

Abstract
Peptides derived from heptad repeat regions adjacent to the fusion peptide and transmembrane domains of many viral fusion proteins form stable helical bundles and inhibit fusion specifically. Paramyxovirus SV5 fusion (F) protein-mediated fusion and its inhibition by the peptides N-1 and C-1 were analyzed. The temperature dependence of fusion by F suggests that thermal energy, destabilizing proline residues and receptor binding by the hemagglutinin–neuraminidase (HN) protein collectively contribute to F activation from a metastable native state. F-mediated fusion was reversibly arrested by low temperature or membrane-incorporated lipids, and the resulting F intermediates were characterized. N-1 inhibited an earlier F intermediate than C-1. Co-expression of HN with F lowered the temperature required to attain the N-1-inhibited intermediate, consistent with HN binding to its receptor stimulating a conformational change in F. C-1 bound and inhibited an intermediate of F that could be detected until a point directly preceding membrane merger. The data are consistent with C-1 binding a pre-hairpin intermediate of F and with helical bundle formation being coupled directly to membrane fusion.
Keywords: membrane fusion/paramyxovirus/peptide inhibition/viral entry/viral fusion glycoprotein
Introduction
Fusion of the membrane of enveloped viruses with cellular membranes is a prerequisite for viral entry and is thought to include the following events: (i) receptor binding by the viral attachment protein; (ii) activation of the viral fusion protein; (iii) insertion of a hydrophobic fusion peptide into the target membrane; (iv) refolding of the fusion protein; and (v) membrane merger and fusion pore dilation (reviewed in Hernandez et al., 1996; Skehel and Wiley, 2000). The viral fusion-mediating glycoproteins (fGps) from influenza virus (HA), HIV/SIV (gp160), retroviruses (Env) and paramyxoviruses (F) share several structural and functional features. These fGps are synthesized as single polypeptide chains that form trimers before being cleaved by host cell proteases to yield membrane-distal and membrane-anchored subunits (for paramyxoviruses, the F0 precursor is cleaved to F1+F2; see Figure 1). The new N-terminal region of the membrane-anchored subunit, termed the fusion peptide, contains hydrophobic residues, which insert into target membranes during fusion (reviewed in Hernandez et al., 1996). The ectodomains of many of the membrane-anchored subunits also contain two conserved 4-3 heptad repeat (HR) regions, designated HRA and HRB, which are located near the fusion peptide and the transmembrane (TM) domains. These HR regions refold into helical bundles with HRA forming an interior, trimeric coiled coil and HRB binding in the grooves of HRA in an anti-parallel fashion, resulting in the fusion peptides and TM domains being proximal (reviewed in Skehel and Wiley, 2000). The similarities in the structures of the core trimers suggest a common fusion mechanism (Skehel and Wiley, 1998). However, unresolved issues include the individual mechanisms of fGp activation, the characteristics of fusion protein intermediates, and the relationship between helix bundle formation and membrane fusion.

Fig. 1. Schematic diagram of the SV5 F protein. The positions of the signal sequence (ss), cleavage site (*), fusion peptide, heptad repeats A and B (HRA and HRB), transmembrane domain (TM) and cytoplasmic tail are shown. The locations of the N-1 and C-1 peptides within the domain structure of F are indicated.
The triggering events that initiate membrane fusion by different viruses vary substantially for different fGps. For example, cleaved HA is thought to be in a metastable state, which is triggered by exposure to an acidic environment to refold into its lowest energy state (Ruigrok et al., 1986; Carr et al., 1997). HIV gp41 is triggered at neutral pH by gp120 binding to the cell surface proteins CD4 and a chemokine co-receptor (Moore et al., 1990; Furuta et al., 1998). The triggering mechanism of paramyxovirus F at neutral pH is not clear. One model for a tightly regulated trigger for paramyxovirus fusion is that, upon binding sialic acid, the hemagglutinin–neuraminidase (HN) glycoprotein undergoes a receptor-induced conformational change, which in turn triggers a conformational change in F (Lamb, 1993; Sergel et al., 1993). For many paramyxoviruses, the F proteins only cause syncytium formation when co-expressed with their homotypic HN protein, and a type-specific interaction required for fusion may occur between the HN and F proteins (reviewed in Lamb and Kolakofsky, 2001). In contrast, the SV5 F protein causes syncytia formation in the absence of its homotypic HN protein (Paterson et al., 1985). Moreover, SV5 F mediates fusion using uncleaved HA as a binding protein in fusion assays that require two different cell types to be bound together (Paterson et al., 2000). For paramyxovirus F proteins that do not absolutely require HN co-expression for fusion, an F conformational change may be triggered by contact with the target membrane or by binding to an unidentified F receptor (Lamb, 1993).
Following activation, fGps may form intermediate conformations before and during membrane fusion. Triggering of HA, Env and F has been shown to result in the insertion of the fusion peptides into target membranes (Novick and Hoekstra, 1988; Tsurudome et al., 1992; Hernandez et al., 1997; Damico et al., 1998). For the paramyxovirus respiratory syncytial virus, storage of soluble F may trigger the release of the fusion peptide as a new morphology is observed by electron microscopy (Calder et al., 2000). Peptides derived from the HRA and HRB regions (designated N and C peptides, respectively) have been used to inhibit intermediates of HIV gp41 and paramyxovirus F (reviewed in Chan and Kim, 1998; Lamb and Kolakofsky, 2001). For example, after a receptor-activated conformational change, C peptides derived from HIV gp41 inhibit membrane fusion in a dominant-negative manner by binding the HRA regions of pre-hairpin intermediates of gp41 (Chan et al., 1998; Furuta et al., 1998). The N and C peptides are thought to inhibit fusion by binding analogous regions in fGp intermediates, and thereby preventing the proteins from forming helical bundles required for fusion.
After fusion protein activation and the formation of fusion protein intermediates, many viral fusion proteins refold into a six-helix bundle structure. However, the role of helix bundle formation in membrane fusion is not clear. The helical bundles may bring the cellular and viral membranes in proximity, with higher order clustering of fusion protein trimers facilitating membrane fusion (Chernomordik et al., 1998). Alternatively, helix bundle formation may be directly coupled to membrane fusion, with the energy released upon protein refolding being used to perform the work of membrane fusion (Baker et al., 1999; Melikyan et al., 2000). The distinguishing features between the above models are the temporal relationship between six-helix bundle formation and membrane merger. Recent experiments on the peptide inhibition of HIV gp41-mediated fusion have suggested that N and C peptides inhibit a fusion intermediate that directly precedes membrane merger, consistent with the formation of the six-helix bundle structure in HIV gp41 occurring concurrently with membrane merger (Melikyan et al., 2000).
Here we describe several determinants of the activation of paramyxovirus SV5 F protein and characterize fusion intermediates captured by peptide inhibitors. Co-expression of HN with F promotes the formation of an F intermediate susceptible to N-1 inhibition, suggesting that receptor binding by HN induces a conformational change in F. The data are also consistent with C-1 binding a transient pre-hairpin intermediate of F and inhibiting fusion by preventing six-helix bundle formation, which may be directly coupled to membrane fusion.
Results
HN co-expression contributes to fusion by SV5 W3A strain F
The SV5 WR strain F protein requires co-expression of its homotypic HN protein for syncytia formation (Ito et al., 1997), whereas the SV5 W3A strain F protein does not (Paterson et al., 1985; Horvath et al., 1992). The amino acid sequences of the F proteins from the WR and W3A strains differ at positions 22, 443 and 516 (Paterson et al., 1984; Ito et al., 1997). While the identity of the amino acid residue at position 516 has been shown to have minimal effects on cell–cell fusion, proline residues at positions 22 and 443 appear to contribute to faster fusion kinetics, a lower temperature required for fusion activation, and HN-independent fusion (Ito et al., 1997; Paterson et al., 2000). To study further the contribution of HN co-expression to membrane fusion mediated by W3A F, the temperature dependence of fusion was measured for W3A F co-expressed either with its homotypic HN or with influenza virus HA, which served as a binding protein but did not mediate fusion. Target erythrocytes (RBCs) co-labeled with the lipidic dye octadecyl rhodamine B chloride (R18) and the aqueous dye 6-carboxyfluorescein (CF) were bound at 4°C to effector CV-1 cells expressing F and either HN or HA. At 4°C, neither lipid mixing nor contents mixing occurred for any of the F mutant samples. The effector–target cell complexes were then incubated at various temperatures for 5 min, and the extents of R18 and CF dye transfer were measured by confocal microscopy. For all of the samples, the extents of lipid mixing versus contents mixing did not differ significantly (data not shown). HN co-expression decreased the temperature required to activate F for fusion and increased the extent of F-mediated fusion (Figure 2A). Thus, while not absolutely required for W3A F-mediated fusion, HN co-expression clearly promotes F-mediated fusion. The contribution of HN co-expression to the temperature dependence of fusion mediated by a cleavage mutant of W3A F (designated FR3), which was used in subsequent peptide inhibition studies (see below), was also determined. In the FR3 cleavage mutant, the furin consensus sequence was converted to a cleavage site that is only cleaved and biologically activated for fusion by addition of exogenous trypsin (Paterson et al., 1989). FR3 was co-expressed with either HN or HA, and FR3 was activated by trypsin cleavage immediately before beginning the dye transfer assays. Cleaved FR3 (W3A) required less thermal energy to mediate fusion in the presence of its homotypic HN (Figure 2B), indicating that HN also promotes W3A F-mediated fusion in the context of the cleaved FR3 protein.

Fig. 2. Temperature dependence of fusion by the F protein of SV5 strain W3A co-expressed with either SV5 HN (open squares) or influenza virus HA (closed squares). R18/CF double-labeled RBCs were bound at 4°C to CV-1 cells co-expressing the F proteins and either HN or HA. The samples were subsequently incubated at the reported temperatures for 5 min. Dye transfer was assayed by confocal microscopy. Fusion is expressed as contents mixing events per microscopic field, averaged over 4–6 fields. Lipid mixing (R18 transfer) and contents mixing (CF transfer, reported here) were coincident. (A) W3A F-mediated fusion is promoted by HN co-expression. (B) Fusion mediated by cleaved W3A strain FR3 (the cleavage site mutant) is also promoted by HN co-expression. Expression of HN, uncleaved HA or cleaved HA alone did not cause detectable fusion, and F+HN and F+HA fusion were inhibited by addition of the F-specific antibody F1a.
N-1 inhibits an earlier intermediate than C-1
The results of several studies on the modes of fusion inhibition by HR peptides from paramyxoviruses are inconsistent. The N-1 peptide derived from SV5 F has been reported to inhibit membrane fusion at the hemifusion stage (Joshi et al., 1998), whereas an N peptide derived from Newcastle disease virus (NDV) F only inhibited subsequent fusion if the peptide was added prior to proteolytic cleavage of F0 into F1 and F2 subunits (Young et al., 1999). Moreover, a preformed complex of N and C peptides derived from Sendai virus F has been reported to dissociate in the presence of phospholipid vesicles, and the peptides have been reported to partition preferentially into vesicles over the aqueous phase (Ben-Efraim et al., 1999). To study further fusion inhibition by the N-1 and C-1 peptides derived from SV5 F, a series of experiments was performed to determine the timing and binding mode of peptide inhibition.
The SV5 F-derived HR peptides inhibited cell–cell fusion when present during the entirety of incubation of target cells with effector cells expressing W3A F and HN, with N-1 and C-1 having IC50 values of ∼16 µM and 35 nM, respectively (data not shown). To study the chronology of fusion inhibition, the cleavage mutant FR3 was co-expressed with HN, and the transfer of both lipidic (R18) and aqueous (CF) dye from co-labeled RBCs was used as a readout for fusion. In the assay, either N-1 or C-1 was incubated with effector cells at four distinct stages of the fusion assay: (i) before F0 cleavage; (ii) after F0 cleavage but before target cell binding; (iii) during target cell binding at 4°C; or (iv) during 37°C incubation. N-1, but not C-1, inhibited cell–cell fusion mediated by effector cells expressing F and HN, if present during target cell binding at 4°C (Figure 3C). These data are consistent with N-1 inhibiting a fusion intermediate that evolves earlier and has a lower thermal energy requirement than the intermediate that C-1 inhibits. Neither N-1 nor C-1 inhibited fusion when present either before or after cleavage (data not shown), or when present at times both before and also after cleavage (Figure 3B). Both peptides inhibited fusion if added at the beginning of the 37°C incubation (Figure 3D). N-1 and C-1 inhibited both lipid and contents mixing simultaneously if they inhibited at all. Previous work reported that N-1 inhibited contents mixing measured by β-galactosidase expression in one assay, but did not inhibit lipid mixing measured by R18 dye transfer in a different assay (Joshi et al., 1998). The most likely explanation for the discrepancy is that the previous study performed separate assays for lipid and contents mixing, and N-1 has a tendency to aggregate after freezing and thawing and after long-term storage at 4°C (C.J.Russell, unpublished observations). Light scattering measurements showed that N-1 samples used in the present study were not aggregated (data not shown), and dye transfer assays using co-labeled RBCs showed that N-1 inhibited both lipid and contents mixing.

Fig. 3. Inhibition of cell–cell fusion by N-1 and C-1. CV-1 cells co-expressing SV5 HN and FR3 were incubated with TPCK–trypsin for 1 h to cleave the F0 precursor to the F1 and F2 subunits. RBCs co-labeled with R18 (red) and CF (green) were bound to the CV-1 cells at 4°C for 1 h. The samples were incubated at 37°C for 10 min, re-incubated at 4°C and analyzed for dye transfer by confocal microscopy. Either the N-1 or C-1 peptides were incubated with the samples at the following stages of the assay: (A) no peptide at any stage; (B) peptide before and after cleavage; (C) peptide during RBC binding at 4°C; and (D) peptide during the 37°C incubation. The samples were washed three times with PBS between each stage. The peptide concentrations were 40 µM.
HN co-expression promotes N-1 inhibition at low temperature
To study the role of HN in formation of the F protein intermediate susceptible to N-1 peptide inhibition at low temperature, peptide order-of-addition experiments were performed using effector CV-1 cells co-expressing W3A F and either HN or uncleaved HA. The peptides N-1 or C-1 were added to samples during incubation at either 4, 15 or 37°C. N-1 inhibited subsequent fusion if it was added to the effector cells co-expressing F and HN during the 4, 15 or 37°C incubations (Figure 4). Moreover, the fusion inhibition by N-1 occurred to a greater extent at increasing incubation temperatures, consistent with a thermal requirement for the formation of an F intermediate susceptible to N-1 inhibition. In contrast, N-1 added to effector cells co-expressing F and HA during the 4°C incubation did not inhibit fusion, and N-1 added at 15°C had little inhibitory effect (Figure 4C). N-1 inhibited cell–cell fusion in cells co-expressing F and HA if added during the 37°C incubation, indicating that the formation of an intermediate susceptible to N-1 does not absolutely require HN co-expression. C-1 inhibited both HN+F and HA+F fusion only if present during the 37°C incubation, most likely by binding a later intermediate of F than the F intermediate with which N-1 presumably interacts (see below). The inhibition by N-1 and C-1 of fusion mediated by W3A F in the absence of HN implies that, upon activation, W3A F proceeds through all of the intermediates in the absence of HN co-expression, albeit with higher incubation temperatures required.

Fig. 4. Quantification of N-1 and C-1 inhibition of fusion mediated by SV5 W3A F. The conditions of the assay were similar to those in Figure 3 except that cleavage by TPCK–trypsin was not needed for W3A F and the samples were incubated at 15°C for 30 min between the 4°C binding of RBCs for 1 h and the 37°C incubation for 10 min. (A) Timeline of peptide order-of-addition experiment. Arrows denote the addition of peptide at either primary, secondary or tertiary stages. Samples were washed three times with PBS before each temperature change. (B) For cell–cell fusion by W3A F co-expressed with HN, N-1 inhibits fusion if present during either the 4, 15 or 37°C incubations, and is a more potent inhibitor as the incubation temperature is increased. (C) For cell–cell fusion by W3A F co-expressed with uncleaved influenza virus HA, N-1 does not inhibit fusion at 4°C, is a mild inhibitor of fusion at 15°C, and inhibits if present at 37°C. C-1 inhibits both HN+F and HA+F fusion only after thermal activation at 37°C. The peptide concentrations were 40 µM. The means and standard errors shown are for contents mixing from 3–5 fields.
The F protein intermediate inhibited by C-1 binds target cells independently of HN
When RBCs are bound to cells expressing SV5 HN at 4°C and are warmed to 37°C, most RBCs are released (Dutch et al., 1998). The loss of RBC binding activity by HN at 37°C may be due to either the intrinsic neuraminidase activity of HN or a decrease in the affinity of HN for sialic acid as temperature is increased (R.Paterson, unpublished). The number of bound but unfused RBCs (R18 labeled) was counted after 15 min of 37°C incubation of N-1 or C-1 with effector CV-1 cells (SYTO 11 labeled) expressing HN and F (Figure 5). Cells expressing HN without F did not promote fusion and displayed a substantial loss of RBC binding after incubation at 37°C. After 37°C incubation in the presence of N-1, fusion was inhibited and most of the RBCs were released from the effector cells. In contrast, C-1 inhibited fusion while simultaneously promoting retention of unfused RBCs, suggesting that the F protein intermediate inhibited by C-1 had acquired the ability to interact with target cells directly. The interaction between the F protein and target cells may be due either to binding of F to an unidentified receptor or to the insertion of the fusion peptide region of F into target membranes. If C-1 inhibits fusion by binding to a coiled coil formed by HRA regions of F (see below), then it is possible that the fusion peptide regions adjacent to the central coiled coil could extend the fusion peptides into target membranes. Activated intermediates of influenza virus HA (Chernomordik et al., 1997) and retrovirus Env (Hernandez et al., 1997; Damico et al., 1998) have been shown to interact directly with target membranes, presumably due to insertion of the fusion peptide regions into target membranes.

Fig. 5. Binding and fusion of RBCs to CV-1 cells expressing W3A F and HN. (A) Confocal images of RBCs labeled with R18 (red) and CV-1 cells labeled with SYTO 11 (green). (B) Quantification of RBC binding (black bars) and the number of lipidic dye transfer events (white bars) after 15 min of 37°C incubation. The means and standard errors shown are for three trials.
C-1 binds a transient intermediate of F
C peptides derived from gp41 appear to inhibit HIV fusion by binding to HRA region trimers exposed in a transient pre-hairpin intermediate of gp41 after receptor/co-receptor binding (reviewed in Chan and Kim, 1998; Furuta et al., 1998). To test the conditions under which the C-1 peptide might bind directly to SV5 F, a modified C-1 peptide designated HAt-C1 was synthesized which contained an 11 residue HA tag (YPYDVPDYASL) at the N-terminus of C-1. Like C-1, HAt-C1 inhibited SV5 F-mediated fusion only if present during 37°C incubation of effector and target cells, and had an IC50 of ∼50 nM (data not shown). The HA tag was used for co-immunoprecipitation (coIP) of the F protein using the 12CA5 tag-specific antibody. The F protein was only captured by coIP if HAt-C1 was incubated with the effector cells after target cell binding during a 37°C incubation (Figure 6A). Thus, the HAt-C1 peptide only bound F under the same conditions in which it inhibited fusion. Increasing the amount of HAt-C1 incubated with the samples increased the amount of coIP of F, and the coIP of F was competed away by adding an excess of the untagged C-1 peptide (Figure 6B). To test the notion that paramyxovirus C-1-like peptides partition into membranes (Ben-Efraim et al., 1999), C-1 stock solution was pre-incubated with either RBCs, CV-1 cells or Vero cells. After 1 h incubation at 37°C, the cells were pelleted by centrifugation and the supernatants were collected. The supernatants subsequently inhibited cell–cell fusion with slightly greater IC50 values than C-1 that was not pre-incubated with cells, with >90% of the C-1 peptide remaining in aqueous solution after pre-incubation with the cells (data not shown). Thus, there is no evidence to suggest that C-1 prevents fusion by inhibiting lipidic intermediates rather than binding F protein intermediates.
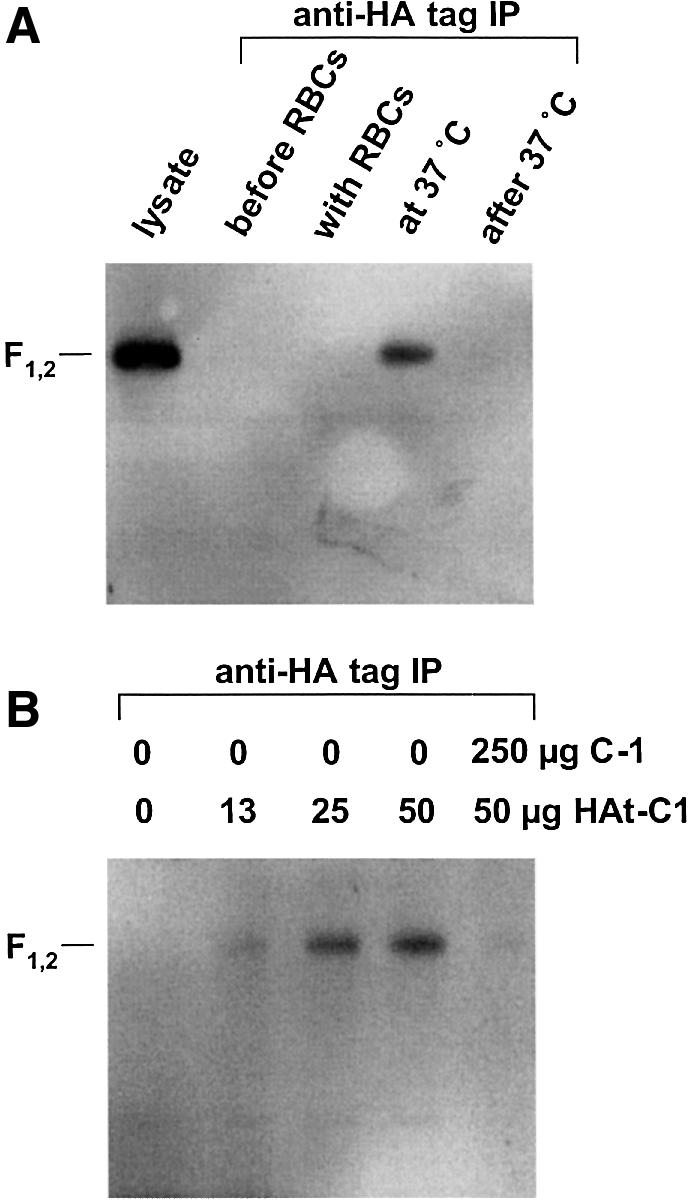
Fig. 6. HA-tagged C-1 peptide (HAt-C1) binds F under the same conditions that it inhibits cell–cell fusion. HAt-C1 inhibited fusion only when added to F-expressing cells bound to target cells during 37°C incubation (data not shown). (A) Lane 1, whole-cell lysate of Vero cells expressing SV5 W3A and HN. Lanes 2–5, immunoprecipitation with the HA tagged peptide HAt-C1. Intact Vero cells transiently expressing F and HN were incubated with human RBCs at 4°C, and the Vero–RBC complexes were subsequently warmed to 37°C. The HAt-C1 peptide was incubated with samples either before RBC incubation, during RBC incubation, during the 37°C incubation or after the 37°C incubation. The F protein was co-immunoprecipitated using the 12CA5 antibody against the HA tag on the peptide. The samples were electrophoresed under non-reducing conditions by SDS–PAGE and a western blot was performed using a rabbit polyclonal peptide antiserum against F2. (B) Binding of HAt-C1 to F is dose dependent and can be competed by an excess of untagged C-1 peptide.
Six-helix bundle formation and membrane merger
To determine the chronological relationship between six-helix bundle formation and membrane merger, experiments were performed to probe for C-1 binding (which occurs prior to bundle formation) to a fusion intermediate arrested at a point directly preceding membrane merger. Membrane-incorporated lipids can promote or inhibit fusion in a manner that correlates with their dynamic molecular shapes either by favoring or preventing the formation of membrane stalk intermediates of fusion (Chernomordik et al., 1997). In the present work, fusion was reversibly arrested by addition of the membrane-binding lipid stearoyl-lysophosphatidylcholine (LPC), which has been shown to inhibit fusion at the point of membrane merger (Chernomordik et al., 1998). The resulting LPC-arrested fusion intermediates were probed for C-1 inhibition. The rationale behind the experiment (Melikyan et al., 2000) was that if six-helix bundle formation was concurrent with membrane merger, then the LPC-arrested intermediate may remain sensitive to C-1; however, if six-helix bundle formation occurred before membrane merger, then the binding site for C-1 would no longer be available and the LPC-arrested intermediate would lose sensitivity to C-1.
SV5 F-mediated fusion was inhibited by LPC and promoted by oleic acid (OA) (Figure 7A), as found previously for fusion mediated by influenza virus HA and HIV gp41 (Chernomordik et al., 1997; Melikyan et al., 2000). After one round of incubation at 37°C in the presence of LPC for 10 min, a 4°C washout of LPC using bovine serum albumin (BSA), and a second round of incubation at 37°C for 10 min, R18 and CF dye transfer from RBCs to effector cells was observed (Figure 7B, first bar pair). When the N-1 or C-1 peptides were added to the LPC-arrested samples at the beginning of the first stage of 37°C incubation, subsequent fusion during the second stage of heating was inhibited (Figure 7B, second and third bar pairs). C-1 also inhibited fusion if added to the samples at 4°C after the first stage of 37°C incubation, but before LPC removal; however, N-1 lost its inhibitory potency after the first stage of heating (Figure 7B, fifth and fourth bar pairs, respectively). The data suggest that C-1 bound F and inhibited fusion up until the point of the LPC arrest directly preceding membrane merger. In contrast, by the time the system evolved to the LPC-arrested stage at 37°C, N-1 lost its inhibitory activity. Membrane fusion may require the concerted action of multiple F oligomers. At the stage of LPC arrest, it is possible that a sub-threshold amount of F molecules may be completely refolded, while others that are not may be sufficient for C-1 inhibition. In another experiment, either N-1 or C-1 was added at various times during the first stage of 37°C incubation for 15 min in the presence of LPC (Figure 7C). C-1 bound F and inhibited fusion if added at any time during the 37°C incubation. C-1 also inhibited both lipid and contents mixing if added after LPC removal at 4°C (data not shown). However, N-1 lost the ability to inhibit fusion as a function of incubation time at 37°C. These data are consistent with N-1 inhibiting an earlier intermediate of the F protein than C-1, and with C-1 binding F during the LPC arrest directly preceding membrane merger. If C-1 binds F before six-helix bundle formation at a point directly preceding membrane merger, then six-helix bundle formation and membrane merger may be concurrent events.

Fig. 7. Reversible arrest of late fusion intermediates using membrane-incorporated lipids. To prevent the release of RBCs from CV-1 cells during 37°C incubations, uncleaved influenza HA was co-expressed with SV5 W3A F. Insets depict timelines of conditions. The means and standard errors are for three trials. White bars denote R18 lipid mixing and black bars denote CF contents mixing. (A) LPC (10 µM) inhibits fusion, OA (10 µM) promotes lipid mixing and an equimolar amount of OA can restore fusion inhibited by LPC (10 µM each). (B) RBC–CV-1 cell complexes were incubated at 37°C in the presence of LPC, washed with a 4°C BSA solution to remove LPC, and warmed a second time at 37°C. Arrows in the inset denote primary and secondary addition of peptide. Bar pairs represent R18 and CF data for each condition. (C) Similar conditions to those reported in (B), except that peptide was added as a function of time after the onset of the first stage of 37°C incubation (arrows in the inset denote addition of peptide). (D) Two models of the relationship between six-helix bundle formation and membrane fusion. Consistent with model (ii), the data suggest that six-helix bundle formation and membrane fusion are concurrent events.
Discussion
Several critical events involved in paramyxovirus membrane fusion are F protein precursor (F0) cleavage, HN binding to target cells, activation of F for fusion and F-mediated fusion (Figure 8). Most paramyxovirus F proteins require co-expression of their homotypic HN protein for fusion, and some biochemical data suggest that protein– protein interactions occur between the HN and F proteins (reviewed in Lamb and Kolakofsky, 2001). In addition, the recently solved crystal structures of HN from the paramyxovirus NDV suggest that the catalytic site for both hemagglutinating and neuraminidase activities is a single site, which is regulated by a conformational switch, and that HN undergoes a limited but significant conformational change upon binding sialic acid (Crennell et al., 2000). Such conformational changes in HN due to receptor binding may then be translated into the triggering of a conformational change of F. The present data for paramyxovirus SV5 W3A strain F-mediated fusion are consistent with the binding of HN to sialic acid promoting a conformational change in F, since co-expression of HN with F promoted the formation of an F intermediate susceptible to N-1 inhibition at 4°C. However, a universal model of the triggering mechanism for paramyxovirus fusion must reconcile the anomalous observation that W3A F can also promote membrane fusion in the absence of its homotypic HN (Paterson et al., 1985).

Fig. 8. Model of the paramyxovirus fusion mechanism. Four steps in SV5 F-mediated fusion are F0 cleavage, HN binding to target cells, activation of F for fusion and F-mediated fusion. The N-1 peptide inhibits a temperature-arrested intermediate that forms after the binding of HN (but not uncleaved HA) to target cells, consistent with the binding of HN to its receptor sialic acid stimulating a conformational change in F. The N-1 binding site becomes unavailable as a function of time after thermal activation. Plausible binding modes of N-1 to F regions are binding HRB as trimers or HRA as monomers/dimers. C-1 binds and inhibits a pre-hairpin intermediate of F that forms after thermal activation and remains until a point directly preceding membrane merger, as probed by LPC arrest. The data presented here suggest that six-helix bundle formation and membrane fusion are concurrent events. F domains: fusion peptide (black); HRA/N-1 (red); intervening domain (gold); HRB/C-1 (blue); TM domain (gray). As F2 and F1 are structurally integral parts of the F protein, these subunits are not shown separately.
Previous studies have shown that temperature can be used as an artificial stimulus to trigger fusion by paramyxovirus F (Paterson et al., 2000; Wharton et al., 2000). Moreover, proline residues at positions 22 and 443 of SV5 F contribute to HN-independent fusion by SV5 W3A strain F (Paterson et al., 2000). The present data show that co-expression of SV5 HN promotes both the formation of the N-1-susceptible intermediate in F and membrane fusion mediated by W3A F. Taken together, the data on SV5 F-mediated fusion show that thermal energy, proline residues at positions 22 and 443 of SV5 F, and co-expression of SV5 HN have an additive effect on the activation of F for fusion, perhaps by lowering a kinetic barrier to refolding F from a metastable, native state into a fusion-active conformation. Further evidence that mutations of critical residues in paramyxovirus F can result in HN-independent fusion activation has been obtained by mutational analyses of the WR and T1 strains of SV5 (Ito et al., 2000) and the AV strain of NDV (Sergel et al., 2000). Cleavage of paramyxovirus F0 into F1 and F2 subunits is required for fusion, and results in both a conformational change and exposure of a new hydrophobic region (reviewed in Dutch et al., 2001; Lamb and Kolakofsky, 2001). In the present work, cleavage of F0 in the absence of activation by target cell binding did not result in the binding of inhibitory peptides to F or the acquisition of HN-independent binding of RBCs by F. While cleavage of F is an essential step in the fusion mechanism of paramyxoviruses, we do not predict that F cleavage alone triggers full activation of F for fusion when co-expressed with HN, although cleavage of F in the absence of HN co-expression may generate a lower-energy conformation.
Peptides derived from conserved HR regions of fGps of various paramyxoviruses and HIV are potent and specific inhibitors of membrane fusion (reviewed in Chan and Kim, 1998; Lamb and Kolakofsky, 2001). Moreover, many of these N and C peptides co-assemble into highly thermostable six-helix bundles, which are thought to correspond to the core structures formed by the fGps in fusogenic or post-fusion states (Chan et al., 1997; Weissenhorn et al., 1997; Baker et al., 1999; Zhao et al., 2000). The peptides most likely inhibit fusion by binding to regions of the glycoproteins analogous to those observed in the crystal structures of the peptide complexes, with the C peptides binding HRA region trimers and the N peptides binding either as trimers to isolated HRB regions or as monomers/dimers to isolated HRA regions. The intermolecular binding of the peptides to the HR regions could be favored over intramolecular association of the two HR regions if fGp intermediates form in which the HR regions are accessible to exogenous peptide binding but not to intra-trimer assembly. For example, C peptides derived from HIV gp41 have been shown to gain their inhibitory activity by binding a pre-hairpin intermediate that forms after receptor-activated conformational changes (reviewed in Chan and Kim, 1998), but before six-helix bundle formation (Melikyan et al., 2000).
The present data suggest that the N-1 and C-1 peptides derived from SV5 F have a mechanism of action similar to those derived from HIV gp41. In contrast to data obtained in peptide inhibition studies on NDV F-mediated fusion (Young et al., 1999), neither N-1 nor C-1 from SV5 F inhibited fusion if present before or after F0 cleavage but before binding of target cells. Both N-1 and C-1 inhibited fusion intermediates that became susceptible to peptide binding after target cell binding (N-1) followed by thermal activation (N-1 and C-1), with N-1 inhibiting an earlier intermediate of the F protein than C-1. The F protein intermediate inhibited by C-1 developed HN-independent RBC binding and remained until the LPC arrest directly preceding membrane fusion. The following results are consistent with C-1 inhibiting fusion by binding to transiently accessible HRA trimers of an SV5 F pre-hairpin intermediate (see Figure 8): (i) C-1 specifically inhibits SV5 F-mediated fusion (Joshi et al., 1998); (ii) C-1 specifically binds in the grooves formed by N-1 trimers (Baker et al., 1999); (iii) the HA-tagged version of C-1 captures F under the same conditions as it inhibits F-mediated fusion; (iv) C-1 partitions predominantly into the aqueous phase after stable exposure to biological cells; and (v) the C-1-inhibited state acquires HN-independent binding of RBCs, perhaps by insertion of fusion peptide regions into target membranes. In contrast to SV5 F-derived N-1 losing inhibitory potency after thermal activation during LPC arrest, an N peptide derived from HIV gp41 inhibited gp41-mediated fusion after thermal activation until a point directly preceding membrane merger, as assessed by LPC arrest and temperature-jump experiments (Melikyan et al., 2000). Thus, the refolding of SV5 F after triggering may differ somewhat from the refolding by HIV gp41, at least with respect to the accessibility of N peptide binding sites in late fGp intermediates. The loss of SV5 N-1 inhibition as the system evolved toward LPC arrest may suggest that monomeric N-1 may bind to HRA regions of F that would become inaccessible upon pre-hairpin intermediate formation, if the binding of oligomeric N-1 to HRB were available until the stage of membrane merger (Figure 8).
Accumulating evidence suggests that six-helix bundle formation by SV5 F is directly coupled to membrane merger. HRA is contiguous with the fusion peptide region, and both regions have a 4-3 repeat of conserved residues that align in register on a helical net plot (Baker et al., 1999). Moreover, the seven-residue sequence between HRB and the TM domain has been shown to be dispensable for fusion activity (Zhou et al., 1997; Baker et al., 1999). The result is a protein conformation in which the conserved HRA and HRB regions directly abut the fusion peptide and TM regions, respectively. The membrane-binding lipid LPC reversibly arrests fusion at a point directly preceding membrane merger by preventing membrane stalk formation (Chernomordik et al., 1997). As C-1 apparently binds transiently exposed HRA trimers in a pre-hairpin intermediate of F and inhibits fusion up until the point directly preceding membrane merger during LPC arrest (see above and Figure 7), the six-helix bundle would not have formed during the LPC arrest. As a result, six-helix bundle formation and membrane merger are most likely concurrent, and the energy required for the work of membrane merger may at least in part be contributed by six-helix bundle formation. The SV5 F peptide complex has a melting temperature >90°C (Joshi et al., 1998), suggesting that a large amount of free energy may be released upon six-helix bundle formation. The free energy released by the formation of a six-helix bundle by HIV gp41-derived peptides has been estimated to be more than sufficient to account for the energy required for membrane fusion (Melikyan et al., 2000).
The formation of coiled coils in HIV gp41 and SNARE complexes may also directly cause membrane fusion. DP178, a C peptide from HIV gp41, inhibits membrane fusion by binding a pre-hairpin intermediate up until a point directly preceding membrane merger, as probed by LPC arrest and temperature-jump experiments (Melikyan et al., 2000). The results are consistent with six-helix bundle formation by HIV gp41 being the rate-limiting step between the arrested intermediate and fusion, and with the free energy released upon bundle formation being directly and immediately used for fusion pore formation. SNARE proteins are involved in eukaryotic intracellular membrane fusion (reviewed in Jahn and Südhof, 1999). Vesicular and target SNARE proteins have been shown to form parallel, heterotrimeric four-helix bundles (Poirier et al., 1998; Sutton et al., 1998), which may mediate membrane fusion by coupling v- and t-SNARE complex formation directly to membrane merger (Chen et al., 1999; Parlati et al., 1999). If SV5 F, HIV gp41 and SNARE proteins all share similar structural architectures and functional mechanisms, then these diverse fusion proteins may either have arisen from a common ancestor or developed a convergent strategy for eliciting membrane merger: coupling coiled coil formation directly to membrane fusion.
Materials and methods
Peptides, cells, viruses and plasmids
The amino acid sequences of the N-1 and C-1 peptides have been reported previously (Joshi et al., 1998). N-1 was expressed and purified as described previously (Joshi et al., 1998). Purified C-1 and HAt-C1 were obtained commercially (Genemed Synthesis, Inc., San Francisco, CA). Monolayer cultures of Vero cells and CV-1 cells were grown as described (Paterson et al., 2000). The recombinant vaccinia virus vTF7-3 was grown in CV-1 cells and the plasmids used have been described previously (Paterson et al., 2000).
Expression and quantification of F, HN and HA proteins
For confocal microscopy, the F, HN and HA proteins were expressed using the recombinant vaccinia virus–T7 RNA polymerase transient expression system (Paterson et al., 2000). For flow cytometry, monolayers of CV-1 cells in 60-mm dishes infected with vTF7-3 and transfected with plasmid DNA were prepared (Paterson et al., 2000). The monoclonal antibody F1a, specific for the SV5 F protein (Randall et al., 1987), was used as the primary antibody, and fluorescein isothio cyanate-conjugated goat anti-mouse immunoglobulin G (Jackson ImmunoResearch Laboratories Inc., West Grove, PA) as the secondary antibody. The cell surface fluorescence of 10 000 cells was analyzed using a FACSCalibur flowcytometer (Becton Dickinson, San Jose, CA).
Analysis of lipid and contents mixing by confocal microscopy
Human RBCs were single labeled with the lipid probe R18 (Molecular Probes, Eugene, OR) or dual labeled with R18 and CF (Molecular Probes) (Paterson et al., 2000). Analysis of lipid and contents mixing for effector cells expressing F and HN or F and HA by scanning confocal microscopy was performed as described previously (Paterson et al., 2000). Fusion was quantified by counting positive events of R18 or CF dye transfer from RBCs to CV-1 cells and averaging the fusion events from 3–5 fields. For the peptide order-of-addition experiments, either N-1 or C-1 (40 µM) was incubated with the CV-1 effector cells either before trypsin cleavage, after trypsin cleavage, during RBC incubation at 4°C or during 37°C incubation. For experiments on HN-independent retention of RBCs, CV-1 effector cells transfected with HN and F were labeled with 1 µM SYTO 11 nucleic acid dye (Molecular Probes) at 37°C for 30 min, single-labeled R18–RBCs (1.0 ml of 0.05% hematocrit) were used as fusion targets, and the CV-1 cells were washed five times with ice-cold phosphate-buffered saline (PBS) to remove unbound RBCs after 37°C incubation.
Capture of an F intermediate
Monolayers of Vero cells in 10-cm dishes were transfected with 3 µg each of pCAGGS F and pCAGGS HN. At 18 h post-transfection, the Vero cells were incubated with 5 ml of 0.1% hematocrit RBCs at 4°C for 1 h. Unbound RBCs were removed by washing the samples five times with ice-cold PBS. The ice-cold PBS was replaced with 37°C PBS and the samples were incubated at 37°C for 45 min. In one experiment, 50 µg of HAt-C1 were added to the Vero cells expressing HN and F either before RBC binding, during RBC binding, during the 37°C incubation or after the 37°C incubation. In another experiment, varying quantities of HAt-C1 and C-1 were incubated with the Vero cells during the 37°C incubation. After the 37°C incubation, the samples were washed three times with PBS, incubated with 5 ml of Dulbecco’s modified Eagle’s medium containing 100 µg of anti-HA monoclonal antibody 12CA5 at 25°C for 3 h and washed five times with PBS. The Vero cells were lysed with 1 ml of ice-cold RIPA lysis buffer containing protease inhibitors (Paterson et al., 2000) and 25 mM iodoacetamide, and the clarified supernatants were incubated with 30 µl of protein A–Sepharose at 4°C overnight. The samples were washed with RIPA buffer three times and analyzed by 15% SDS–PAGE under non-reducing conditions. The gels were then transferred to Immobilon-P PVDF membranes (Millipore, Bedford, MA), immunoblotted with an anti-F2 rabbit anti-peptide primary antibody followed by a horseradish peroxidase-conjugated goat anti-mouse secondary antibody, and detected by chemiluminescence.
Reversible fusion inhibition by lipids
Stock solutions of 10 µM LPC (Avanti Polar Lipids, Birmingham, AL) were prepared freshly as aqueous dispersions in PBS and vortexed until clear. A stock solution of 1 mM oleic acid (OA; Sigma) was prepared in ethanolic solution. After binding of R18/CF-labeled RBCs at 4°C and washing away unbound RBCs (see above), the ice-cold PBS was replaced with ice-cold LPC stock solution and incubated at 4°C for 15 min. The 4°C LPC solution was subsequently replaced with 37°C LPC solution and the samples were incubated at 37°C. For samples containing both LPC and OA, 30 µl of the 1 mM OA stock solution were added to 3 ml of LPC stock solution before the mixed solution was incubated with the samples at 37°C. Either N-1 or C-1 (40 µM) was added to the samples during or after the 37°C incubation. Following the 37°C incubations, the samples were washed with ice-cold PBS. The LPC was removed by multiple washes with an ice-cold 10 mg/ml solution of fatty-acid free BSA (Sigma) and the ice-cold BSA solution replaced with 37°C PBS; the samples were then bathed at 37°C for 10 min, washed with ice-cold PBS and placed on ice until confocal microscopic analysis of dye transfer.
Acknowledgments
Acknowledgements
We thank Reay G.Paterson, Rebecca Ellis Dutch, Frederic S.Cohen and Grigory B.Melikyan for helpful discussions. This work was supported in part by Research Grant AI-23173 from the National Institute of Allergy and Infectious Disease. C.J.R. is an Associate and R.A.L. is an Investigator of the Howard Hughes Medical Institute.
References
- Baker K.A., Dutch,R.E., Lamb,R.A. and Jardetzky,T.S. (1999) Structural basis for paramyxovirus-mediated membrane fusion. Mol. Cell, 3, 309–319. [DOI] [PubMed] [Google Scholar]
- Ben-Efraim I., Kliger,Y., Hermesh,C. and Shai,Y. (1999) Membrane-induced step in the activation of Sendai virus fusion protein. J. Mol. Biol., 285, 609–625. [DOI] [PubMed] [Google Scholar]
- Calder L.J., Gonzalez-Reyes,L., Garcia-Barreno,B., Wharton,S.A., Skehel,J.J., Wiley,D.C. and Melero,J.A. (2000) Electron microscopy of the human respiratory syncytial virus fusion protein and complexes that it forms with monoclonal antibodies. Virology, 271, 122–131. [DOI] [PubMed] [Google Scholar]
- Carr C.M., Chaudhry,C. and Kim,P.S. (1997) Influenza hemagglutinin is spring-loaded by a metastable native conformation. Proc. Natl Acad. Sci. USA, 94, 14306–14313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan D.C. and Kim,P.S. (1998) HIV entry and its inhibition. Cell, 93, 681–684. [DOI] [PubMed] [Google Scholar]
- Chan D.C., Fass,D., Berger,J.M. and Kim,P.S. (1997) Core structure of gp41 from the HIV envelope glycoprotein. Cell, 89, 263–273. [DOI] [PubMed] [Google Scholar]
- Chan D.C., Chutkowski,C.T. and Kim,P.S. (1998) Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proc. Natl Acad. Sci. USA, 95, 15613–15617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.A., Scales,S.J., Patel,S.M., Doung,Y.C. and Scheller,R.H. (1999) SNARE complex formation is triggered by Ca2+ and drives membrane fusion. Cell, 97, 165–174. [DOI] [PubMed] [Google Scholar]
- Chernomordik L.V., Leikina,E., Frolov,V., Bronk,P. and Zimmerberg,J. (1997) An early stage of membrane fusion mediated by the low pH conformation of influenza hemagglutinin depends upon membrane lipids. J. Cell Biol., 136, 81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernomordik L.V., Frolov,V.A., Leikina,E., Bronk,P. and Zimmerberg,J. (1998) The pathway of membrane fusion catalyzed by influenza hemagglutinin: restriction of lipids, hemifusion and lipidic fusion pore formation. J. Cell Biol., 140, 1369–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crennell S., Takimoto,T., Portner,A. and Taylor,G. (2000) Crystal structure of the multifunctional paramyxovirus hemagglutinin– neuraminidase. Nature Struct. Biol., 7, 1068–1074. [DOI] [PubMed] [Google Scholar]
- Damico R.L., Crane,J. and Bates,P. (1998) Receptor-triggered mem brane association of a model retroviral glycoprotein. Proc. Natl Acad. Sci. USA, 95, 2580–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutch R.E., Joshi,S.B. and Lamb,R.A. (1998) Membrane fusion promoted by increasing surface densities of the paramyxovirus F and HN proteins: comparison of fusion reactions mediated by simian virus 5 F, human parainfluenza virus type 3 F and influenza virus HA. J. Virol., 72, 7745–7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutch R.E., Hagglund,R.N., Nagel,M.A., Paterson,R.G. and Lamb,R.A. (2001) Paramyxovirus fusion (F) protein: a conformational change on cleavage activation. Virology, 281, 138–150. [DOI] [PubMed] [Google Scholar]
- Furuta R.A., Wild,C.T., Weng,Y. and Weiss,C.D. (1998) Capture of an early fusion-active conformation of HIV-1 gp41. Nature Struct. Biol., 5, 276–279. [DOI] [PubMed] [Google Scholar]
- Hernandez L.D., Hoffman,L.R., Wolfsberg,T.G. and White,J.M. (1996) Virus–cell and cell–cell fusion. Annu. Rev. Cell Dev. Biol., 12, 627–661. [DOI] [PubMed] [Google Scholar]
- Hernandez L.D., Peters,R.J., Delos,S.E., Young,J.A.T., Agard,D.A. and White,J.M. (1997) Activation of a retroviral membrane fusion protein: soluble receptor-induced liposome binding of the ALSV envelope glycoprotein. J. Cell Biol., 139, 1455–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath C.M., Paterson,R.G., Shaughnessy,M.A., Wood,R. and Lamb,R.A. (1992) Biological activity of paramyxovirus fusion proteins: factors influencing formation of syncytia. J. Virol., 66, 4564–4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M., Nishio,M., Kawano,M., Kusagawa,S., Komada,H., Ito,Y. and Tsurudome,M. (1997) Role of a single amino acid at the amino terminus of the simian virus 5 F2 subunit in syncytium formation. J. Virol., 71, 9855–9858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M., Nishio,M., Komada,H., Ito,Y. and Tsurudome,M. (2000) An amino acid in the heptad repeat 1 domain is important for the haemagglutinin–neuraminidase-independent fusing activity of simian virus 5 fusion protein. J. Gen. Virol., 81, 719–727. [DOI] [PubMed] [Google Scholar]
- Jahn R. and Südhof,T.C. (1999) Membrane fusion and exocytosis. Annu. Rev. Biochem., 68, 863–911. [DOI] [PubMed] [Google Scholar]
- Joshi S.B., Dutch,R.E. and Lamb,R.A. (1998) A core trimer of the paramyxovirus fusion protein: parallels to influenza virus hemagglutinin and HIV-1 gp41. Virology, 248, 20–34. [DOI] [PubMed] [Google Scholar]
- Lamb R.A. (1993) Paramyxovirus fusion: a hypothesis for changes. Virology, 197, 1–11. [DOI] [PubMed] [Google Scholar]
- Lamb R.A. and Kolakofsky,D. (2001) Paramyxoviridae: the viruses and their replication. In Knipe,D.M. and Howley,P.M. (eds), Fields Virology, 4th edn. Lippincott, Williams and Wilkins, Philadelphia, PA, in press.
- Melikyan G.B., Markosyan,R.M., Hemmati,H., Delmedico,M.K., Lambert,D.M. and Cohen,F.S. (2000) Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J. Cell Biol., 151, 413–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore J.P., McKeating,J.A., Weiss,R.A. and Sattentau,Q.J. (1990) Dissociation of gp120 from HIV-1 virions induced by soluble CD4. Science, 250, 1130–1142. [DOI] [PubMed] [Google Scholar]
- Novick S.L. and Hoekstra,D. (1988) Membrane penetration of Sendai virus glycoproteins during the early stage of fusion with liposomes as determined by hydrophobic affinity labeling. Proc. Natl Acad. Sci. USA, 85, 7433–7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parlati F., Weber,T., McNew,J.A., Westermann,B., Sollner,T.H. and Rothman,J.E. (1999) Rapid and efficient fusion of phospholipid vesicles by the α-helical core of a SNARE complex in the absence of an N-terminal regulatory domain. Proc. Natl Acad. Sci. USA, 96, 12565–12570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson R.G., Harris,T.J.R. and Lamb,R.A. (1984) Fusion protein of the paramyxovirus simian virus 5: Nucleotide sequence of mRNA predicts a highly hydrophobic glycoprotein. Proc. Natl Acad. Sci. USA, 81, 6706–6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson R.G., Hiebert,S.W. and Lamb,R.A. (1985) Expression at the cell surface of biologically active fusion and hemagglutinin– neuraminidase proteins of the paramyxovirus simian virus 5 from cloned cDNA. Proc. Natl Acad. Sci. USA, 82, 7520–7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson R.G., Shaughnessy,M.A. and Lamb,R.A. (1989) Analysis of the relationship between cleavability of a paramyxovirus fusion protein and length of the connecting peptide. J. Virol., 63, 1293–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson R.G., Russell,C.J. and Lamb,R.A. (2000) Fusion protein of the paramyxovirus SV5: destabilizing and stabilizing mutants of fusion activation. Virology, 270, 17–30. [DOI] [PubMed] [Google Scholar]
- Poirier M.A., Xiao,W., Macosko,J.C., Chan,C., Shin,Y.K. and Bennett,M.K. (1998) The synaptic SNARE complex is a parallel four-stranded helical bundle. Nature Struct. Biol., 5, 765–769. [DOI] [PubMed] [Google Scholar]
- Randall R.E., Young,D.F., Goswami,K.K.A. and Russell,W.C. (1987) Isolation and characterization of monoclonal antibodies to simian virus 5 and their use in revealing antigenic differences between human, canine and simian isolates. J. Gen. Virol., 68, 2769–2780. [DOI] [PubMed] [Google Scholar]
- Ruigrok R.W.H., Martin,S.R., Wharton,S.A., Skehel,J.J., Bayley,P.M. and Wiley,D.C. (1986) Conformational changes in the hemagglutinin of influenza virus which accompany heat-induced fusion of virus with liposomes. Virology, 155, 484–497. [DOI] [PubMed] [Google Scholar]
- Sergel T., McGinnes,L.W., Peeples,M.E. and Morrison,T.G. (1993) The attachment function of the Newcastle disease virus hemagglutinin– neuraminidase protein can be separated from fusion promotion by mutation. Virology, 193, 717–726. [DOI] [PubMed] [Google Scholar]
- Sergel T.A., McGinnes,L.W. and Morrison,T.G. (2000) A single amino acid change in the Newcastle disease virus fusion protein alters the requirement for HN protein in fusion. J. Virol., 74, 5101–5107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skehel J.J. and Wiley,D.C. (1998) Coiled coils in both intracellular vesicle and viral membrane fusion. Cell, 95, 871–874. [DOI] [PubMed] [Google Scholar]
- Skehel J.J. and Wiley,D.C. (2000) Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem., 69, 531–569. [DOI] [PubMed] [Google Scholar]
- Sutton R.B., Fasshauer,D., Jahn,R. and Brunger,A.T. (1998) Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 Å resolution. Nature, 395, 347–353. [DOI] [PubMed] [Google Scholar]
- Tsurudome M., Gluck,R., Graf,R., Falchetto,R., Schaller,U. and Brunner,J. (1992) Lipid interactions of the hemagglutinin HA2 NH2-terminal segment during influenza virus-induced membrane fusion. J. Biol. Chem., 267, 20225–20232. [PubMed] [Google Scholar]
- Weissenhorn W., Dessen,A., Harrison,S.C., Skehel,J.J. and Wiley,D.C. (1997) Atomic structure of the ectodomain from HIV-1 gp41. Nature, 387, 426–430. [DOI] [PubMed] [Google Scholar]
- Wharton S.A., Skehel,J.J. and Wiley,D.C. (2000) Temperature dependence of fusion by Sendai virus. Virology, 271, 71–78. [DOI] [PubMed] [Google Scholar]
- Young J.K., Li,D., Abramowitz,M.C. and Morrison,T.G. (1999) Interaction of peptides with sequences from the Newcastle disease virus fusion protein heptad repeat regions. J. Virol., 73, 5945–5956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X., Singh,M., Malashkevich,V.N. and Kim,P.S. (2000) Structural characterization of the human respiratory syncytial virus fusion protein core. Proc. Natl Acad. Sci. USA, 97, 14172–14177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J., Dutch,R.E. and Lamb,R.A. (1997) Proper spacing between heptad repeat B and the transmembrane domain boundary of the paramyxovirus SV5 F protein is critical for biological activity. Virology, 239, 327–339. [DOI] [PubMed] [Google Scholar]