

Abstract
Translation initiation region (TIR) of the rpsA mRNA encoding ribosomal protein S1 is one of the most efficient in Escherichia coli despite the absence of a canonical Shine–Dalgarno-element. Its high efficiency is under strong negative autogenous control, a puzzling phenomenon as S1 has no strict sequence specificity. To define sequence and structural elements responsible for translational efficiency and autoregulation of the rpsA mRNA, a series of rpsA′–′lacZ chromosomal fusions bearing various mutations in the rpsA TIR was created and tested for β-galactosidase activity in the absence and presence of excess S1. These in vivo results, as well as data obtained by in vitro techniques and phylogenetic comparison, allow us to propose a model for the structural and functional organization of the rpsA TIR specific for proteobacteria related to E.coli. According to the model, the high efficiency of translation initiation is provided by a specific fold of the rpsA leader forming a non-contiguous ribosome entry site, which is destroyed upon binding of free S1 when it acts as an autogenous repressor.
Keywords: autogenous control/mRNA structure/phylogenetic analysis/ribosomal protein S1/translation initiation
Introduction
Both in eukaryotes and prokaryotes, translational control is exerted predominantly at the initiation step, thus providing a direct and rapid means to change the activity of an mRNA in response to various environmental signals. In prokaryotes, the intrinsic translational efficiency of an mRNA depends mainly on the primary structure of the region recognized by a 30S ribosomal subunit while searching for a translational start (ribosome binding site, RBS). The minimum set of sequence elements required for high translational efficiency of RBS includes the initiation codon, the Shine–Dalgarno (SD) sequence (a stretch of nucleotides complementary to the anti-SD sequence ACCUCCUUA at the 3′-end of 16S rRNA) and an appropriate spacing in between (Gold, 1988; McCarthy and Gualerzi, 1990; Ringquist et al., 1992; Chen et al., 1994). It is commonly assumed that these sequence elements should be embedded in an unstructured (or weakly structured) region since a ribosome can bind RBS only in its unfolded state (de Smit and van Duin, 1990; de Smit, 1998). One of the most intriguing exceptions to these rules is the rpsA mRNA of Escherichia coli encoding ribosomal protein (r-protein) S1. This unique mRNA, albeit responsible for synthesis of an abundant cellular protein, does not contain a canonical SD-element. Whereas SD-elements in other r-protein RBSs comprise at least four contiguous nucleotides complementary to the anti-SD sequence, the rpsA SD-like element GAAG can form only 3 bp. Moreover, in earlier works (Dunn et al., 1978; Schwartz et al., 1981), the GAAG sequence was shown to be very inefficient in driving translation. Nevertheless, with this vestigial SD the rpsA translation initiation region (TIR) has an extremely high intrinsic activity in vivo, far above the activity of many E.coli RBSs containing canonical SD-elements (Boni et al., 2000).
The product of the rpsA mRNA, r-protein S1, is essential for cell viability in Gram-negative bacteria and has two well documented functions in translation: it promotes binding of ribosomes to mRNA and, like several other r-proteins, it regulates its own synthesis as an autogenous repressor (Subramanian, 1983, 1984; Roberts and Rabinovitz, 1989; Skouv et al., 1990; Boni et al., 1991, 2000; Sorensen et al., 1998). S1 is an unusual r-protein (reviewed by Subramanian, 1983, 1984): it is the largest (61 kDa) and the longest r-protein of E.coli (its length of ∼23 nm is comparable to the longest dimension of 30S subunit itself), with a complex molecular design. Its globular N-terminal domain is responsible for protein– protein interactions, including binding to the ribosome, whereas the central and C-terminal parts form an elongated RNA-binding domain comprising four highly homologous repeats of the so-called S1-motif (Subramanian, 1983, 1984; Bycroft et al., 1997). S1 interacts with mRNA via this RNA-binding domain during initiation of protein synthesis and, most likely, during the overall translation process (see Subramanian, 1984; Boni et al., 1991, for models).
S1 belongs to the OB fold family of proteins highly specific for single-stranded nucleic acids (Draper and Reynaldo, 1999), but it has no strict sequence specificity and binds polyU, polyC and polyA as well as various heterogenous RNAs (Subramanian, 1983, 1984). Its known extremely high affinity to the polypyrimidines was attributed to the cooperativity of binding (Draper and von Hippel, 1978). At the same time, no cooperativity was found for polyA, and the possibility of cooperative binding of S1 to any natural RNA has never been studied. However, specificity of S1–RNA interactions may have been underestimated. Thus, earlier studies on Qβ phage RNA revealed only two regions bound by S1 (Goelz and Steitz, 1977), one of which, situated upstream of the coat protein RBS, was shown to be responsible for S1-mediated recognition of Qβ RNA by both ribosome and Qβ replicase (Boni et al., 1991; Miranda et al., 1997). Footprinting experiments on various mRNAs have revealed site-specific interaction of S1 with U- or A/U-rich single-stranded regions within mRNA leaders (Boni et al., 1991; Tzareva et al., 1994), and SELEX experiments (Ringquist et al., 1995) have shown that S1 (either free or within the 30S subunit) can bind specific RNA aptamers with very high affinity. Recent in vitro data of Mogridge and Greenblatt (1998) indicate that S1 binds the rrn anti-terminator, BoxA, more tightly than some BoxA mutants or the reversed BoxA sequence. All these data suggest the existence of a hierarchy of RNA targets with respect to their affinity for S1. The most prominent illustration of this hierarchy is the ability of S1 to control its own synthesis in vivo, i.e. to distinguish its own mRNA among all the others and to act as a highly specific translational repressor (Skouv et al., 1990; Rasmussen et al., 1993; Boni et al., 2000).
The mechanism for S1 autogenous control, although still unsolved, seems to differ in some important aspects from other examples of r-protein-mediated translational repression (for review see Zengel and Lindall, 1994). Other regulatory r-proteins bind specifically to 16S or 23S rRNA during ribosome assembly, so that their primary target in a cell is rRNA, and only in the absence of free rRNA can they bind to the cognate mRNA and inhibit translation. In contrast, S1 binds to ribosome late in assembly by means of protein–protein interactions, and it is directly involved in mRNA binding (see above). The question of how excess S1 can inhibit its own mRNA without affecting all the others remains a puzzle. The goal of the present study was to gain a new insight into this problem.
Recently, we have shown that both the exceptional efficiency of translation initiation in vivo and autogenous control of the rpsA mRNA strictly require the presence of at least 90 nucleotides upstream of the rpsA start codon (Boni et al., 2000). Here, we propose a structural and functional organization of this region specific for E.coli and related proteobacteria. By site-directed mutagenesis we have defined sequence and structure elements responsible for the rpsA translational control. Based on these in vivo data, as well as those obtained by in vitro techniques and phylogenetic analysis, we propose that in the absence of canonical SD interactions, a specific fold of the rpsA TIR plays a major role by providing optimal spatial arrangement of the sequence elements involved in direct contacts with the ribosome. We suggest that autogenous repression of S1 synthesis works mainly by a steric perturbation in the rpsA TIR fold upon binding of free S1, with a negative effect on the efficiency of the 30S-rpsA mRNA recognition.
Results
Secondary structure of the rpsA translation initiation region
Recently, we created a series of rpsA′–′lacZ chromosomal fusions in which β-galactosidase synthesis was driven at the transcription level by the lac promoter–operator region, and at the translational level by the rpsA TIR progressively shortened from the 5′ side (Boni et al., 2000). The longest rpsA region fused to the lacZ gene comprised nucleotides from –145 to +57 with respect to the rpsA start codon. Truncation down to the –91 position does not bring any significant change in efficiency of β-galactosidase synthesis, which for both constructions is remarkably high (Table I). Moreover, in the presence of S1-expressing plasmid pSP261 (Pedersen et al., 1984), this high translation activity is inhibited >20-fold, indicating strong autogenous repression. Slightly larger 5′-deletion (down to –82) impairs both properties, whereas further truncations partially restore translation activity but not autoregulation. Thus, the shortest rpsA leader (29 nt) active in translation is as efficient as the genuine lacZ RBS (5600 U, see Boni et al., 2000) but can not be repressed by excess S1 (Table I). We conclude that the rpsA TIR bearing the 91-nt leader contains all of the information necessary and sufficient for both high efficiency and autogenous control of S1 synthesis.
Table I. Effect of 5′ deletions upon the activity of the rpsA TIR and its autocontrol by S1.
| Length of rpsA leader (nt) | β-galactosidase activitya |
Repressionb | |
|---|---|---|---|
| pControlc | pS1c | ||
| 145 | 18 500 ± 1500 | 900 ± 150 | 21 |
| 91 | 18 900 ± 800 | 800 ± 100 | 24 |
| 82 | 1900 ± 200 | 500 ± 100 | 3.8 |
| 66 | 4500 ± 200 | 2200 ±100 | 2 |
| 45 | 6600 ± 800 | 5300 ± 800 | 1.2 |
| 29 | 5450 ± 150 | 5200 ± 200 | 1.05 |
aNanomoles of ONPG hydrolyzed per min and per mg of total protein. Average of three or more independent assays.
bRatio of the β-galactosidase expression in cells carrying pControl and pS1, respectively.
c‘pControl’ and ‘pS1’ are plasmids pACYC184 and its S1-expressing derivative pSP261, correspondingly (Pedersen et al., 1984).
Earlier, the S1 mRNA secondary structure in the vicinity of the start codon was studied in vitro by enzymatic probing (Tzareva et al., 1993). Taking into account the probing results and computer modeling data obtained with the mfold program of M.Zuker (http://bioinfo.math.rpi.edu/∼mfold), we propose here a model for structural organization of the rpsA TIR extending to position –91 (Figure 1). Secondary structure of the TIR is represented by three hairpins (I–II–III) separated by A/U-rich single-stranded regions (ss-1 and ss-2). Hairpins I and II are very stable (–11.7 and –11.9 kcal/mol), whereas the third (III), which contains an initiator AUG in a large apical loop and a degenerate SD-like element (GAAG) in a stem, has a rather low stability (–3.2 kcal/mol). By itself, this location should not prevent ribosome binding because the hairpin is weak (see de Smit and van Duin, 1990), but GAAG is known to be inefficient as an SD-element in initiation (Dunn et al., 1978; Schwartz et al., 1981) and thus, it can not provide the unusually high translation activity of the rpsA TIR (Table I). Since all 5′-deletions downstream of position –91 disrupt the I–II–III structure (Figure 1) and impair both activity and autocontrol, we suggest that it is the specific fold of the rpsA TIR that plays a crucial role in both processes.

Fig. 1. Folding of the E.coli rpsA TIR predicted from in vitro enzymatic probing and computer modeling data. Single-stranded A/U-rich regions separating three consecutive hairpin-loop structures (I, II, III) are denoted ss-1 and ss-2. The sequence is numbered from the first base of the initiator codon (in bold). The location of the site-directed mutations and 5′ deletions described in the text are indicated by arrows. Double mutations are boxed, deletions of individual nucleotides are marked by ‘Δ’.
Phylogenetic analysis
RNA secondary structures predicted by computer modeling or by in vitro probing do not necessarily exist in vivo. To gain phylogenetic support for the proposed rpsA TIR fold, we used the first 100 nt of the E.coli rpsA coding sequence as a searching probe for homologous sequences from other bacteria (‘microbial genomes: finished and unfinished’ at NCBI server http://www.ncbi.nlm.nih.gov/BLAST). The best ‘hits’ were found to belong to the same group of eubacteria as E.coli—gamma subdivision of proteobacteria. Analysis of the sequences upstream from the start codons of these rpsA genes revealed that despite significant sequence divergence (except for the Salmonella group which has the same rpsA leader sequences for all representatives and shows the highest homology with E.coli), all of the rpsA TIRs can be folded into the same I–II–III structure as that of E.coli (Figure 2). The only exception is the rpsA TIR of Haemophilus influenzae (the genome of which is highly reduced: 1.83 versus 4.67 Mb for E.coli), where hairpin I and region ss-1 are absent.

Fig. 2. Phylogenetic conservation of the rpsA TIR folding in proteobacteria from gamma subdivision. Conserved GGA-motifs in loops I and II are boxed, the degenerate SD-like sequences are indicated by vertical bars.
The rpsA TIRs found share not only remarkable structural similarity but also some conserved sequence features (Figure 2). First, all of them contain a degenerate SD sequence (mostly GAAG) in a stem of the weak hairpin III with the start AUG codon on the top. Secondly, in all cases, the hairpins are separated by extended A/U-rich regions. Thirdly, in all cases, hairpins I and II contain internal loops or bulges, which could participate in tertiary structure formation; in particular, an internal loop at the bottom of hairpin II is highly conserved among all the species. Finally, in all cases, apical loops of the strong hairpins I and II comprise GGA sequence (GAA in loop II of Buchnera aphidicola and Buchnera sp. APS).
The phylogenetic similarity (Figure 2) suggests that the proposed folding of the E.coli rpsA TIR (Figure 1) has functional validity and, most likely, is essential for translational control of the rpsA gene expression within at least five branches of the gamma-proteobacteria (Enterobacteriaceae, Pasterellaceae, Vibrionaceae, Buchnera, Shewanella).
Site-directed mutagenesis of the E.coli rpsA leader
To define sequence and structure elements required for high translation activity and autocontrol, we subjected the rpsA TIR bearing the 91-nt leader to site-directed mutagenesis, and then transferred the mutated rpsA′–′lacZ fusions onto the E.coli chromosome. To monitor autogenous control, the resulting strains were transformed with the S1-expressing plasmid pSP261 or with the control vector pACYC184, and β-galactosidase activity was measured in each case (Table II).
Table II. Effect of site-directed mutations within the 91-nt rpsA leader upon the activity of the rpsA TIR and its autocontrol by S1a.
| Mutation | β-galactosidase activity |
Repression | |
|---|---|---|---|
| pControl | pS1 | ||
| WT | 18 900 ± 800 | 800 ± 100 | 24 |
| GG→AU at –79, –78 | 4600 ± 300 | 2500 ± 200 | 1.8 |
| GG→AU at –40, –39 | 3700 ± 300 | 2100 ± 250 | 1.8 |
| –39G→A | 19 500 ± 1000 | 4400 ± 400 | 4.5 |
| ΔG loopI | 11 300 ± 200 | 1100 ± 100 | 10.3 |
| –78G→A | 6800 ± 800 | 1250 ± 50 | 5.4 |
| –80 ΔU | 17 000 ± 2000 | 3000 ± 500 | 5.7 |
| –77A→C | 11 000 ± 500 | 700 ± 100 | 16 |
| –76A→C | 18 800 ± 1000 | 800 ± 100 | 24 |
| –9A→G | 40 000 ± 3500 | 26 000 ± 2500 | 1.5 |
| –8G→C, +11C→G | 13 500 ± 500 | 700 ± 200 | 19 |
| –27C→G | 37 000 ± 2000 | 28 000 ± 2000 | 1.3 |
| –52C→G | 38 000 ± 3000 | 13 000 ± 1000 | 2.9 |
| –26G→C | 4100 ± 500 | 300 ± 50 | 13.7 |
| –26G→C, –53C→G | 20 500 ± 2000 | 3100 ± 500 | 6.7 |
| –14, –13 ΔCC | 18 000 ± 500 | 700 ± 50 | 26 |
aSee Table I for legend.
The mutations were designed with the aim of elucidating the role of evolutionary conserved elements found within the rpsA TIR (Figures 1 and 2). Two double mutations (GG→AU) and three point mutations were introduced to check the significance of highly conserved GG-sequences in loops I and II (Figure 1). Three point mutations were also introduced in loop I to examine the role of the context around the GG motif. The β-galactosidase assay (Table II) showed that each GG→AU mutation affects translation very negatively. An analogous effect was obtained by a complete deletion of hairpin I (the 66-nt rpsA leader; Table I). Point mutations in the GG-elements as well as around them had lower effects (Table II) if any (–76A→C). Thus, the simultaneous presence of both GG-sequences in apical loops I and II appears crucial for the high translation activity of the rpsA TIR.
A second group included a point mutation (–9A→G) that transforms the degenerate SD-element (GAAG) into a conventional one (GAGG), and a double mutation (–8G→C, +11C→G) that should eliminate the residual complementarity with the anti-SD sequence while maintaining the stability of the hairpin. The –9A→G mutation increased translation activity of the rpsA TIR (2-fold), but also, and most prominently, it nearly abolished autogenous repression (Table II). The double mutation (–8G→C, +11C→G) slightly reduced translation activity but barely affected autocontrol. We cannot completely exclude that this small decrease results from elimination of the residual complementarity with the anti-SD, but it seems more likely that it is caused by reorganization of hairpin III into a more stable local structure where the initiation codon is partially base paired (Figure 3). We conclude that the degeneration of the rpsA SD-element is required to provide a strong autogenous control of S1 synthesis, without significantly compromising the intrinsic rpsA TIR activity. Thus, the high rpsA TIR activity is not based upon canonical SD–anti-SD interactions.

Fig. 3. Structural reorganization of the rpsA TIR hairpin III resulting from the double mutation –8G→C, +11C→G, as predicted by mfold algorithm. Mutated nucleotides are boxed, energy parameters are shown above each structure.
A third group included mutations that strengthen or weaken the local structure at the bottom of hairpin II (Figure 1). Two point C→G mutations at positions –27 and –52 are predicted to strengthen hairpin II by closing the conserved internal loop (Figures 1 and 2). The β-galactosidase assay showed that these mutations largely relieve autogenous control and increase translation activity of the TIR, just like the –9A→G mutation discussed above (Table II). The quantitative difference in effects of –27 and –52 mutations on autorepression may be ascribed either to the fact that the –27C→G mutation stabilizes the lower part of the stem to a higher extent, or to the specific importance of C at position –27 for repressor complex formation. One can expect that the –26G→C mutation should have an opposite effect: it must disrupt the bottom helix and destabilize hairpin II. Indeed, this mutation dramatically decreased the rpsA TIR activity both in the absence and presence of S1 in trans (Table II). When a second mutation (–53C→G) was introduced to restore the bottom helix by creating a G–C base pair, the wild-type activity was regained (with some loss in the efficiency of autorepression). The results obtained with this group of mutations (Table II) indicate that an internal loop and the bottom helix of hairpin II serve to maintain the proper balance between translation efficiency and autorepression.
An additional mutation (–13, –14 ΔCC) was introduced to evaluate the possible role of alternative base pairings, e.g. between (–35)CAGGU and (–15)ACCUG regions (see Figure 1). This mutation did not bring significant changes (Table II), indicating that neither alternative base pairing nor the exact length of ss-2 is important.
S1 binding to the rpsA TIR in vitro
Prior to searching for the S1 target(s) on its own messenger by conventional in vitro techniques, it was reasonable to examine whether the specificity of the rpsA translational autorepression observed in vivo can be reproduced in vitro. Ternary initiation complex formation on cognate (rpsA) and non-cognate (ssb) mRNAs in the absence or presence of increasing concentrations of S1 was studied by toeprinting (Figure 4). In the absence of S1, no stop signals (toeprints) at the position +16 (from the A + 1 of the initiation codon, see Hartz et al., 1988) were observed, clearly indicating that the 30S lacking S1 is unable to form a ternary initiation complex on either rpsA or ssb mRNAs. In both cases, addition of free S1 up to a 1:1 protein:30S ratio restored the ability of 30S to bind mRNA and hence to generate a toeprint signal, but excess S1 was inhibitory (Figure 4). Thus, qualitatively, both mRNAs respond similarly to the absence or presence of S1. But what is remarkable is that S1 concentrations that are already inhibitory for initiation complex formation on the S1 mRNA are not yet inhibitory for the ssb mRNA (Figure 4A and B). Other natural mRNAs (atpE, ompA, rplL, T7 gene10, T4 gp32) gave similar results in analogous assays, indicating that gene-non-specific inhibition of ternary complex formation requires a much higher concentration of free S1 than necessary for the rpsA mRNA (our unpublished results). Thus, toeprinting data show that autogenous repression occurs at the level of 30S initiation complex formation; it can be reproduced in vitro, and therefore, does not require additional trans-acting factors. On the other hand, the fact that free S1 at a high S1/30S molar ratio is able to inhibit translation initiation in a gene-non-specific manner suggests that regulation of S1 production should be essential for a bacterial cell.

Fig. 4. S1 dependence of ternary initiation complex formation on the rpsA and ssb mRNAs (toeprint analysis). (A) Autoradiograms of 8% sequencing gels showing inhibition of the extension reactions (see Materials and methods). Concentration of individual components was: mRNA, 0.04 µM; 30S subunits lacking S1, 0.4 µM; uncharged initiator tRNA, 4 µM; free S1 as indicated over lanes. FT is the signal corresponding to full-length reverse transcript, +16 is the position of the toeprint signal. (B) Quantification of the toeprint results by densitometric scanning. Relative toeprint is a toeprint/(FT + toeprint) ratio, i.e. the percentage of the mRNA involved in initiation complex formation.
Direct interaction of free S1 with the rpsA TIR was shown in vitro by mobility shift assay (Figure 5). The results indicate that all truncated variants of the rpsA TIR (Table I; Figure 1) contain targets for S1 binding, including the shortest ones, which are not autoregulated in vivo (Figure 5A). It is noteworthy that free RNAs corresponding to the 66- and 45-nt leaders have similar mobilities in a native gel, most likely because of the presence (66-nt, hairpin II) and the absence (45-nt) of stable secondary structure. In contrast, their bandshifting patterns are clearly distinct: there are two shifted bands in the case of the 66-nt leader, with the position of the lower band being the same as for the 45-nt leader (Figure 5A). It is reasonable to suggest that the 66-nt leader forms two types of complex with S1 differing in stoichiometric composition. Consistently, the 91-nt leader forms mainly the complex with higher stoichiometry. Mobility shift assay at progressively decreased S1 concentrations revealed that S1 binds to the longest rpsA TIR more tightly than to the short one (Figure 5B). This unequal affinity of rpsA leaders for S1 can account for the observed different susceptibility to autogenous control in vivo (Table I). As the 91- and 145-nt leaders show the same repression level in vivo (Table I), we believe that the specific fold proposed for the rpsA TIR (Figure 1) plays a major role in tight repressor complex formation. The fact that higher stoichiometry of S1 binding provides higher apparent binding affinity (Figure 5B) suggests that this tight repressor complex can be formed by cooperative binding of several S1 molecules to the rpsA leader.
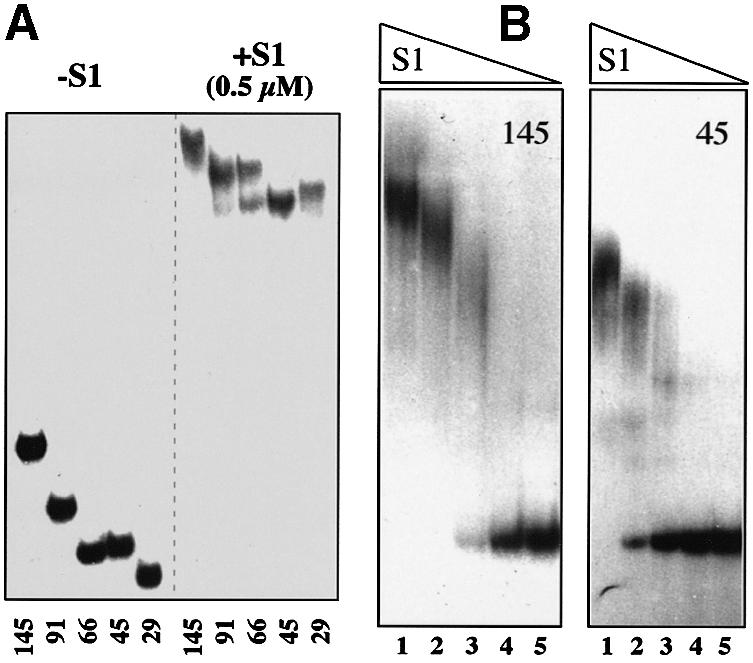
Fig. 5. Gel mobility shift assay of the S1-rpsA TIR complexes. Concentration of high specific activity RNA in each assay is 2–5 nM. (A) Results of band shifting (5% native gel) showing that all truncated rpsA leaders carry targets for S1 binding. The length of the rpsA leader is indicated below each lane. Formation of complexes with different stoichiometry in the case of 66-nt leader is clearly visible on the rightmost half of the gel. (B) Band shift assay for the 145- (left) and 45-nt (right) rpsA leaders at progressively decreased S1 concentration showing their different affinity for S1. Concentration of S1 (µM) in lanes: 1, 0.5; 2, 0.25; 3, 0.125; 4, 0.062; 5, 0.031.
To define the S1 targets within the rpsA TIR, chemical and enzymatic footprinting techniques were used (Figure 6). Modification with diethylpyrocarbonate (DEPC) revealed that bound S1 protects A-residues within ss-1 and ss-2 regions, on the left of the bottom helix of hairpin II, and within the sequence preceding the start codon (including the A + 1) in a loop III (Figure 6A and C). Maybe significantly, all three protected regions bear homologous motifs: UUAAACAA (ss-1), UUAAAU AUAAAC (ss-2) and UUAAACA (loop III). The same regions were protected by S1 against RNases PhyM and T2 (not shown). No protection against RNase T1 was observed, indicating that S1 does not interact with unpaired G-residues (Figure 6B). Whereas the T1 digestion pattern is consistent with the proposed secondary structure model (Figure 1), it should be noted that the GG-sequences in apical loops I and II become even more accessible to T1 in the presence of S1, and G-26 below the internal loop becomes accessible only at higher S1 concentration. The latter suggests that the lower part of hairpin II is melted within the S1–rpsA TIR complex. It is also significant that DEPC probing revealed not only protection of several regions by S1 but also the appearance of new modification sites: thus, A-residues 3′ to the conserved GG-motifs in loops I and II become reactive only in the presence of S1. It is known that at neutral pH (our binding conditions) DEPC allows the mapping of single-stranded adenines (carbethoxylation of N7) and monitoring of the involvement of N7-A in tertiary interactions (see Ehresmann et al., 1987). Thus, the footprinting results not only show where S1 binds to the TIR (protection against DEPC and RNases PhyM and T2) but also imply the presence of certain spatial (tertiary) structure that can be changed upon S1 binding (e.g. S1-mediated conformational changes in loops I and II).

Fig. 6. Footprint analysis of S1 binding to the rpsA TIR. (A) Diethyl pyrocarbonate (DEPC) modification of the A-residues within the rpsA mRNA in the absence (–) or presence (+) of S1 (2.4 µM). U, G, C, A lanes, dideoxy-sequencing of in vitro RNA transcript by primer extension. Structural elements (vertical bars) and certain positions of the rpsA TIR are indicated on the left and on the right of the panel. The A-residues that are modified only in the presence of S1 are indicated by arrows. (B) Partial digestion of the rpsA mRNA with RNase T1 in the absence (lane 1) or presence of increasing concentration of S1: lane 2, 0.5 µM; lane 3, 1.0 µM; lane 4, 2.0 µM. (C) The rpsA TIR structure with indication of positions protected by S1 against DEPC (black triangles), and positions with increased reactivity towards DEPC (gray triangles) or T1 digestion (gray circles) in the presence of S1.
Taken together, the in vitro data allow us to conclude that extended ss-regions of the rpsA TIR, except loops I and II, are involved in interaction with free S1 upon repressor complex formation; this interaction is able to change the rpsA TIR conformation, on the one hand, and to prevent 30S initiation complex formation, on the other.
Discussion
Structural and functional organization of the rpsA TIR
Here, we have shown that both the unusually high translation activity of the E.coli rpsA TIR and its strong autocontrol, preventing accumulation of free S1 in a cell, are mediated by the specific folding of the TIR into three hairpins I–II–III. The predicted fold has a strong phylogenetic support, indicating that it is a characteristic feature of a wide group of proteobacteria related to E.coli (Figure 2). Despite large sequence divergences, the rpsA TIRs within this group share some remarkable features, one of which is the absence of a canonical SD-element upstream of the start codon. The latter appears to be specific for species from gamma subdivision only, because the rpsA leaders from other groups of proteobacteria contain a ‘normal’ SD-element, e.g. the rpsA RBS of Rhizobium meliloti (alpha subdivision) comprises classical AGGAG sequence properly spaced (5 nt) from the initiator AUG (DDBJ/EMBL/GenBank accession No. X07528). It is likely that these organisms have evolved a different way to control S1 synthesis.
It should be noted that within the gamma subdivision there exists branches that have developed the rpsA TIR structure differing from that of E.coli. Thus, in our search for rpsA TIRs within accessible microbial genomes, we used the beginning of the E.coli rpsA coding sequence as a probe, assuming that this region is important for TIR folding, and we did not find the rpsA TIR from Pseudomonas species, although the corresponding sequences are available. Indeed, it appears that the rpsA TIRs of Pseudomonas aeruginosa (AE004740) and Pseudomonas putida (unfinished genome, data provided by TIGR website) comprise ‘normal’ SD-elements (AGGU in P.aeruginosa, AGGA in P.putida) and share a folding pattern that is completely different from that shown on Figure 2. As recently reported, specific features of the S10 leader essential for L4-mediated autogenous control in E.coli are also conserved in several branches of gamma-proteobacteria, but the S10 leader from P.aeruginosa shows completely different folding (Allen et al., 1999). Altogether, these observations indicate that fine mechanisms for regulation of r-protein synthesis likely evolved later than Pseudomonas species and other gamma-proteobacteria had diverged.
As shown here for the E.coli rpsA TIR, the degeneration of the SD-domain is strictly necessary for autogenous control. The absence of a consensus SD should help free S1 to compete with the 30S subunit for rpsA TIR binding. The alteration of the GAAG sequence for a canonical GAGG element relieves autocontrol (30S wins) and increases the apparent activity of the TIR (Table II). We believe that this increase is mainly a consequence of the substantial loss of autogenous repression (at the steady-state cellular concentration of S1, the wild-type rpsA TIR is still repressed ∼3-fold, see Boni et al., 2000), as mutations –27C→G and –52C→G also cause increases in activity and a similar loss of autocontrol (Table II). Although the sequence around position –27 resulting from –27C→G mutation is reminiscent of a classical SD-element (GGAGG), the 26-nt region separating it from the start codon is too long to represent an appropriate spacing (5–11 nt according to Ringquist et al., 1992), and can not form a hairpin structure able to bring SD and AUG in a closer proximity.
Two of the above mutations introduced here by site-directed mutagenesis (–27C→G and –9A→G) were previously selected as increasing the expression of the rpsA–lacZ fusion from a plasmid, and this increase was ascribed to creation of ‘strong’ SD sequences enhancing the rpsA TIR translational activity (Rasmussen et al., 1993). Here, we directly show that effect of the –27C→G mutation is not related to creation of a new SD sequence, because the symmetrical –52C→G mutation has an analogous effect. Both mutations strengthen the bottom part of the hairpin II, which was shown to be involved in S1 binding and melted by free S1 (Figure 6). We suppose that strengthening of the bottom part of hairpin II hampers the formation of a tight repressor complex and hence causes the observed loss of autogenous control (and apparent increase in TIR activity) by disfavoring S1 in its competition with 30S. Disruption of the bottom helix (–26G→C) has an opposite effect, presumably by favoring S1 binding and increasing its ability to compete with 30S. A substantial drop in translation efficiency in this mutant can be completely eliminated by the second mutation (–26G→C, –53C→G) restoring the bottom helix (Table II). The effects of these mutations on the TIR activity and autocontrol not only support the proposed structure (Figure 1) but also emphasize its important role in regulation of S1 synthesis.
GG-motifs in apical loops I and II may form a non-contiguous SD-element
The most striking result of mutagenic analysis is that two highly conserved GG-motifs in apical loops I and II, far upstream from the start codon, are necessary for high translation activity of the rpsA TIR (Table II). Both GG-elements should be present simultaneously, as each GG→AU mutation (Table II) or deletion of hairpin I (Table I, 66-nt leader) causes approximately the same drop in translation of the rpsA–lacZ fusion. Point mutations of G-residues in loop I also have a negative effect, albeit less pronounced (Table II). Aside from results of site-directed mutagenesis, the functional role of GG-sequences in the apical loops could be predicted from their strict conservation in the rpsA leaders of bacteria related to E.coli (Figure 2).
The fact that short sequence elements situated far upstream of the canonical RBS (estimated to comprise the nucleotides from –20 to +15 around initiation codon, see Gold, 1988) affect translation efficiency so significantly, suggests that they could be involved in direct interactions with a ribosome. According to SELEX results (Ringquist et al., 1995), there are only two components of 30S ribosomal subunit that directly participate in mRNA selection—the anti-SD sequence at the 3′-end of 16S rRNA, and protein S1. As footprinting of S1 bound to the rpsA TIR argues against direct interaction of S1 with loops I and II (Figure 6A and B), one may suppose that GG(A)-sequences represent targets for the anti-SD sequence, i.e. they may form a spatial, non-contiguous SD-element.
The rpsA TIR as an example of IRES-like prokaryotic element
Although a common model for translation initiation in bacteria (Figure 7A) considers the presence of stable secondary structure in the vicinity of the initiator codon as an inhibitory factor (de Smit, 1998 and references herein), data have already accumulated showing the existence of non-inhibitory or even positive structured elements within TIRs of prokaryotic mRNAs (Gold, 1988; Sacerdot et al., 1998; Nivinskas et al., 1999). According to these data, the ribosomal mRNA binding track can tolerate the presence of rather long stable hairpins that bring together and optimize the arrangement of crucial mRNA sequence elements involved in initiation complex formation (Figure 7B and C). Thus, the RBS of the T4 gene 38 includes an extremely stable hairpin between the SD domain and start codon that was suggested to increase the speed at which the AUG is positioned in contact with the anticodon without interfering with joining of the 50S particle (Gold, 1988). The RBS of the T4 gene 25 was also proposed to form a hairpin that brings the SD-sequence into proximity with the start codon (Nivinskas et al., 1999; Figure 7B). The threonyl-synthetase mRNA is another example of non-contiguous (‘split’) RBS: its efficiency is provided not only by a conventional RBS but also by an upstream ss-region (‘enhancer’ on Figure 7C) separated from the SD-element by a strong hairpin structure (Sacerdot et al., 1998).

Fig. 7. Models for ternary initiation complex formation on mRNAs with canonical (A) or non-contiguous ribosome binding sites (B–D). Models (B)–(D) demonstrate that the ribosomal mRNA binding track can tolerate the presence of stable secondary structures which properly arrange essential sequence elements involved in ribosome binding (see text for comments). The enhancer (open boxes) is a single-stranded region found within 5′-leaders of many mRNAs that positively affects their translation efficiency (‘enhancing effect’). This enhancing effect can result from additional complementarity to the 16S RNA (as proposed by Olins and Rangwala, 1989) or from preferential binding by S1 within the 30S subunit (Boni et al., 1991; Zhang and Deutscher, 1992).
We believe that the rpsA TIR also represents a non-contiguous ribosome-binding region (Figure 7D). In contrast to the examples discussed above, where stable stem–loop structures do not form functional contacts with ribosome, the rpsA TIR illustrates a situation where the apical loops of stable hairpins, by forming a non-contiguous SD-element, might directly interact with the ribosome mRNA binding centre. An alternative is that 30S recruitment proceeds via interaction of S1 (within 30S) with the ss-regions in between the hairpins (which could serve as an ‘enhancer’ or ribosome entry site; see Figure 7D). In this case, the GG-motifs in loops I and II might be necessary to create an optimal tertiary structure of the TIR, allowing the ss-regions 1 and 2 to adopt a proper conformation for efficient recognition by S1 when it acts as a key mRNA binding component of 30S subunit.
Both variants (Figure 7D) imply that the rpsA TIR spatial (tertiary) structure is well adjusted to the 30S ribosome surface and, in this aspect, it can be regarded as a prokaryotic IRES-like element, reminiscent of the IRESes (internal ribosome entry sites) of picornavirus RNAs with their highly developed structure. According to the current model, the specific tertiary structure of IRESes serves to properly arrange short sequences for efficient contacts with translational apparatus, thus giving kinetic advantages for translation of downstream coding sequences (Jackson, 1996). A similar prediction (kinetic effect) can be made for the rpsA TIR structure that may facilitate transition from initiation to elongation by reducing ‘the clearing time’ (the time taken for the ribosome to clear the RBS for the entry of the subsequent ribosome). Although the rpsA TIR described here represents a so-far unique example of a stable secondary (or even tertiary) structure required for translation efficiency in bacteria, such a system seems to be adopted by chloroplasts, where long highly folded mRNA leaders are able to provide efficient translation initiation in the absence of SD interactions (Fargo et al., 1999 and references herein).
The model for translational control of ribosomal protein S1 synthesis
Taken together, the results obtained allow us to propose a mechanism for translation regulation of S1 synthesis. According to our model, the rpsA TIR has a very high intrinsic activity because of the optimal spatial arrangement of crucial elements involved in direct interaction with 30S ribosomal subunit (see Figure 7D). As no conventional SD-element is present, the recruitment of ribosomes proceeds mainly via this specifically folded TIR.
The mechanism underlying autogenous repression of S1 synthesis involves the specific binding of free S1 (most likely several molecules) to A/U-rich ss-regions, in between the hairpins, that disrupts optimal spatial arrangement of the TIR (Figure 8). This disruption is likely to be based on the so-called melting activity of S1, which directly follows from its exclusive binding to single-stranded RNA. Earlier studies on S1 binding to the RNA fragment, which forms the hairpin comprising two helices separated by the bulge, gave compelling evidence that under physiological conditions S1 can melt out only the weak bottom helix (which is partially unfolded) but not the strong one (Yuan et al., 1979). Analogously, our T1 footprinting results imply that only the bottom helix of hairpin II is melted by free S1 (G-26 becomes accessible), but accessibility of G-residues in the strong helices (hairpin I and an upper helix in hairpin II) does not change (Figure 6B). Moreover, strengthening of the bottom helix II (mutations –27C→G and –52C→G) almost abolishes autogenous control, most likely because free S1 can not melt the helix and perturb the TIR conformation. In contrast, an opening of the bottom helix in the –26G→C mutant has a strong negative effect on the TIR activity by disrupting its active conformation and favoring repressor complex formation. As expected, this negative effect can be reversed by a second mutation (–53C→G) restoring the bottom helix (Table II).
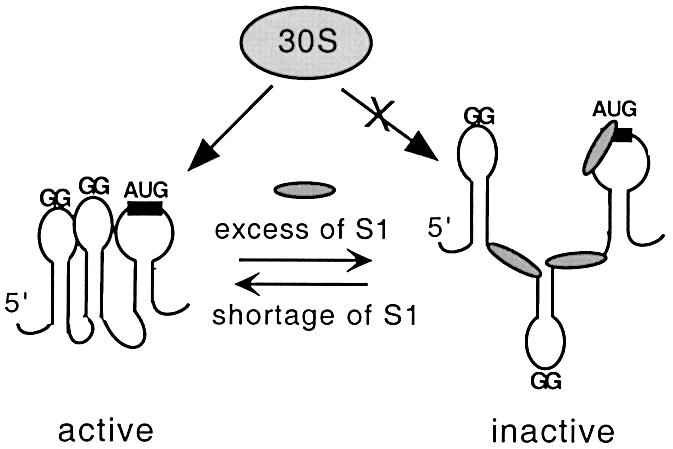
Fig. 8. Model for autogenous regulation of protein S1 synthesis. Active conformation of the rpsA TIR can be bound either by 30S subunit, thus providing efficient translation of the rpsA mRNA, or by free S1 when it is present in excess over ribosomes. Free S1 disturbs the active conformation of its cognate TIR, thereby acting as a specific translational repressor.
S1 is able to serve as a repressor only in the absence of conventional SD. Once the vestigial GAAG is changed for the canonical GAGG element (or vice versa, the anti-SD CCUC is mutated to CUUC; see Jacob et al., 1987), free S1 cannot compete with 30S for the TIR, just like it cannot serve as a repressor in vivo for any canonical mRNA (see Boni et al., 2000). Equilibrium between the repressed (inactive) state of the rpsA mRNA and its active conformation involved in initiation complex formation can explain how a steady-state amount of S1 in a cell could be continuously adjusted to the ribosome level, since only S1-containing ribosomes are active in translation initiation. We believe that in vivo and in vitro results obtained here are consistent with this scheme (Figure 8). At the same time, more work is required to define which of the two alternative ways (Figure 7D) is used by the rpsA TIR for primary ribosome recruitment.
Materials and methods
Bacterial strains
Strain XL1-blue (Stratagen) was used for plasmid propagation. Strain ENSO (formerly HfrG6Δlac12; Dreyfus, 1988) carries a deletion covering the promoter and RBS of the lacZ gene and was used for construction of chromosomal rpsA–lacZ fusions. Strain RZ1032 [Hfr KL16 PO/45 (lysA61-62) dut1ung1 thi1 relA1 supE44 zbd-279::Tn10] was used for preparation of uridilated single-stranded DNA for site-directed mutagenesis (Kunkel et al., 1987).
Construction of the rpsA′–′lacZ translational fusions
The in-frame cloning of DNA fragments corresponding to the rpsA TIR and its 5′-truncated variants in pEMBLΔ46 (a derivative of pEMBL8+ lacking the lacZ RBS) between the lac operator and the lacZ coding sequence was described earlier (Boni et al., 2000). The resulting plasmids pES1145, pES191, pES182, pES166, pES145 and pES129 (where the numbers 145, 91, etc. correspond to the length of the rpsA leader) were then used to transfer the rpsA–lacZ fusions onto the chromosome of the ENSO strain of E.coli by homologous recombination (Dreyfus, 1988). The strains obtained were then transformed either by the S1-expressing plasmid pSP261 (Pedersen et al., 1984) or by the parent vector pACYC184, and β-galactosidase activities were measured essentially as described (Boni et al., 2000).
Site-directed mutagenesis of the rpsA regulatory region
Plasmid pES191 bearing 91-nt rpsA leader was prepared as uridilated single-stranded DNA, and various point or double mutations were introduced using appropriate oligonucleotides according to Kunkel et al., (1987). Selected plasmids, shown by sequencing to carry the desired mutations, were used to transfer the mutated rpsA TIRs onto the chromosome of ENSO as described above. The resulting strains, in which synthesis of β-galactosidase was driven by mutated rpsA TIRs, were then transformed either by pSP261 or pACYC184 and tested for β-galactosidase activity.
Phylogenetic analysis
To find out whether the specific features of the E.coli rpsA TIR are conserved in other bacteria, we used the first 100 nt of the E.coli rpsA coding sequence (U00096) as a searching probe for homologous regions in ‘microbial genomes: finished and unfinished’ at NCBI BLAST server (http://www.ncbi.nim.nih.gov./blast). The best alignments for the very beginning of the coding sequence were obtained for species from gamma subdivision of proteobacteria: Enterobacteriaceae—Salmonella typhimurium LT2 (unfinished fragment WUGSC_99287/stmlt2-E2.Contig574); Salmonella typhi CT18 (unfinished fragment Sanger_601/ S.typhi_CT18); Salmonella paratyphi A (unfinished fragment WUGSC_32027/spara_B_SPA.0.955); Klebsiella pneumoniae (unfinished fragment WUGSC_573/kpneumo_B_KPN.Contig891); Yersenia pestis (unfinished fragment Sanger_632/Y.pestis_Contig1001); Vibrionaceae—Vibrio cholerae chromosome 1 (AE003852); Pasteurellaceae—Pasteurella multocida PM70 (unfinished fragment CBCUMN_747/pmultocida_Contig); Actinobacillus actinomycetemcomitans (unfinished fragment OUACGT_714/A.actin_182); Haemophilus influenzae Rd (L42023); Alteromonadaceae—Shewanella putrefaciens (unfinished fragment TIGR_24/sputr_6412). Sequence data on the Buchnera species were from DDBJ/EMBLGenBank: B.aphidicola (L43549) and Buchnera sp. APS (AP001118). For each organism, the sequence upstream from the rpsA start codon was found on the websites provided by the BLAST server or in DDBJ/EMBL/GenBank, and rpsA TIR folding was performed using the E.coli rpsA TIR structure (Figure 1) as a model. All the TIRs can be folded in a similar manner (Figure 2), including Pasteurella multocida and Buchnera sp. APS, which are not shown.
Toeprinting assay
Extension inhibition analysis of 30S initiation complex formation, or toeprinting (Hartz et al., 1988), was performed with the rpsA and ssb mRNAs essentially as described (Boni et al., 1991; Tzareva et al., 1993, 1994). The linearized plasmids pMBS1-8 (a derivative of pGEM-3Z; Tzareva et al., 1993) and pGS 2.3 (a derivative of pGEM2; Boni et al., 1991) bearing the rpsA and ssb genes of E.coli, respectively, were used for in vitro mRNA synthesis with T7 RNA-polymerase (Riboprobe Gemini System II; Promega). Preparations of 30S subunits lacking S1 (30S-S1) and free S1 were described (Boni et al., 1991). The binding buffer used for ternary initiation complex formation and for other in vitro experiments (see below) was 20 mM Tris–HCl pH 7.6, 10 mM MgCl2, 100 mM NH4Cl, 6 mM 2-mercaptoethanol. Each reaction (10 µl) contained 4 pmol of 30S-S1 subunits, 40 pmol of uncharged initiator tRNA of E.coli (Sigma), ∼0.4 pmol of the mRNA annealed with the cognate 5′-labeled primer, and increasing concentration of free S1 (or an equal volume of binding buffer). The 17-nt ssb primer 5′-GGC AACTGCGCCACCAT is complementary to the region (+76 to +92) of the ssb mRNA, and the 18-nt rpsA primer 5′-TAGCAACAA CAACGCCAC to the region (+74 to +91) of the rpsA mRNA. Reaction mixes were incubated at 37°C for 10 min and analyzed by primer extension with AMV reverse transcriptase (Promega).
Gel mobility shift assay
To prepare high specific activity RNA probes, the BamHI–HindIII fragments comprising the rpsA TIRs with truncated rpsA leaders were recloned from pEMBL-derivatives (pES1145-pES129 series, see above) into the BglII–HindIII sites of pSP73 (Promega) under the control of the pSP6 promoter. The resulting plasmids (pSS1145–pSS129 series) were linearized at HindIII site and used for run-off transcription in vitro in the presence of [32P]αUTP (3000 Ci/mmol, Amersham). The labeled RNA fragments were incubated for 10 min at 37°C in the binding buffer with or without addition of free S1, then chilled on ice and separated at 10°C on 5% native polyacrylamide gel.
Footprinting of S1 bound to the rpsA mRNA
The rpsA mRNA (0.3–0.4 pmol, obtained as described for toeprinting experiments) was preincubated with or without purified S1 for 10 min at 37°C in 8 µl of binding buffer, then 3 µg of carrier tRNA and 1 µl of DEPC (undiluted stock) or ribonuclease solution were added, followed by incubation at 37°C. Incubation with DEPC was for 3 min; partial digestions were performed with RNases PhyM (A/U-specific, 0.5 U per assay, 7 min), T2 (non-specific with preference to A residues, 10–4 U, 7 min) and T1 (G-specific, 0.02 U, 30 s). Reactions were stopped by addition of 20 µl of stop-solution (0.4 M sodium acetate pH 5.2, 20 mM EDTA, 4 µg of carrier tRNA). RNA was extracted with phenol, precipitated with ethanol, recovered from pellets, then annealed with 5′-labeled rpsA primer and analyzed by primer extension as in the toeprinting assay.
Acknowledgments
Acknowledgements
We thank S.Pedersen for pSP261, and I.Iost, M.Springer, M.de Smit and J.van Duin for fruitful discussions and comments. We acknowledge The Institute for Genomic Research (TIGR) for providing preliminary sequence data on its website at http://www.tigr.org. This work was supported by the RFBR grants 97-04-48834 and 00-04-48115 to I.V.B. and by grants from ARC (no. 5474) and MENRT (program ‘Microbiologie’) to M.D. I.V.B. has been supported by a ‘Chercheur Associé’ fellowship from CNRS, France.
References
- Allen T., Shen,P., Samsel,L., Liu,R., Lindahl,L. and Zengel,J.M. (1999) Phylogenetic analysis of L4-mediated autogenous control of the S10 ribosomal protein operon. J. Bacteriol., 181, 6124–6132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boni I.V., Issaeva,D.M., Musychenko,M.L. and Tzareva,N.V. (1991) Ribosome-messenger recognition: mRNA target sites for ribosomal protein S1. Nucleic Acids Res., 19, 155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boni I.V., Artamonova,V.S. and Dreyfus,M. (2000) The last RNA-binding repeat of the Escherichia coli ribosomal protein S1 is specifically involved in autogenous control. J. Bacteriol., 182, 5872–5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bycroft M., Hubbard,T.J., Proctor,M., Freund,S.M. and Murzin,A.G. (1997) The solution structure of the S1 RNA binding domain: a member of an ancient nucleic acid-binding fold. Cell, 88, 235–242. [DOI] [PubMed] [Google Scholar]
- Chen H., Bjerknes,M., Kumar,R. and Jay,E. (1994) Determination of the optimal aligned spacing between the Shine–Dalgarno sequence and the translation initiation codon of Escherichia coli mRNAs. Nucleic Acids Res., 22, 4953–4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Smit M.H. (1998) Translational control by mRNA structure in eubacteria: molecular biology and physical chemistry. In Simons,R.W. and Grunberg-Manago,M. (eds), RNA Structure and Function. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 495–540.
- de Smit M.H. and van Duin,J. (1990) Control of prokaryotic translation initiation by mRNA secondary structure. Prog. Nucleic Acid Res. Mol. Biol., 38, 1–35. [DOI] [PubMed] [Google Scholar]
- Draper D.E. and Reynaldo,L.P. (1999) RNA binding strategies of ribosomal proteins. Nucleic Acids Res., 27, 381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper D.E. and von Hippel,P.H. (1978) Nucleic acid binding properties of Escherichia coli ribosomal protein S1. II. Co-operativity and specificity of binding site II. J. Mol. Biol., 122, 339–359. [DOI] [PubMed] [Google Scholar]
- Dreyfus M. (1988) What constitutes the signal for the initiation of protein synthesis on Escherichia coli mRNAs? J. Mol. Biol., 204, 79–94. [DOI] [PubMed] [Google Scholar]
- Dunn J.J., Buzash-Pollert,E. and Studier,F.W. (1978) Mutations of bacteriophage T7 that affect initiation of synthesis of the gene 0.3 protein. Proc. Natl Acad. Sci. USA, 75, 2741–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehresmann C., Baudin,F., Mougel,M., Romby,P., Ebel,J.-P. and Ehresmann,B. (1987) Probing the structure of RNAs in solution. Nucleic Acids Res., 15, 9109–9128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fargo D.C., Boynton,J.E. and Gillham,N.W. (1999) Mutations altering the predicted secondary structure of a chloroplast 5′ untranslated region affect its physical and biochemical properties as well as its ability to promote translation of reporter mRNAs both in the Chlamydomonas reinhardtii chloroplast and in Escherichia coli. Mol. Cell. Biol., 19, 6980–6990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goelz S. and Steitz,J. (1977) Escherichia coli ribosomal protein S1 recognizes two sites in bacteriophages Qβ RNA. J. Biol. Chem., 252, 5177–5179. [PubMed] [Google Scholar]
- Gold L. (1988) Post-transcriptional regulatory mechanisms in Escherichia coli. Annu. Rev. Biochem., 57, 199–233. [DOI] [PubMed] [Google Scholar]
- Hartz D., McPheeters,D.S., Traut,R. and Gold,L. (1988) Extension inhibition analysis of translation initiation complexes. Methods Enzymol., 164, 419–425. [DOI] [PubMed] [Google Scholar]
- Jackson R.J. (1996) Comparative view of initiation site selection mechanisms. In Hershey,J.W.B., Mathews,M.B. and Sonenberg,N. (eds), Translational Control. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 71–112.
- Jacob W.F., Santer,M. and Dahlberg,A.E. (1987) A single base change in the Shine–Dalgarno region of 16S rRNA of Escherichia coli affects translation of many proteins. Proc. Natl Acad. Sci. USA, 84, 4757–4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel T.A., Roberts,J.D. and Zakour,R.A. (1987) Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol., 154, 367–382. [DOI] [PubMed] [Google Scholar]
- McCarthy J.E.G. and Gualerzi,C. (1990) Translational control of prokaryotic gene expression. Trends Genet., 6, 78–85. [DOI] [PubMed] [Google Scholar]
- Miranda G., Schuppli,D., Barrera,I., Hausherr,C., Sogo,J.M. and Weber,H. (1997) Recognition of bacteriophage Qβ plus strand RNA as a template by Qβ replicase: role of RNA interactions mediated by ribosomal protein S1 and host factor. J. Mol. Biol., 267, 1089–1103. [DOI] [PubMed] [Google Scholar]
- Mogridge J. and Greenblatt,J. (1998) Specific binding of Escherichia coli ribosomal protein S1 to boxA transcriptional antiterminator RNA. J. Bacteriol., 180, 2248–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nivinskas R., Malys,N., Klausa,V., Vaikunaite,R. and Gineikiene,E. (1999) Post-transcriptional control of bacteriophage T4 gene 25 expression: mRNA secondary structure that enhances translational initiation. J. Mol. Biol., 288, 291–304. [DOI] [PubMed] [Google Scholar]
- Olins P.O. and Rangwala,S.H. (1989) A novel sequence derived from bacteriophage T7 mRNA acts as an enhancer of translation of the lacZ gene of Escherichia coli. J. Biol. Chem., 264, 16973–16976. [PubMed] [Google Scholar]
- Pedersen S., Skouv,J., Kajitani,M. and Ishihama,A. (1984) Transcriptional organization of the rpsA operon of Escherichia coli. Mol. Gen. Genet., 196, 135–140. [DOI] [PubMed] [Google Scholar]
- Rasmussen M.D., Sorensen,M.A. and Pedersen,S. (1993) Isolation and characterization of mutants with impaired regulation of rpsA, the gene encoding ribosomal protein S1 of Escherichia coli. Mol. Gen. Genet., 240, 23–28. [DOI] [PubMed] [Google Scholar]
- Ringquist S., Shinedling,S., Barrick,D., Green,L., Binkley,J., Stormo,G.D. and Gold,L. (1992) Translation initiation in Escherichia coli: sequences within the ribosome binding site. Mol. Microbiol., 6, 1219–1229. [DOI] [PubMed] [Google Scholar]
- Ringquist S., Jones,T., Snyder,E., Gibson,T., Boni,I. and Gold,L. (1995) High-affinity RNA ligands to Escherichia coli ribosomes and ribosomal protein S1: comparison of natural and unnatural binding sites. Biochemistry, 34, 3640–3648. [DOI] [PubMed] [Google Scholar]
- Roberts M.W. and Rabinovitz,J.C. (1989) The effect of Escherichia coli ribosomal protein S1 on the translational specificity of bacterial ribosomes. J. Biol. Chem., 264, 2228–2235. [PubMed] [Google Scholar]
- Sacerdot C., Caillet,J., Graffe,M., Eyermann,F., Ehresmann,B., Ehresmann,C., Springer,M. and Romby,P. (1998) The Escherichia coli threonyl-tRNA synthetase gene contains a split ribosomal binding site interrupted by a hairpin structure that is essential for autoregulation. Mol. Microbiol., 29, 1077–1090. [DOI] [PubMed] [Google Scholar]
- Schwartz M., Roa,M. and Debarbouille,M. (1981) Mutations that affect lamB gene expression at a posttranscriptional level. Proc. Natl Acad. Sci. USA, 78, 2937–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skouv J., Schnier,J., Rasmussen,M.D., Subramanian,A.R. and Pedersen,S.(1990) Ribosomal protein S1 of Escherichia coli is the effector for regulation of its own synthesis. J. Biol. Chem., 265, 17044–17049. [PubMed] [Google Scholar]
- Sorensen M.A., Fricke,J. and Pedersen,S. (1998) Ribosomal protein S1 is required for translation of most, if not all, natural mRNAs in Escherichia coli in vivo. J. Mol. Biol., 280, 561–569. [DOI] [PubMed] [Google Scholar]
- Subramanian A.R. (1983) Structure and functions of ribosomal protein S1. Prog. Nucleic Acid Res. Mol. Biol., 28, 101–142. [DOI] [PubMed] [Google Scholar]
- Subramanian A.R. (1984) Structure and functions of the largest Escherichia coli ribosomal protein. Trends Biochem. Sci., 9, 491–494. [Google Scholar]
- Tzareva N.V., Musychenko,M.L. and Boni,I.V. (1993) Secondary structure analysis of the regulatory region of the Escherichia coli rpsA mRNA. Bioorg. Khim., 19, 968–977. [PubMed] [Google Scholar]
- Tzareva N.V., Makhno,V.I. and Boni,I.V. (1994) Ribosome-messenger recognition in the absense of the Shine-Dalgarno interactions. FEBS Lett., 337, 189–194. [DOI] [PubMed] [Google Scholar]
- Yuan R.C., Steitz,J.A., Moore,P.B. and Crothers,D.M. (1979) The 3′ terminus of 16S rRNA: secondary structure and interaction with ribosomal protein S1. Nucleic Acids Res., 7, 2399–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengel J.M. and Lindahl,L. (1994) Diverse mechanisms for regulating ribosomal protein synthesis in Escherichia coli. Prog. Nucleic Acid Res. Mol. Biol., 47, 331–370. [DOI] [PubMed] [Google Scholar]
- Zhang J. and Deutscher,M.P. (1992) A uridine-rich sequence required for translation of prokaryotic mRNA. Proc. Natl Acad. Sci. USA, 89, 2605–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]