

Abstract
Folding to completion of actin and tubulin in the eukaryotic cytosol requires their interaction with cytosolic chaperonin CCT [chaperonin containing tailless complex polypeptide 1 (TCP-1)]. Three-dimensional reconstructions of nucleotide-free CCT complexed to either actin or tubulin show that CCT stabilizes both cytoskeletal proteins in open and quasi-folded conformations mediated through interactions that are both subunit specific and geometry dependent. Here we find that upon ATP binding, mimicked by the non-hydrolysable analog AMP-PNP (5′-adenylyl-imido-diphosphate), to both CCT–α-actin and CCT– β-tubulin complexes, the chaperonin component undergoes concerted movements of the apical domains, resulting in the cavity being closed off by the helical protrusions of the eight apical domains. However, in contrast to the GroE system, generation of this closed state does not induce the release of the substrate into the chaperonin cavity, and both cytoskeletal proteins remain bound to the chaperonin apical domains. Docking of the AMP-PNP–CCT-bound conformations of α-actin and β-tubulin to their respective native atomic structures suggests that both proteins have progressed towards their native states.
Keywords: actin/chaperonin/electron microscopy/protein folding/tubulin
Introduction
In the past decade it has become clear that, under normal growth conditions, the folding of many proteins requires the help of other proteins, named molecular chaperones, which interact with non-native structures to help them overcome kinetic barriers on the pathway to their native folds (Bukau and Horwich, 1998). Chaperonins, one of the major classes of folding-assisting proteins, have been well characterized. They are found in all living cells, being strictly required for viability. Chaperonins comprise two subclasses: Group I, found in eubacteria and endosymbiotic organelles (Bukau and Horwich, 1998; Ellis and Hartl, 1999) and Group II, found in archaea and in the cytosol of eukaryotic cells (Gutsche et al., 1999; Willison, 1999). Among the Group I chaperonins, GroEL from Escherichia coli has been most extensively studied. Its atomic structure is known (Braig et al., 1994; Xu et al., 1997) and also the low-resolution structures of many of its reaction intermediates have been obtained by cryo-electron microscopy (cryo-EM) (Roseman et al., 1996). GroEL is made up of two identical back-to-back stacked homo-heptameric rings built with a 60 kDa subunit. Apical, intermediate and equatorial domains are the main functional components of each monomer (Braig et al., 1994). GroEL requires a cofactor, GroES, to fold substrates efficiently. GroES, a homo-heptameric ring of a 10 kDa subunit (Hunt et al., 1996), interacts with the apical domains of GroEL in the presence of nucleotide. Current models for the function of GroEL propose that unfolded substrates, displaying exposed hydrophobic regions, can be bound by the hydrophobic sites of GroEL apical domains (Chen and Sigler, 1999). Upon ATP binding to the equatorial domains, GroES interacts with the substrate-bound GroEL ring and displaces it into the now hydrophilic cavity, where it has a chance to fold correctly (Bukau and Horwich, 1998; Ellis and Hartl, 1999).
However, less is known about the structures and mechanisms of action of Group II chaperonins (Gutsche et al., 1999). One of its main representatives is the cytosolic chaperonin CCT [chaperonin containing tailless complex polypeptide 1 (TCP-1)], also termed TRiC. CCT rings consist of eight different subunits positioned in a precise arrangement (Liou and Willison, 1997; Llorca et al., 1999b; Willison, 1999; Grantham et al., 2000). The sequence differences among the CCT subunits are located mainly in their apical domains (Kim et al., 1994), suggesting some degree of specificity towards substrate binding, which is reinforced by the fact that two proteins, actin and tubulin, have been found to be the major substrates of CCT. It has been hypothesized that a link exists between the evolution of the eukaryotic cell and the folding of actins and tubulins, and that it has involved the transformation of a more simple archaeabacterial chaperonin to the highly complex CCT (Willison, 1999; Leroux and Hartl, 2000; Llorca et al., 2000; Willison and Grantham, 2001). Recently, the three dimensional (3D) reconstruction of CCT–α-actin and CCT–β-tubulin complexes has shed some light on certain structural aspects of CCT substrate recognition (Llorca et al., 1999b, 2000); CCT seems to be recognizing actin and tubulin in native-like conformations. The eukaryotic chaperonin binds each of the two topological domains of the two cytoskeletal proteins (Kabsch et al., 1990; Nogales et al., 1998) using opposite regions of the CCT ring, thus stabilizing extended and open conformations. The CCT subunits involved in this binding mode have been mapped directly by immunoelectron microscopy. The actin small domain binds to the CCTδ subunit and the large C-terminal domain to either CCTβ or CCTε, the pair of latter subunits binding actin with high affinity (Llorca et al., 1999b; Hynes and Willison, 2000). On the other hand, tubulin uses two possible binding arrangements involving two sets of five subunits, but with CCTβ and CCTε again being the subunits binding the C-terminal domain of tubulin with the highest affinity in each of the two arrangements (Llorca et al., 2000). Biochemical studies have corroborated the identity of the actin and tubulin domains involved in the binding to specific CCT subunits (Hynes and Willison, 2000; Llorca et al., 2000; Ritco-Vonsovici and Willison, 2000).
In this work, 3D reconstructions of the 5′-adenylyl-imido-diphosphate (AMP-PNP)-bound forms of CCT– α-actin and CCT–β-tubulin complexes, combined with the docking of atomic structures of the two cytoskeletal proteins and immunolabelling experiments, have shed new light on the molecular mechanism of actin and tubulin folding. The results suggest that the eukaryotic chaperonin has evolved from its precursor to fold these complex eukaryotic proteins by coupling sequential changes in the apical domains of the chaperonin subunits that occur upon nucleotide binding, to concerted movements in the substrate molecules that lead to their successful folding.
Results
Three-dimensional structures of the AMP-PNP–CCT and AMP-PNP–CCT–α-actin complexes
α-actin was chemically denatured, incubated with CCT in a diluting buffer, and then the free actin molecules were removed by size-exclusion chromatography. The non-hydrolysable ATP analog AMP-PNP was added to a concentration high enough (10 mM) to saturate all the nucleotide binding sites and mimic ATP binding (but not hydrolysis). Aliquots of this solution were vitrified and images were obtained at low temperature (–170°C). Tilted top views of CCT were processed (Figure 1A) and the particles were classified by two different independent methods (see Materials and methods) into two populations: those with an empty cavity and those having a mass inside the chaperonin cavity. The 3D reconstruction carried out with the substrate-free CCT particles incubated with AMP-PNP (AMP-PNP–CCT from now on; Figure 1B–D) shows closure of both CCT cavities compared with the open conformation of the apo-CCT structure (Llorca et al., 1999a, 2000). The conformation obtained here is very similar to that of the thermosome X-ray structure (Ditzel et al., 1998), and the docking of the two structures reveals a good correlation (Figure 1E and F), which supports the notion that the conformation obtained for the thermosome crystal structure might be the one generated upon ATP binding (Ditzel et al., 1998; Gutsche et al., 2000). The conformation of both CCT rings is similar to one of the rings of the 3D reconstruction of the ATP-bound form of CCT [see the top ring in Figure 3A–C in Llorca et al. (1999a)], albeit at higher resolution. In the present CCT structure, the mass corresponding to the entire helical protrusions is now visualized (revealed in the docking top view; Figure 1E). In the former volumes (Llorca et al. 1999a), only the base of the apical domains was visualized, resulting in an apparent wider hole, whereas the structure revealed here shows the real magnitude of the closure [compare Figure 5A of Llorca et al. (1999a) with Figure 1E of this work]. The differences between the two 3D reconstructions have to do not only with the improvement in the image processing, but also to the fact that the latter reconstruction has been carried out using a more homogeneous population (generated with the non-hydrolysable analog AMP-PNP) than the previous one (generated using ATP and possibly having a mixture of conformations).

Fig. 1. Three-dimensional reconstruction of CCT in the presence of AMP-PNP. (A) A gallery of ice-embedded CCT particles in the presence of AMP-PNP. (B–D) Different views of the 3D structure of the AMP-PNP–CCT complex, generated after the processing of 1057 particles. (E and F) Top and side views, respectively, of the docking between the thermosome X-ray structure (in purple and white; Ditzel et al., 1998) and the 3D reconstruction of AMP-PNP–CCT (yellow grid).

Fig. 3. Three-dimensional reconstruction of the CCT–β-tubulin complex in the presence of AMP-PNP. (A and B) Different views of the AMP-PNP–CCT–β-tubulin complex, generated after the processing of 3501 particles. In (B), a cut along the longitudinal axis shows the tubulin molecule in the interior of the cavity. (C) Side view of the docking between the thermosome X-ray structure (in purple and white; Ditzel et al., 1998) and the 3D reconstruction of AMP-PNP–CCT–tubulin (yellow grid). (D and E) The transparent AMP-PNP–CCT–β-tubulin volume with the tubulin molecule, calculated by subtracting the volumes of the tubulin-containing ring and the substrate-free ring, shaded in red. (F) Two views of the extracted tubulin volume.

Fig. 5. Docking of the atomic coordinates of actin and tubulin with their corresponding EM volumes extracted from the AMP-PNP–CCT–α-actin and AMP-PNP–CCT–β-tubulin complexes. (A and B) Two views of the docking with actin. (C and D) Two views of the docking with tubulin. In both cases, the envelope of the EM volume has been drawn as a yellow grid. Different domains of the actin and tubulin structures have been coloured as in Llorca et al. (2000). For α-actin, N-terminal domain residues D1–R177 are coloured red and the C-terminal residues Q263–R372 and L178–F262 yellow and white, respectively. For β-tubulin, the N-terminal residues M1–I204 are coloured red, and the C-terminal residues D205–L265, H266–I384 and Q385–D437 are coloured green, white and yellow, respectively. Nucleotides (ATP for actin and GTP for tubulin) are coloured blue.
The 3D structure of the CCT–α-actin complex in the presence of AMP-PNP (AMP-PNP–CCT–α-actin complex from now on) is very similar to the substrate-free structure described above, but shows an extra density close to the apical regions of one of the rings (Figure 2C). As occurs during the 3D reconstruction of the nucleotide-free CCT–α-actin complex (Llorca et al., 1999b), no particles of CCT have been found containing substrate molecules in both rings. The reconstructed volume of the actin molecule can be determined from the bulk of the reconstruction after calculation of the difference map between the actin-bound and actin-free ring of the 3D reconstruction (red shading in Figure 2D and E). A similar volume is obtained after calculating the difference map between the actin-bound ring of the AMP-PNP–CCT–α-actin complex and either of the two rings of the CCT–AMP-PNP complex (not shown). The volume of the actin molecule extracted from the 3D reconstruction (top and side view in Figure 2F and G, respectively) shows a heart-like shape (Figure 2F) similar to the atomic structure of actin (Kabsch et al., 1990) when filtered at low resolution. Parenthetically, the actin molecule seems attached to the apical domains and not liberated into the cavity.

Fig. 2. Three-dimensional reconstruction of CCT–α-actin complex in the presence of AMP-PNP. (A–C) Different views of the 3D structure of the AMP-PNP–CCT–α-actin complex generated after the processing of 1640 particles. (C) A cut along the longitudinal axis whereby the substrate in the interior of the cavity is revealed. (D and E) The transparent AMP-PNP–CCT–α-actin volume showing the actin molecule, calculated by subtracting the volumes of the actin-containing ring and the substrate-free ring, shaded in red. (F) Top and side views of the extracted actin volume.
Three-dimensional structure of the AMP-PNP–CCT–β-tubulin complex
Recombinant human β-tubulin was chemically denatured, incubated with CCT in a diluting buffer, and unbound tubulin was subsequently removed by size-exclusion chromatography. Upon incubation with AMP-PNP, cryo-EM of the CCT particles was performed. Tilted top views of CCT were processed and classified according to the presence or absence of mass inside the chaperonin cavity. The 3D reconstruction of the β-tubulin-bound CCT particles shows a very symmetrical, closed structure (Figure 3A and B) with the tubulin molecule placed off-centre and hanging from the apical domains of one of the rings (Figure 3B and red shading in D and E). The degree of closure of both rings is greater than that observed for the 3D reconstructions of both the apo-CCT and the CCT–α-actin complex in the presence of AMP-PNP (Figures 1 and 2). It has been shown previously that the nucleotide-free conformation of the CCT–β-tubulin complex reveals a more closed disposition of the rings than in the case of the apo-CCT or the CCT–α-actin complex (Llorca et al., 2000), and this behaviour seems to be continued in the nucleotide-bound state. This is confirmed when a docking between the atomic structure of the thermosome and the 3D structure of the AMP-PNP– CCT–β-tubulin complex is performed (Figure 3C), which again reveals a good fitting between the two structures, except that, in contrast to the docking carried out with the AMP-PNP–CCT structure (Figure 1E and F) or the AMP-PNP–CCT–α-actin complex (not shown), the apical domains of the AMP-PNP–CCT–β-tubulin complex are pulled inwards and downwards (∼8 Å; see top and bottom of Figure 3C). The most likely explanation for this difference in the CCT conformations generated by the two cytoskeletal proteins is that whereas actin interacts with CCT subunits through two delimited regions (Llorca et al., 1999b), tubulin binds to five different CCT subunits (Llorca et al., 2000) via apparently numerous binding sites (Ritco-Vonsovici and Willison, 2000), thus generating a more compact structure.
Another notable structural feature observed in the 3D reconstruction of the AMP-PNP–CCT-β-tubulin complex presented here and in the nucleotide-free CCT–β-tubulin complex shown in Llorca et al. (2000) is that the downward and inward movements of the apical domains take place simultaneously in both rings, despite the fact that only one of the rings contains tubulin. This symmetry is also found in the atomic structure of the thermosome (Ditzel et al., 1998) and in the structures of the thermosome and TF55 determined by cryo-EM by Schoehn et al. (2000a,b), and may reflect the very strong negative co-operativity between CCT rings upon ATP binding, since the Hill coefficient is practically equivalent to the number of subunits in the ring (7.2; Kafri et al., 2001). A possible explanation for this behaviour may be due to the differences in the inter-ring subunit arrangement between Group I chaperonins, in which every subunit interacts with two opposite subunits (Braig et al., 1994), and Group II chaperonins, in which each subunit opposes only one subunit (Ditzel et al., 1998).
As mentioned above, the tubulin mass is found hanging from the apical domains of one side of the CCT ring. The tubulin mass can be extracted from the bulk of the CCT volume by calculating the difference map between the tubulin-loaded ring and the substrate-free ring of the CCT–β-tubulin complex. The extracted tubulin (Figure 3F and red shading in D and E) has a compact shape and occupies approximately one-third of the cavity volume (Figure 3B). The fact that the reconstructed volume of tubulin is located off-centre, interacting with one side of the CCT cavity and only partially occupying it, precludes the possibility of the reconstructed substrate coming from the averaging of tubulin molecules randomly placed within the cavity. As in the case of actin, the structural rearrangements occurring in the CCT–β-tubulin complex upon nucleotide binding close the chaperonin cavity, but tubulin continues to be bound to the apical domains.
Biochemical correlates of the closure of the CCT cavity upon nucleotide binding
We wanted to confirm the closure of the CCT cavity upon nucleotide binding by using a biochemical technique, and for this we used differential immunoprecipitation of CCT by binding monoclonal antibodies reacting to different regions of the chaperonin (Figure 4). Immunoprecipitation of [35S]CCT in the absence or presence of AMP-PNP with the monoclonal antibody 23C, which binds to the very C-terminus of CCTα, located inside the chaperonin cavity (Grantham et al., 2000), shows that CCT–AMP-PNP virtually does not immunoprecipitate with 23C. This is consistent with the model that the helical protrusions obscure the CCT cavity upon nucleotide binding and block the access of the antibody to the chaperonin cavity. On the other hand, the variation in immunoprecipitation yield of [35S]CCT in the absence or presence of AMP-PNP with the monoclonal antibody εAD1 is much less perturbed. εAD1 recognizes an epitope on the outer surface of the helical protrusion of the CCTε apical domain, when compared with the atomic structure of the thermosome (both α- and β-subunits) (Ditzel et al., 1998), and would be expected to be able to bind CCT in the presence and absence of nucleotide.

Fig. 4. Immunoprecipitation of CCT in the presence of AMP-PNP. Sucrose gradient (20S) samples containing [35S]CCT were incubated in the presence and absence of 10 mM AMP-PNP, and immunoprecipitated under non-denaturing conditions with either a C-terminus anti-CCTα or an anti-CCTε apical domain monoclonal antibody. Recovered proteins were analysed by SDS–PAGE followed by autoradiography. The starting material (S) and background (minus antibody) controls are indicated.
Docking of the actin and tubulin X-ray coordinates within their reconstructed volumes
The volumes of α-actin and β-tubulin are not averaged out during the 3D reconstruction procedure, clearly indicating that the two cytoskeletal proteins maintain their interaction with CCT after nucleotide binding, each probably in a native or quasi-native conformation. To test this hypothesis, the atomic coordinates of the native structures of actin (Kabsch et al., 1990) and tubulin (Nogales et al., 1998) were fitted, using the SITUS quantitative algorithm (Wriggers et al., 1999), into the volumes of the EM counterparts extracted from their respective complexes. The reconstructed volume of the α-actin molecule bound to CCT fits well with its native X-ray structure (Figure 5A and B), and there is a good agreement between the different actin subdomains and the lobular structure of the reconstructed volume (see Figure 2F and G). The two best solutions in terms of root-mean square (r.m.s.) generated by the docking algorithm, and both of them being significantly better than any of the others obtained, place the atomic structure of actin horizontally into the flat shape of the reconstructed volume of actin and with one of the tips of the actin molecule (Kabsch et al., 1990) interacting with an apical domain of CCT. This uncertainty probably has to do with the low resolution of the reconstructed actin. One of the solutions places the actin molecule with the tip of the large domain interacting with the CCT apical domains. This actin region is the same one that has been shown to bind to the eukaryotic chaperonin in its nucleotide-free form [white domain in Figure 5A and B; see the same domain in Llorca et al. (2000)]. The second solution, however, places this actin region pointing towards the CCT cavity and not interacting with the apical domains. If this second solution were correct, this would imply that after nucleotide binding to CCT, the actin molecule would lose all its contacts with the apical domains of CCT and rebind to the chaperonin using completely different regions. Since the first solution maintains the tip of the large domain, the region that has been shown to bind CCT with the highest affinity (Rommelaere et al., 1999; Hynes and Willison, 2000; Llorca et al., 2000), interacting with CCT, it accounts, in a simpler and more reasonable way, for the transition between the nucleotide-free and the nucleotide-bound conformation by preserving the interactions between the chaperonin and the respective substrate.
The atomic structure of native tubulin also fits quite well into the volume of the reconstructed tubulin complexed to nucleotide-bound CCT (Figure 5C and D). The two best solutions provided by the docking algorithm, both significantly better than any others, place the atomic structure of tubulin in a vertical position, hanging from the underside of the apical domains. In the first of the two solutions, the region that the docking algorithm places as interacting with CCT is the one possessing the highest affinity for apo-CCT [white domain in Figure 5C and D; see the same domain in Llorca et al. (2000) and Ritco-Vonsovici and Willison (2000)]. The other solution places the tubulin molecule upside down with the highest affinity binding region pointing to the centre of the cavity. This uncertainty may again have to do with the low resolution of the reconstructed tubulin. As discussed previously in the case of AMP-PNP–CCT–α-actin, the first solution seems to maintain the tubulin molecule bound to the chaperonin using the same CCT-binding regions before and after the nucleotide binding, and therefore has been chosen as the most likely possibility. This assumption correlates well with the immunolabelling experiments described below.
Immunoelectron microscopy of the CCT–β-tubulin complex in the presence of AMP-PNP
After careful inspection of the tubulin location in the AMP-PNP–CCT–β-tubulin complex (Figure 3), it becomes clear that the substrate is placed asymmetrically in the chaperonin cavity (see in Figure 6A the projection of the AMP-PNP–CCT–β-tubulin complex along the longitudinal axis). Based on the previous findings, which locate the tubulin molecule bound to specific CCT subunits before nucleotide binding to the chaperonin (Llorca et al., 2000), we reasoned that tubulin could also be placed in a defined position within the chaperonin cavity after nucleotide binding. To prove this hypothesis, we labelled the AMP-PNP–CCT–β-tubulin complex with a monoclonal antibody specifically recognizing the CCTδ subunit. The average image obtained after the processing of two-dimensional (2D), negatively stained, top views of the complexes (Figure 6B) clearly shows that the tubulin density is placed almost opposite to the labelled CCTδ subunit and near the CCTβ/ε region. It has previously been shown (Llorca et al., 2000) that tubulin binds to CCT in two different geometrical arrangements involving the interaction of the C-terminal region of tubulin with the CCTε/ζ/β or CCTβ/γ/θ subunits, and the N-terminal domain with the diametrically opposite subunits (CCTθ/δ or CCTη/α subunits, respectively). The average image of the immunocomplex presented here clearly suggests that, after nucleotide binding, the tubulin molecule is displaced away from the CCTδ subunit and moves towards the CCTβ/ε subunits, the subunits involved in binding tubulin with the highest affinity in the previous step of the folding cycle (Llorca et al., 2000; Ritco-Vonsovici and Willison, 2000).
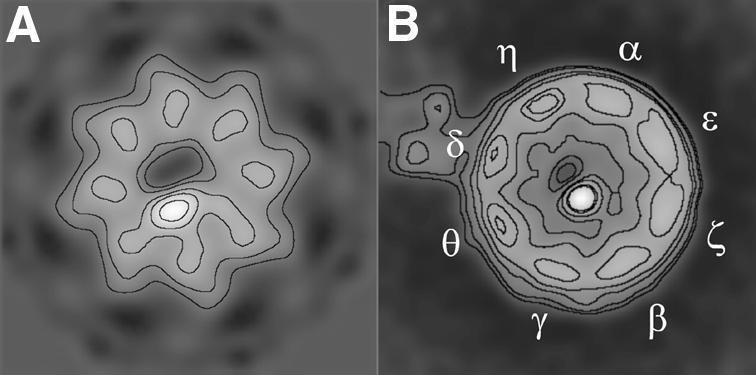
Fig. 6. Immunomicroscopy of the AMP-PNP–CCT–β-tubulin complex incubated with anti-CCTδ. (A) Projection of the 3D reconstruction of the AMP-PNP–CCT–β-tubulin complex along the z-axis. (B) Two-dimensional average image of the top views obtained from negatively stained immunocomplexes of AMP-PNP–CCT–β-tubulin–8g (anti-CCTδ) (405 particles). Note that the different shape of both images is due to the fact that (A) is the projection of the whole CCT structure, whereas (B) is the projection of the apical domains of the grid-interacting ring.
A similar approach could not be carried out with actin because the reconstructed molecule is placed horizontally, filling the upper part of the cavity completely, making it impossible to distinguish different locations within the cavity by 2D immunomicroscopy.
Discussion
Conformational changes of CCT upon ATP binding and hydrolysis
As with Group I chaperonins, ATP-driven conformational changes are essential to the mechanism of the eukaryotic CCT (Gutsche et al., 1999; Willison, 1999). Despite their great mechanistic importance, there are still discrepancies and uncertainties about the effects of different nucleotides on Group II chaperonin structure. For the archaeal chaperonins, open, closed and bullet-shaped conformations have been found by cryo-EM and 3D reconstruction (Schoehn et al., 2000a,b), but their occurrence is, according to these authors, independent of the presence or absence of nucleotides. However, small-angle neutron scattering experiments carried out on the thermosome suggest that ATP binding induces an even wider opening of the cavity, which is closed only after nucleotide hydrolysis (ADP-Pi conformation; Gutsche et al., 2000). The X-ray structure of the nucleotide-free thermosome also shows a closed conformation (Ditzel et al., 1998), but it has been argued that this conformation is generated by the sulfate ions present in the crystallization experiments (Gutsche et al., 2000). Regarding the eukaryotic chaperonin CCT, previous EM studies have revealed that in the presence of ATP, CCT closes one cavity, which adopts a conformation resembling the X-ray structure of the thermosome, although the precise nucleotide state of the rings of that 3D reconstruction is unknown (Llorca et al., 1999a).
Here we have incubated CCT with a high concentration (10 mM) of the non-hydrolysable ATP analog AMP-PNP and the 3D reconstruction obtained (both in the absence and presence of substrate) reveals a closed, symmetrical structure. The absence of bullet-like particles, previously described for the archaeal chaperonins (Schoehn et al., 2000a,b) and for CCT in the presence of ATP (Llorca et al., 1999a), indicates a complete saturation of the nucleotide binding sites under these conditions, which is consistent with an apparent ATP binding constant for the second ring of CCT of 533 µM (Kafri et al., 2001). The 3D reconstructions thus generated reveal that nucleotide binding to CCT is necessary and sufficient to close the CCT cavity. The almost perfect fitting between the 3D structure of AMP-PNP–CCT and the atomic structure of the thermosome clearly indicates that the apical domains point inwards, closing the cavity (see the fitting in Figure 1E between the helical protrusions of the thermosome and the envelope of the EM structure of CCT).
We now have an extensive set of 3D reconstructions ranging from substrate-free apo-CCT to its complexes with actin and tubulin in different nucleotide states (Llorca et al., 1999a,b, 2000 and this work). A model of the conformational changes undergone by CCT upon nucleotide and substrate binding can be elaborated (Figure 7) utilizing all the CCT structures obtained so far, whereby apo-CCT (Figure 7.1) is able to bind ATP and undergo structural changes leading to the closure of the cavity (Figure 7.2), or to interact with non-native forms of either actin (Figure 7.3) or tubulin (Figure 7.5). In the cell, substrate loading onto CCT may take place either concomitantly with chain elongation in the ribosomes (McCallum et al., 2000), or transferred from prefoldin/GimC (Vainberg et al., 1998; Siegers et al., 1999), or even directly from solution, although the exact order of events is not yet clear. During any of these pre-chaperonin folding steps, both cytoskeletal proteins must fold their domains to adopt native-like structures that are recognized by CCT. In each CCT-binding mode, actin binds to the apical domains of two CCT subunits (Llorca et al., 1999b), whereas tubulin adopts a more complex interaction with CCT, encompassing both the base and helical protrusions of the apical domains of five different subunits (Llorca et al., 2000), thus generating a tighter conformation of the apical domains. ATP binding (mimicked by AMP-PNP) to the CCT–α-actin (Figure 7.4) and CCT–β-tubulin (Figure 7.6) complexes induces movements of the apical domains that seal the cavity, although tubulin seems to induce a more closed conformation of the chaperonin.

Fig. 7. Model of the structural changes undergone by CCT during its functional cycle. The nucleotide-free, substrate-free structure (1) shows an open conformation of its apical domains, which undergoes large structural changes upon ATP binding such that the cavity is closed (2). The substrate-free structure is able to bind unfolded actin (3) and tubulin (5) molecules in a quasi-native conformation. Binding of tubulin generates a more closed conformation of the apical domains than observed without substrate or after actin binding. ATP binding to the CCT–α-actin (4) or CCT–β-tubulin (6) complexes induces conformational changes of the chaperonin apical domains that seal the cavity using their helical extensions. The more downward and inward distribution of the apical domains in the CCT–β-tubulin complex compared with the CCT–α-actin complex is maintained after ATP binding. The model proposed here is based on the results published by Llorca et al. (1999b, 2000) and the results shown in this work.
Finally, the closed conformation induced upon ATP binding is maintained after nucleotide hydrolysis, as revealed by the 3D reconstruction carried out with CCT–β-tubulin in the presence of high concentrations of ADP and Pi (results not shown). This conformational state does not induce liberation of substrate into the cavity. Perhaps this occurs only after nucleotide release and the subsequent return of the eukaryotic chaperonin to the open, nucleotide-free state, or with the help of other cellular cofactors that may be required to facilitate effective substrate release.
Molecular model for the CCT-mediated folding of actin and tubulin: the ‘sequential allosteric ring’ mechanism
The folding mechanism generally accepted for the Group I chaperonins is based predominantly on numerous biochemical and structural results performed with GroEL, and is a non-specific one. GroEL binds non-native forms of a large number of proteins mainly through reciprocal hydrophobic interactions (Chen and Sigler, 1999), and the bound molecules are partially unfolded by mechanical stretching during the ATP and GroES-induced conformational changes (Shtilerman et al., 1999). Substrate is then released within the GroEL–GroES chamber, where it is given a new chance to fold in a hydrophilic environment, hence the name ‘Anfinsen or folding cage’ (Ellis, 1994). Several rounds of binding, encapsulation and release are required for the folding of a protein, and the inefficiency of this process is traded for the folding of an ample set of proteins.
On the other hand, CCT is known from biochemical (Tian et al., 1995a) and structural studies (Llorca et al., 1999b, 2000) to bind actin and tubulin folding intermediates whose subdomains seem to contain a high degree of folded structure. In the cell, the ATP-driven chaperonin-mediated folding reaction takes place quite rapidly, without the existence of multiple rounds of binding and release (Siegers et al., 1999), although there are discrepancies on this issue (Farr et al., 1997). Nevertheless, although GroEL is able to bind actin and tubulin, and undergo multiple cycles of release and rebinding, only CCT is able to generate the native conformations of these two cytoskeletal proteins (Tian et al., 1995b). This all suggests that CCT must deal with very specific folding problems encountered by these two proteins.
What then is the fate of actin and tubulin during these concerted movements in CCT described above? Combining previous data (Llorca et al., 1999b, 2000) and the results presented here, a new model for the mechanism of CCT-assisted folding of actin and tubulin is proposed (Figure 8). CCT captures, directly from translating ribosomes or from prefoldin, actin and tubulin folding intermediates already having significant native-like domain structures. In the case of actin, the quasi-folded structure is then stabilized in an open conformation by CCT binding to the tips of the two domains with opposite regions of the chaperonin ring (Figure 8.1). The N-terminal domain of actin (coloured red) binds to CCTδ, and the C-terminus (coloured white) to either CCTβ or CCTε (only the first alternative is shown). In the case of tubulin, the N- and the C-terminal domains, which interact with CCT after reaching quasi-folded conformations, bind to opposite regions of the chaperonin ring (Figure 8.3). The N-terminal domain (coloured red) binds to the CCTθ/δ or CCTη/α subunits and the C-terminal domain to the CCTβ/γ/θ or CCTε/ζ/β subunits (only the first alternative is shown). The two cytoskeletal proteins, now each bound to CCT in an open, quasi-native conformation, are ready to undergo the final stage of their folding process, which only the eukaryotic chaperonin seems able to facilitate. Although the nature of the folding-limiting process is not known, it has been suggested that it could be related to the binding or loading of the nucleotide to the two cytoskeletal proteins (Llorca et al., 2000).

Fig. 8. Model of the structural changes undergone by the actin and tubulin molecules during the CCT functional cycle. Docking models with the atomic structures of actin and tubulin have been overimposed to the volume of the same proteins complexed with CCT. Quasi-folded actin (1) and tubulin (3) molecules bind to apo-CCT using the domains described in Llorca et al. (1999b, 2000). The N-terminal domains of both cytoskeletal proteins (coloured red) bind to CCT with less affinity than the corresponding C-terminal domains. ATP binding induces large movements of the CCT apical domains that seal the cavity. These movements occur sequentially, starting in CCTα, and move in an anti-clockwise direction (see yellow arrows in 1 and 3). Following this sequence, the N-terminal domains of both actin and tubulin molecules are the first ones to respond to the rearrangement of the apical domains, resulting in their release and movement towards the C-termini, giving rise to a more native conformation (2 and 4). The C-terminal domains bind to CCT with a higher affinity than the N-terminal domains, and this interaction is maintained after nucleotide binding and hydrolysis. Nucleotide release induces the return to the nucleotide-free, open state and the liberation of the folded substrate. The model proposed here is based on the results published by Liou and Willison (1997), Lin and Sherman (1997), Llorca et al. (1999b, 2000) and the results shown in this work. Only one of the possibilities for actin and tubulin binding to CCT is shown, although the other fits equally well with the model proposed.
Nucleotide binding to CCT induces large conformational changes in its apical domains and in the shape of the actin and tubulin bound to the chaperonin (Figure 8.2 and 4). The 3D reconstructions of AMP-PNP–CCT– α-actin and AMP-PNP–CCT–β-tubulin obtained in this study, and the good fitting between the reconstructed volume of actin and tubulin and their corresponding atomic structures, support the view that the two proteins have completed their folding process, but remain bound to the apical region of CCT rather than being released in the cavity. This is surprising, but neverthless reveals a completely different assisted-folding mechanism compared with the general one previously proposed for all chaperonins (Farr et al., 1997). The passive mechanism of GroEL, in which the most critical step in the folding cycle is the liberation of the unfolded substrate into the cavity to undergo auto-folding, has, in the case of CCT, evolved into a more active, physical mechanism whereby the movements of the apical domains upon ATP binding are coupled to the folding movements of actin and tubulin.
How does this mechanism operate? We believe that the mechanism of actin and tubulin folding by CCT is specific, and comprises not only the structural rearrangements undergone by the chaperonin upon nucleotide binding, which are probably similar to those of other chaperonins, but also involves the special kinetics of this chaperonin and the particular geometrical constraints imposed by the two cytoskeletal proteins through interaction with specific CCT subunits. A recent study suggests that the eukaryotic chaperonin has positive intra-ring cooperativity and negative inter-ring cooperativity in ATP hydrolysis, as has been observed for GroEL (Kafri et al., 2001). The authors also propose that the kinetic behaviour of CCT, with respect to nucleotide binding and hydrolysis, could be explained by differences in the intrinsic affinities for ATP of the different intra-ring subunits. This supports a model, based on genetic analysis of ATP-site mutants of yeast CCT, in which catalytic cooperativity of ATP binding and hydrolysis in CCT takes place in a sequential manner (Lin and Sherman, 1997). According to this model, ATP binding/hydrolysis would proceed through the following steps (translated to the Greek letter code of the murine CCT): CCTα → CCTγ → CCTβ → CCTζ (this is the order obtained with the mutated CCT subunits; see arrows in Figure 8.1 and 3 for a more comprehensive sequence). Our results undoubtedly show that ATP binding to CCT generates large movements of the apical domains (∼70° clockwise, viewed from the top; Nitsch et al., 1998; Llorca et al., 1999a) that seal the cavity. If this process were to take place sequentially, as described above, then the N-terminally located domains of actin and tubulin would be the first ones to undergo the structural rearrangements induced by the movements of the apical domains they are interacting with. Biochemical (Hynes and Willison, 2000; Llorca et al., 2000; Ritco-Vonsovici and Willison, 2000) and in vivo (McCallum et al., 2000) data show that these domains bind to CCT with low affinity, so we suggest at this stage that the conformational changes of the apical domains must be large enough to permit the movement of the N-terminal domain of both actin or tubulin towards the C-terminus. Simultaneously, and as a consequence of this movement, the weak interactions between the CCT apical domains and the N-terminal domains of the two cytoskeletal proteins are broken and the N-terminal domains are liberated. The C-terminal regions of actin and tubulin are bound mainly to either the CCTβ or CCTε subunits, which, according to the sequential model shown above, would be among the last ones to undergo the conformational change induced by ATP binding. The 3D reconstruction of AMP-PNP–CCT–β-tubulin and the immunomicroscopy carried out with the same complexes show that the tubulin molecule is located, after nucleotide binding, near the CCTβ/ε subunits. These results support biochemical data suggesting that the interactions between the C-terminal domains of actin and tubulin and CCT are the strongest (Hynes and Willison, 2000; Llorca et al., 2000; Ritco-Vonsovici and Willison, 2000). Taken all together, these data lead us to suggest that the sequential conformational changes undergone by CCT upon ATP binding would induce the movements of the N-terminal domains of actin and tubulin towards their respective C-terminal domains in order to reach their native conformations (Figure 8.2 and 4), which would be acquired finally only after the incorporation of the structural nucleotide. In fact, Tian et al. (1995a) have found GTP binding to tubulin within the chaperonin complex in the presence of ATP. Similarly, Farr et al. (1997) have shown that tubulin can bind GTP when complexed to CCT in the presence of AMP-PNP. We have tested the ability of the purified CCT–β-tubulin complexes used for EM to incorporate radiolabelled GTP in the absence or presence of different nucleotides. The results obtained (not shown) clearly indicate that whereas in the absence of any adenine nucleotide GTP is not able to bind to CCT, the presence of either ATP or AMP-PNP induces the loading of GTP into the tubulin molecule complexed to CCT. This finding corroborates the above mentioned experiments and further supports the notion that tubulin in the AMP-PNP– CCT–β-tubulin complex is already folded to a state that is competent for GTP binding.
Surprisingly, the structural changes undergone by the CCT subunits involved in interactions with the C-terminal domains do not release the cytoskeletal proteins into the chaperonin cavity. It is tempting to suggest, at this stage, a physical explanation for this behaviour that is again related to the sequential mechanism of ATP binding/hydrolysis. We hypothesize that the sequential anti-clockwise conformational changes undergone by the apical domains upon ATP binding (Lin and Sherman, 1997) reach first the N-terminal domains of actin and tubulin, causing their liberation from the chaperonin and thus freeing them to move towards the C-terminal domains in order to attain the native (or quasi-native) conformation. When the conformational changes of the apical domains reach the CCT subunits interacting with the C-terminal domain, being the last remaining regions interacting with CCT, their movements do not liberate the folded molecules into the cavity. However, a substrate liberation step must occur after nucleotide hydrolysis, perhaps after nucleotide release and the subsequent return of the eukaryotic chaperonin to the open, nucleotide-free state, either by the movements of the chaperonin apical domains or with the help of other cellular cofactors that may be required to facilitate effective substrate release.
The results presented in this study raise an important question about the role of the chaperonin cavity in assisted folding. It has become clear for Group I chaperonins like GroEL that the cavity enclosed underneath the GroES cap serves as a folding chamber into which substrates are released and given a new chance to fold. The walls of the cavity are strongly hydrophilic, thus mimicking conditions of infinite dilution for the substrate, which could then try to refold using the information contained in its primary sequence, as postulated by Anfinsen many years ago (Anfinsen, 1973).
Our results indicate a different role for the ring-shape structure of CCT, since the substrate remains bound to the apical domains throughout most of the folding cycle of the eukaryotic chaperonin. The CCT ring is composed of eight different subunits placed in a precise arrangement so that the toroidal structure can sustain a vectorial mechanism that couples the sequential changes occurring in the apical domains of the chaperonin subunits to concerted movements in the substrate molecules that lead to their successful folding. Nevertheless, a role for the cavity in isolating the substrate from non-desired interactions during the folding process cannot be ruled out, and may also be a useful mechanistic adjunct in vivo.
The present work gives new insights into the function of the eukaryotic chaperonin in actin and tubulin folding, but also opens more interesting questions. How are other CCT substrates dealt with by the chaperonin? If the suggested molecular mechanism has evolved concomitantly to eukaryotic evolution and the appearance of actin and tubulin, what were the original functions of the archaeal chaperonins and what is their mechanism of action?
Materials and methods
CCT purification and generation of antibodies
Murine CCT was purified as described by Liou and Willison (1997). The anti-CCTδ 8g monoclonal antibody was prepared as described by Llorca et al. (1999b). The anti-CCTε rat monoclonal antibody εAD1 (epsilon apical domain 1; clone PK/29/23/8d/6b) was raised using E.coli produced recombinant mouse CCTε apical domain (residues E220–N391 in pET11d plus His6 tag at the C-terminus).
Preparation of CCT for immunoprecipitation
Swiss 3T3 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) [10% fetal calf serum (FCS)]. 35S labelling was carried out for 20 h in methionine-deficient DMEM (10% FCS) supplemented with 100 µCi/T75 flask in vitro translation grade methionine and a 1/20th volume of non-deficient DMEM (10% FCS). Cells from two T75 flasks were trypsinized, washed once in phosphate-buffered saline, and the cell pellet resuspended in 200 µl of breaking buffer (50 mM HEPES pH 7.2, 90 mM KCl) and 0.5% NP-40. The cell suspension was mixed by pipetting, and centrifuged at top speed in a bench top centrifuge for 5 min. The resulting cell extract was applied to a 10–40% sucrose gradient and centrifuged at 85 000 g for 18 h at 4°C.
Eighteen microlitres of 20S sucrose gradient fraction containing [35S]CCT were incubated in the presence and absence of 10 mM AMP-PNP for 10 min at room temperature. The respective anti-CCT monoclonal antibody (4.5 µg) was added and the volume increased to 200 µl with breaking buffer with a final concentration of 0.5% NP-40. Samples were incubated on ice for 2 h and 100 µl of 1:1 suspension of protein G beads added for a further 1 h with constant mixing. Beads were washed three times in breaking buffer with 0.5% NP-40. Samples were prepared for SDS–PAGE by the addition of 50 µl of 2× gel loading buffer to the packed, washed beads, and samples resolved on 10% polyacrylamide gels followed by autoradiography.
Preparation of complexes
Commercial α-actin (from rabbit muscle; Sigma) and recombinant β-tubulin were prepared as described (Llorca et al., 2000), denatured in 7 M guanidinium chloride and incubated in a diluting buffer containing 0.4 µM purified murine CCT, as described by Liou and Willison (1997). Complexes of CCT–α-actin and CCT–β-tubulin were prepared by incubation for 15 min at room temperature at a 1:10 molar ratio. The unbound substrate was removed by gel filtration chromatography in Sepharose 6B. Then, the column peak containing CCT–α-actin or CCT–β-tubulin complexes was incubated with AMP-PNP (10 mM). Immunolabelling of AMP-PNP–CCT–β-tubulin complexes was carried out by incubating with anti-CCTδ monoclonal antibody, 8g, as described (Llorca et al., 1999b).
Electron microscopy
For cryo-EM, aliquots of the samples were applied to the grids for 1 min, blotted for 5 s and frozen quickly in liquid ethane at –180°C. Images were recorded at 20° tilt in a JEOL 1200EX-II electron microscope equipped with a Gatan cold stage operated at 120 kV and recorded on Kodak SO-163 film at a 60 000× nominal magnification and ∼1.5 µm underfocus. For the immunomicroscopy, aliquots of the immunocomplexes were applied to carbon grids, negatively stained with 2% uranyl acetate, and recorded at 0° tilt.
Image processing and 3D reconstruction
In both negative stain or frozen–hydrated studies, top views were selected and 2D processing was carried out as described by Llorca et al. (1999a). Substrate-bound and -free particles were separated by a self-organizing map algorithm as described by Marabini and Carazo (1994). When 3D reconstructions were carried out, a second classification procedure was used, as follows: because the conformational changes generated in CCT after nucleotide binding can make it more difficult to distinguish unambiguously between side and top views as well as between substrate-free or substrate-loaded CCT particles, classification of complex mixtures was also carried out by separating homogeneous data sets using the correlation coefficients of each particle with the model. In each round of refinement, particles with a high correlation coefficient were grouped and used for independent reconstructions. By performing this process iteratively, final reconstructions were made of homogeneous populations. Two-dimensional averaging of negatively stained immunocomplexes and 3D reconstructions were carried out as described (Llorca et al., 1999b). The volumes were generated using ART with blobs (Algebraic Reconstruction Techniques; Marabini et al., 1998). Eight-fold symmetry was applied to the CCT volume except in the inner cylinder comprising the CCT cavity where substrate is located, as described previously (Llorca et al., 1999b). The final resolution was calculated by Fourier ring correlation of two independent reconstructions, and the value obtained (∼27 Å for all the complexes) was used to low-pass filter the volume.
Docking of the cryo-EM structures of actin and tubulin bound to CCT with their atomic counterparts
Docking was performed using SITUS (Wriggers et al., 1999) and visualized using VMD (Humphrey et al., 1996). The volume corresponding to actin or tubulin was extracted from the cryo-EM 3D reconstructions of the CCT–substrate complexes by volume difference, and quantized using a self-organizing algorithm, which reduces the volume to a small number of codebook vectors. The atomic structures of native actin (Kabsch et al., 1990) and tubulin (Nogales et al., 1998) were independently quantized using the same number of codebook vectors as for the cryo-EM volume. Among the several docking solutions generated by the docking program, the criteria to select the best fit were based on the r.m.s. deviation between the codebook vectors from the high-resolution data and the low-resolution EM data.
Acknowledgments
Acknowledgements
This work was partially supported by grants from the DGICYT (J.M.V. and J.L.C.). O.L. is a fellow from the Comunidad Autónoma de Madrid. The UK laboratory is supported by the Cancer Research Campaign (CRC). We thank the CRC Hybridoma, Sutton Unit for antibodies.
References
- Anfinsen C.B. (1973) Principles that govern the folding of protein chains. Science, 181, 223–230. [DOI] [PubMed] [Google Scholar]
- Braig K. Otwinowski,Z., Hegde,R., Boisvert,D.C., Joachimiak,A., Horwich,A.L. and Sigler,P.B. (1994) The crystal structure of the bacterial chaperonin GroEL at 2.8 Å. Nature, 371, 578–586. [DOI] [PubMed] [Google Scholar]
- Bukau B. and Horwich,A.L. (1998) The hsp70 and hsp60 chaperone machines. Cell, 92, 351–366. [DOI] [PubMed] [Google Scholar]
- Chen L. and Sigler,P.B. (1999) The crystal structure of a GroEL/peptide complex: plasticity as a basis for substrate diversity. Cell, 99, 757–768. [DOI] [PubMed] [Google Scholar]
- Ditzel L., Löwe,J., Stock,D., Stetter,K.O., Huber,H., Huber,R. and Steinbacher,S. (1998) Crystal structure of the thermosome, the archaeal chaperonin and homologue of CCT. Cell, 93, 125–138. [DOI] [PubMed] [Google Scholar]
- Ellis R.J. (1994) Opening and closing the Anfinsen cage. Curr. Biol., 4, 633–635. [DOI] [PubMed] [Google Scholar]
- Ellis R.J. and Hartl,F.U. (1999) Principles of protein folding in the cellular environment. Curr. Opin. Struct. Biol., 9, 102–110. [DOI] [PubMed] [Google Scholar]
- Farr G.W., Scharl,E.C., Schumacher,R.J., Sondek,S. and Horwich,A.L. (1997) Chaperonin-mediated folding in the eukaryotic cytosol proceeds through rounds of release of native and nonnative forms. Cell, 89, 927–937. [DOI] [PubMed] [Google Scholar]
- Grantham J., Llorca,O., Valpuesta,J.M. and Willison,K.R. (2000) Partial occlusion of both cavities of CCT with antibody has no effect upon the rates of β-actin or α-tubulin folding. J. Biol. Chem., 275, 4587–4591. [DOI] [PubMed] [Google Scholar]
- Gutsche I., Essen,L.O. and Baumeister,W. (1999) Group II chaperonins: new TRIC(k)s and turns of a protein folding machine. J. Mol. Biol., 293, 295–312. [DOI] [PubMed] [Google Scholar]
- Gutsche I., Holzinger,J., Röble,M., Heumann,H., Baumeister,W. and May,R.P. (2000) Conformational rearrangements of an archaeal chaperonin upon ATPase cycling. Curr. Biol., 10, 405–408. [DOI] [PubMed] [Google Scholar]
- Humphrey W., Dalke,A. and Schulten,K. (1996) VMD—visual molecular dynamics. J. Mol. Graph., 14, 33–38. [DOI] [PubMed] [Google Scholar]
- Hunt J.F., Weaver,A.J., Lanfry,S., Gierasch,L. and Deisenhofer,J. (1996) The crystal structure of the GroES co-chaperonin at 2.8 Å resolution. Nature, 381, 571–580. [DOI] [PubMed] [Google Scholar]
- Hynes G.M. and Willison,K.R. (2000) Individual subunits of the chaperonin containing TCP-1 (CCT) mediate interactions with binding sites located on subdomains of β-actin. J. Biol. Chem., 275, 18985–18994. [DOI] [PubMed] [Google Scholar]
- Kabsch W., Mannherz,H.G., Suck,D., Pai,E.F. and Holmes,K.C. (1990) Atomic structure of the actin:DNase I complex. Nature, 347, 37–44. [DOI] [PubMed] [Google Scholar]
- Kafri G., Willison,K.R. and Horovitz,A. (2001) Nested allosteric interactions in the cytoplasmic chaperonin containing TCP-1 from bovine testis. Protein Sci., 10, 445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S., Willison,K.R. and Horwich,A.L. (1994) Cytosolic chaperonin subunits have a conserved ATPase domain but diverged polypeptide-binding domains. Trends Biochem. Sci., 19, 543–548. [DOI] [PubMed] [Google Scholar]
- Leroux M.R. and Hartl,F.U. (2000) Protein folding: versatility of the cytosolic chaperonin TRiC/CCT. Curr. Biol., 10, R260–R264. [DOI] [PubMed] [Google Scholar]
- Lin P. and Sherman,F. (1997) The unique hetero-oligomeric nature of the subunits in the catalytic cooperativity of the yeast Cct chaperonin complex. Proc. Natl Acad. Sci. USA, 94, 10780–10785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou A.K.F. and Willison,K.R. (1997) Elucidation of the subunit orientation in CCT (chaperonin containing TCP1) from the subunit composition of micro-complexes. EMBO J., 16, 4311–4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorca O., Smyth,M.G., Carrascosa,J.L., Willison,K.R., Radermacher, M., Steinbacher,S. and Valpuesta,J.M. (1999a) 3D reconstruction of the ATP-bound form of CCT reveals the asymmetric folding conformation of a type II chaperonin. Nature Struct. Biol., 6, 639–642. [DOI] [PubMed] [Google Scholar]
- Llorca O., McCormack,E., Hynes,G., Grantham,J., Cordell,J., Carrascosa,J.L., Willison,K.R., Fernández,J.J. and Valpuesta,J.M. (1999b) Eukaryotic type II chaperonin CCT interacts with actin through specific subunits. Nature, 402, 693–696. [DOI] [PubMed] [Google Scholar]
- Llorca O., Martín-Benito,J., Ritco-Vonsovici,M., Grantham,J., Hynes,G., Willison,K.R., Carrascosa,J.L. and Valpuesta,J.M. (2000) Eukaryotic chaperonin CCT stabilizes actin and tubulin folding intermediates in open quasi-native conformations. EMBO J., 19, 5971–5979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marabini R. and Carazo,J.M. (1994) Pattern recognition and classification of images of biological macromolecules using artificial neural networks. Biophys. J., 66, 1804–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marabini R., Herman,G.T. and Carazo,J.M. (1998) 3D reconstruction in electron microscopy using ART with smooth spherically symmetric volume elements (blobs). Ultramicroscopy, 72, 53–65. [DOI] [PubMed] [Google Scholar]
- McCallum C.D., Do,H., Johnson,A.E. and Frydman,J. (2000) The interaction of the chaperonin tailless complex polypeptide 1 (TCP1) ring complex (TRiC) with ribosome-bound nascent chains examined using photo-cross-linking. J. Cell Biol., 149, 591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitsch M., Walz,J., Typke,D., Klumpp,M., Essen,L.O. and Baumeister,W. (1998) Group II chaperonin in an open conformation examined by electron tomography. Nature Struct. Biol., 5, 855–857. [DOI] [PubMed] [Google Scholar]
- Nogales E., Wolf,S.G. and Downing,K.H. (1998) Structure of the αβ-tubulin dimer by electron crystallography. Nature, 391, 199–203. [DOI] [PubMed] [Google Scholar]
- Ritco-Vonsovici M. and Willison,K.R. (2000) Defining the eukaryotic chaperonin-binding sites in human tubulins. J. Mol. Biol., 304, 81–98. [DOI] [PubMed] [Google Scholar]
- Rommelaere H., De Neve,M., Melki,R., Vandekerckhove,J. and Ampe,C. (1999) The cytosolic class II chaperonin CCT recognizes delineated hydrophobic sequences in its target proteins. Biochemistry, 38, 3246–3257. [DOI] [PubMed] [Google Scholar]
- Roseman A.M., Chen,S., White,H., Braig,K. and Saibil,H.R. (1996) The chaperonin ATPase cycle: mechanism of allosteric switching and movements of substrate-binding domains in GroEL. Cell, 87, 241–251. [DOI] [PubMed] [Google Scholar]
- Schoehn G., Quaite-Randall,E., Jimenez,J.L., Joachimiak,A. and Saibil,H.R. (2000a) Three conformations of an archaeal chaperonin, TF55 from Sulfolobus shibatae. J. Mol. Biol., 296, 813–819. [DOI] [PubMed] [Google Scholar]
- Schoehn G., Hayes,M., Cliff,M., Clarke,A.R. and Saibil,H.R. (2000b) Domain rotations between open, closed and bullet-shaped forms of the thermosome, an archael chaperonin. J. Mol. Biol., 301, 323–332. [DOI] [PubMed] [Google Scholar]
- Shtilerman M., Lorimer,G.H. and Englander,S.W. (1999) Chaperonin function: folding by forced unfolding. Science, 284, 822–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegers K., Waldmann,T., Leroux,M.R., Grein,K., Schevchenko,A., Schiebel,E. and Hartl,F.U. (1999) Compartmentation of protein folding in vivo: sequestration of non-native polypeptide by the chaperonin–GimC system. EMBO J., 18, 6730–6743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian G., Vainberg,I.E., Tap,W.D., Lewis,S.A. and Cowan,N.J. (1995a) Quasi-native chaperonin-bound intermediates in facilitated protein folding. J. Biol. Chem., 270, 23910–23913. [DOI] [PubMed] [Google Scholar]
- Tian G., Vainberg,I.E., Tap,W.D., Lewis,S.A. and Cowan,N.J. (1995b) Specificity in chaperonin-mediated protein folding. Nature, 375, 250–253. [DOI] [PubMed] [Google Scholar]
- Vainberg I.E., Lewis,S.A., Rommelaere,H., Ampe,C., Vandekerckhove, J., Klein,H.L. and Cowan,N.J. (1998) Prefoldin, a chaperone that delivers unfolded proteins to cytosolic chaperonin. Cell, 93, 863–873. [DOI] [PubMed] [Google Scholar]
- Willison K.R. (1999) Composition and function of the eukaryotic cytosolic chaperonin-containing TCP1. In Bukau,B. (ed.), Molecular Chaperones and Folding Catalysts. Harwood Academic Publishers, Amsterdam, The Netherlands, pp. 555–571.
- Willison K.R. and Grantham,J. (2001) The roles of cytosolic chaperonin, CCT, in normal eukaryotic cell growth. In Lund,P. (ed.), Molecular Chaperones: Frontiers in Molecular Biology. Oxford University Press, Oxford, UK, 90–118.
- Wriggers W., Milligan,R.A. and McCammon,J.A. (1999) SITUS: a package for docking crystal structures into low-resolution maps from electron microscopy. J. Struct. Biol., 125, 185–195. [DOI] [PubMed] [Google Scholar]
- Xu Z., Horwich,A.L. and Sigler,P.B. (1997) The crystal structure of the asymmetric GroEL–GroES–(ADP)7 chaperonin complex. Nature, 388, 741–750. [DOI] [PubMed] [Google Scholar]