

Abstract
HPr kinase/phosphatase (HprK/P) is a key regulatory enzyme controlling carbon metabolism in Gram- positive bacteria. It catalyses the ATP-dependent phosphorylation of Ser46 in HPr, a protein of the phosphotransferase system, and also its dephosphorylation. HprK/P is unrelated to eukaryotic protein kinases, but contains the Walker motif A characteristic of nucleotide-binding proteins. We report here the X-ray structure of an active fragment of Lactobacillus casei HprK/P at 2.8 Å resolution, solved by the multiwavelength anomalous dispersion method on a seleniated protein (PDB code 1jb1). The protein is a hexamer, with each subunit containing an ATP-binding domain similar to nucleoside/nucleotide kinases, and a putative HPr-binding domain unrelated to the substrate-binding domains of other kinases. The Walker motif A forms a typical P-loop which binds inorganic phosphate in the crystal. We modelled ATP binding by comparison with adenylate kinase, and designed a tentative model of the complex with HPr based on a docking simulation. The results confirm that HprK/P represents a new family of protein kinases, first identified in bacteria, but which may also have members in eukaryotes.
Keywords: catabolite repression/HPr phosphorylation/Lactobacillus casei/P-loop/protein kinase
Introduction
Bacteria are highly adaptive organisms capable of growing under a great variety of environmental conditions. A key to adaptability is the large number of catabolic genes enabling bacteria to use many different kinds of carbon sources. Most bacteria possess a variety of metabolic pathways to optimize the metabolism or de novo synthesis of carbohydrates. The expression of the catabolic genes can be turned on and off in response to the composition of the environment, i.e. the available carbon and energy sources. To do so, signals must be sensed and converted via signal transduction systems into cellular responses, such as gene activation or repression. Carbon catabolite repression (CCR) is one of the ways bacteria respond to metabolic and environmental conditions (Stülke and Hillen, 2000; Deutscher et al., 2001). In Bacillus subtilis, the expression of ∼10% of the genome is regulated by CCR, including genes coding for enzymes of central metabolic pathways (Miwa et al., 2000; Moreno et al., 2001).
In Gram-positive bacteria, the first step in the CCR signal transduction cascade is the ATP-dependent phosphorylation of the phosphocarrier protein HPr. HPr is phosphorylated on a histidine as part of the phosphoenolpyruvate:glycose phosphotransferase system (PTS), which transports carbohydrates (Postma et al., 1993). In the presence of high concentrations of glycolytic intermediates (e.g. fructose-1,6-bisphosphate; FBP), HPr can also be phosphorylated on Ser46 by the bifunctional HPr kinase/phosphatase (HprK/P; Figure 1) (Deutscher and Saier, 1983; Deutscher et al., 1986). HprK/P is also capable of slowly phosphorylating Ser46Thr mutant HPr (Reizer et al., 1989). High concentrations of inorganic phosphate (Pi) stimulate the reverse reaction, i.e. dephosphorylation of Ser46, catalysed by the same protein. Gram-positive bacteria taking up a rapidly metabolizable carbohydrate have a high concentration of FBP and a low amount of Pi (Thompson and Torchia, 1984), favouring the kinase function. Under these conditions, HprK/P produces serine-phosphorylated HPr (P-Ser-HPr), which interacts with the catabolite control protein A (CcpA), a member of the LacI/GalR repressor family (Henkin et al., 1991), and allows CcpA to bind to the catabolite response elements cre (Weickert and Chambliss, 1990; Fujita et al., 1995; Galinier et al., 1999). Thus, P-Ser-HPr is a co-repressor in CCR.

Fig. 1. Alignment of bacterial HprK/P sequences. The secondary structure on top is defined by DSSP (Kabsch and Sander, 1983) for the truncated L.casei subunit. Blue frames are for conserved residues, white characters in red boxes for strict identity, and red characters in white boxes for similarity. Accession numbers for Gram-positive bacteria: Lactobacillus casei SwissProt Q9RE09, Enterococcus faecalis SwissProt O07664, Streptococcus mutans SwissProt Q9ZA56, Streptococcus bovis SwissProt Q9WXK7, Streptococcus pyogenes orf1717 (http://pedant.mips.biochem.mpg.de), Streptococcus salivarius SwissProt Q9ZA98, Lactococcus lactis MOLOKO database (http://spock.jouy.inra.fr), Bacillus subtilis Swiss-Prot O34483, Bacillus halodurans TrEMBL P82557, Staphylococcus xylosus SwissProt Q9S1H5, Clostridium acetobutylicum (http://www.genomecorp.com/htdocs/sequences/clostridium/clospage.html), Mycoplasma genitalium SwissProt P47331, Mycoplasma pneumoniae Swiss-Prot P75548, Ureaplasma parvum SwissProt Q9PR69. Gram-negative bacteria: Treponema pallidum SwissProt O83600, Neisseria gonorrhoeae (http://www.ncbi.nlm.nih.gov/Microb_blast/unfinishedgenome.html), Neisseria meningitidis TrEMBL Q9K6Y4, Xylella fastidiosa SwissProt Q9PDH3. Figure realized with ESPript (Gouet et al., 1999).
Proteins related to eukaryotic Ser/Thr protein kinases do exist in bacteria, along with the histidine kinases of two-component systems and tyrosine kinases (Grangeasse et al., 1997; Hanks et al., 1988; Ilan et al., 1999; Inouye et al., 2000). The sequence of B.subtilis HprK/P displays no significant similarity to eukaryotic Ser/Thr protein kinases (Galinier et al., 1998). It does not contain the highly conserved sequence motifs characteristic of the eukaryotic protein kinase family. Instead, it contains the Walker motif A (G/AxxxxGKT/S), which is present in many nucleotide-binding proteins (Walker et al., 1982), forming a typical loop, called the P-loop, where the phosphate moiety of ATP binds. The presence of this motif in HprK/P suggests that it belongs to a new family of Ser/Thr protein kinases.
We describe here the three-dimensional structure of a truncated form of the 319 residue Lactobacillus casei HprK/P. The deletion of residues 1–127 yielded a catalytically active fragment, which could be expressed in Escherichia coli and was found to crystallize readily. The X-ray structure was solved by the multiwavelength anomalous dispersion (MAD) method on a selenomethionine-substituted protein. The resulting atomic model was refined to 2.8 Å resolution. It shows the protein to be a symmetrical hexamer where each subunit contains an ATP-binding domain similar to that of adenylate or cytidylate kinase, and a putative HPr substrate-binding domain. Inorganic phosphate was found to be present in the P-loop, helping in modelling bound ATP. Docking simulations allowed us to build a tentative model of the interaction with the HPr substrate. The present structure, the first of a prokaryotic Ser/Thr protein kinase, definitely establishes HprK/P as a paradigm for a new family of protein kinases related to nucleoside/nucleotide kinases.
Results
Protein purification and characterization
In an attempt to determine the function of the poorly conserved N-terminal part of HprK/P, we amplified by PCR a truncated L.casei hprK gene missing the 5′ part encoding amino acids 2–127 (Figure 1). The protein product exhibited in vitro enzymatic activities identical to those of the full-length protein (Materials and methods). The truncated gene was inserted into a His tag expression vector. The encoded truncated protein was overproduced in E.coli and purified as described in Materials and methods. The expression level was higher than for the full-length protein and the truncated form could be subjected to crystallization. Electrospray mass spectrometry gave a mass of 22 654 Da, which is in agreement with the calculated value (His tag included). Size-exclusion chromatography showed that the truncated form forms a stable and homogeneous oligomer of ∼140 kDa, possibly a hexamer. An identical experiment performed with the full-length L.casei HprK/P (37 kDa per monomer) also suggested that it is composed of six subunits. Equilibrium sedimentation confirmed that truncated L.casei HprK/P is a homogeneous hexamer in solution (results not shown).
We produced HprK/P containing selenomethionine in order to grow crystals suitable for MAD phasing. The substituted protein was overproduced in amounts similar to normal HprK/P, but it was found in the cell pellet and was recovered by resolubilization in 0.5% Triton X-100. Mass spectrometry using the MALDI-TOF method confirmed that all six methionines (including two in the His tag) were fully seleniated.
Structure determination and model quality
Truncated HprK/P crystallized in ammonium phosphate (Materials and methods) in space group P6322 with cell parameters a = b = 107.74 Å and c = 66.50 Å. The crystals contained one monomer per asymmetric unit and 44% solvent. Diffraction data collected near the selenium edge led to the location of four of the six expected selenium atoms. MAD phasing yielded an electron density map that could be interpreted at 3.0 Å resolution. An atomic model of HprK/P was built and refined further to 2.8 Å resolution. Residues 128–134 and 308–319 at the N- and C-terminal ends, and residues 241–252 in an internal loop, are missing from the model because the electron density was weak and could not be interpreted reliably. In addition, 14 residues had weak side chain density. The present structure (PDB code 1jb1) contains 161 residues, 47 water molecules and one phosphate ion. The crystallographic R-factor is 23.5% for the data in the 20.0–2.8 Å resolution range. The stereochemistry is correct, with 81% of the residues being in the most favoured region of a Ramachandran plot, and only one (Asp179) in a disallowed region. A summary of the crystallographic data is given in Table I.
Table I. Data collection and processing statistics.
| λ1 (inflection) | λ2 (remote) | λ3 (peak) | High resolution | |
|---|---|---|---|---|
| Data collection | ||||
| λ (Å) | 0.9792 | 0.9393 | 0.9393 | |
| resolution (Å) | 3.0 | 3.0 | 3.0 | 2.8 |
| observations | 56 655 | 57 474 | 54 528 | 94 476 |
| unique reflections | 4872 | 4873 | 4730 | 5926 |
| completeness (%) | 99.6 | 99.6 | 99.7 | 99.3 |
| I/σ | 9.9 (2.2) | 10.7 (3.6) | 10.4 (2.6) | 5.1 (1.8) |
| Rsym (%) | 5.3 (31.8) | 4.5 (20.7) | 5.3 (28.7) | 9.3 (42.6) |
| phasing power | ||||
| centric iso | – | 2.23 | 2.52 | |
| acentric iso/ano | –/2.04 | 3.22/1.36 | 2.99/2.24 | |
| Overall figure of merit | (20–3.0 Å) | |||
| centric | 0.503 | |||
| acentric | 0.578 | |||
| Refinement | ||||
| Rcryst (%) | 23.5 | |||
| Rfree (%) | 28.6 | |||
| no. of reflections used | 5557 | |||
| protein atoms | 1170 | |||
| r.m.s. deviation from ideal | ||||
| bonds (Å) | 0.01 | |||
| angles (°) | 3.05 | |||
| Ramachandran plot | ||||
| most favoured (%) | 81.0 | |||
| allowed (%) | 18.4 | |||
| disallowed (%) | 0.6 |
Values in parentheses are for the outer resolution shell.
Rsym (I) = ΣhklΣi|Ihkl,i – <Ihkl>|/Σhkl Σi|Ihkl,i|, where <Ihkl> is the mean intensity of the multiple Ihkl,i observations for symmetry-related reflections.
Rcryst = Σhkl|Fobs – Fcalc|/Σhkl|Fobs|. Rfree is for a test set including 5.6% of the data.
The side chains of R137, H140, Y184, E204, R206, W237, D240, L254, F256, D257, K268, R271, E298 and H307 are truncated at Cβ.
Subunit structure
The HprK/P monomer forms a globular α/β unit from which the C-terminal helix α4 protrudes (Figure 2). The globular unit contains 11 β-strands, βA–βK, and three α-helices, α1–α3. It is built around a central five-stranded β-sheet of topology DBCJK, where βB runs antiparallel to the four other strands. Residues G155DSGVGKS162 forming the Walker motif A are in the loop that connects βC to α1, the putative P-loop of HprK/P. Residues 241–252 connecting βJ to βK are disordered, but the 14 Å gap between Asp240 and Gln253 could be closed by building these residues in a helical conformation. One face of the β-sheet is covered by helix α1 connecting βC to βD, and helix α3 following βK. The other face of the central β-sheet packs against a second β-sheet oriented perpendicular to it, which is four-stranded and antiparallel with topology AEFI. An unusual structural motif made of a β-hairpin (strands GH), the short helix α2 and two single turns of 310 helices labelled η1 and η2 caps the top edge of the central β-sheet and will be referred to as the capping motif.
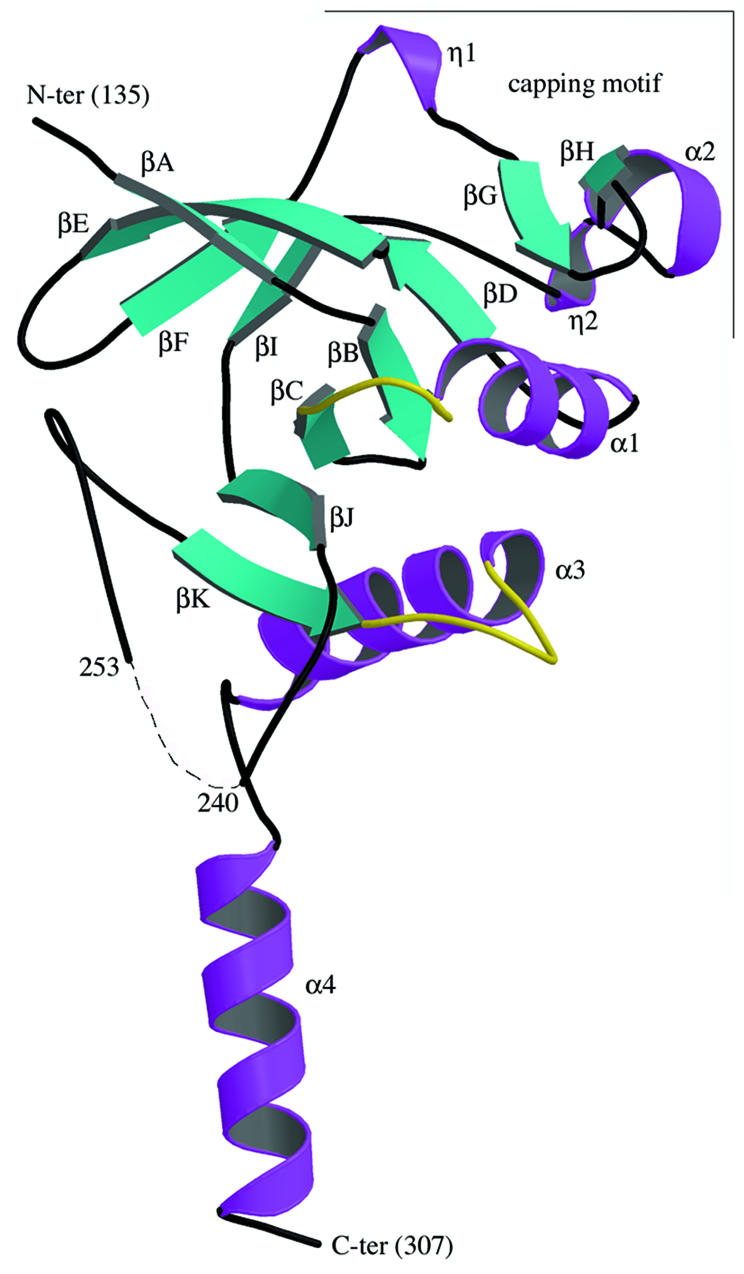
Fig. 2. The L.casei HprK/P fold: a ribbon diagram of the truncated 128–319 monomer. The N- and C-terminal residues and residues 241–252 (dashes) are disordered and missing from the model. The monomer contains 11 β-strands βA–βK and four α-helices α1–α4. The ‘capping motif’ on top includes βG, βH, α2 and two single-turn 310-helices labelled η1 and η2. The P-loop (residues 155–160), linking α1 to βC, and the K3 loop (residues 267–272), linking βK to α3, are highlighted in yellow. The P-loop contains the Walker A motif and the phosphate-binding site; the K3 loop is involved in trimer contacts. This and the following figures were drawn using MOLSCRIPT (Esnouf, 1997) and RASTER 3D (Merritt and Bacon, 1997), except Figures 4B and 6A drawn with BOBSCRIPT (Esnouf, 1999).
Quaternary structure
As observed with HprK/P in solution, it also formed hexamers in the crystal and exhibited the dihedral 3-fold symmetry of space group P6322 (Figure 3). It is composed of two trimers in two layers, with extensive contacts within trimers and weaker ones between trimers. In the hexamer, subunit contacts bury 2600 Å2 per monomer, i.e. 27% of its solvent-accessible surface area. The trimer contacts represent 2000 Å2 of this. Viewed along the 3-fold axis, each subunit seems to put an arm formed by helices α3 and α4 around the capping motif and helix α1 of a neighbouring subunit in the trimer (Figure 4A). Additional contacts at the centre of the hexamer and near the 3-fold axis involve the putative P-loops, and also the conserved P266xxxGR271 sequence. This sequence forms the loop connecting βK to α3, which we call the K3 loop. Residues 159–160 of the P-loop of one subunit and 269–270 of the K3 loop of another subunit are in direct contact.

Fig. 3. The HprK/P hexamer. The hexamer forms a 55 Å thick two-layered structure with a trimer of 80 Å diameter in each layer. The bottom trimer is in mauve. Each subunit of the top trimer is in a different colour, with the P-loop and the K3 loop highlighted in yellow. (A) View along the 3-fold axis. The six K3 loops are at the centre, the N- and C-terminal ends at the periphery of the hexamer. (B) Orthogonal view along the 2-fold axis. The P-loops, K3 loops and the disordered 241–252 residues are all located at the surface of the trimers.
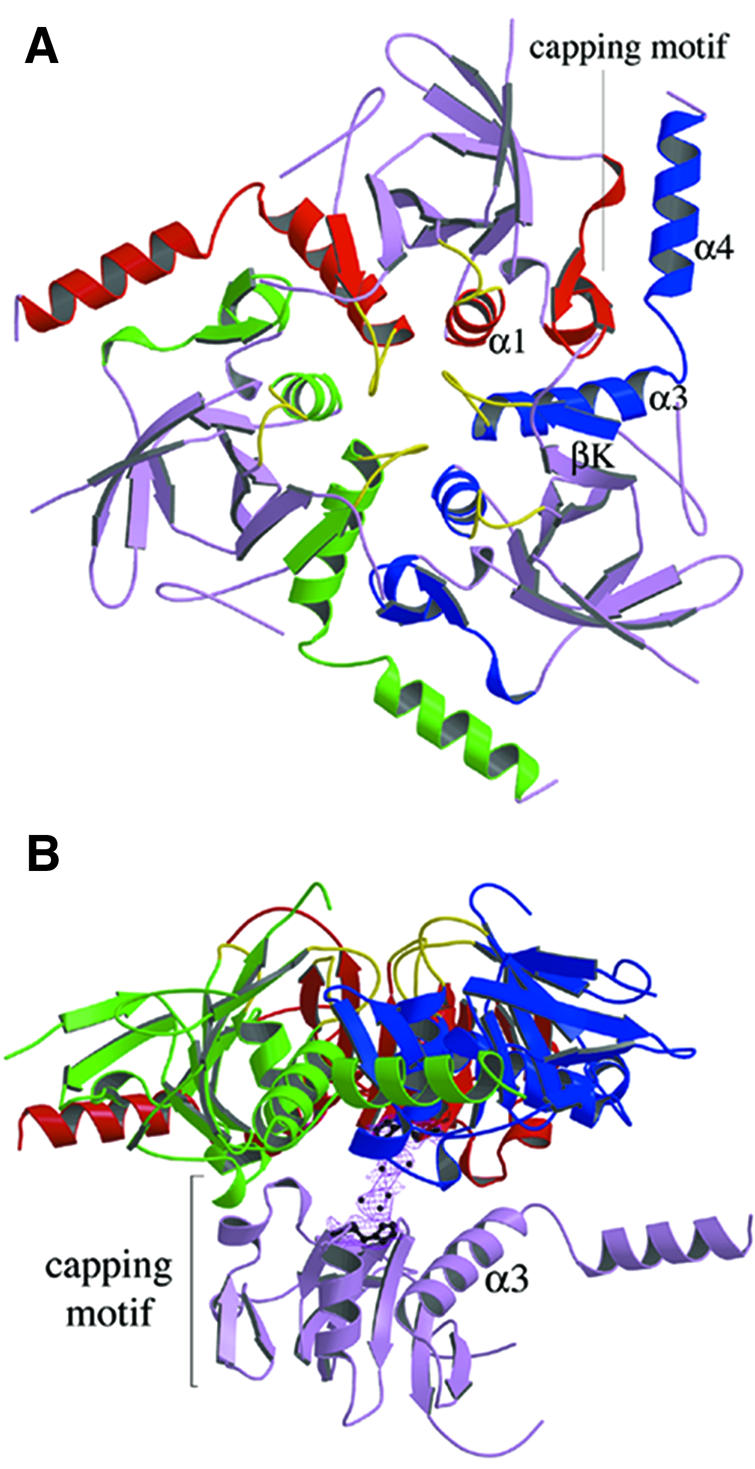
Fig. 4. Subunit contacts in the hexamer. (A) The trimer. Regions involved in the contacts are highlighted in red, green and blue, respectively, for each subunit. C-terminal helices α3 and α4 from the blue subunit pack against helix α1 and the capping motif of the red subunit. Additional contacts involve the K3 and P-loops in yellow. (B) Dimer contacts. One subunit of the bottom trimer makes contact with all three subunits of the top trimer, mostly through loops at the edge of the central β-sheet, the C-terminus of the capping motif and the C-terminus of helix α3. On the 2-fold axis, density in a (2Fo – Fc) map countoured at 1σ is interpreted as a set of water molecules. It bridges His173 from the bottom and top subunits.
The dimer interface buries only 640 Å2 per monomer, much less than the trimer interface. Nevertheless, each subunit of a trimer is in contact with all three subunits of the other trimer, mostly via loops at the N-terminal edge of the central β-sheet, the C-terminal ends of the capping motif and of helix α3 (Figure 4B). Near the 2-fold symmetry axis, a large spot of uninterpreted electron density bridges the side chain of His173 in one subunit to its counterpart in the 2-fold symmetry-related subunit. Due to the limited resolution, the density has been modelled as a cluster of water molecules, but the shape and intensity rather suggest the presence of a pair of metal ions.
Comparison with other proteins
We performed a search for structural homologues of the HprK/P subunit in a representative sample of the Protein Data Bank using program DALI (Holm and Sander, 1996). The search confirmed that HprK/P has no similarity to eukaryotic Ser/Thr protein kinases. It returned only two proteins with significant structural similarity showing a Z-score above 4. They were E.coli cytidylate kinase (Briozzo et al., 1998) and Bacillus stearothermophilus adenylate kinase (Berry and Phillips, 1998) (Figure 5). When the structures of cytidylate kinase and HprK/P were superimposed, 90 residues occupied identical positions (within a 3.8 Å Cα r.m.s. deviation), although only 14% of them were identical in the two sequences. Adenylate kinase scored almost as high.

Fig. 5. Comparison of HprK/P with cytidylate and adenylate kinases. The conserved α/β structural motif is highlighted in yellow ribbon in the HprK/P subunit and the two proteins with the highest DALI scores, E.coli cytidine monophosphate kinase (CMPK, PDB code 1cke) and B.stearothermophilus adenylate kinase (ADK, PDB code 1zin). The conserved nucleotide-binding motif includes a five-stranded parallel β-sheet and two α-helices (with the P-loop highlighted in red). However, in HprK/P, βB (in blue) is antiparallel to the other four strands whilst all strands in CMPK and ADK are parallel and the topology differs.
In the SCOP (Murzin et al., 1995) structural classifications, both HprK/P analogues belong to the superfamily of P-loop-containing nucleotide-binding proteins and the family of nucleotide and nucleoside kinases. In these kinases, the domain that binds the phosphate donor nucleotide has a α/β/α fold with a parallel five-stranded β-sheet and a distinct topology. The donor nucleotide (usually ATP or GTP complexed to Mg2+) binds at the C-terminal edge of the β-sheet with the phosphate groups in the so-called P-loop (Saraste et al., 1990). The region of the HprK/P structure that resembles cytidylate and adenylate kinases includes the central β-sheet DBCJK and helices α1 and α3 (Figure 5). Although the β-sheet topology is different, it clearly belongs to the same structural family. The remainder of the HprK/P structure, i.e. β-sheet AEFI and the capping motif, has no equivalent in other kinases or any other protein of known structure, suggesting that it is specific for the HPr substrate.
Nucleotide-binding site
In adenylate kinase and related P-loop-containing proteins, the phosphate groups of the donor nucleotide interact with main chain NH groups of the P-loop. The loop contains the Walker motif A (Walker et al., 1982), which is the only sequence motif conserved within this family. In L.casei HprK/P, the putative P-loop connects βC to α1. In this region, a high electron density is observed next to the main chain (Figure 6A). It indicates the presence of a phosphate ion interacting with the main chain nitrogens of residues 158–162 (Figure 6B), which stems from the 400 mM Pi present in the crystallization mixture. As in other nucleotide-binding proteins of this family (Via et al., 2000), the phosphate interacts with the side chains of Lys161 and Ser162. Interestingly, it is within hydrogen bonding distance of the carboxylate of Glu163, which is strictly conserved among HprK/P sequences (Figure 1). This implies that the phosphate ion is protonated on the oxygen involved in this interaction. Additional density suggests that a water molecule, or possibly a metal ion, bridges the phosphate ion to the Glu306 side chain of an adjacent hexamer. Glu306 belongs to α4 and is the penultimate residue of HprK/P visible in the electron density. The water-mediated interaction is part of the crystal packing, which may contribute to ordering α4.


Fig. 6. The P-loop and bound phosphate. (A) The P-loop connecting strand βC to helix α1. A bound phosphate ion (in red) is shown in its 2Fo – Fc electron density map contoured at 1σ. The ADP molecule in green is positioned by comparison with Sulfolobus acidocaldarius adenylate kinase (PDB code 1nks-F) (Vonrhein et al., 1998). Its β-phosphate overlaps the phosphate ion of HprK/P. (B) Stereo view of interactions made by the phosphate ion. Hydrogen bonds (dashes) link oxygen atoms of the phosphate ion to the four NH groups of P-loop residues 158–162 and to the side chains of Lys161, Ser162 and Glu163.
The bound phosphate further confirms the presence of a P-loop in HprK/P and helps in building a model of the HprK/P–nucleotide complex by comparison with other P-loop-containing proteins. A superposition with adenylate kinase in complex with ADP (Figure 6A) shows that the phosphate in HprK/P occupies the position of the β-phosphate of the nucleotide. Superposition with other members of the family confirms this result. However, HprK/P is oligomeric and, if we assume that the nucleotide has the same conformation as in adenylate kinase, the adenine base overlaps with atoms belonging to the K3 loop of a neighbouring subunit in the hexamer. Thus, either the nucleotide or the protein must change conformation to avoid overlaps. Fluorescence studies suggest that the B.subtilis HprK/P, which is also oligomeric, binds ATP cooperatively (Jault et al., 2000). This favours the hypothesis that nucleotide binding is accompanied by a protein conformational change, possibly affecting the quaternary structure.
Modelling the HPr–HprK/P interaction
In the absence of a nucleotide and the presence of Pi, the truncated form of L.casei HprK/P catalyses the dephosphorylation of P-Ser-HPr like the full-length protein (Dossonnet et al., 2000). The form of the protein present in phosphate-grown crystals should therefore be capable of binding P-Ser-HPr as its substrate. We performed a docking simulation to assess possible modes of HPr–HprK/P interaction. Several structures of HPr have been solved by crystallography and NMR. All are very similar, and the recent X-ray structure of P-Ser-HPr from Enterococcus faecalis (Audette et al., 2000) shows that phosphorylation leads only to small structural changes. HPr is made of a four-stranded antiparallel β-sheet on which the three α-helices ABC pack (Jia et al., 1994). In the PTS, HPr is phosphorylated on His15 by enzyme I (EI) at the expense of phosphoenolpyruvate, and then transfers its phospho group to the sugar-specific enzyme IIA (EIIA) (Postma et al., 1993). The two phosphorylatable residues of HPr cap α-helices: His15 is at the N-terminus of helix A and Ser46 at the N-terminus of the short helix B.
Docking of HPr on the truncated HprK/P hexamer was performed by simulated annealing (Cherfils et al., 1991). Among the docked complexes that buried the largest surface area (1770 Å2), one had a distance of 5.4 Å between Ser46 of HPr and Ser157 in the P-loop of HprK/P (Figure 7). This docked complex provides a plausible model of the mode of binding and delineates regions of the enzyme and substrate which may interact. In the model, HPr sits on top of one trimer and makes contacts with two neighbouring subunits of that trimer. The phosphate transfer reaction should take place on the subunit whose P-loop approaches Ser46. This subunit also contacts helix αA of HPr through its capping motif, which we assumed to be part of the substrate-binding domain. In the other subunit, the C-terminal helix α4 stacks parallel to helices αA and αC of HPr.

Fig. 7. A model of the interaction of HPr with HprK/P. The serine-phosphorylated form of E.faecalis HPr (Audette et al., 2000) is drawn in yellow in the position and orientation determined by the docking simulation described in the text. Green van der Waals spheres represent the phosphorylated Ser46 and phosphorylable His15 side chains. HPr is predicted to sit on the top of a HprK/P trimer and interact with both the red and the blue subunits. Its phosphoserine is near the P-loop of the red subunit, which also interacts with HPr via its capping motif. The C-terminal helix α4 of the blue subunit sits in between αA and αC of HPr. An ADP molecule is drawn in ball-and-stick bound to the red subunit as described in Figure 6.
Discussion
Although missing the first 127 amino acids, the truncated L.casei HprK/P described here carries out both catalytic functions of HprK/P. The structure, the first of a bacterial Ser/Thr protein kinase, definitely establishes that it is unrelated to eukaryotic kinases (Hanks et al., 1988; Taylor and Radzio-Andzelm, 1994). Instead, it belongs to the P-loop-containing family of nucleotide-binding proteins, with adenylate kinase as a paradigm (Walker et al., 1982). Like other proteins of this family, HprK/P has an ATP-binding domain and a domain specific for the phosphate acceptor, HPr in this case. The family has many members in both eukaryotes and prokaryotes. All members with a known function phosphorylate low molecular weight compounds: nucleosides, nucleoside monophosphates and intermediates in biosynthetic pathways. However, many others have no established function and, if they are kinases, their substrates have not been identified. The example of HprK/P suggests that some may have proteins rather than small molecules as substrates.
The N-terminal fragment missing in the truncated protein is poorly conserved in HprK/Ps from different bacteria (Figure 1). Its function remains to be determined, but it could form an independent domain, likely to be on the surface of the hexamer. The C-terminal 12 residues, present but disordered in the crystal, are also poorly conserved or absent from other HprK/Ps. In contrast, residues 135–307, which form the putative ATP- and HPr-binding domains, are highly conserved and must have the same tertiary structure in the protein from different organisms. It is less certain that the quaternary structure is conserved. Available data, based mostly on size-exclusion chromatography, suggest the presence of oligomers ranging from the dimer (Kravanja et al., 1999) to the decamer (Brochu and Vadeboncoeur, 1999) in HprK/P of different bacteria. The L.casei HprK/P is a hexamer in solution, in both the absence and presence of its ligands HPr, ATP and/or FBP (data not shown). The X-ray structure predicts the binding sites for both the nucleotide and HPr to be at subunit interfaces. Mutation experiments show that the disruption of subunit interactions leads to aggregation and loss of HPr phosphorylation (data not shown). Thus, the hexamer seems needed for structural stability and activity. Nevertheless, we cannot exclude that other HprK/Ps are not hexameric. Homologous proteins with closely related tertiary structures and different quaternary structures are not unprecedented. Thus, nucleoside diphosphate kinases, which share >45% sequence identity, exist as either hexamers or tetramers (Giartosio et al., 1996).
We performed a docking simulation to define a possible mode of HPr binding. The simulation provided a rough model, which suggests that the binding site is formed by two adjacent subunits of the HprK/P hexamer. Whilst the binding stoichiometry is unknown, the model will fit up to six HPr molecules per hexamer. On the other hand, the surface of HPr that is predicted to contact the kinase overlaps with regions known from structural studies to interact with the PTS components EI and EIIAGlc (Wang et al., 2000). This surface includes the two phosphorylatable residues Ser46 and His15. Our docking simulation places the latter not far from the N-terminus of the truncated HprK/P chain. Thus, the missing residues 1–134 of the full-length protein could make further contacts with HPr. The model fits with the observation that His15-phosphorylated HPr is a very poor substrate for Ser46 phosphorylation by HprK/P and, conversely, P-Ser46-HPr is a very poor substrate for the phosphorylation of His15 catalysed by EI of the PTS (Reizer et al., 1984).
P-Ser-HPr, the product of the HprK/P kinase reaction, is the central regulator of carbon metabolism in Gram-positive bacteria. It controls the activity of the PTS (Deutscher et al., 1994) and of several transcriptional regulators containing a PTS phosphorylation domain (Stülke et al., 1998). It participates in inducer exclusion (Dossonnet et al., 2000) and acts as co-repressor in CCR (Fujita et al., 1995). When carbon sources are plentiful, P-Ser-HPr stimulates the expression of enzymes of several metabolic pathways, including glycolysis, and prevents the synthesis of enzymes catabolizing less efficient carbon sources. The first signal seems to be an increase in FBP and a decrease in the Pi concentration (Thompson and Torchia, 1984; Neves et al., 1999). FBP and Pi are the main regulators of HprK/P, with FBP stimulating and Pi inhibiting the kinase activity. Direct binding of FBP to B.subtilis HprK/P has been demonstrated in fluorescence experiments (Jault et al., 2000). In the crystal, we observe bound Pi, but the FBP-binding site remains to be characterized. No structural feature resembling the effector binding site in l-lactate dehydrogenase (Piontek et al., 1990) or pyruvate kinase (Jurica et al., 1998), two FBP-controlled enzymes, is detected in the truncated C-terminal domain of HprK/P.
As the phosphate ion in the P-loop occupies the expected position of the β-phosphate of ATP, the X-ray structure suggests that direct competition between ATP and Pi causes the inhibitory effect of Pi on the kinase activity. The hydrogen bond with the conserved Glu163, a peculiar feature of HprK/P, recalls a similar interaction in bacterial periplasmic phosphate receptor proteins, which strongly favours binding of monobasic phosphate over deprotonated sulfate (Quiocho, 1996). The ATPase domain of muscle myosin has a P-loop with a conserved glutamate in an equivalent position. Instead of participating in phosphate binding, its carboxylate points away from the nucleotide and interacts with two conserved lysines (Smith and Rayment, 1996). In HprK/P, the P-loop is at a subunit interface. Glu163 of one P-loop is close to the conserved Arg271 of the K3 loop of another subunit. A polar interaction is conceivable in the absence of phosphate.
The mutant HprK/P proteins described by Monedero et al. (2001), which exhibit almost normal kinase activity, have lost the stimulating effect of Pi on the dephosphorylation activity. The mechanism of this reaction is not understood, but it does not appear to be simply the reverse kinase reaction, i.e. the transfer of the phosphoryl group from P-Ser-HPr back to ADP. Adding ADP does not stimulate dephosphorylation of P-Ser-HPr, and no ATP is formed when the reaction is carried out in the presence of equimolar amounts of ADP and Pi (Monedero et al., 2001). Among the mutants, G160F and E163K affect residues of the P-loop that interact with the bound phosphate. They are likely to lower the affinity for Pi, but the biochemical data imply that they do so without drastically changing the affinity for ATP. The other mutations (V267F, G270E, G270R and N272I) all affect residues of the K3 loop, which is in contact with the P-loop of a neighbouring subunit. These mutations therefore may have an indirect effect on the P-loop conformation.
HprK/P is not the only prokaryotic protein kinase to contain the Walker motif A in its sequence. Another is the B.subtilis protein PrkA, a Ser/Thr protein kinase that phosphorylates a 60 kDa protein of unknown function (Fischer et al., 1996). In addition, autophosphorylating tyrosine kinases with a sequence related to the Walker motif A have been detected in several bacteria (Grangeasse et al., 1997; Ilan et al., 1999; Vincent et al., 2000). Protein ChvG of Agrobacterium tumefaciens contains both the sequence motifs characteristic of a sensor histidine kinase and the Walker motif A (Charles and Nester, 1993). These proteins probably all contain a domain belonging to the same family of nucleotide-binding proteins. We suggest that this new type of protein kinases, which are relatively abundant in bacteria, also exist in eukaryotes where they may have been mistaken for nucleoside/nucleotide kinases in view of their sequences.
Materials and methods
Cloning of the hprK fragment encoding L.casei Δ127HprK/P
A DNA fragment encoding amino acid residues 128–319 of L.casei HprK/P was amplified by PCR using chromosomal L.casei DNA as template. The forward primer used for the PCR contained an ATG start codon preceded by a BamHI site, whereas the reverse primer contained a KpnI site following the stop codon. The PCR product was cut with BamHI–KpnI and cloned into the expression vector pQE30 (Qiagen) digested with the same enzymes. The resulting plasmid pHPRKLcΔ127 expressing the His-tagged truncated protein was used to transform the E.coli strain NM522.
Protein purification
Escherichia coli NM522 transformed with pHPRKLcΔ127 was grown in LB medium and the synthesis of the truncated L.casei HprK/P was induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 4 h at 37°C. Cells were harvested subsequently by centrifugation (10 min, 5000 g, 4°C) and resuspended in lysis buffer (50 mM Tris–HCl pH 8.0, 100 mM NaCl, 200 µg/ml pefabloc, 100 µM benzamidine, 1 µg/ml leupeptin, 1 µg/ml aprotinin, 1 µg/ml pepstatin A, 0.05% NP-40). After addition of 200 µM lysozyme, the cells were incubated for 15 min at room temperature and disrupted by sonication using a Fisher Scientific sonifier. Cell debris was removed by centrifugation (30 min, 20 000 g, 4°C) and His-tagged HprK/P was purified at room temperature on an Ni-NTA–agarose column (Qiagen) as previously described for intact HprK/P (Galinier et al., 1998). For crystallization purposes, the protein was purified further by ion exchange chromatography. The sample was dialysed overnight at 4°C against buffer A [20 mM bisTris propane pH 7.3, 150 mM NaCl, 1 mM EDTA, 0.1 mM dithiothreitol (DTT)] and applied at 4°C onto a MonoQ HR 5/5 (Pharmacia) column. Elution was performed with a 20 ml gradient from 0 to 30% of buffer B (20 mM bisTris propane pH 7.3, 1 M NaCl, 1 mM EDTA, 0.1 mM DTT). Fractions were analysed by SDS–PAGE followed by Coomassie Blue staining. HprK/P-containing fractions were pooled, concentrated using an Amicon YM-10 membrane (exclusion size 10 kDa) and stored at –80°C in elution buffer. The protein concentration was estimated from the calculated extinction coefficient ε280 = 0.48/M/cm.
To obtain seleniated HprK/P, E.coli NM522x pHPRKLcΔ127 was grown overnight at 28°C in 1 l of modified M9 minimal medium containing 50 µg/ml methionine until an OD600 between 0.7 and 1.3 was reached. As this strain is not auxotroph for methionine, induction was performed in a culture medium devoid of this amino acid, but containing selenomethionine and excess amino acids known to inhibit methionine biosynthesis (van Duyne et al., 1993). The cells were resuspended in 1 l of fresh M9 minimal medium supplemented with 50 µg/ml selenomethionine. After 30 min of shaking at 28°C, 1 mM IPTG was added and the culture was incubated for an additional 2 h at 28°C. After cell lysis and centrifugation, the protein was found in the pellet. The seleniated sample was then resuspended in 40 ml of 20 mM Tris–HCl pH 8.0, 500 mM NaCl, 0.5% Triton X-100. After centrifugation (15 min, 20 000 g, 4°C), half of the protein was recovered in the supernatant. Additional resolubilized protein was obtained by washing the pellet with 20 mM Tris–HCl pH 8.0, 500 mM NaCl. The solubilized sample was purified as described above.
Activity assay
The kinase and phosphatase activities of the purified truncated enzyme were tested following the published protocol (Monedero et al., 2001). The different forms of HPr were separated by electrophoresis on non-denaturing 12.5% polyacrylamide gels, which were stained with Coomassie Blue. The effect of inorganic phosphate on kinase and phosphatase activity of HprK/P is illustrated in Figure 8.

Fig. 8. Activity assays with truncated L.casei HprK/P. (A) HPr kinase assay. HPr at 12 µM was incubated for 10 min at 37°C with 400 nM HprK/P and increasing concentrations of Pi (0, 2, 5 and 20 mM) in the presence of 5 mM MgCl2, 10 mM FBP and 1 mM ATP. (B) P-Ser-HPr phosphatase assay. P-Ser-HPr at 12 µM was incubated for 5 min at 37°C with 88 nM HprK/P and increasing concentrations of Pi (0, 0.2, 1 and 3 mM) in the presence of 5 mM MgCl2.
Size-exclusion chromatography
Gel permeation experiments were performed on a Superose-12 HR 10/30 column (Pharmacia) equilibrated with 20 mM HEPES pH 7.0, 150 mM NaCl. Samples at 3 mg/ml protein concentration were centrifuged (10 min, 20 000 g, 4°C) prior to loading 200 µl onto the column, which had been calibrated with molecular weight markers (Bio-Rad gel filtration standard kit).
Crystallization of Δ127HprK/P
Crystallization conditions were screened using the hanging-drop vapour diffusion method and the Hampton Research sparse matrix. Crystals were obtained at 18°C with 1 M mono-ammonium dihydrogen phosphate as precipitant and 100 mM tri-sodium citrate dihydrate pH 5.6 as buffer. The drop was formed by mixing 2 µl of a solution containing 5 mg/ml protein and 2 µl of the crystallization solution. Crystal quality was improved by lowering the precipitant concentration to 400 mM and the pH to 5.2. The selenomethionine-containing protein crystallized under the same conditions.
X-ray diffraction data collection
All data collection was performed on station ID14-H4 at the ESRF (Grenoble, France) using cryoprotected crystals maintained at 100 K using the Oxford Cryosystem Cryostream device. Cryoprotection was achieved by soaking the crystals for 2 min in a solution containing 1.2 M ammonium phosphate, 100 mM sodium citrate pH 5.2 and 30% glycerol prior to freezing. Diffraction data were collected from a single crystal of seleniated protein at three wavelengths close to the selenium K-edge: 0.9393, 0.9792 and 0.9790 Å. Images were processed with the software DENZO (Otwinowski and Minor, 1997) to 3.0 Å resolution. An additional data set collected from another single crystal at wavelength 0.9393 Å was processed to 2.8 Å resolution. Scaling of the MAD data was performed using SCALA (CCP4, 1994) and the positions of 48 selenium atoms were found in the unit cell (four atoms per asymmetric unit) using SOLVE (Terwilliger and Berendzen, 1999). After heavy atom sites refinement, the phases were calculated using the program SHARP (De la Fortelle and Bricogne, 1997) and the resulting density map was subjected to solvent flattening using the program SOLOMON (De la Fortelle and Bricogne, 1997). Further scaling of the data was performed with the CCP4 program suite (CCP4, 1994). The data collection, phasing and refinement statistics are reported in Table I.
Structure determination and refinement
At this stage, the electron density map was of sufficient quality for a model to be built using TURBO-FRODO (Roussel and Cambillau, 1989). In a first step, calculated phases were combined with the original MAD phases in a refinement by simulated annealing at 3 Å resolution using the program CNS (Brünger et al., 1998). The model was refined further to 2.8 Å resolution using the single data set by manual rebuilding combined with further refinement using only calculated phases. Quality control of the model was performed with the program PROCHECK (Laskowski et al., 1993). Buried surfaces were calculated using the program ASA (A.M.Lesk, Cambridge, probe size 1.4 Å).
Docking
The docking calculation was performed with the algorithm DOCK (Cherfils et al., 1991). The procedure explores the position and orientation of the two proteins, bringing them into van der Waals contact and evaluating the buried surface area. It operates on simplified protein models where each residue is represented by a sphere centred on the centre of gravity of its side chain. Thus, the calculation is insensitive to the small differences observed between HPr structures. We used the E.coli HPr crystal structure (Jia et al., 1993). All degrees of rotational/translational freedom, in steps of 5°, were set free for simulated annealing. The docked complexes with a buried surface over 1000 Å2 were sorted and clustered, keeping the average position in each cluster for visual inspection. Those where Ser46 of HPr faced the P-loop of the kinase were selected, and a second docking simulation was performed in steps of 2°, limiting the search to the neighbourhood of these preferred orientations.
Acknowledgments
Acknowledgements
We thank G.Leonard at ESRF (Grenoble) beamline ID14-H4 for his help with data collection, and H.Belrhali for access to beamline ID14-H1. The MALDI-TOF and electrospray mass spectrometry experiments were performed by P.Le Maréchal (Orsay) and O.Laprévote (Gif-sur-Yvette). We are grateful to L.Bousset (LEBS, Gif/Yvette) for his help in data processing. S.F. acknowledges support by the Ligue Nationale contre le Cancer.
References
- Audette G.F., Engelmann,R., Hengstenberg,W., Deutscher,J., Hayakawa,K., Quail,J.W. and Delbaere,L.T. (2000) The 1.9 Å resolution structure of phospho-serine 46 HPr from Enterococcus faecalis. J. Mol. Biol., 303, 545–553. [DOI] [PubMed] [Google Scholar]
- Berry M.B. and Phillips,G.N.,Jr (1998) Crystal structures of Bacillus stearothermophilus adenylate kinase with bound Ap5A, Mg2+ Ap5A and Mn2+ Ap5A reveal an intermediate lid position and six coordinate octahedral geometry for bound Mg2+ and Mn2+. Proteins, 32, 276–288. [DOI] [PubMed] [Google Scholar]
- Briozzo P., Golinelli-Pimpaneau,B., Gilles,A.M., Gaucher,J.F., Burlacu-Miron,S., Sakamoto,H., Janin,J. and Barzu,O. (1998) Structures of Escherichia coli CMP kinase alone and in complex with CDP: a new fold of the nucleoside monophosphate binding domain and insights into cytosine nucleotide specificity. Structure, 6, 1517–1527. [DOI] [PubMed] [Google Scholar]
- Brochu D. and Vadeboncoeur,C. (1999) The HPr(Ser) kinase of Streptococcus salivarius: purification, properties and cloning of the hprK gene. J. Bacteriol., 181, 709–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger A.T. et al. (1998) Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- CCP4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Charles T.C. and Nester,E.W. (1993) A chromosomally encoded two-component sensory transduction system is required for virulence of Agrobacterium tumefaciens. J. Bacteriol., 175, 6614–6625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherfils J., Duquerroy,S. and Janin,J. (1991) Protein–protein recognition analyzed by docking simulation. Proteins, 11, 271–280. [DOI] [PubMed] [Google Scholar]
- De la Fortelle E. and Bricogne,G. (1997) Maximum likehood heavy atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Methods Enzymol., 276, 472–494. [DOI] [PubMed] [Google Scholar]
- Deutscher J. and Saier,M.H.,Jr (1983) ATP-dependent protein kinase-catalyzed phosphorylation of a seryl residue in HPr, a phosphate carrier protein of the phosphotransferase system in Streptococcus pyogenes. Proc. Natl Acad. Sci. USA, 80, 6790–6794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutscher J., Pevec,B., Beyreuther,K., Kiltz,H.H. and Hengstenberg,W. (1986) Streptococcal phosphoenolpyruvate–sugar phosphotransferase system: amino acid sequence and site of ATP-dependent phosphorylation of HPr. Biochemistry, 25, 6543–6551. [DOI] [PubMed] [Google Scholar]
- Deutscher J., Reizer,J., Fischer,C., Galinier,A., Saier,M.H.,Jr and Steinmetz,M. (1994) Loss of protein kinase-catalyzed phosphorylation of HPr, a phosphocarrier protein of the phosphotransferase system, by mutation of the ptsH gene confers catabolite repression resistance to several catabolic genes of Bacillus subtilis. J. Bacteriol., 176, 3336–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutscher J., Galinier,A. and Martin-Verstraete,I. (2001) Carbohydrate uptake and metabolism. In Sonenschein,A.L., Hoch,J.A. and Losick,R. (eds) Bacillus subtilis and its Closest Relatives: From Genes to Cells. American Society for Microbiology, Washington, DC, pp. 137–158.
- Dossonnet V., Monedero,V., Zagorec,M., Galinier,A., Pérez-Martinez,G. and Deutscher,J. (2000) Phosphorylation of HPr by the bifunctional HPr kinase/P-Ser-HPr phosphatase from Lactobacillus casei controls catabolite repression and inducer exclusion, but not inducer expulsion. J. Bacteriol., 182, 2582–2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esnouf R.M. (1997) An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J. Mol. Graph. Model., 15, 112–133, 132–134. [DOI] [PubMed] [Google Scholar]
- Esnouf R.M. (1999) Further additions to MolScript version 1.4, including reading and contouring of electron-density maps. Acta Crystallogr. D, 55, 938–940. [DOI] [PubMed] [Google Scholar]
- Fischer C., Geourjon,C., Bourson,C. and Deutscher,J. (1996) Cloning and characterization of the Bacillus subtilis prkA gene encoding a novel serine protein kinase. Gene, 168, 55–60. [DOI] [PubMed] [Google Scholar]
- Fujita Y., Miwa,Y., Galinier,A. and Deutscher,J. (1995) Specific recognition of the Bacillus subtilis gnt cis-acting catabolite-responsive element by a protein complex formed between CcpA and seryl-phosphorylated HPr. Mol. Microbiol., 17, 953–960. [DOI] [PubMed] [Google Scholar]
- Galinier A., Kravanja,M., Engelmann,R., Hengstenberg,W., Kilhoffer,M.C., Deutscher,J. and Haiech,J. (1998) New protein kinase and protein phosphatase families mediate signal transduction in bacterial catabolite repression. Proc. Natl Acad. Sci. USA, 95, 1823–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galinier A., Deutscher,J. and Martin-Verstraete,I. (1999) Phosphoryl ation of either crh or HPr mediates binding of CcpA to the Bacillus subtilis xyn cre and catabolite repression of the xyn operon. J. Mol. Biol., 286, 307–314. [DOI] [PubMed] [Google Scholar]
- Giartosio A., Erent,M., Cervoni,L., Morera,S., Janin,J., Konrad,M. and Lascu,I. (1996) Thermal stability of hexameric and tetrameric nucleoside diphosphate kinases. Effect of subunit interaction. J. Biol. Chem., 271, 17845–17851. [DOI] [PubMed] [Google Scholar]
- Gouet P., Courcelle,E., Stuart,D.I. and Metoz,F. (1999) ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics, 15, 305–308. [DOI] [PubMed] [Google Scholar]
- Grangeasse C., Doublet,P., Vaganay,E., Vincent,C., Deleage,G., Duclos,B. and Cozzone,A.J. (1997) Characterization of a bacterial gene encoding an autophosphorylating protein tyrosine kinase. Gene, 204, 259–265. [DOI] [PubMed] [Google Scholar]
- Hanks S.K., Quinn,A.M. and Hunter,T. (1988) The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science, 241, 42–52. [DOI] [PubMed] [Google Scholar]
- Henkin T.M., Grundy,F.J., Nicholson,W.L. and Chambliss,G.H. (1991) Catabolite repression of α-amylase gene expression in Bacillus subtilis involves a trans-acting gene product homologous to the Escherichia coli lacl and galR repressors. Mol. Microbiol., 5, 575–584. [DOI] [PubMed] [Google Scholar]
- Holm L. and Sander,C. (1996) Mapping the protein universe. Science, 273, 595–603. [DOI] [PubMed] [Google Scholar]
- Ilan O., Bloch,Y., Frankel,G., Ullrich,H., Geider,K. and Rosenshine,I. (1999) Protein tyrosine kinases in bacterial pathogens are associated with virulence and production of exopolysaccharide. EMBO J., 18, 3241–3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inouye S. et al. (2000) A large family of eukaryotic-like protein Ser/Thr kinases of Myxococcus xanthus, a developmental bacterium. Microb. Comp. Genomics, 5, 103–120. [DOI] [PubMed] [Google Scholar]
- Jault J.-M., Fieulaine,S., Nessler,S., Gonzalo,P., Di Pietro,A., Deutscher,J. and Galinier,A. (2000) The HPr kinase from Bacillus subtilis is a homo-oligomeric enzyme which exhibits strong positive cooperativity for nucleotide and fructose 1,6-bisphosphate binding. J. Biol. Chem., 275, 1773–1780. [DOI] [PubMed] [Google Scholar]
- Jia Z., Quail,J.W., Waygood,E.B. and Delbaere,L.T. (1993) The 2.0 Å resolution structure of Escherichia coli histidine-containing phosphocarrier protein HPr. A redetermination. J. Biol. Chem., 268, 22490–22501. [DOI] [PubMed] [Google Scholar]
- Jia Z., Quail,J.W., Delbaere,L.T. and Waygood,E.B. (1994) Structural comparison of the histidine-containing phosphocarrier protein HPr. Biochem. Cell Biol., 72, 202–217. [DOI] [PubMed] [Google Scholar]
- Jurica M.S., Mesecar,A., Heath,P.J., Shi,W., Nowak,T. and Stoddard,B.L. (1998) The allosteric regulation of pyruvate kinase by fructose-1,6-bisphosphate. Structure, 6, 195–210. [DOI] [PubMed] [Google Scholar]
- Kabsch W. and Sander,C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers, 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- Kravanja M. et al. (1999) The hprK gene of Enterococcus faecalis encodes a novel bifunctional enzyme: the HPr kinase/phosphatase. Mol. Microbiol., 31, 59–66. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., MacArthur,M.W., Moss,D.S. and Thornton,J.M. (1993) PROCHECK—a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr., 26, 283–291. [Google Scholar]
- Merritt E.A. and Bacon,D.J. (1997) Raster3D: photorealistic molecular graphics. Methods Enzymol., 277, 505–524. [DOI] [PubMed] [Google Scholar]
- Miwa Y., Nakata,A., Ogiwara,A., Yamamoto,M. and Fujita,Y. (2000) Evaluation and characterization of catabolite-responsive elements (cre) of Bacillus subtilis. Nucleic Acids Res., 28, 1206–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monedero V., Poncet,S., Mijakovic,I., Fieulaine,S., Dossonnet,V., Martin-Verstraete,I., Nessler,S. and Deutscher,J. (2001) Mutations lowering the phosphatase activity of HPr kinase/phosphatase switch off carbon metabolism. EMBO J., 20, 3928–3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno M.S., Schneider,B.L., Maile,R.R., Weyler,W. and Saier,M.H.,Jr (2001) Catabolite repression mediated by CcpA protein in Bacillus subtilis: novel modes of regulation revealed by whole-genome analyses. Mol. Microbiol., 39, 1366–1381. [DOI] [PubMed] [Google Scholar]
- Murzin A.G., Brenner,S.E., Hubbard,T. and Chothia,C. (1995) SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol., 247, 536–540. [DOI] [PubMed] [Google Scholar]
- Neves A.R., Ramos,A., Nunes,M.C., Kleerebezem,M., Hugenholtz,J., de Vos,W.M., Almeida,J. and Santos,H. (1999) In vivo nuclear magnetic resonance studies of glycolytic kinetics in Lactococcus lactis. Biotechnol. Bioeng., 64, 200–212. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor,W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Piontek K., Chakrabarti,P., Schar,H.P., Rossmann,M.G. and Zuber,H. (1990) Structure determination and refinement of Bacillus stearothermophilus lactate dehydrogenase. Proteins, 7, 74–92. [DOI] [PubMed] [Google Scholar]
- Postma P.W., Lengeler,J.W. and Jacobson,G.R. (1993) Phosphoenolpyruvate:carbohydrate phosphotransferase systems of bacteria. Microbiol. Rev., 57, 543–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiocho F.A. (1996) Atomic basis of the exquisite specificity of phosphate and sulfate transport receptors. Kidney Int., 49, 943–946. [DOI] [PubMed] [Google Scholar]
- Reizer J., Novotny,M.J., Hengstenberg,W. and Saier,M.H.,Jr (1984) Properties of ATP-dependent protein kinase from Streptococcus pyogenes that phosphorylates a seryl residue in HPr, a phosphocarrier protein of the phosphotransferase system. J. Bacteriol., 160, 333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reizer J., Sutrina,S.L., Saier,M.H., Stewart,G.C., Peterkofsky,A. and Reddy,P. (1989) Mechanistic and physiological consequences of HPr(ser) phosphorylation on the activities of the phosphoenolpyruvate:sugar phosphotransferase system in Gram-positive bacteria: studies with site-specific mutants of HPr. EMBO J., 8, 2111–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussel A. and Cambillau,C. (1989) TURBO-FRODO. In Silicon Graphics Geometry Partners Directory. Silicon Graphics, Mountain View, CA, pp. 77–78.
- Saraste M., Sibbald,P.-R. and Wittinghofer,A. (1990) The P-loop, a common motif in ATP and GTP binding proteins. Trends Biochem. Sci., 15, 430–434. [DOI] [PubMed] [Google Scholar]
- Smith C.A. and Rayment,I. (1996) Active site comparisons highlight structural similarities between myosin and other P-loop proteins. Biophys. J., 70, 1590–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stülke J., Arnaud,M., Rapoport,G. and MartinVerstraete,I. (1998) PRD—a protein domain involved in PTS-dependent induction and carbon catabolite repression of catabolic operons in bacteria. Mol. Microbiol., 28, 865–874. [DOI] [PubMed] [Google Scholar]
- Stülke J. and Hillen,W. (2000) Regulation of carbon catabolism in bacillus species. Annu. Rev. Microbiol., 54, 849–880. [DOI] [PubMed] [Google Scholar]
- Taylor S.S. and Radzio-Andzelm,E. (1994) Three protein kinase structures define a common motif. Structure, 2, 345–355. [DOI] [PubMed] [Google Scholar]
- Terwilliger T.C. and Berendzen,J. (1999) Automated MAD and MIR structure solution. Acta Crystallogr. D, 55, 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J. and Torchia,D.A. (1984) Use of 31P nuclear magnetic resonance spectroscopy and 14C fluorography in studies of glycolysis and regulation of pyruvate kinase in Streptococcus lactis. J. Bacteriol., 158, 791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Duyne G.D., Standaert,R.F., Karplus,P.A., Schreiber,S.L. and Clardy,J. (1993) Atomic structure of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J. Mol. Biol., 229, 105–124. [DOI] [PubMed] [Google Scholar]
- Via A., Ferre,F., Brannetti,B., Valencia,A. and Helmer-Citterich,M. (2000) Three-dimensional view of the surface motif associated with the P-loop structure: cis and trans cases of convergent evolution. J. Mol. Biol., 303, 455–465. [DOI] [PubMed] [Google Scholar]
- Vincent C., Duclos,B., Grangeasse,C., Vaganay,E., Riberty,M., Cozzone,A.J. and Doublet,P. (2000) Relationship between exopolysaccharide production and protein-tyrosine phosphorylation in Gram-negative bacteria. J. Mol. Biol., 304, 311–321. [DOI] [PubMed] [Google Scholar]
- Vonrhein C., Bönisch,H., Schäfer,G. and Schulz,G.E. (1998) The structure of a trimeric archaeal adenylate kinase. J. Mol. Biol., 282, 167–179. [DOI] [PubMed] [Google Scholar]
- Walker J.E., Saraste,M., Runswick,M.J. and Gay,N.J. (1982) Distantly related sequences in the α- and β-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J., 1, 945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G., Sondej,M., Garrett,D.S., Peterkofsky,A. and Clore,G.M. (2000) A common interface on histidine-containing phosphocarrier protein for interaction with its partner proteins. J. Biol. Chem., 275, 16401–16403. [DOI] [PubMed] [Google Scholar]
- Weickert M.J. and Chambliss,G.H. (1990) Site-directed mutagenesis of a catabolite repression operator sequence in Bacillus subtilis. Proc. Natl Acad. Sci. USA, 87, 6238–6242. [DOI] [PMC free article] [PubMed] [Google Scholar]