

Abstract
Inhibitory Smads, i.e. Smad6 and Smad7, are potent antagonists of the BMP–Smad pathway by interacting with activated bone morphogenetic protein (BMP) type I receptors and thereby preventing the activation of receptor-regulated Smads, or by competing with activated R-Smads for heteromeric complex formation with Smad4. The molecular mechanisms that underlie the regulation of I-Smad activity have remained elusive. Here we report the identification of a cytoplasmic protein, previously termed associated molecule with the SH3 domain of STAM (AMSH), as a direct binding partner for Smad6. AMSH interacts with Smad6, but not with R- and Co-Smads, upon BMP receptor activation in cultured cells. Consistent with this finding, stimulation of cells with BMP induces a co-localization of Smad6 with AMSH in the cytoplasm. Ectopic expression of AMSH prolongs BMP-induced Smad1 phosphorylation, and potentiates BMP-induced activation of transcriptional reporter activity, growth arrest and apoptosis. The data strongly suggest that the molecular mechanism by which AMSH exerts its action is by inhibiting the binding of Smad6 to activated type I receptors or activated R-Smads.
Keywords: AMSH/BMP/signaling/Smad/TGF-β
Introduction
Bone morphogenetic proteins (BMPs) are cytokines that were identified originally by their ability to induce formation of ectopic cartilage and bone, and subsequently were shown to be multifunctional proteins that regulate a wide spectrum of functions such as proliferation, differentiation and apoptosis of a large variety of cell types, including osteoblasts, epithelial cells, neurons and immune cells (Reddi, 1997; Wozney, 1998). BMPs belong to a large family of structurally and functionally related proteins, known as the transforming growth factor-β (TGF-β) superfamily (Derynck and Feng, 1997; Massagué, 1998). Like other members of the TGF-β superfamily, BMPs elicit their cellular effects through activation of specific combinations of type I and type II serine/threonine kinase receptors (Kawabata et al., 1998a). Type I receptors act downstream of type II receptors, and have been shown to determine signaling specificity within the heteromeric receptor complex (Derynck and Feng, 1997; Massagué, 1998). Activated type I receptors propagate the signal through phosphorylation of specific receptor-regulated Smads (R-Smads). Whereas BMP type I receptors activate Smad1, Smad5 and Smad8, Smad2 and Smad3 are phosphorylated by the TGF-β type I receptor. Activated R-Smads assemble into heteromeric complexes with common partner (Co)-Smad, i.e. Smad4, which translocate into the nucleus, where they regulate the transcription of target genes (Derynck et al., 1998; Massagué and Wotton, 2000). It is of note that activated BMP receptors have also been found to initiate other intracellular pathways, distinct from the Smad pathway, e.g. the activation of JNK and p38 MAP kinase pathways (Heldin et al., 1997).
The inhibitory Smads (I-Smads), i.e. Smad6 and Smad7, prevent the activation of R- and Co-Smads. They do so by interacting efficiently with the activated type I receptors thereby preventing access of R-Smads to, and phosphorylation by, the activated type I receptors (Hayashi et al., 1997; Imamura et al., 1997; Nakao et al., 1997, Souchelnytskyi et al., 1998; Lebrun et al., 1999). Smad6 has also been found to exert its inhibitory effect on signaling by competing with Smad4 for heteromeric complex formation with activated Smad1 (Hata et al., 1998). Recently, another mechanism by which I-Smads antagonize TGF-β signaling has been described. Smad7 was found to interact with Smurfs, which are HECT-domain ubiquitin ligases that target the TGF-β receptor for proteasomal and lysosomal degradation (Kavsak et al., 2000; Ebisawa et al., 2001).
Whereas Smad6 appears to inhibit BMP signaling preferentially, Smad7 acts as a general inhibitor of TGF-β family signaling (Itoh et al., 1998; Souchelnytskyi et al., 1998; Ishisaki et al., 1999). Smad6 has been reported to occur in two forms, possibly as a result of differential promoter usage or alternative splicing; a short form mainly consisting of the MH2 domain and a long form containing an N-terminal sequence with weak similarity to MH1 domains of R- and Co-Smads (Riggins et al., 1996; Imamura et al., 1997; Topper et al., 1997; Hata et al., 1998). Herein, we have designated the Smad6 short and long forms Smad6S and Smad6L, respectively. Structure–function analysis has revealed that the C-terminal domain of I-Smads is sufficient and required for its inhibitory activity (Hayashi et al., 1997; Souchelnytskyi et al., 1998). The inhibitory effect of Smad6S on the inhibition of BMP signaling is stronger than that of Smad6L (Hata et al., 1998). The N-terminal regions of I-Smads may function to target specific pathways for inhibition (Souchelnytskyi et al., 1998).
The expression of I-Smads is induced quickly upon stimulation by members of the TGF-β family; thus, I-Smads may be part of negative feedback control mechanisms (Nakao et al., 1997; Afrakhte et al., 1998; Takase et al., 1998; Ishisaki et al., 1999). In addition, other factors, including interferon-γ and tumor necrosis factor-α, have been found to interfere with TGF-β signaling through up-regulation of I-Smad expression (Ulloa et al., 1999; Bitzer et al., 2000). Differential compartmentalization of I-Smads appears to be another mechanism to regulate the activity of I-Smads; in the absence of ligand, I-Smads are located predominantly in the nucleus, but are exported rapidly into cytoplasm after ligand stimulation (Itoh et al., 1998; Nakayama et al., 1998; Zhu et al., 1999). The mechanism for nuclear export of I-Smads is not known.
To advance our understanding of how the action of I-Smads is controlled, we have screened for I-Smad-binding proteins using the yeast two-hybrid interaction assay. We identified associated molecule with the SH3 domain of STAM (AMSH) as an I-Smad-binding protein. Previously, AMSH was identified as a signal-transducing adaptor molecule (STAM)-interacting protein, and implicated in interleukin-2 (IL-2) and granulocyte–macrophage colony-stimulating factor (GM-CSF) signaling (Tanaka et al., 1999). We found that AMSH inhibits the antagonistic effects of Smad6 on the BMP–Smad pathway. Our results suggest that the AMSH expression level plays a critical role in determining the potency of BMP-induced responses.
Results
Identification of an I-Smad-interacting protein
To obtain insight into the mechanism of action of I-Smads, we isolated proteins that specifically interact with Smad6 using the yeast two-hybrid system. We used a fragment of Smad6 as a bait in a screen of an amplified human fetal brain library (with an original complexity of 4 × 106 independent cDNA clones). We obtained 75 positive clones which we could classify into 12 groups of distinct cDNAs. The clones in the majority group (39 positives) were derived from partial cDNAs encoding AMSH. AMSH was isolated originally as a binding partner of the SH3 domain of STAM (Tanaka et al., 1999), and it has been suggested to play a role in intracellular signal transduction of cytokines. Subsequently, we investigated whether there is specificity of AMSH in the interaction with R-, Co- and I-Smads using the yeast two-hybrid system. We found that AMSH could bind Smad6S and Smad7, but not Smad2 and Smad4 (data not shown). To analyze the interaction between Smads and AMSH further, we analyzed the interaction in COS7 cells transfected with differentially epitope-tagged Smads and AMSH in the absence or presence of a constitutively active (ca) form of BMP type I receptor, also termed activin receptor-like kinase (ALK)6. Western blotting of immunoprecipitates from extracts of transfected COS cells revealed that AMSH interacted with Smad6S and Smad6L, and that this interaction was promoted upon co-expression with caALK6 (Figure 1A). Smad7 also associated strongly with AMSH, but did so even in the absence of caALK6. Smad1 and Smad4, however, did not interact with AMSH even in the presence of caALK6 (Figure 1A). Moreover, we could not detect any interaction between AMSH and Smad5 or Smad8 in cultured mammalian cells (data not shown). Taken together, our data show that AMSH interacts directly with I-Smads, but not with R- and Co-Smads.
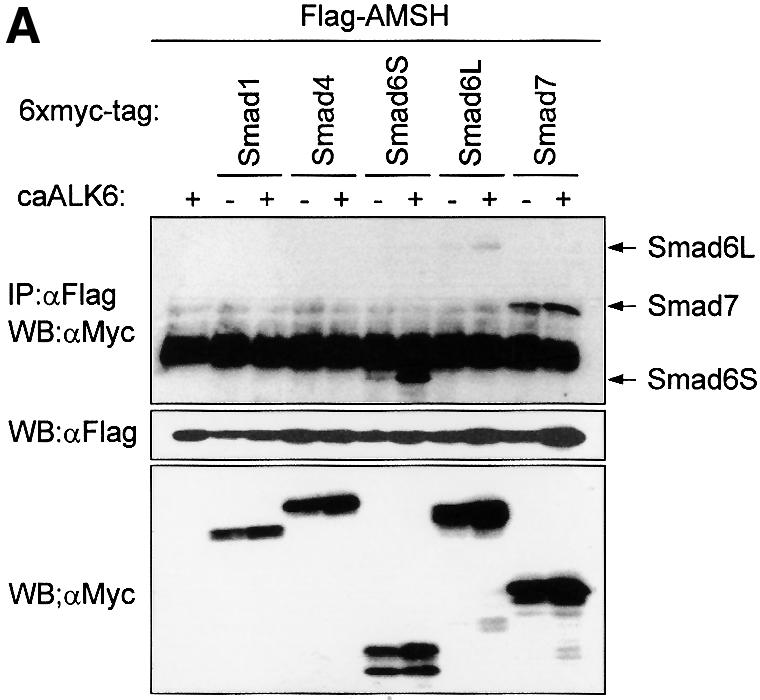

Fig. 1. BMP receptor activation induces interaction and co-localization of AMSH with I-Smads. (A) Interaction of AMSH with I-Smads. Flag-AMSH was co-transfected with 6×Myc-Smads with or without caALK6 in COS7 cells. Immunoprecipitations were performed with Flag M5 antibody, and co-immunoprecipitated Smads were detected by western blotting with Myc antibody (upper panel). The expression of Flag-AMSH and 6×Myc-Smads was measured by applying total cell lysate (1:50) on an SDS–polyacrylamide gel followed by western blotting with Flag M5 (middle panel) or Myc antibody (lower panel). (B) I-Smads and AMSH are localized in the cytoplasm upon BMP-7 stimulation. C2C12 cells were transiently transfected with Myc-tagged AMSH, and Flag-tagged Smad6L or Flag-tagged Smad7. Then, the cells were stimulated for 2 h with BMP-7. Myc-AMSH was visualized with rabbit polyclonal Myc antibody followed by Texas red-conjugated goat anti-rabbit IgG (red), and Flag-I-Smads were detected with mouse monoclonal Flag M5 antibody followed by FITC-conjugated goat anti-mouse IgG (green). Co-localization of AMSH and I-Smads appears as yellow.
I-Smads and AMSH are both localized in cytoplasm after BMP-7 stimulation
The subcellular location of AMSH is unknown. The observed BMP-dependent interaction between Smad6 and AMSH made us investigate the subcellular distribution of AMSH in the absence or presence of BMP-7. C2C12 cells were co-transfected with Myc-tagged AMSH and Flag-tagged I-Smads, and cells were stimulated with BMP-7. Subsequently, the subcellular location of AMSH and I-Smads was determined by confocal laser scanning microscopy after staining AMSH with Tex red-conjugated goat anti-rabbit IgG antibody and I-Smads with fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG antibody, respectively (see also Materials and methods). In the absence or presence of BMP-7, AMSH was present predominantly in the cytoplasm, and found to be localized in particular surrounding the nuclear membrane. Consistent with the previous report on TGF-β-induced nuclear export of Smad7 (Itoh et al., 1998), Smad6L and Smad7 were present mainly in nucleus before BMP-7 stimulation, and a 2 h treatment with BMP-7 induced an accumulation of I-Smads in the cytoplasm (Figure 1B). The diffusely stained I-Smads in the cytoplasm overlapped in part with punctuately stained AMSH. After 8 h of BMP-7 treatment, I-Smads were again localized predominantly in the nucleus, whereas AMSH remained in the cytoplasm (data not shown). Thus, consistent with their physical interaction, AMSH and I-Smads co-localize in cytoplasm upon BMP-7 stimulation.
AMSH promotes BMP-induced transcriptional reporter activity
BMP activates a Smad-binding element-driven reporter plasmid (SBE)4-luc (Jonk et al., 1998). Ectopic expression of I-Smads inhibits the BMP-induced activation of (SBE)4-luc reporter (Hata et al., 1998; Imamura et al., 1997; Nakao et al., 1997). Since AMSH interacts with I-Smads upon BMP type I receptor activation, we investigated the effect of AMSH on activation of the (SBE)4-luc reporter induced by BMP or R- or Co-Smads in the absence or presence of I-Smads. HepG2 cells were transfected with AMSH alone or with AMSH together with combinations of Smads, and stimulated with BMP-7. AMSH increased the activity of luciferase upon BMP-7 stimulation. In addition, AMSH was found to cooperate with Smad1 to potentiate the BMP-mediated activation of (SBE)4-luc reporter. Interestingly, ectopic expression of AMSH also increased the basal reporter activity. As previously reported (Hata et al., 1998; Imamura et al., 1997; Nakao et al., 1997), Smad6L and Smad7 were found to inhibit BMP-7-induced (SBE)4-luc activity. AMSH was found to mitigate the inhibitory effect of Smad6L, but was not efficient in antagonizing the potent inhibitory effect of Smad7 (Figure 2A).

Fig. 2. AMSH potentiates the BMP/Smad-induced transcriptional response. (A) AMSH stimulates BMP/Smad1-induced transcription. HepG2 cells were transfected with (SBE)4-luc, AMSH and Smads in the absence (solid bars) or presence (open bars) of 100 ng/ml BMP-7. (B) AMSH requires Smad4 to potentiate BMP-7-induced transcription. MDA-MB468 cells were transfected with (SBE)4-luc, AMSH and Smad4 without (solid bars) or with (open bars) BMP-7.
Smad4 is known to recruit R-Smads after ligand stimulation, whereafter the complex is translocated to the nucleus. To examine whether AMSH can activate the BMP-7-induced (SBE)4-luc activity in the absence of Smad4, MDA-MB468 cells that are deficient in Smad4 were transfected with AMSH in the absence or presence of Smad4. In the absence of Smad4, AMSH did not affect the activity of (SBE)4-luc even in the presence of BMP-7. However, in the presence of Smad4, AMSH enhanced the BMP-7-induced luciferase activity (Figure 2B).
AMSH enhances BMP-7-induced growth inhibition and apoptosis
Mouse B-cell hybridoma HS-72 cells are highly responsive to BMP-induced growth inhibition and apoptosis (Ishisaki et al., 1999). A Myc-AMSH expression plasmid containing a hygromycin B dominant selection marker was stably transfected into HS-72 cells to investigate the effect of AMSH on BMP-7-induced responses in this cell line. After the selection by hygromycin B, we obtained three transformants of HS-72 cells, AM101, AM103 and AM111. The expression of Myc-AMSH in AM111 cells was higher than that in AM101, whereas no expression of Myc-AMSH was detected in AM103 (Figure 3A). Using these cell lines, we first investigated the effect of Myc-AMSH on BMP-7-induced growth inhibition. Among the cell lines examined, the growth of AM111 cells was inhibited most potently by BMP-7 with an ID50 of 40 ng/ml, compared with 75 ng/ml for AM101, 150 ng/ml for AM103 and 150 ng/ml for wild-type (Figure 3B). Thus, HS-72 cells gained more sensitivity to BMP-7 upon higher expression of AMSH protein. To analyze the effect of AMSH on BMP-7-induced apoptosis, we stained transformants with propidium iodide after treatment of cells with BMP-7 and determined the number of apoptotic cells by fluorescent-activated cell sorting (FACS). BMP-7-induced apoptosis in transformants was potentiated upon elevated expression of AMSH. Whereas 38 and 14% of AM111 and AM101 cells, respectively, became apoptotic in the presence of BMP-7, only 9 and 10% of parental HS-72 and AM103 cells treated with BMP-7 were apoptotic, respectively (Figure 3C). The G1 arrest induced by BMP-7 followed a similar pattern in response to ectopic Myc-AMSH expression; in the presence of BMP-7, 90% of AM111 cells were in the G1 phase of the cell cycle, compared with 82% of HS-72 or AM103 cells (Figure 3D). Taken together, these data strongly suggest that AMSH promotes BMP-7-induced biological responses in a strictly dose-dependent manner.


Fig. 3. AMSH enhances the sensitivity to BMP-7 in HS-72 plasma cells. (A) Expression of AMSH in HS-72 transformants. Total lysate from each transformant (2 × 105 cells) was applied on the SDS–polyacrylamide gel, and the expression of AMSH was detected by western blotting using anti-Myc antibody. (B) AMSH potentiates BMP-7-mediated growth inhibition. The potentiation of growth inhibition by AMSH in three independent clones (AM101, AM103 and AM111) is shown. As a control, the effect of BMP-7 on parental HS-72 cells is shown. The relative growth compared with control is plotted against the concentration of BMP-7. All data shown are means ± SD. The percentage viability was calculated as follows: percentage viability = 100 × (A570–620 nm with BMP-7/A570–620 nm without BMP-7). (C) AMSH induces apoptosis in HS-72 cells in the presence of BMP-7. The cells were left unstimulated (solid bars) or stimulated with 500 ng/ml BMP-7 for 24 h (open bars), then the apoptotic cells in 1 × 104 cells analyzed were counted by FACScan. (D) AMSH increases the number of G1-arrested cells in the presence of BMP-7. Twenty-four hours after the treatment of each transformant with 500 ng/ml BMP-7, the cells were fixed and analyzed by FACScan. The data show the percentages of G1, S and G2/M phases in 1 × 104 cells. The experiments were performed at least three times, and the results from representative experiments are shown.
AMSH prolongs the BMP-7-induced Smad phosphorylation
BMP R-Smads, i.e. Smad1, Smad5 and Smad8, are phosphorylated in response to BMP-mediated activation of BMP type I receptors. Using an antibody that specifically detects phosphorylated BMP R-Smads, we compared the BMP-7-induced phosphorylation of endogenous BMP R-Smads in HS-72 cells with that in AM111 cells (Figure 4). The phosphorylation of BMP R-Smads in HS-72 cells peaked at 1 h after BMP-7 stimulation and declined rapidly thereafter. BMP-7-induced phosphorylation in AM111 cells was induced to similar peak levels as in HS-72 cells but, in contrast to parental HS-72 cells, the signal remained high between 1 and 4 h of BMP-7 treatment. Thereafter, the phosphorylation declined but continued at a higher level after 12 h of BMP-7 stimulation (Figure 4). Thus, although AMSH does not interact directly with BMP R-Smads, it stimulates the phosphorylation of BMP R-Smads by BMP-7.

Fig. 4. AMSH prolongs the BMP-7-induced Smad phosphorylation. HS-72 and AM111 cells were stimulated with 500 ng/ml BMP-7 for 0, 1, 4, 8 and 12 h. Total lysates from the cells (2 × 105 cells) were blotted and stained with anti-pS1 (upper panel) or anti-Smad1/5 (lower panel) antibody. Similar results were obtained in three independent experiments.
AMSH antagonizes the inhibitory effects of Smad6 on the BMP–Smad pathway
Smad6L inhibits BMP signaling mediated by Smads in the cytoplasm by interacting with activated BMP type I receptors, and thereby preventing the activation of R-Smads (Imamura et al., 1997), and by binding activated R-Smads, thereby competing with the formation of heteromeric complex of R- and Co-Smads (Hata et al., 1998). To examine whether AMSH affects the ability of Smad6L to associate with the activated BMP type I receptor, Myc-AMSH was co-transfected with Flag-Smad6L and hemagglutinin (HA)-caALK6 in COS7 cells, immunoprecipitated with HA antibody and then blotted with Flag M5 antibody. As previously reported (Imamura et al., 1997), we found that Smad6L interacted with caALK6 (Figure 5A) and less efficiently with wild-type ALK6 (data not shown). Importantly, AMSH was found to inhibit the interaction between Smad6L and caALK6 (Figure 5A). In addition, consistent with the notion that the inhibitory effect of AMSH is mediated through its association with Smad6L and not by binding to BMP type I receptor, AMSH was found to be incapable of binding directly to the BMP receptor complex, nor did it bind indirectly via Smad6L (data not shown).
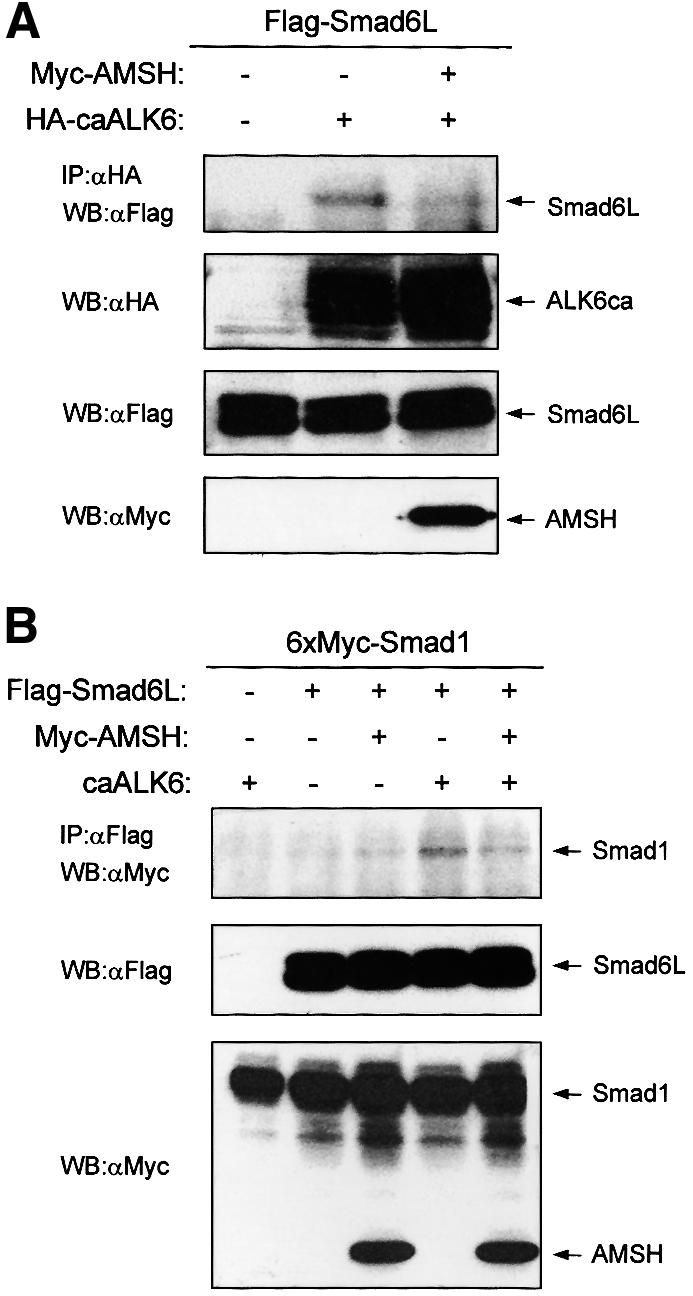
Fig. 5. AMSH inhibits interactions of Smad6L with activated BMP type I receptor and Smad4. (A) AMSH inhibits the association between Smad6L and ALK6. Myc-AMSH was co-transfected with Flag-Smad6L and HA-caALK6 in COS7 cells. Immunoprecipitations were performed with HA 12C5 (Boehringer Mannheim) antibody, and co-immuno precipitated Smad6L was detected by western blotting with Flag M5 antibody (upper panel). The expression of HA-caALK6, Flag-Smad6L and Myc-AMSH was measured by applying total cell lysate (1:50) on an SDS–polyacrylamide gel followed by western blotting with HA (second panel), Flag M5 (third panel) or Myc antibody (lower panel). (B) The interaction between Smad6L and activated Smad1 upon BMP type I receptor activation is inhibited by AMSH. Myc-AMSH was co-transfected with 6×Myc-Smad1 and Flag-Smad6L with or without caALK6 in COS7 cells. Immunoprecipitations were performed with Flag M5 antibody, and co-immunoprecipitated Smad6L was detected by western blotting with Myc antibody (upper panel). The expression of Flag-Smad6L, 6×Myc-Smad1 and Myc-AMSH was measured by applying total cell lysate (1:50) on an SDS–polyacrylamide gel followed by western blotting with Flag M5 (middle panel) or Myc antibody (lower panel).
We next investigated the effect of AMSH on the association of Smad6L with Smad1 in the presence or absence of caALK6 in COS7 cells. We observed that Smad6L interacted with Smad1 upon ALK6 activation; this interaction was, however, less efficient than the association of Smad6L with caALK6. Importantly, AMSH was found to interfere with the interaction between Smad6L and Smad1 (Figure 5B). Thus, AMSH inhibits the interaction between Smad6L and both activated BMP type I receptor and activated Smad1. A molecular mechanism is hereby provided by which AMSH potentiates BMP signaling, i.e. by antagonizing the inhibitory action of I-Smads.
BMP type I receptor activation stimulates AMSH phosphorylation
The export of Smad6 from the nucleus in response to receptor activation may be sufficient to induce the receptor-dependent interaction between AMSH and Smad6. However, in order for these proteins to interact, a BMP-induced post-translational modification leading to a conformational change in AMSH might be needed. Therefore, we examined whether AMSH becomes phosphorylated upon BMP receptor activation. AMSH was transfected into COS7 cells in the absence or presence of caALK6, metabolically labeled with [32P]orthophosphate and immunoprecipitated. AMSH was found to be weakly constitutively phosphorylated. Interestingly, caALK6 stimulated AMSH phosphorylation (Figure 6A, lane 1 versus lane 2). Preliminary experiments in which cells were stimulated with BMP-7 suggested that the phosphorylation of AMSH occurred relatively late (after 3 h) and therefore was likely to be indirect. It is known that BMP or TGF-β family signaling is mediated in part through MAP kinase pathways (Atfi et al., 1997; Engel et al., 1999; Hanafusa et al., 1999; Hocevar et al., 1999; Ravanti et al., 1999; Sano et al., 1999; Kimura et al., 2000). Therefore, we analyzed the effect of MAP kinase inhibitors on the caALK6-induced phosphorylation of AMSH. SB203580, a p38 inhibitor, inhibited the phosphorylation of AMSH in a dose-dependent manner (Figure 6A, lanes 5 and 6), whereas PD98059, an ERK inhibitor, had no effect (Figure 6A, lanes 3 and 4). In addition, dominant-negative kinases for JNK and p38 efficiently blocked caALK6-induced phosphorylation of AMSH (Figure 6B), suggesting that JNK and/or p38 mediates the phosphorylation of AMSH after BMP type I receptor activation.

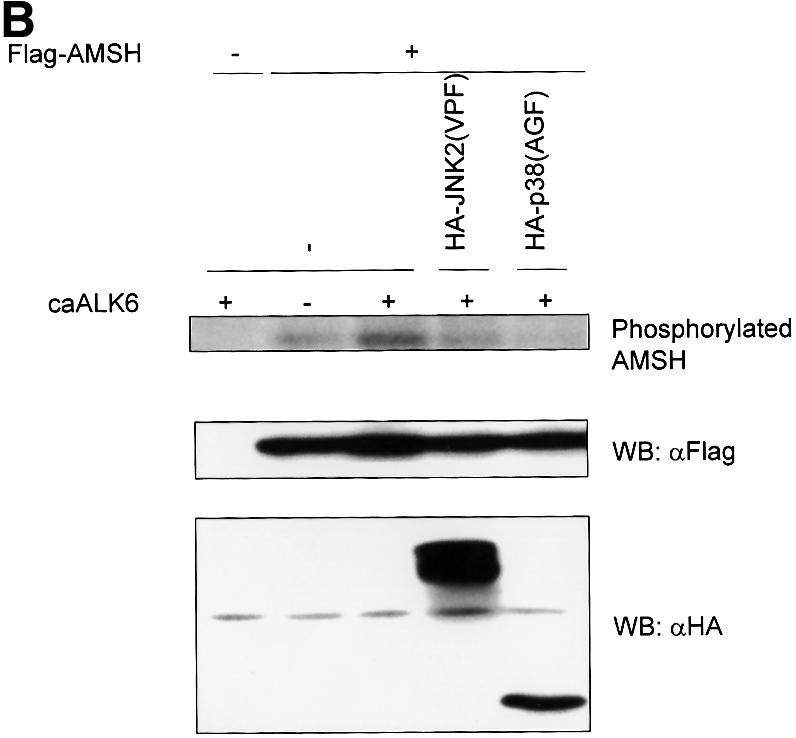
Fig. 6. BMP type I receptor-induced AMSH phosphorylation. (A) SB203580 inhibits the phosphorylation of AMSH by caALK6. Flag-AMSH was transfected with or without caALK6. Then, PD98059 or SB203580 was added to culture medium 1 h before the addition of [32P]orthophosphate. Immunoprecipitations were performed with Flag M5 antibody (upper panel). The expression of Flag-AMSH in COS7 cells was visualized by western blotting using Flag M5 antibody (lower panel). (B) Dominant-negative forms of JNK2 and p38 block the phosphorylation of AMSH by caALK6. COS7 cells were transfected with the combinations of Flag-AMSH, HA-JNK2(VPF), HA-p38(AGF) and caALK6 and then metabolically labeled with [32P]orthophosphate. Immunoprecipitations were performed with Flag M5 antibody (upper panel). The expression of Flag-AMSH, HA-JNK2(VPF) and HA-p38(AGF) in COS7 cells was visualized by western blotting using Flag M5 (middle panel) or HA antibody (lower panel).
Subsequently, we determined the major phosphorylation sites in AMSH stimulated with caALK6. We observed four major phosphopeptides (Figure 7, spots a–d) which were induced potently by caALK6. Phosphoamino acid analysis revealed that only serine residues in each spot were phosphorylated (data not shown). The exact position of phosphoserine residues in four phosphopeptides was identified by Edman degradation analysis; spot a for Ser243, Ser245 and Ser247, spot b for Ser2, and spots c and d for Ser48. To confirm the position of the phosphoserine residues, the serine residue(s) in each phosphopeptide was replaced by alanine residues. Then, each mutant as well as wild-type AMSH was transfected into COS7 cells in the absence or presence of caALK6, and tryptic phosphopeptide mapping of each mutant was performed. As seen in Figure 7, each spot corresponding to the phosphopeptide containing phosphoserine disappeared in the tryptic phosphopeptide mapping.

Fig. 7. Phosphopeptide mapping in AMSH. Flag-AMSH or its mutants were co-transfected in the absence or presence of caALK6 in COS cells. After metabolic labeling with [32P]orthophosphate, proteins were immunoprecipitated with Flag M5 antibody. After trypsinization, two-dimensional electrophoresis was carried out. Four major spots, surrounded with broken lines, are shown as a–d.
Next we examined the effect of phosphorylation on BMP receptor-induced interaction of AMSH and Smad6L. To our surprise, we found that the interaction between Smad6L and the AMSH (S243A,S245A,S247A) phosphorylation-defective mutant was stronger than with wild-type AMSH (Figure 8A). On the other hand, the other two mutants, AMSH(S2A) and AMSH(S48A), interacted with Smad6L with efficiency similar to wild-type AMSH (data not shown). Consistent with these observations, AMSH (S243A,S245A,S247A) potentiated BMP-7-induced transcriptional reporter activity more efficiently than wild-type AMSH (Figure 8B). Furthermore, we investigated whether dominant-negative p38 or JNK affected the interaction of AMSH with Smad6L upon BMP receptor activation. Consistent with the result using the AMSH mutant, the interaction between AMSH and Smad6L by caALK6 obviously increased in the presence of dominant-negative p38 or JNK (Figure 8C). Taken together, our experiments suggest that p38- or JNK-induced phosphorylation of AMSH, which can be induced by caALK6, decreases the negative effect of AMSH on Smad6 function.


Fig. 8. AMSH (S243A,S245A,S247A) mutant is more active than wild-type AMSH. (A) Interaction of the AMSH (S243A,S245A,S247A) mutant with Smad6L upon BMP type I receptor activation. Immuno precipitations were performed with Flag M5 antibody, and co-immunoprecipitated Smad6L was detected by western blotting with Myc antibody (upper panel). The expression of Flag-AMSH or Flag-AMSH (S243A,S245A,S247A), and 6×Myc-Smad6L was measured by applying total cell lysate (1:50) on an SDS–polyacrylamide gel followed by western blotting with Flag M5 (middle panel) or Myc antibody (lower panel). (B) AMSH (S243A,S245A,S247A) is more active than wild-type AMSH in potentiating BMP-7-induced transcriptional responses in HepG2 cells. Experiments were performed as described in the legend to Figure 2A. (C) Dominant-negative p38 and JNK augment the interaction of AMSH with Smad6L upon BMP type I receptor activation. Immunoprecipitations were performed with Flag M5 antibody, and co-immunoprecipitated Smad6L was detected by western blotting with Myc antibody (upper panel). The expression of Flag-AMSH, 6×Myc-Smad6L and HA-JNK2(VPF) or HA-p38(AGF) was measured by applying total cell lysate (1:50) on an SDS–polyacrylamide gel followed by western blotting with Flag M5 (second panel), Myc (third panel) and HA antibodies (lower panel), respectively.
Discussion
I-Smads are inhibitors of the BMP–Smad pathway by inhibiting the access of R-Smads to the activated receptor (Hayashi et al., 1997; Imamura et al., 1997; Nakao et al., 1997) and by preventing heteromeric complex formation of R-Smads with Smad4 (Hata et al., 1998). To obtain insight into the mechanisms that regulate I-Smad function, we identified proteins that associate with I-Smads. We identified AMSH as a binding partner for Smad6 using the yeast two-hybrid system; the two proteins were found to interact in response to BMP receptor activation in mammalian cells. Importantly, BMP receptor activation induced a nuclear export of Smad6, and a subsequent co-localization of Smad6 and AMSH in the cytoplasm. Ectopic expression of AMSH potentiated BMP/Smad-induced transcriptional responses, as well as BMP-mediated growth arrest and apoptosis. Analysis of the kinetics of BMP-induced Smad1 phosphorylation revealed that ectopic AMSH expression prolonged the time period when Smad1 was phosphorylated after BMP-7 stimulation. Consistent with this finding, AMSH was found to inhibit the interaction between Smad6 and the activated BMP type I receptor, thereby allowing more efficient BMP receptor-induced phosphorylation of R-Smads. In addition, AMSH was found to interfere with the interaction between Smad6 and the activated R-Smad. Thus, AMSH promotes BMP signaling by negatively regulating the function of I-Smads.
The yeast two-hybrid interaction assay implicates the N-terminal half of the Smad6 MH2 domain in the direct binding with AMSH. Smad6 associates via its MH2 domain with the BMP type I receptor and Smad1 (Imamura et al., 1997; Hata et al., 1998). It is possible that the N-terminal half of the MH2 domain in Smad6 is also involved in the interaction with BMP type I receptor and Smad1.
AMSH was identified previously as a STAM-associated protein which regulates the response to IL-2 and GM-CSF in BAF-B03 cells (Tanaka et al., 1999). AMSH is ubiquitously expressed in human tissues (Tanaka et al., 1999), and database searches reveal AMSH-related sequences in Drosophila, Caenorhabditis elegans and Schizosaccharomyces pombe. Similarity with proteins in Drosophila and S.pombe is confined mainly to the C-terminal part of AMSH. In addition to its regulatory role in the BMP–Smad pathway, AMSH might thus play a controlling role in other signaling pathways in a large variety of species. In fact, we have observed that ectopic expression of AMSH in HS-72 cells resulted in an increased Fas-induced apoptosis (data not shown).
AMSH was found not only to potentiate BMP-induced effects, but also to increase the basal levels of BMP-mediated R-Smad activation as determined by SBE-driven reporter activity and Smad1 phosphorylation; as AMSH does not interact with R-Smads, it is possible that AMSH potentiated endogenously produced BMP. Other studies have also shown that basal transcriptional reporter activity is caused by endogenously produced active ligand, as this activity could be inhibited by adding neutralizing antibodies to TGF-β family ligands to cells (de Caestecker et al., 1997), or by transfecting dominant-negative type II receptors into cells (Zhang et al., 1996).
BMP stimulated a transient nuclear export of I-Smad although the localization of AMSH remained unchanged in the cytoplasm upon BMP stimulation. AMSH thereby is given an opportunity to associate with I-Smad. In our analysis of whether there are specific post-translational modifications of AMSH in response to BMP stimulation, we found that BMP receptor activation mediated AMSH phosphorylation, indirectly via activation of p38 and JNK. We cannot exclude that another kinase is involved which can be activated by p38 or JNK. Contrary to what we had expected, phosphorylation of AMSH via p38 or JNK inhibited the interaction between I-Smads and AMSH. We cannot exclude the possibility that phosphorylation at another residue(s) or other alterations may occur in AMSH in response to BMP which promotes its interaction with I-Smads. Another possibility is that Smad6 undergoes a modification in response to BMP stimulation. Consistent with previous observations (Imamura et al., 1997), we found Smad6 to be highly constitutively phosphorylated. BMP receptor simulation weakly induced Smad6 phosphorylation (S.Itoh and P.ten Dijke, unpublished result), but whether this plays a role in the association with AMSH remains to be investigated.
We demonstrated that AMSH can interact with Smad7. Smad7 is known to inhibit not only BMP, but also TGF-β signaling. We therefore investigated whether AMSH affected the TGF-β signaling pathway. As expected, AMSH could interact with Smad7 in cultured cells upon TGF-β receptor activation and augmented TGF-β-induced luciferase reporter activity (data not shown). In addition, we found that caALK5 induced the phosphorylation of AMSH (data not shown). It is thus likely that AMSH regulates the TGF-β–Smad pathway in a similar way as the BMP–Smad pathway.
BMP type I receptor-mediated activation of p38 and JNK may induce a phosphorylated AMSH which is less efficient in binding Smad6 and thus less efficient in promoting BMP signaling. In addition, the expression of the positive regulator AMSH was found to be down-regulated upon prolonged treatment with BMP (data not shown). Both events occur after BMP-induced Smad1 phosphorylation and I-Smad nuclear export (events that occur within 1 h of BMP stimulation), and may thus be part of a negative feedback mechanism. The intensity and duration of the signal are important parameters for morphogens such as BMP and determine how a cell will respond to BMP. Moreover, stimuli that regulate AMSH expression will provide a means whereby signaling via the BMP pathway may be affected. A search for such stimuli is under way.
Negative regulation of BMP-induced biological responses has been shown to occur at nearly every step in the BMP signal transduction pathway, e.g. BMP-binding proteins noggin and chordin prevent binding to receptors (Piccolo et al., 1996; Zimmerman et al., 1996), BAMBI acts as a pseudo type I receptor and inhibits type I receptor homomeric complex formation (Onichtchouk et al., 1999), I-Smads prevent formation of active Smad complexes (Hata et al., 1998; Imamura et al., 1997; Souchelnytskyi et al., 1998), and ERK MAP kinase-induced phosphorylation of Smad1 abrogates its nuclear accumulation (Kretzschmar et al., 1997). There are fewer examples of positive regulators of the BMP–Smad pathway, e.g. factors that stimulate the expression of TGF-β family ligands or their signaling receptors, and through interaction of Smads with transcriptional (co-) activators (Miyazono, 2000). In the present study, we have identified a novel mechanism through which BMP–Smad signaling can be promoted by inhibiting the function of I-Smads through their interaction with AMSH.
Materials and methods
Expression plasmids
pEG-Smad6S(1–158), containing the 158 N-terminal amino acid residues of Smad6S, was made by PCR and inserted into pEG202 (Golemis et al., 1992). Flag-Smad6L, 6×Myc-Smad1, 6×Myc-Smad2 and 6×Myc-Smad3 were provided by Dr K.Miyazono (Imamura et al, 1997; Nishihara et al., 1998). 6×Myc-Smad4, 6×Myc-Smad6S, 6×Myc-Smad6L and 6×Myc-Smad7 were constructed using 6×Myc-pcDNA3 (Nishihara et al., 1998). Myc-AMSH was made using Myc-pcDNA3 (Kawabata et al., 1998b). Expression constructs for HA-tagged caALK6, (SBE)4-luc, Flag-Smad1, Flag-Smad4, Flag-Smad7 and Flag-AMSH have been described previously (Nakao et al, 1997; Jonk et al., 1998; Persson et al., 1998; Tamaki et al., 1998; Tanaka et al, 1999). Dominant-negative forms for HA-p38(AGF) and HA-JNK2(VPF) were obtained from Dr E.Nishida. Myc-AMSH in pcDNA3.1/Hygro (+) (Invitrogen) was used to isolate stable transformants expressing Myc-AMSH.
Cell culture and transfections
HepG2 and HS-72 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Sigma) containing 10% fetal calf serum (FCS; Sigma) and 1× MEM non-essential amino acids (Sigma). C2C12, MDA-MB468 and COS7 cells were maintained in DMEM containing 10% FCS. For selection of stable transformants, HS-72 cells were maintained in DMEM containing 10% FCS and 400 U/ml of hygromycin B (Calbiochem).
Yeast two-hybrid screening
pEG-Smad6S(1–158) and a human fetal brain library in the pJG4-5 vector were provided by Dr R.Brent. Library screens were carried out using Leu2 and β-galactosidase reporters (pSH18–34) within the yeast strain, EGY48. In brief, EGY48 cells were transformed with pEG-Smad6S(1–158), pSH18–34 and the library, and plated in galactose-containing medium without leucine. Positive colonies were picked 3–5 days after plating (Golemis et al., 1992). Subsequently, positive colonies were tested again and confirmed as real positive clones.
Transcriptional reporter assays
One day prior to transfection, HepG2 and MDA-MB468 cells were seeded at 2.5 × 105 cells/well in 6-well plates. The cells were transfected using the calcium phosphate co-precipitation method, as previously described (Jonk et al., 1998). In all experiments, β-galactosidase (pCH110, Pharmacia) activity was measured to normalize for transfection efficiency. Each transfection was carried out in triplicate and repeated at least twice.
Immunoprecipitation and western blotting
Combinations of Smads and AMSH in the presence or absence of caALK6 were transfected in COS7 cells at 1.2 × 106 cells/10 cm dish using Fugene 6 (Boehringer Mannheim). At 40 h post-transfection, the cells were lysed in 1 ml of lysis buffer [20 mM Tris, pH 7.4, 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride (PMSF), 5 µg/ml leupeptin, 2.5 µg/ml aprotinin, 2 mM sodium vanadate, 40 mM NaF and 20 mM β-glycerophosphate]. The cell lysates were pre-cleared with protein G–Sepharose beads (Pharmacia) and incubated with Flag M5 antibody (Sigma) for 2 h at 4°C. Subsequently, protein G–Sepharose beads were added to the reaction mixture and incubated for 30 min at 4°C. After washing the immunoprecipitates with high salt buffer (20 mM Tris, pH 7.4, 500 mM NaCl, 1% Triton X-100, 1 mM PMSF, 5 µg/ml leupeptin, 2.5 µg/ml aprotinin, 2 mM sodium vanadate, 40 mM NaF and 20 mM β-glycerophosphate) three times and with lysis buffer once, the immunoprecipitates and aliquots of total cell lysates were separated by SDS–PAGE and transferred to a Hybond-C Extra membrane (Amersham). The membrane was probed subsequently with Myc 9E10 (Santa Cruz) or Flag M5 monoclonal antibody. Primary antibodies were detected with horseradish peroxidase-conjugated goat anti-mouse antibody (Amersham) and chemiluminescent substrate. For detection of non-phosphorylated and phosphorylated BMP–Smads, HS-72 or its transformant was lysed with lysis buffer, and total lysates were applied on a 7% SDS–polyacrylamide gel. After transfer of proteins on the membrane, the membrane was probed with anti-BMP-Smad (QWL) (Tamaki et al., 1998) or anti-phosphorylated BMP-Smad antibody (pS1) (Persson et al., 1998), and then bands that were recognized by the antibodies were detected by chemiluminescent reaction. The detection of the interactions between Smad1 and Smad6L or between HA-caALK6 and Smad6L was performed according to the above method except that TNE buffer (10 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 1 mM PMSF, 5 µg/ml leupeptin, 2.5 µg/ml aprotinin, 2 mM sodium vanadate, 40 mM NaF and 20 mM β-glycerophosphate) was used to lyse the cells. In order to detect the expression of endogenous AMSH in HS-72 cells, monoclonal anti-AMSH antibody (Tanaka et al., 1999) was used.
Immunofluorescence
C2C12 cells were grown in 8-well permanox slides (LAB-TEK) at 5 × 103 cells/well 1 day before transfection. Transfection with Myc-tagged AMSH and Flag-tagged I-Smads was performed with Fugene 6. After 24 h, the cells were starved in DMEM containing 0.3% FCS for 14 h, and then stimulated with 500 ng/ml BMP-7. After treatment, the slides were washed once with phosphate-buffered saline (PBS), fixed for 10 min with 2% paraformaldehyde, washed three times with PBS, subsequently permeabilized with 0.5% Triton X-100 in PBS for 5 min and washed again three times with PBS. Slides were blocked with 5% normal swine serum (DAKO) in PBS at 37°C for 1 h and incubated with 5% normal swine serum (in PBS) containing 10 µg/ml mouse monoclonal Flag M5 and 10 µg/ml rabbit polyclonal Myc (Upstate Biotechnology) antibodies at 4°C overnight. The slides were then washed three times with PBS, incubated with 5% normal swine serum (in PBS) including both FITC-conjugated goat anti-mouse IgG antibody (diluted 1:1000) (DAKO) and Texas red-conjugated goat anti-rabbit IgG antibody (diluted 1: 200) (Molecular Probes) at room temperature for 1 h, and washed three times with PBS. To visualize the fluorescence, a confocal laser scanning microscope (Leica) was used.
MTT assay
To observe the growth inhibition of the cells by BMP-7, the cells (2 × 104 cells/well in a 96-well plate) were incubated with DMEM containing 5% FCS in the presence of various concentrations of BMP-7 for 48 h and then examined for cell viability by a calorimetric assay with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma) (Nishihara et al., 1993). Absorbance was determined at a wavelength of 570 nm with background subtraction at 620 nm. The experiments were performed at least three times. Representative results are shown.
Detection of cell cycle arrested and apoptotic cells
To analyze cell cycle arrest and apoptotic cells induced by BMP-7, the cells (1 × 106/12-well) in DMEM containing 5% FCS were stimulated with 500 ng/ml BMP-7 for 24 h. Subsequently, the cells were washed once with PBS, resuspended in hypotonic buffer (0.1% sodium citrate, 0.1% Triton X-100) and stained with 50 µg/ml propidium iodide. The cell suspension was kept at 4°C overnight, and then analyzed using FACScan (Becton Dickinson).
[32P]orthophosphate labeling of cells, tryptic phosphopeptide mapping and two-dimensional phosphoamino acid analysis
COS7 cells were cultured in phosphate-free medium for 3 h. Subsequently, 37 MBq/ml [32P]orthophosphate was added in the culture medium. After 40 min, the cells were lysed with the lysis buffer, immunoprecipitated with Flag M5 antibody, separated by SDS–PAGE and transferred to a Hybond-C Extra membrane. For tryptic phosphopeptide mapping, AMSH bands were localized by exposure on a FujiX Bio-Imager (Fuji), excised from the filter and digested in situ with trypsin (modified sequencing grade; Promega). Two-dimensional phosphopeptide mapping was done using the Hunter thin-layer electrophoresis apparatus (HTLE-7000; CBS Scientific), essentially as described by Boyle et al. (1991). First dimension electrophoresis was performed in pH 1.9 buffer (formic acid:glacial acetic acid:water; 44:156:1800) for 23 min at 2000 V, and second dimension ascending thin-layer chromatography in isobutyric acid buffer (isobutyric acid:n-butanol:pyridine:glacial acetic acid:water; 1250:38:96:58:558). After exposure, phosphopeptides were eluted from the plates in the pH 1.9 buffer and lyophilized. The fractions were then subjected to two-dimensional phosphoamino acid analysis, and Edman degradation analyses were performed to determine the position of phosphoamino acid(s) in the phosphopeptide (Souchelnytskyi et al., 1996).
Acknowledgments
Acknowledgements
We thank Dr M.Kawabata for insightful discussions on the use of the yeast two-hybrid assay, Dr K.Miyazono for Smad cDNAs, Dr T.Nishihara for HS-72 cells, Dr R.Brent for the yeast two-hybrid system, Dr S.Souchelnytskyi for critical discussion of phosphopeptide mapping, Dr.E.Nishida for dominant-negative JNK and p38 constructs, and Dr K.Sampath for recombinant BMP-7. We are grateful to Dr L.Oomen and Ms L.Brocks for expert assistance with confocal analysis, to Ms S.Grimsby for DNA sequencing, and to Mr C.Wernstedt for Edman degradation. This work was supported by the Dutch Cancer Society (NKI 2000-2217)
References
- Afrakhte M., Morén,A., Jossan,S., Itoh,S., Sampath,K., Westermark,B., Heldin,C.-H., Heldin,N.-E. and ten Dijke,P. (1998) Induction of inhibitory Smad6 and Smad7 mRNA by TGF-β family members. Biochem. Biophys. Res. Commun., 249, 505–511. [DOI] [PubMed] [Google Scholar]
- Atfi A., Djelloul,S., Chastre,E., Davis,R. and Gespach,C. (1997) Evidence for a role of Rho-like GTPases and stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK) in transforming growth factor β-mediated signaling. J. Biol. Chem., 272, 1429–1432. [DOI] [PubMed] [Google Scholar]
- Bitzer M., von Gersdorff,G., Liang,D., Dominguez-Rosales,A., Beg,A.A., Rojkind,M. and Böttinger,E.P. (2000) A mechanism of suppression of TGF-β/SMAD signaling by NF-κB/RelA. Genes Dev., 14, 187–197. [PMC free article] [PubMed] [Google Scholar]
- Boyle W.J., van der Geer,P. and Hunter,T. (1991) Phosphopeptide mapping and phosphoamino acid analysis by two-dimensional separation on thin-layer cellulose plates. Methods Enzymol., 201, 110–149. [DOI] [PubMed] [Google Scholar]
- de Caestecker M.P., Hemmati,P., Larisch-Bloch,S., Ajmera,R., Roberts,A.B. and Lechleider,R.J. (1997) Characterization of functional domains within Smad4/DPC4. J. Biol. Chem., 272, 13690–13696. [DOI] [PubMed] [Google Scholar]
- Derynck R. and Feng,X.-H. (1997) TGF-β receptor signaling. Biochim. Biophys. Acta, 1333, 105–150. [DOI] [PubMed] [Google Scholar]
- Derynck R., Zhang,Y. and Feng,X.-F. (1998) Smads: transcriptional activators of TGF-β responses. Cell, 95, 737–740. [DOI] [PubMed] [Google Scholar]
- Ebisawa T., Fukuchi,M., Murakami,G., Chiba,T., Tanaka,K., Imamura,T. and Miyazono,K. (2001) Smurf1 interacts with transforming growth factor-β type I receptor through Smad7 and induces receptor degradation. J. Biol. Chem., 276, 12477–12480. [DOI] [PubMed] [Google Scholar]
- Engel M.E., McDonnell,M.A., Law,B.K. and Moses,H.L. (1999) Interdependent SMAD and JNK signaling in transforming growth factor-β-mediated transcription. J. Biol. Chem., 274, 37413–37420. [DOI] [PubMed] [Google Scholar]
- Golemis E.A. and Brent,R. (1992) Fused protein domains inhibit DNA binding by LexA. Mol. Cell. Biol., 12, 3006–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanafusa H., Ninomiya-Tsuji,J., Masuyama,N., Nishita,M., Fujisawa, J.-i., Shibuya,H., Matsumoto,K. and Nishida,E. (1999) Involvement of the p38 mitogen-activated protein kinase pathway in transforming growth factor-β-induced gene expression. J. Biol. Chem., 274, 27161–27167. [DOI] [PubMed] [Google Scholar]
- Hata A., Lagna,G., Massagué,J. and Hemmati-Brivanlou,A. (1998) Smad6 inhibits BMP/Smad1 signaling by specifically competing with the Smad4 tumor suppressor. Genes Dev., 12, 186–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi H. et al. (1997) The MAD-related protein Smad7 associates with the TGFβ receptor and functions as an antagonist of TGFβ signaling. Cell, 89, 1165–1173. [DOI] [PubMed] [Google Scholar]
- Heldin C.-H., Miyazono,K. and ten Dijke,P. (1997) TGF-β signalling from membrane to nucleus through SMAD proteins. Nature, 390, 465–471. [DOI] [PubMed] [Google Scholar]
- Hocevar B.A, Brown,T.L. and Howe,P.H. (1999) TGF-β induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J., 18, 1345–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura T., Takase,M., Nishihara,A., Oeda,E., Hanai,J., Kawabata,M. and Miyazono,K. (1997) Smad6 inhibits signalling by the TGF-β superfamily. Nature, 389, 622–626. [DOI] [PubMed] [Google Scholar]
- Ishisaki A., Yamato,K., Hashimoto,S., Nakao,A., Tamaki,K., Nonaka,K., ten Dijke,P., Sugino,H. and Nishihara,T. (1999) Differential inhibition of Smad6 and Smad7 on bone morphogenetic protein- and activin-mediated growth arrest and apoptosis in B cells. J. Biol. Chem., 274, 13637–13642. [DOI] [PubMed] [Google Scholar]
- Itoh S., Landström,M., Hermansson,A., Itoh,F., Heldin,C.-H., Heldin, N.-E. and ten Dijke,P. (1998) Transforming growth factor β1 induces nuclear export of inhibitory Smad7. J. Biol. Chem., 273, 29195–29201. [DOI] [PubMed] [Google Scholar]
- Jonk L.J.C., Itoh,S., Heldin,C.-H., ten Dijke,P. and Kruijer,W. (1998) Identification and functional characterization of a Smad binding element (SBE) in the JunB promoter that acts as a transforming growth factor-β, activin, and bone morphogenetic protein-inducible enhancer. J. Biol. Chem., 273, 21145–21152. [DOI] [PubMed] [Google Scholar]
- Kavsak P., Rasmussen,R.K., Causing,C.G., Bonni,S., Zhu,H., Thomson,G.H. and Wrana,J.L. (2000) Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGFβ receptor for degradation. Mol. Cell, 6, 1365–1375. [DOI] [PubMed] [Google Scholar]
- Kawabata M., Imamura T. and Miyazono K. (1998a) Signal transduction by bone morphogenetic proteins. Cytokine Growth Factor Rev., 9, 49–61. [DOI] [PubMed] [Google Scholar]
- Kawabata M., Inoue,H., Hanyu,A., Imamura,T. and Miyazono,K. (1998b) Smad proteins exist as monomers in vivo and undergo homo- and hetero-oligomerization upon activation by serine/threonine kinase receptors. EMBO J., 17, 4056–4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura N., Matsuo,R., Shibuya,H., Nakashima,K. and Taga,T. (2000) BMP2-induced apoptosis is mediated by activation of the TAK1–p38 kinase pathway that is negatively regulated by Smad6. J. Biol. Chem., 275, 17647–17652. [DOI] [PubMed] [Google Scholar]
- Kretzschmar M., Doody,J. and Massagué,J. (1997) Opposing BMP and EGF signalling pathways converge on the TGF-β family mediator Smad1. Nature, 389, 618–622. [DOI] [PubMed] [Google Scholar]
- Lebrun J.J., Takabe,K., Chen,Y. and Vale,W. (1999) Roles of pathway-specific and inhibitory Smads in activin receptor signaling. Mol. Endocrinol., 13, 15–23. [DOI] [PubMed] [Google Scholar]
- Massagué J. (1998) TGF-β signal transduction. Annu. Rev. Biochem., 67, 753–791. [DOI] [PubMed] [Google Scholar]
- Massagué J. and Wotton,D. (2000) Transcriptional control by the TGF-β/Smad signaling system. EMBO J., 19, 1745–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazono K. (2000)Positive and negative regulation of TGF-β signaling. J. Cell Sci., 113, 1101–1109. [DOI] [PubMed] [Google Scholar]
- Nakao A. et al. (1997) Identification of Smad7, a TGFβ-inducible antagonist of TGF-α signalling. Nature, 389, 631–635. [DOI] [PubMed] [Google Scholar]
- Nakayama T., Gardner,H., Berg,L.K. and Christian,J.L. (1998) Smad6 functions as an intracellular antagonist of some TGF-β family members during Xenopus embryogenesis. Genes Cells, 3, 387–394. [DOI] [PubMed] [Google Scholar]
- Nishihara A., Hanai,J.-i., Okamoto,N., Yanagisawa,J., Kato,S., Miyazono,K. and Kawabata,M. (1998) Role of p300, a transcriptional coactivator, in signalling of TGF-β. Genes Cells, 3, 613–623. [DOI] [PubMed] [Google Scholar]
- Nishihara T., Okahashi,N. and Ueda,N. (1993) Activin A induces apoptotic cell death. Biochem. Biophys. Res. Commun., 197, 985–991. [DOI] [PubMed] [Google Scholar]
- Onichtchouk D., Chen,Y.G., Dosch,R., Gawantka,V., Delius,H., Massagué,J. and Niehrs,C. (1999) Silencing of TGF-β signalling by the pseudoreceptor BAMBI. Nature, 401, 480–485. [DOI] [PubMed] [Google Scholar]
- Persson U., Izumi,H., Souchelnytskyi,S., Itoh,S., Grimsby,S., Engström,U., Heldin,C.-H., Funa,K. and ten Dijke,P. (1998) The L45 loop in type I receptors for TGF-β family members is a critical determinant in specifying Smad isoform activation. FEBS Lett., 434, 83–87. [DOI] [PubMed] [Google Scholar]
- Piccolo S., Sasai,Y., Lu,B. and De Robertis,E.M. (1996) Dorsoventral patterning in Xenopus: inhibition of ventral signals by direct binding of chordin to BMP-4. Cell, 86, 589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravanti L., Häkkinen,L., Larjava,H., Saarialho-Kere,U., Foschi,M., Han,J. and Kähäri,V.-M. (1999) Transforming growth factor-β induces collagenase-3 expression by human gingival fibroblasts via p38 mitogen-activated protein kinase. J. Biol. Chem., 274, 37292–37300. [DOI] [PubMed] [Google Scholar]
- Reddi A.H. (1997) Bone morphogenetic proteins: an unconventional approach to isolation of first mammalian morphogens. Cytokine Growth Factor Rev., 8, 11–20. [DOI] [PubMed] [Google Scholar]
- Riggins G.J. et al. (1996) Mad-related genes in the human. Nature Genet., 13, 347–349. [DOI] [PubMed] [Google Scholar]
- Sano Y., Harada,J., Tashiro,S., Gotoh-Mandeville,R., Maekawa,T. and Ishii,S. (1999) ATF-2 is a common nuclear target of Smad and TAK1 pathways in transforming growth factor-β signaling. J. Biol. Chem., 274, 8949–8957. [DOI] [PubMed] [Google Scholar]
- Souchelnytskyi S., ten Dijke,P., Miyazono,K. and Heldin,C.-H. (1996) Phosphorylation of Ser165 in TGF-β type I receptor modulates TGF-β1-induced cellular responses. EMBO J., 15, 6231–6240. [PMC free article] [PubMed] [Google Scholar]
- Souchelnytskyi S., Nakayama,T., Nakao,A., Morén,A., Heldin,C.-H., Christian,J.L. and ten Dijke,P. (1998) Physical and functional interaction of murine and Xenopus Smad7 with bone morphogenetic protein receptors and transforming growth factor-β receptors. J. Biol. Chem., 273, 25364–25370. [DOI] [PubMed] [Google Scholar]
- Takase M., Imamura,T., Sampath,T.K., Takeda,K., Ichijo,H., Miyazono,K. and Kawabata,M. (1998) Induction of Smad6 mRNA by bone morphogenetic proteins. Biochem. Biophys. Res. Commun., 244, 26–29. [DOI] [PubMed] [Google Scholar]
- Tamaki K., Souchelnytskyi,S., Itoh,S., Nakao,A., Sampath,K., Heldin,C.-H. and ten Dijke,P. (1998) Intracellular signaling of osteogenic protein-1 through Smad5 activation. J. Cell Physiol., 177, 355–363. [DOI] [PubMed] [Google Scholar]
- Tanaka N., Kaneko,K., Asao,H., Kasai,H., Endo,Y., Fujita,T., Takeshita,T. and Sugamura,K. (1999) Possible involvement of a novel STAM-associated molecule ‘AMSH’ in intracellular signal transduction mediated by cytokines. J. Biol. Chem., 274, 19129–19135. [DOI] [PubMed] [Google Scholar]
- Topper J.N. et al. (1997) Vascular MADs: two novel MAD-related genes selectively inducible by flow in human vascular endothelium. Proc. Natl Acad. Sci. USA, 94, 9314–9319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulloa L., Doody,J. and Massagué,J. (1999) Inhibition of transforming growth factor-β/SMAD signalling by the interferon-γ/STAT pathway. Nature, 397, 710–713. [DOI] [PubMed] [Google Scholar]
- Wozney J.M. (1998) The bone morphogenetic protein family: multifunctional cellular regulators in the embryo and adult. Eur. J. Oral Sci., 106 (Suppl.), 160–166. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Feng,X.-F., We,R. and Derynck,R. (1996) Receptor-associated Mad homologues synergize as effectors of the TGF-β response. Nature, 383, 168–172. [DOI] [PubMed] [Google Scholar]
- Zhu H.-J., Iaria,J. and Sizeland,A.M. (1999) Smad7 differentially regulates transforming growth factor β-mediated signaling pathways. J. Biol. Chem., 274, 32258–32264. [DOI] [PubMed] [Google Scholar]
- Zimmerman L.B., De Jesus-Escobar,J.M. and Harland,R.M. (1996) The Spemann organizer signal noggin binds and inactivates bone morphogenetic protein 4. Cell, 86, 599–606. [DOI] [PubMed] [Google Scholar]