencing reaction that was then processed in a Lycor automated sequencer (Lycor, Lincoln, NE).
Expression and Purification of Protein Fusions
The p24 coding region was amplified by using PCR primers (5′-CCAAAAATCTCTTGGATCCATGTCC and 5′-CCAGAACTCGAGATTCCATTC) that inserted a BamHI site immediately preceding the ATG and a XhoI site after the stop codon, respectively. The PCR product was purified and inserted into the BamHI and XhoI sites of the pET-21a vector (Novagen, Madison, WI), creating a fusion protein with a T7 tag at the N terminus and a histidine tag at the C terminus. The truncated form of the p24 protein was produced by using the same XhoI primer and a PCR primer (5′-TTAACATGTCGCGGATCCGATTATTTTG) inserting a BamHI site 67 amino acids from the N terminus. These constructs were made in XL-1 blue E. coli cells (Stratagene) and then transferred in the expression strain BL21 pLysS (Novagen). A single colony from the latter was then grown at 37°C, and the culture was induced for 3 hr by isopropyl-β-d-thiogalactopyranoside. The cells were then harvested and resuspended in 0.1 volume of START buffer (20 mM Na2HPO4, pH 7.2, and 500 mM NaCl). After treatment with a freeze/thaw cycle and sonication, the samples were centrifuged at 11,000g for 1 hr at 4°C. One volume of 50% PEG 8000 was added to the supernatants, and the precipitated proteins were centrifuged for 1 hr at 11,000g. Pellets were washed and resuspended in START buffer. Fusion proteins were purified by using the HiTrap affinity columns and a fast protein liquid chromatography apparatus (Amersham Pharmacia Biotech), according to manufacturer's instructions. The columns were charged with 100 mM NiSO4, and the samples were loaded in START buffer. The fusion proteins were eluted in START buffer containing 50 mM EDTA. Ten percent glycerol was added to the samples for long-term storage at –80°C. For EMSA, the eluted samples were first diluted 1:1 in EMSA buffer without NaCl.
Electrophoretic Mobility Shift Assays
Double-stranded ERE was prepared as described previously (Després et al., 1995). Single-stranded synthetic oligonucleotides for the –130 to –105 region of the ERE were labeled by using T4 polynucleotide kinase. Specific activities ranged from 5 × 104 to 105 counts min−1 (cpm) ng−1 and were adjusted by adding cold DNA to give all samples the same value for use in comparing the different mutant oligonucleotides. Reaction mixtures contained 1 μL (20,000 cpm) of end-labeled oligonucleotide and 40 μL of DNA affinity or 10 μL of Q Sepharose extract or 10 μg of nuclear proteins of the crude extract with a final EDTA concentration of 50 mM. Reactions were performed at room temperature for 15 min and subsequently loaded onto 5.4% polyacrylamide gels (29:1 acrylamide:bisacrylamide in 100 mM Tris-HCl, pH 8.0, 100 mM borate, and 2 mM EDTA). After electrophoresis, the gels were blotted onto Whatman 3MM paper and autoradiographed at −80°C on Kodak (Rochester, NY) XAR films. Quantitation of DNA bound by PBF-2 was assessed by liquid scintillation counting of the retarded band excised from the gel or by measuring the density of the shifted probe on developed films by using a densitometry package from Molecular Dynamics (Sunnyvale, CA).
UV Cross-Linking
Bluescript plasmids (Stratagene) containing wild-type oligonucleotides were internally labeled with Klenow and radioactive α-32P-dTTP. The cloned oligonucleotide was excised from the plasmid by digestion with XbaI and purified. The gel-purified oligonucleotide was heat-denatured and used as a probe in EMSA. Reaction mixtures were irradiated for 30 min with a UV transilluminator (Fotodyne, San Diego, CA) before EMSA. The retarded complexes were excised from the gel and subjected to electroelution followed by digestion with DNase I according to standard protocol (Sambrook et al., 1989). Cross-linked proteins were then separated by 12% SDS-PAGE. The gels were subsequently blotted onto Whatman 3MM paper and autoradiographed at −80°C.
Protein Gel Blot of PBF-2
A synthetic peptide corresponding to the sequence SPEFSPLDSGAFK of the p24 protein was synthesized (Sheldon Biotechnology Centre, Montréal, Québec). An antiserum was prepared by immunizing female New Zealand white rabbits with the synthesized p24 peptide. For protein gel blots, DNA affinity-purified proteins from 250 g of potato tubers were precipitated with 5 volumes of cold acetone, resuspended in SDS sample buffer, and separated by SDS-PAGE. Protein gel blots were performed as described previously (Constable and Brisson, 1992) with p24 antibody used at 1000-fold dilution. The blots were developed with the electrochemiluminescent detection system (Amersham Pharmacia Biotech) according to the manufacturer's instructions.
β-Glucuronidase Reporter Gene Constructs
All DNA manipulations were performed according to standard procedures (Sambrook et al., 1989). The mutant EREs were constructed from the vector pLP9, which contains the –135 to –47 region of the ERE coupled to the –27 to +145 region of the PR-10a gene by the sequence 5′-TCGAATTCCTGCAGG. To create the pLP9 vector, the plasmid pDPM686 (Matton et al., 1993) was cut with the restriction enzyme PstI in the 15-bp linker region, blunt-ended, and ligated to an 8-bp oligonucleotide at the vector, resulting in a 19-bp full-length sequence from –47 to –27 (5′-TCGAATTCCGGTCGACCGG).
PCR site-directed mutagenesis was used to create the ERE mutants. The two wild-type primers were 5′-GCGAAGCTTGATTCTAGATAAAATGACACAAATGTCAAAAATGG and 5′-CCACCCGGG-GATCCAGCTTTGAAC. The reaction mixtures contained 50 ng of template DNA (pLP9), 25 pmol of each primer, 200 μM dNTPs, 50 mM KCl, 10 mM Tris-Cl, pH 9.0, 0.01% gelatin, 0.1% Triton X-100, 2.5 mM MgCl2, and 0.4 units of Taq DNA polymerase (Roche Molecular Biochemicals, Laval, Québec) in a total reaction mixture volume of 30 μL. PCR reactions were performed at an annealing temperature of 50°C, and the PCR products were digested with HindIII and BamHI and ligated into the digested pLP9 vector. The entire insert of each mutant was sequenced to ensure that no additional mutations had been introduced in the sequence.
Protoplast Isolation and Electroporation
Leaf mesophyll protoplasts were electroporated with CsCl-purified supercoiled plasmid DNA as previously described (Matton et al., 1993). Luciferase plasmid pWB216 (Barnes, 1990) containing the luciferase gene under control of the cauliflower mosaic virus 35S promoter was coelectroporated with 40 μg of reporter plasmids. Protoplasts were pelleted, resuspended in 100 μL of cell culture lysis reagent (Promega, Nepean, Ontario), and frozen immediately. Luciferase activity was determined by adding 50 μL of Luciferase Assay Reagent (Promega) to 5 μL of freshly thawed protoplast extracts and without delay measuring photon emission (EG and G Berthold Lumat LB9507 Luminometer, Bad Wildbad, Germany) for 1 min. β-Glucuronidase (GUS) activity was measured as described previously (Matton et al., 1993) and standardized by using luciferase activity as an indicator of transformation efficiency.
Acknowledgments
We thank Louise Paquet for preparation of the pLP9 reporter plasmid and optimizing the GUS reporter gene assays. We also thank Louise Cournoyer for preparing in vitro grown plants and Barbara Otrysko for the gift of certified potato tubers. We are also very grateful to Franz Lang and his group for sequencing the p24 cDNA in its entirety. This work was supported by research grants from the Natural Science and Engineering Research Council (NSERC) of Canada and from the Fonds pour la Formation de Chercheurs et l'Aide à la Recherche, Québec. D.D., C.D., and A.J. were recipients of NSERC postgraduate scholarships.
References
- Altschul, S.F., Madden, T.L., Schäffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. (1997). Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes, W.M. (1990). Variable patterns of expression of luciferase in transgenic tobacco leaves. Proc. Natl. Acad. Sci. USA 87, 9183–9187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann, U., Frank, R., and Blöcker, H. (1989). Probing hairpin structures of small DNAs by nondenaturing polyacrylamide gel electrophoresis. Anal. Biochem. 183, 152–158. [DOI] [PubMed] [Google Scholar]
- Bazar, L., Meighen, D., Harris, V., Duncan, R., Levens, D., and Avignan, M. (1995). Targeted melting and binding of a DNA regulatory element by a transactivator of c-myc. J. Biol. Chem. 270, 8241–8248. [DOI] [PubMed] [Google Scholar]
- Breiteneder, H., Pettenburger, K., Bito, A., Valenta, R., Kraft, D., Rumpold, H., Scheiner, O., and Breitenbach, M. (1989). The gene coding for the major birch pollen allergen, Bet vI, is highly homologous to a pea disease resistance response gene. EMBO J. 8, 1935–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bufe, A., Spangfort, M.D., Kahlert, H., Schlaak, M., and Becker, W.M. (1996). The major birch pollen allergen, Bet v1, shows ribonuclease activity. Planta 199, 413–415. [DOI] [PubMed] [Google Scholar]
- Constable, C.P., and Brisson, N. (1992). The defense-related STH-2 gene product of potato shows race-specific accumulation after inoculation with low concentrations of Phytophthora infestans zoospores. Planta 188, 289–295. [DOI] [PubMed] [Google Scholar]
- Dai, X., Greizerstein, M.B., Nadas-Chinni, K., and Rothman-Denes, L.B. (1997). Supercoil-induced extrusion of a regulatory DNA hairpin. Proc. Natl. Acad. Sci. USA 94, 2174–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayne, A., Malkhosyan, S., and Mirkin, S.M. (1992). Transcriptionally driven cruciform formation in vivo. Nucleic Acids. Res. 20, 5991–5997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Després, C., Subramaniam, R., Matton, D.P., and Brisson, N. (1995). The activation of the potato PR-10a gene requires the phosphorylation of the nuclear factor PBF-1. Plant Cell 7, 589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dröge, P., and Nordheim, A. (1991). Transcription-induced conformational change in a topologically closed DNA domain. Nucleic Acids Res. 19, 2941–2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan, R., Bazar, L., Michelotti, G., Tomonaga, T., Krutzsch, H., Avignan, M., and Levens, D. (1994). A sequence specific, single-strand binding protein activates the far upstream element of c-myc and defines a new DNA-binding motif. Genes Dev. 8, 465–480. [DOI] [PubMed] [Google Scholar]
- Euglem, T., Rushton, P.J., Schmelzer, E., Hahlbrock, K., and Somssich, I.E. (1999). Early nuclear events in plant defense signalling: Rapid gene activation by WRKY transcription factors. EMBO J. 18, 4689–4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster, R., Izawa, T., and Chua, N.-H. (1994). Plant bZIP proteins gather at ACGT elements. FASEB J. 8, 192–200. [DOI] [PubMed] [Google Scholar]
- Gerber, H.-P., Seipel, K., Georgiev, O., Höfferer, M., Hug, M., Rusconi, S., and Schaffner, W. (1994). Transcriptional activation modulated by homopolymeric glutamine and proline stretches. Science 263, 808–811. [DOI] [PubMed] [Google Scholar]
- Giaver, G.N., and Wang, J.C. (1988). Supercoiling of intracellular DNA can occur in eukaryotic cells. Cell 55, 849–856. [DOI] [PubMed] [Google Scholar]
- Haas, S., Gordon, J., and Khalili, K. (1993). A developmentally regulated DNA-binding protein from mouse brain stimulates myelin basic protein gene expression. Mol. Cell. Biol. 13, 3103–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas, S., Steplewski, A., Siracusa, L., Amini, S., and Khalili, K. (1995). Identification of a sequence specific single-stranded DNA binding protein that suppresses transcription of the mouse myelin basic protein gene. J. Biol. Chem. 270, 12503–12510. [DOI] [PubMed] [Google Scholar]
- Horwitz, M.S.Z., and Loeb, L.A. (1988). An E. coli promoter that regulates transcription by DNA superhelix-induced cruciform extrusion. Science 241, 703–705. [DOI] [PubMed] [Google Scholar]
- Israel, D.I. (1993). A PCR-based method for high-stringency screening of DNA libraries. Nucleic Acids Res. 21, 2627–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izawa, T., Foster, R., and Chua, N.-H. (1993). Plant bZIP protein DNA binding specificity. J. Mol. Biol. 230, 1131–1144. [DOI] [PubMed] [Google Scholar]
- Jupin, I., and Chua, N.-H. (1996). Activation of the CaMV as-1 cis-element by salicylic acid: Differential DNA-binding of a factor related to TGA1a. EMBO J. 15, 5679–5689. [PMC free article] [PubMed] [Google Scholar]
- Kahn, J.D., Yun, E., and Crothers, D.M. (1994). Detection of localized DNA flexibility. Nature 368, 163–166. [DOI] [PubMed] [Google Scholar]
- Kamada, S., and Miwa, T. (1992). A protein binding to CarG box motifs and to single-stranded DNA functions as a transcriptional repressor. Gene 119, 229–236. [DOI] [PubMed] [Google Scholar]
- Kawata, T., Imada, T., Shiraishi, H., Okada, K., Shimura, Y., and Iwabuchi, M. (1992). A cDNA encoding HBP-1b homologue in Arabidopsis thaliana. Nucleic Acids Res. 20, 1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keen, N.T. (1992). The molecular biology of disease resistance. Plant Mol. Biol. 19, 109–122. [DOI] [PubMed] [Google Scholar]
- Korfhage, U., Trezzini, G.F., Meier, I., Hahlbrock, K., and Somssich, I.E. (1994). Plant homeodomain protein involved in transcriptional regulation of a pathogen defense-related gene. Plant Cell 6, 695–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalski, D., Natale, D.A., and Eddy, M.J. (1988). Stable DNA unwinding, not “breathing,” accounts for single-strand–specific nuclease hypersensitivity of specific A+T-rich sequences. Proc. Natl. Acad. Sci. USA 85, 9464–9468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb, C.J., Lawton, M.A., Dron, M., and Dixon, R.A. (1989). Signals and transduction mechanisms for activation of plant defenses against microbial attack. Cell 56, 863–867. [DOI] [PubMed] [Google Scholar]
- Lamb, P., and McKnight, S.L. (1991). Diversity and specificity in transcriptional regulation: The benefits of heterotypic dimerization. Trends Biochem. Sci. 16, 417–422. [DOI] [PubMed] [Google Scholar]
- Lebel, E., Heifetz, P., Thorne, L., Uknes, S., Ryals, J., and Ward, E. (1998). Functional analysis of regulatory sequences controlling PR-1 gene expression in Arabidopsis. Plant J. 16, 223–233. [DOI] [PubMed] [Google Scholar]
- Linthorst, H.J.M. (1991). Pathogenesis-related proteins of plants. Crit. Rev. Plant Sci. 10, 123–150. [Google Scholar]
- Liu, L.F., and Wang, J.C. (1987). Supercoiling of the DNA template during transcription. Proc. Natl. Acad. Sci. USA 84, 7024–7027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnien, E., Dalschaert, X., and Devreux, M. (1980). Different radiosensitivities of Nicotiana plumbaginifolia leaves and regenerating protoplasts. Plant Sci. Lett. 19, 231–241. [Google Scholar]
- Marineau, C., Matton, D.P., and Brisson, N. (1987). Differential accumulation of potato mRNAs during the hypersensitive response induced by arachidonic acid elicitor. Plant Mol. Biol. 9, 335–342. [DOI] [PubMed] [Google Scholar]
- Matton, D.P., and Brisson, N. (1989). Cloning, expression, and sequence conservation of pathogenesis-related gene transcripts of potato. Mol. Plant Microbe Interact. 2, 325–331. [DOI] [PubMed] [Google Scholar]
- Matton, D.P., Prescott, G., Bertrand, C., Camirand, A., and Brisson, N. (1993). Identification of cis-acting elements involved in the regulation of the pathogenesis-related gene STH-2 in potato. Plant Mol. Biol. 22, 279–291. [DOI] [PubMed] [Google Scholar]
- Michelotti, G., Tomonaga, T., Krutzsh, H., and Levens, D. (1995). Cellular nucleic acid binding protein regulates the CT element of the human c-myc protooncogene. J. Biol. Chem. 270, 9494–9499. [DOI] [PubMed] [Google Scholar]
- Michelotti, G., Michelotti, E.F., Pullner, A., Duncan, R.C., Eick, D., and Levens, D. (1996). Multiple single-stranded cis elements are associated with activated chromatin of the human c-myc gene in vivo. Mol. Cell. Biol. 16, 2656–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiseyev, G.P., Beintema, J.J., Fedoreyeva, L.I., and Yakovlev, G.I. (1994). High sequence similarity between a ribonuclease from ginseng calluses and fungus-elicited proteins from parsley indicates that intracellular pathogenesis-related proteins are ribonucleases. Planta 193, 470–472. [DOI] [PubMed] [Google Scholar]
- Moons, A., Prinsen, E., Bauw, G., and Van Montagu, M. (1997). Antagonistic effects of abscisic acid and jasmonates on salt stress–inducible transcripts in rice shoots. Plant Cell 9, 2243–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralidharan, V., Tretiakova, A., Steplewski, A., Haas, S., Amini, S., Johnson, E., and Khalili, K. (1997). Evidence for inhibition of MyEF-2 binding to MBP promoter by MEF-1/Pur α. J. Cell. Biochem. 66, 524–531. [DOI] [PubMed] [Google Scholar]
- Niderman, T., Genetet, I., Bruyere, T., Gees, R., Stinzi, A., Legrand, M., Fritig, B., and Mosinger, E. (1995). Pathogenesis-related PR-1 proteins are antifungal: Isolation and characterization of three 14-kilodalton proteins of tomato and of a basic PR-1 of tobacco with inhibitory activity against Phytophthora infestans. Plant Physiol. 108, 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley, B.R., Kirsch, D.R., and Morris, N.R. (1980). A simplified ultrasensitive silver stain for detecting proteins in polyacrylamide gels. Anal. Biochem. 105, 361–363. [DOI] [PubMed] [Google Scholar]
- Osmark, P., Boyle, B., and Brisson, B. (1998). Sequential and structural homology between intracellular pathogenesis-related proteins and a group of latex proteins. Plant Mol. Biol. 38, 1243–1246. [DOI] [PubMed] [Google Scholar]
- Ponstein, A.S., Bres-Vloemans, S.A., Sela-Buurlage, M.B., van den Elzen, P.J., Melchers, L.S., and Cornelissen, B.J. (1994). A novel pathogen- and wound-inducible tobacco (Nicotiana tabacum) protein with antifungal activity. Plant Physiol. 104, 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rushton, P.J., Torres, J.T., Parniske, M., Wernert, P., Hahlbrock, K., and Sommsich, I.E. (1996). Interaction of elicitor-induced DNA binding proteins with elicitor response elements in the promoters of parsley PR1 genes. EMBO J. 15, 5690–5700. [PMC free article] [PubMed] [Google Scholar]
- Sambrook, J., Fritsch, E.F., and Maniatis, T. (1989). Molecular Cloning: A Laboratory Manual. (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press).
- Schlumbaum, A., Mauch, F., Voegeli, U., and Boller, T. (1986). Plant chitinases are potent inhibitors of fungal growth. Nature 324, 365–367. [Google Scholar]
- Schwechheimer, C., Smith, C., and Bevan, M.W. (1998). The activities of acidic and glutamine-rich transcriptional activation domains in plant cells: Design of modular transcription factors for high-level expression. Plant Mol. Biol. 36, 195–204. [DOI] [PubMed] [Google Scholar]
- Seipel, K., Georgiev, O., and Schaffner, W. (1992). Different activation domains stimulate transcription from remote (‘enhancer’) and proximal (‘promoter’) positions. EMBO J. 11, 4961–4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smidt, M.P., Russchen, B., Snippe, L., Wijnholds, J., and Ab, G. (1995). Cloning and characterization of a nuclear, site specific ssDNA binding protein. Nucleic Acids Res. 23, 2389–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somssich, I.E., Schmelzer, E., Kawalleck, P., and Hahlbrock, K. (1988). Gene structure and in situ transcript localization of pathogenesis-related protein 1 in parsley. Mol. Gen. Genet. 213, 93–98. [DOI] [PubMed] [Google Scholar]
- Subramaniam, R., Després, C., and Brisson, N. (1997). A functional homolog of mammalian protein kinase C participates in the elicitor-induced defense response in potato. Plant Cell 9, 653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swoboda, I., Hoffmannsommergruber, K., Oriordain, G., Scheiner, O., Heberlebors, E., and Vicente, O. (1996). BET v1 proteins, the major birch pollen allergens and members of a family of conserved pathogenesis-related proteins, show ribonuclease activity in vitro. Physiol. Plant. 96, 433–438. [Google Scholar]
- Takimoto, M.T., Tomonaga, T., Matunis, M., Avignan, M., Krutzsch, H., Dreyfuss, G., and Levens, D. (1993). Specific binding of heterogeneous ribonucleoprotein particle K to the human c-myc promoter, in vitro. J. Biol. Chem. 268, 18249–18258. [PubMed] [Google Scholar]
- Van Loon, L.C., Pierpoint, W.S., Boller, T., and Conejero, V. (1994). Recommendations for naming plant pathogenesis-related proteins. Plant Mol. Biol. Rep. 12, 245–264. [Google Scholar]
- Vigers, A.J., Roberts, W.K., and Selitrennikoff, C.P. (1991). A new family of plant antifungal proteins. Mol. Plant-Microbe Interact. 4, 315–323. [DOI] [PubMed] [Google Scholar]
- Walter, M.H., Liu, J.-W., Grand, C., Lamb, C., and Hess, D. (1990). Bean pathogenesis-related (PR) proteins deduced from elicitor-induced transcripts are members of a ubiquitous new class of conserved PR proteins including pollen allergens. Mol. Gen. Genet. 222, 353–360. [DOI] [PubMed] [Google Scholar]
- Warner, S.A.J., Scott, R., and Draper, J. (1992). Characterization of a wound-induced transcript from the monocot asparagus that shares similarity with a class of pathogenesis-related (PR) proteins. Plant Mol. Biol. 19, 555–561. [DOI] [PubMed] [Google Scholar]
- Wilkins, R.C., and Lis, J.T. (1999). DNA distortion and multimerization: Novel functions of the glutamine-rich domain of GAGA factor. J. Mol. Biol. 285, 515–525. [DOI] [PubMed] [Google Scholar]
- Xiang, C., Miao, Z., and Lam, E. (1997). DNA-binding proteins, genomic organization and expression pattern of TGA6, a new member of the TGA family of bZIP transcription factors in Arabidopsis thaliana. Plant Mol. Biol. 34, 403–415. [DOI] [PubMed] [Google Scholar]
- Zhang, Y., Fan, W., Kinkema, M., Li, X., and Dong, X. (1999). Interaction of NPR1 with basic leucine zipper protein transcription factors that bind sequences required for salicylic acid induction of the PR-1 gene. Proc. Natl. Acad. Sci. USA 96, 6523–6528. [DOI] [PMC free article] [PubMed] [Google Scholar]