


Abstract
Mitochondrial DNA (mtDNA) defects cause debilitating metabolic disorders for which there is no effective treatment. Patients suffering from these diseases often harbour both a wild-type and a mutated subpopulation of mtDNA, a situation termed heteroplasmy. Understanding mtDNA repair mechanisms could facilitate the development of novel therapies to combat these diseases. In particular, mismatch repair activity could potentially be used to repair pathogenic mtDNA mutations existing in the heteroplasmic state if heteroduplexes could be generated. To date, however, there has been no compelling evidence for such a repair activity in mammalian mitochondria. We now report evidence consistent with a mismatch repair capability in mammalian mitochondria that exhibits some characteristics of the nuclear pathway. A repair assay utilising a nicked heteroduplex substrate with a GT or a GG mismatch in the β-galactosidase reporter gene was used to test the repair potential of different lysates. A low level repair activity was identified in rat liver mitochondrial lysate that showed no strand bias. The activity was mismatch-selective, bi-directional, ATP-dependent and EDTA-sensitive. Western analysis using antibody to MSH2, a key nuclear mismatch repair system (MMR) protein, showed no cross-reacting species in mitochondrial lysate. A hypothesis to explain the molecular mechanism of mitochondrial MMR in the light of these observations is discussed.
INTRODUCTION
Mitochondria are ubiquitous organelles found in all nucleated mammalian cells. They house the enzyme complexes responsible for coupling respiration to the generation of ATP and play a central role in several apoptotic pathways (1). In addition to harbouring metabolic enzymes, mitochondria also contain numerous copies of their own genome, mtDNA, that encode components of the oxidative phosphorylation machinery. Diseases arising from mtDNA defects are varied and debilitating (2). There is no effective treatment for these disorders and new therapies are actively being sought. Many patients harbour populations of both mutated and wild-type mtDNA (a condition termed heteroplasmy), with the mutation often being recessive. One possible way of treating these patients would therefore be to manipulate the balance between mutated and wild-type molecules, allowing the wild-type to propagate at the expense of the mutated genome. A method for the selective targeting of mutated mtDNA has been designed, with the aim of inhibiting replication (3,4). An additional long-term approach would be to import wild-type single-stranded DNA oligomers targeted to the mtDNA mutation, which, on hybridisation to the single-stranded mtDNA during replication, would generate a heteroduplex. Endogenous DNA mismatch repair machinery would then recognise the mismatch, substituting the pathogenic nucleotide with the wild-type counterpart.
But does such a repair activity exist in mammalian mitochondria? Nuclear DNA is protected by a variety of repair systems (5,6). Some pathways, such as the nucleotide excision repair (NER) pathway, are non-specific; they detect lesions arising from distortions of the DNA helix (7). Alternatively, other enzymes target and repair particular lesions. Base excision repair (BER) uses a specific glycosylase, then an AP-endonuclease or AP-lyase to excise the lesion (8). For example, thymine-DNA glycosylase repairs T/G mispairs arising from the spontaneous deamination of cytosine to uracil, and 5-methylcytosine to thymine (9). Errors of replication that result in a mismatch or small loop are detected and corrected by the mismatch repair system (MMR), which also functions in homologous recombination. The best- characterised MMR pathway is the bacterial methyl-directed MutHLS system (10). Homologues of MutS and MutL (but not MutH) have been identified in most eukaryotes as several isoforms (11). In the yeast Saccharomyces cerevisiae, one of the six MutS isoforms, MSH1, encodes a mitochondrial protein that binds base mismatches (12). Null strains show gross mtDNA rearrangements, but a homologue of MSH1 has not been found in mammals.
mtDNA is close to the respiratory chain, a major source of reactive oxygen intermediates, and is unbound by histones, consistent with reports suggesting it is subject to more damage than nuclear DNA (13). Several enzymes involved in mitochondrial BER have been reported (14), however NER has not been demonstrated in mammalian mitochondria. To date, it is still unclear whether mammalian mitochondria harbour a functional MMR activity. Here, we seek to determine whether mitochondrial extracts can repair mismatches using a well-characterised, robust, repair assay. Heteroduplexes were engineered from bacteriophage DNA carrying the reporter gene β-galactosidase, which, when coupled to a bacterial read-out system, was used to measure repair activity for various extracts. A significant MMR activity was detected in purified rat mitochondrial lysates, which repaired a single G-T or G-G mismatch. This activity was sensitive to EDTA, required ATP, was insensitive to aphidicolin and did not require the ubiquitous mismatch repair enzyme, MSH2. No strand bias could be detected. One hypothesis is presented to explain the potential role of MMR in mitochondria.
MATERIALS AND METHODS
Materials
Escherichia coli strains NR9099, NR9162 and CSH50 relevant genotypes are described in the repair assay (15). M13mp2 phage derivatives carrying TGA89 and TCA89 mutations in the β-galactosidase gene were a kind gift from T. A. Kunkel (N.I.E.H.S., NC, USA). Restriction enzymes were from Roche or New England Biolabs.
Specific reagents
Aphidicolin and ATP sulfurylase were from Sigma, complete EDTA-free protease inhibitor tablets from Roche, kaleidoscope pre-stained protein markers from BioRad, MSH2 polyclonal antisera from Santa Cruz and plasmid-safe ATP-dependent DNase from Epicentre. Other reagents were of the highest grade obtainable.
Cell lines and cell lysates
Human HeLa (ATCC no. CCL-2) and LoVo (ATCC no. CCL-229) cell lines were grown in DMEM with 10% (v/v) fetal calf serum. Cell cytoplasmic extracts were prepared as detailed in the repair assay (15).
Mitochondrial lysate preparation
The liver of a freshly killed male Wistar rat (200–250 g) was removed and placed into cold MSE buffer (220 mM mannitol, 70 mM sucrose, 5 mM MOPS, pH 7.0, 2 mM EGTA), prior to preparation and percoll purification as previously reported (16). All preparations were analysed for citrate synthase and adenylate kinase activity and aliquots subjected to western analysis with antibodies to glutamate dehydrogenase and actin to confirm minimal contamination. After purification, mitochondria were washed thoroughly in isotonic EDTA-free buffer, and were lysed essentially as for the cytoplasmic extracts, with an additional liquid nitrogen freeze/thaw before homogenisation to promote bursting. Protein concentrations of the lysates were calculated (Bradford) before snap freezing in liquid nitrogen and storage in small (250–500 µg) aliquots at –80°C.
Preparation of heteroduplexes
Heteroduplex was prepared as reported by Thomas et al. (15). In brief, bacteriophage M13mp2 was propagated in the E.coli strain NR9099 and both the replicative form (RF) and single-stranded positive-strand viral DNA collected. Nicked double-stranded RF DNA was degraded by the addition of plasmid-safe DNase (Epicentre) and the intact RF digested with AvaII or Bsu36I. Linearised RF was melted in the presence of single-stranded viral DNA carrying the desired mutation and the mixture left to anneal. Heteroduplex containing a mismatch with a flanking nick either 3′ or 5′ to the mismatch was therefore generated, depending on which restriction enzyme was used. The heteroduplex was purified on a 0.8% agarose gel followed by electroelution and dialysis to concentrate the sample.
Mismatch repair assays
Each sample (total volume 25 µl) contained: 30 mM HEPES pH 7.8; 15 mM sodium phosphate pH 7.5; 7 mM MgCl2; 4 mM rATP; 200 µM each rCTP, rGTP, rUTP; 100 µM each dATP, dCTP, dGTP and TTP; 40 mM creatine phosphate; 100 µg/ml creatine phosphokinase; 1 fmol (5 ng) purified heteroduplex substrate DNA; and 50 µg of crude cytoplasmic or mitochondrial lysate. The reaction was incubated for 20 min at 37°C, then terminated by the addition of a STOP mix [final concentrations of 2 mg/ml Proteinase K, 2% (w/v) SDS and 50 mM EDTA] and re-incubation at 37°C for 60 min. The substrate was recovered by ammonium acetate precipitation with 0.71 mg/ml tRNA as carrier, a chloroform extraction, and re-precipitation with isopropanol. DNA was resuspended in 5 µl of double-distilled water. When present, aphidicolin was added to a final concentration of 120 µM. In the competition experiments, 1 pmol (1000× molar excess) of either homo- or heteroduplex was used.
Recovered DNA was electroporated into NR9162 bacteria (25 µF, 400 Ω, 2010 V/cm) and plated with a host strain, CSH50, onto minimal media with X-Gal (70 µg/ml) and IPTG (1 mM). Plaques were scored after at least 16 h incubation at 37°C.
ATP experiments
Samples were prepared as above, unless stated. When ATP sulfurylase was used, the ATP-regenerating system was omitted, but included crude lysate, 2.5 mM guanosine 5′-monophosphate (GMP) and 10 mM Na2MoO4. ATP sulfurylase was added to a concentration of 25 µg/ml and the samples incubated at 37°C for 30 min. Substrate heteroduplex was added and the assay allowed to proceed as normal. As control, all buffer components were added in the absence of the ATP sulfurylase.
Competitor duplex preparation
A 789 bp fragment of human mtDNA spanning nucleotides 367–1155 was PCR-amplified from DNA isolated from two fibroblast cell lines encoding either the standard sequence or carrying the A606G polymorphism (17). PCR product from the standard sequence was used as homoduplex competitor after purification. For the heteroduplex competitor generation, each DNA preparation was amplified using one biotinylated primer and one unbiotinylated primer such that the unbiotinylated strands contained a G and T, respectively. PCR products were retained with magnetic streptavidin beads (Dynabeads), and the unbiotinylated strands recovered (following the manufacturer’s recommendation) and annealed to give a GT heteroduplex.
Scoring and statistical analysis
Recovered substrate was plated to give approximately 100–400 plaques per 9 cm dish, with two dishes per sample per experiment. In all cases, experiments were repeated at least three times. In most cases, scoring was performed blind; plaques were scored and the results used to generate repair percentages. Sample variance for this assay has been reported at 10% (15). Two-tailed T-test analyses were performed, assuming a normal distribution for both sample populations.
MSH2 western blot
Crude lysates (7.5–30 µg) were sonicated, boiled for 3 min and separated through a 7.5% (w/v) SDS–PAGE at 30 mA for 150 min or until the ∼50 kDa marker reached the bottom of the gel. Gels were either stained with Coomassie brilliant blue dye and photographed, or transferred onto PVDF membrane [25 mM Tris pH 8.55, 200 mM glycine and 20% (v/v) methanol] for 18 h at 30 V. Membranes were blocked in 1× TBS-T, 5% (w/v) milk powder for 1 h, then incubated with a rabbit polyclonal anti-MSH2 antibody in blocking buffer for 3 h at room temperature. After washing, blots were incubated with HRP-conjugated anti-rabbit secondary antibody in 1× TBS-T, 0.5% (v/v) milk powder (1 h). Blots were washed 5× in TBS-T and exposed to X-ray film following addition of ECL-PLUS reagent as recommended. Murine liver lysates used as controls were prepared essentially as nuclear lysates in (18).
RESULTS
The mismatch repair assay uses the analysis of plaque colour to calculate the percentage repair of nicked heteroduplex substrate after treatment with various cellular lysates and bacterial transfection. The substrates contain various mismatches in the blue/white reporter gene β-galactosidase. When the mismatch repair-deficient E.coli strain NR9162 (MutS) is transfected and plated in the presence of IPTG and X-Gal to allow blue/white scoring, unrepaired heteroduplex gives rise to plaques of mixed blue and clear (white) coloration. Repaired heteroduplex forms plaques of a single (pure-burst) colour. In the following experiments, GT substrates (5′ and 3′ nicked) give a wild-type (blue) phenotype after nick-directed repair, and form clear plaques if repair is directed to the opposite strand. The GG substrate is nick-directed to repair to clear (mutant phenotype). Repair is calculated as (1 – the ratio of the percentages of mixed burst plaques obtained from treated and untreated ‘mock’ substrates) and is expressed as a percentage.
Lysate from percoll-purified rat mitochondria repairs a GT mismatch
Mitochondria were obtained from male Wistar rat livers and purified by percoll gradient centrifugation as detailed in Materials and Methods. Following lysis, the extract was used to test whether mitochondria possessed mismatch repair capability. HeLa cytoplasmic lysate was used as a positive control to define efficient repair, and cytoplasmic lysate derived from the MMR-deficient human cell line LoVo represented a negative control. The GT substrate heteroduplex was tested first as this mismatch is repaired efficiently by extracts from many species. The percoll-purified rat mitochondrial lysate (designated ‘Percoll’) shows 29 ± 2% repair efficiency for the GT-mismatched heteroduplex carrying a nick 3′ to the T (GT3′). In comparison, the HeLa-positive lysate was 65 ± 3% repair efficient, with the negative control LoVo lysate giving 5 ± 2.5% repair (Fig. 1A). For the GT5′ substrate, figures were similar; the Percoll mitochondrial lysate supported 25.5 ± 2% repair, with HeLa and LoVo levels being, respectively, 63 ± 1% and 7 ± 0.5%. LoVo has a deletion in the MSH2 gene encompassing exons 3–8 (19) and is therefore mismatch repair deficient. Comparisons of repair efficiencies for the Percoll and the LoVo lysate returned a highly significant P-value of 5 × 10–4. Nuclear repair in the assay is dependent on the concentration of repair proteins (E. Matheson, unpublished data). As the proteins used by the mitochondrial lysate have not been elucidated, the lower level of Percoll repair may be due either to a lower concentration of repair proteins in the lysate, or an inherently lower efficiency of repair.
Figure 1.

Repair activity in mitochondrial lysate. Nicked heteroduplex template carrying a single mismatch (either GT5′ or GT3′) was prepared and MMR assays performed with HeLa whole cell and percoll-purified rat liver mitochondrial lysate (‘Percoll’) as detailed in Materials and Methods. Following transfection, resultant plaques were scored as mixed or pure burst and the repair efficiency calculated. P-values for the statistical comparisons are shown. (A) Mitochondrial lysate repairs GT heteroduplexes. Repair activity is compared between MMR-proficient (HeLa) and -deficient (LoVo) cytoplasmic lysates and rat mitochondrial lysate (Percoll). For the GT3′: HeLa replicates, n = 6, total plaques counted, t = 2740; Percoll, n = 6, t = 2873; LoVo, n = 3, t = 1599. For the GT5′: HeLa, n = 6, t = 2751; Percoll, n = 6, t = 837; LoVo, n = 3, t = 570. (B) Mitochondrial lysate shows no strand bias for repair. The ratio of blue to clear pure burst plaques indicates strand bias, as is demonstrated by the HeLa lysate. The mock that represents the untreated heteroduplex shows no significant difference when subjected to repair by the mitochondrial lysate. Data is derived from the same experiments as (A). (C) Aphidicolin does not impair repair activity in mitochondrial lysate. Aphidicolin (120 µM), an inhibitor of nuclear polymerases α, δ and ε, was added to various lysates and repair of the GT3′ heteroduplex was measured. Data is from three separate experiments. HeLa ± aphidicolin, P = 0.005; Percoll ± aphidicolin, P =0.975.
Interestingly, although nuclear repair is strongly nick-directed, leading to a large excess of blue plaques over clear for both GT substrates, mitochondrial repair gives approximately equal amounts of blue and clear pure-burst plaque (Fig. 1B). When tested against the blue to clear ratios produced by the mock samples, to which no lysate is added, the bias exhibited by Percoll lysates were not significantly different (P-values of 0.95 and 0.27 for GT5′ and GT3′, respectively).
Aphidicolin is an inhibitor of the major nuclear DNA polymerases α, δ and ε, but has no effect on the mtDNA polymerase γ. It was important to test whether this mismatch repair activity was mitochondrial or whether the lysate had been contaminated by other polymerases during sub- fractionation. Mismatch repair in HeLa cell extracts has been shown to be aphidicolin-sensitive when using a similar genetic read-out assay (20). Further studies suggest the active polymerase is α rather than β, δ or ε (21). Thus, aphidicolin should only allow repair using polymerase γ, with perhaps a small amount of bacterially derived resynthesis. Repair of GT3′ by HeLa lysate drops to below 10%, whereas Percoll repair is unaffected by aphidicolin (Fig. 1C, P-values, respectively, 0.005 and 0.975).
Repair is EDTA-sensitive
EDTA, which chelates divalent cations, is routinely used in mitochondrial preparation; however, the EDTA sensitivity of the mismatch repair assay is well attested, due to the absolute requirement for magnesium (21). Percoll lysates were prepared with and without EDTA to a concentration of 1.5 mM (the EDTA levels present after typical mitochondrial isolation). After dilution of lysate in the assay reaction, final concentrations ranged from 150 to 200 µM. As predicted, EDTA reduced both HeLa and Percoll repair activities of GT3′ (Fig. 2) to 5–10%, close to that recorded in the LoVo control.
Figure 2.

Mitochondrial repair activity is highly sensitive to EDTA. EDTA sensitivity of repair for both nuclear and mitochondrial lysates was assessed on the GT5′ heteroduplex. Data is from three separate experiments. A concentration of EDTA much lower than is often used for isolating mitochondria is shown to have a severely inhibitory effect on repair activity. HeLa ± EDTA (200 µM), P = 0.005; Percoll, ± EDTA, P = 0.0035.
A GG mismatch is also repaired
Very short patch repair activities have been reported. These activities often involve MutY-like proteins and show a preference for GT or GA mismatches (22,23). Although these activities do show biased repair to GC, it is possible that a related activity is responsible for the GT repair in the mitochondrial lysate. To test this, a different substrate was constructed from a phage derivative carrying a different mutation in the β-galactosidase gene. The new heteroduplex contained a GG mismatch with a nick 5′ to this site. This GG5′ mismatch was repaired with a similar efficiency to the GT substrates, 33 ± 2.5%, compared to 58 ± 2% for the HeLa control (Fig. 3). Again, repair bias was similar to the mock, although in this case the mock exhibited a pre-existing bias towards clear plaques. Nonetheless, mock/percoll bias was statistically similar, matching the GT substrates. Thus, repair activity in the mitochondrial lysate showed little bias towards a GT or GG mismatch and, strikingly, did not show any nick-directed bias in repair, as compared to the unrepaired substrate bias.
Figure 3.

A GG5′ mismatch is also repaired. Repair activities (A) and strand bias (B) are shown for a GG5′ heteroduplex in HeLa and Percoll lysates (50 µg). As shown for the GT mispair, GG is also repaired by the mitochondrial lysate. Data is accumulated from four separate experiments: mock, t = 2102; HeLa, t = 2288; Percoll, t = 2084. T-test analysis for strand bias: mock/HeLa, P = 0.012; mock/Percoll, P = 0.64.
Repair activity is not due to nick translation
As the repair activity exhibited by mitochondrial lysate does not show any strand bias, it is a formal possibility that the assay is merely measuring nick translation from nicks close to the mismatch introduced randomly during incubation of the substrate with the lysate. To our knowledge, nick translation activity of mammalian DNA polymerase γ has not been reported. However, it is still important to try and exclude this possibility.
Nick translation does not require ATP. Therefore, to test the ATP-dependence of the mitochondrial activity, samples were prepared in which both ATP and its regenerating system were omitted (Table 1). As crude mitochondrial lysate may contain significant ATP levels, samples were also treated with ATP-sulfurylase to remove all endogenous ATP (24). When lysates were forced to attempt repair without ATP, the repair capability from both nuclear and mitochondrial lysates was severely reduced, although the variability was greater between replicates than those for the basic assay (Table 1). Since the sulfurylase needs molybdate as a co-factor, which could adversely affect repair, two types of controls were performed. The first was a buffer control without sulfurylase (SB, Table 1). Second, after the lysates were ATP-depleted by sulfurylase, ATP was replenished to former levels (4 mM, + sulfurylase + ATP, Table 1). In both cases, significant levels of repair were attained; for the GT5′ substrate: HeLa +/– ATP P = 6.1 × 10–5; Percoll +/– ATP P = 0.00172, where + ATP denotes repair by lysate untreated with sulphurylase (normal repair).
Table 1. Mitochondrial MMR requires ATP.
| Lysate ± components | % Repair heteroduplex template | |
|---|---|---|
| GT5′ | GT3′ | |
| HeLa | 67.8 ± 5 (n = 7) | 62.1 ± 3.8 (n = 7) |
| HeLa + SBa | 74.9 (n = 2) | ND |
| HeLa – ATPb | –8.4 ± 10.1 (n = 3) | –4.1 ± 11.3 (n = 3) |
| HeLa + sulfurylase +ATPc | 34.9 ± 3.8 (n = 6) | ND |
| Percoll | 28.7 ± 2.4 (n = 8) | 26.1 ± 3.1 (n = 8) |
| Percoll + SBa | 23.1 (n = 2) | ND |
| Percoll – ATPb | –10.2 ± 14.1 (n = 3) | 0.85 ± 2.9 (n = 3) |
| Percoll + sulfurylase + ATPc | 29.7 ± 4.1 (n = 3) | ND |
ND, not determined.
aSB indicates that activity was assessed after the addition of sulfurylase buffer without the addition of the enzyme.
bConditions identical to those in footnote a, except for the addition of ATP sulfurylase, and the non-inclusion of ATP in reaction buffers.
cConditions identical to those in footnote b, but following sulfurylase treatment the ATP is replenished to 4 mM.
Adding competitor heteroduplex reduces repair
As a separate test to determine whether nick translation or mismatch repair was responsible for resolution of the substrate heteroduplex, competitor homo- and heteroduplexes were added to the assay in excess of the substrate. A 789 bp heteroduplex incorporating a single GT mismatch was made along with the corresponding GC homoduplex. A 1000-fold molar excess of either species was added to samples in an attempt to sequester mitochondrial proteins specifically responsible for mismatch recognition. The addition of heteroduplex had a marked effect on both HeLa and Percoll repair activity with the GT3′ substrate. For each lysate, repair diminished below LoVo negative levels (Fig. 4). Adding homoduplex to the same excess also reduces activity, particularly with HeLa lysate, although this is not statistically significant. This decrease in activity may have been due to partial sequestration of the scanning component of MMR in the HeLa lysate, but the differential result between HeLa and Percoll lysates is again consistent with a different mechanism of MMR.
Figure 4.

Mitochondrial repair activity is inhibited by competitor heteroduplex. Repair activities of the GT5′ substrate were measured after supplementing various lysates with either a 789 bp competitor hetero- or homoduplex. Activities of both lysates are substantially inhibited by the addition of the heteroduplex (1000-fold excess), whilst the homoduplex had only a marginal effect on mitochondrial repair. Data was from three separate experiments. T-test statistical analysis: HeLa/homoduplex P = 0.060, HeLa/heteroduplex P = 0.001; Percoll/homoduplex P = 0.549, Percoll/heteroduplex P = 0.017.
MSH2 is not detectable in rat liver mitochondrial lysates
Since the above data strongly suggest the presence of mismatch repair in rat mitochondria, the logical progression was to investigate whether any common mismatch protein isoforms are detectable in the Percoll lysate. Since MSH2, an essential mismatch repair protein, has been reported in rat liver mitochondrial preparations (25), western analysis of the lysates used in the above repair assays was carried out using a polyclonal anti-hMSH2 antibody that detects rat, mouse and human MSH2. Repeated testing showed MSH2 in HeLa cytoplasmic lysates, with LoVo giving no signal, as expected (26). However, whilst rat and murine liver whole cell extract showed a cross-reacting species of the expected mass, percoll-purified rat mitochondrial lysate did not (Fig. 5).
Figure 5.
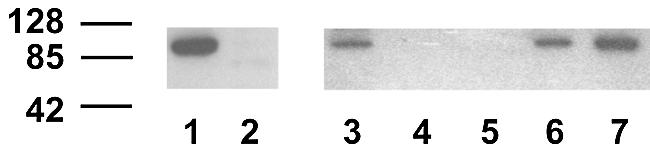
MSH2 is not detectable in rat liver mitochondria. Whole cell lysates and cellular fractions were prepared, separated on a 7.5% polyacrylamide denaturing gel, and transferred to a solid support as detailed in Materials and Methods. Polyclonal antisera were used to detect MSH2 and bound antibody visualised with ECL reagents (Amersham). Lane 1, HeLa cell lysate (7.5 µg); lane 2, LoVo cell lysate (15 µg); lane 3, rat liver cell lysate (15 µg); lanes 4 and 5, purified rat liver mitochondria (7.5 and 30 µg, respectively); lane 6, murine liver cell lysate (7.5 µg); lane 7, murine nuclear extract (7.5 µg). Relevant molecular weight size markers (kDa) are shown to the left of the image.
DISCUSSION
The results set out above are consistent with a mismatch repair activity in mammalian mitochondria. Percoll-purified rat liver mitochondria show a highly reproducible and significant repair capability with both GT and GG mismatches. Consistent with these observations, our preliminary data show a similar repair activity in human HeLa mitochondrial lysates (data not shown). Several experiments indicate this activity is unlikely to be due to nuclear contamination. First, inhibition of most nuclear polymerases by aphidicolin gave no decrease in repair activity. Second, using a variety of markers, contamination of the mitochondrial lysate by other components was negligible. Third, repair activity did not show nick-dependent strand or base repair bias, unlike other known activities. Repair was extremely sensitive to EDTA and it was important to wash mitochondrial preparations in isotonic EDTA-free buffer prior to preparation of the lysate.
Mitochondrial repair is not directed to the nicked strand like its nuclear counterpart; this suggests that the activity is either unbiased, or the method of discrimination is not revealed by this assay. Potentially, nick translation could account for unbiased repair, especially as the mitochondrial lysate exhibits appreciable nuclease activity. Two observations refute this. First, both polymerase and nuclease activities in nick translation are ATP-independent, yet the repair activity in mitochondria is strongly ATP-dependent. Second, the addition of linear competitor DNA carrying a mismatch strongly inhibits mitochondrial MMR efficiencies whereas the fully complementary DNA does not.
What is the mechanism of mitochondrial MMR? At present, our data is unable to provide the answer, but it does give some clues. Several DNA glycosylases have been identified in mammalian mitochondria that function in the removal of base adducts such as oxidised purines [hOGG1, hMYH1 (27)] or thymine glycol [mtTGendo (28)] and uracil [UNG1 (29)]. These glycosylases are postulated to function by short patch BER. To date, there has been no report of NER in mammalian mitochondria, an observation based on the difficulties in repairing large DNA lesions in mtDNA (30). Although it is tempting to suggest that the BER pathway may be intimately involved in mismatch resolution, the absence of repair bias is surprising. In all cases, mitochondrial BER pathways show a clear bias to removing the unwanted base or adduct. Furthermore, although the GT mismatch is likely to be recognised by the BER machinery, the GG mismatch would not be expected to be recognised and resolved with equal or greater efficiency than the GT. Also, the BER pathway needs no ATP until the final ligation step, yet the mitochondrial lysate repair is largely ATP-dependent. A number of mismatch proteins possess ATPase domains, as does DNA helicase, used to unwind the DNA prior to degradation and resynthesis of the lesional strand. It is therefore possible that a small percentage of the repair in the mitochondrial lysate is either BER or nick translation; however, the bulk of repair is clearly via another method.
So, does mitochondrial MMR utilise homologues of the classic MutHLS repair system? MSH1 clearly has a mitochondrial function in yeast, but no homologue is apparent in higher eukaryotes. It is possible that some of the MutL or MutS homologues known to function in MMR may pick up mitochondrial targeting sequences by alternative transcription initiation or splicing, as has been reported for the UNG gene product (29). The most central player is MSH2, which has apparently been identified in rat mitochondrial lysate (25). As reported here, we were unable to detect MSH2 in our rat liver lysates. Protease inhibitors were used in preparing these lysates and western analysis using several mitochondrial markers was unable to detect any evidence of proteolytic degradation. Irrespective, the same lysate clearly demonstrated MMR activity. So, although we cannot conclude that mitochondrial MMR is MSH2-independent, if a mitochondrial form of MSH2 exists, it must be present at a low level in the organelle.
If mitochondrial MMR is MSH2-independent, why is there such low-level activity in LoVo cell lysate, which is only known to carry an msh2 mutation? In mammals, MSH2 forms two heterodimers: with MSH6 it forms MutSα, which repairs base–base mismatches, whilst with MSH3 (MutSβ) it repairs short loops. When MSH2 protein is absent, both MSH3 and MSH6 protein partners are unstable and rapidly degraded (31). Thus, LoVo lysates are functionally lacking in both nuclear MutS complexes; however, a mitochondrially targeted isoform of MSH3 that worked in the absence of MSH2 would not be predicted to be destabilised. Whole cell lysate is used at similar concentrations to the mitochondrial lysates for our assays. Assuming standard 5-fold protein enrichment if mitochondria were purified from LoVo cells, it is interesting to note that the apparent low level of activity in the LoVo lysate would then be increased to the similar levels demonstrated in rat liver mitochondria. Therefore, it is possible that the minimal activity in the LoVo cells could be due to mitochondrial MMR protein in the lysate.
Whatever mechanism is postulated to explain mitochondrial MMR, it is unusual that the repair shows no strand bias in our assay. Why would mitochondria possess a MMR activity that does not show any strand bias? If mitochondria were unable to bias repair, an incorrectly added nucleotide could become mutagenic, acting as a template to drive mutation of the mismatch. This could explain the significantly high level of mtDNA mutation as compared to nuclear DNA (32). However, why retain such an activity when the mismatch would be resolved after a single round of replication, without the need for any form of repair? One intriguing explanation is that the MMR activity in mitochondria has not been retained to repair single base mismatches, but for the resolution of loops. The number of short repeat sequences in the mitochondrial genome is remarkable (see MITOMAP at http://www.mitomap.org). It is well known that large deletions of the genome are common, and believed to be nucleated at repeat sequences that cause slipped replication. Small deletions are relatively uncommon. Mitochondrial MMR may have been retained to repair short palindrome-containing loop mismatches, which must also occur during replication but rarely cause deletions. Taghian et al. (33) have demonstrated such an activity in the nucleus of a mutant CHO cell line. They report that short loops containing palindromic repeats are repaired and are retained, irrespective of nicks, whilst large loops are lost from a heteroduplex substrate. This cell line was able to repair single base mismatches at lower frequency and is reported to be MSH2-deficient. The enzyme responsible for this repeat retention is currently unknown but a mitochondrial homologue may be important to retain the integrity of the mammalian mitochondrial genome.
Acknowledgments
ACKNOWLEDGEMENTS
Paul M. Smith is thanked for his help in mitochondrial preparation, Brian Wilson for providing the murine extracts and Janet Quinn for her encouragement. P.A.M. is supported by an MRC research studentship (G78/6389). This work was partly funded by a grant from the Newcastle Hospitals Special Trustees (R.N.L.) and The Wellcome Trust (R.N.L.)
REFERENCES
- 1.Sastre J., Borras,C., Garcia-Sala,D., Lloret,A., Pallardo,F.V. and Vina,J. (2002) Increasing Healthy Life Span: Conventional Measures and Slowing the Innate Aging Process. Ann. NY Acad. Sci., 959, 1–525. [PubMed] [Google Scholar]
- 2.Chinnery P.F. and Turnbull,D.M. (2001) Epidemiology and treatment of mitochondrial disorders. Am. J. Med. Genet., 106, 94–101. [DOI] [PubMed] [Google Scholar]
- 3.Chrzanowska-Lightowlers Z.M.A., Lightowlers,R.N. and Turnbull,D.M. (1995) Gene-therapy for mitochondrial-DNA defects—is it possible? Gene Ther., 2, 311–316. [PubMed] [Google Scholar]
- 4.Taylor R.W., Chinnery,P.F., Turnbull,D.M. and Lightowlers,R.N. (1997) Selective inhibition of mutant human mitochondrial DNA replication in vitro by peptide nucleic acids. Nature Genet., 15, 212–215. [DOI] [PubMed] [Google Scholar]
- 5.Bohr V.A. and Hanawalt,P.C. (1988) DNA-repair in genes. Pharmacol. Ther., 38, 305–319. [DOI] [PubMed] [Google Scholar]
- 6.Lehman I.R. (1997) Minireview prologue—eukaryotic DNA repair mini review series. J. Biol. Chem., 272, 23463–23463. [Google Scholar]
- 7.Hoeijmakers J.H.J. (1993) Nucleotide excision repair. 2. From yeast to mammals. Trends Genet., 9, 211–217. [DOI] [PubMed] [Google Scholar]
- 8.Parikh S.S., Mol,C.D. and Tainer,J.A. (1997) Base excision repair enzyme family portrait: integrating the structure and chemistry of an entire DNA repair pathway. Structure, 5, 1543–1550. [DOI] [PubMed] [Google Scholar]
- 9.Tomilin N.V. and Aprelikova,O.N. (1989) Uracil DNA glycosylases and DNA uracil repair. Int. Rev. Cytol., 114, 125–179. [DOI] [PubMed] [Google Scholar]
- 10.Jiricny J. (1998) Replication errors: cha(lle)nging the genome. EMBO J., 17, 6427–6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kolodner R.D. and Marsischky,G.T. (1999) Eukaryotic DNA mismatch repair. Curr. Opin. Genet. Dev., 9, 89–96. [DOI] [PubMed] [Google Scholar]
- 12.Chi N.W. and Kolodner,R.D. (1994) Purification and characterization of Msh1, a yeast mitochondrial protein that binds to DNA mismatches. J. Biol. Chem., 269, 29984–29992. [PubMed] [Google Scholar]
- 13.Richter C., Park,J.W. and Ames,B.N. (1988) Normal oxidative damage to mitochondrial and nuclear-DNA is extensive. Proc. Natl Acad. Sci. USA, 85, 6465–6467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Croteau D.L. and Bohr,V.A. (1997) Repair of oxidative damage to nuclear and mitochondrial DNA in mammalian cells. J. Biol. Chem., 272, 25409–25412. [DOI] [PubMed] [Google Scholar]
- 15.Thomas D.C., Umar,A. and Kunkel,T.A. (1995) Measurement of heteroduplex repair in human cell extracts. Methods, 7, 187–197. [Google Scholar]
- 16.McGregor A., Temperley,R., Chrzanowska-Lightowlers,Z.M.A. and Lightowlers,R.N. (2001) Absence of expression from RNA internalised into electroporated mammalian mitochondria. Mol. Genet. Genomics, 265, 721–729. [DOI] [PubMed] [Google Scholar]
- 17.Chinnery P.F., Johnson,M.A., Taylor,R.W., Lightowlers,R.N. and Turnbull,D.M. (1997) A novel mitochondrial tRNA phenylalanine mutation presenting with acute rhabdomyolysis. Ann. Neurol., 41, 408–410. [DOI] [PubMed] [Google Scholar]
- 18.Nilsen H., Haushalter,K.A., Robins,P., Barnes,D.E., Verdine,G.L. and Lindahl,T. (2001) Excision of deaminated cytosine from the vertebrate genome: role of the SMUG1 uracil-DNA glycosylase. EMBO J., 20, 4278–4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu B., Nicolaides,N.C., Markowitz,S., Willson,J.K.V., Parsons,R.E., Jen,J., Papadopolous,N., Peltomaki,P., Delachapelle,A., Hamilton,S.R. et al. (1995) Mismatch repair gene defects in sporadic colorectal cancers with microsatellite instability. Nature Genet., 9, 48–55. [DOI] [PubMed] [Google Scholar]
- 20.Glaser P.M., Sarkar,S.N., Chisholm,G.E. and Summers,W.C. (1987) DNA mismatch repair detected in human cell extracts. Mol. Cell. Biol., 7, 218–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thomas D.C., Roberts,J.D. and Kunkel,T.A. (1991) Heteroduplex repair in extracts of human HeLa cells. J. Biol. Chem., 266, 3744–3751. [PubMed] [Google Scholar]
- 22.Miller E.M., Hough,H.L., Cho,J.W. and Nickoloff,J.A. (1997) Mismatch repair by efficient nick-directed and less efficient mismatch-specific, mechanisms in homologous recombination intermediates in Chinese hamster ovary cells. Genetics, 147, 743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takao M., Zhang,Q.-M., Yonei,S. and Yasui,A. (1999) Differential subcellular localization of human MutY homolog (hMYH) and the functional activity of adenine:8-oxoguanine DNA glycosylase. Nucleic Acids Res., 27, 3638–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.James A.M., Sheard,P.W., Wei,Y.H. and Murphy,M.P. (1999) Decreased ATP synthesis is phenotypically expressed during increased energy demand in fibroblasts containing mitochondrial tRNA mutations—implications for neurodegenerative and mitochondrial DNA diseases. Eur. J. Biochem., 259, 462–469. [DOI] [PubMed] [Google Scholar]
- 25.Chen Z.Y., Felsheim,R., Wong,P., Augustin,L.B., Metz,R., Kren,B.T. and Steer,C.J. (2001) Mitochondria isolated from liver contain the essential factors required for RNA/DNA oligonucleotide-targeted gene repair. Biochem. Biophys. Res. Commun., 285, 188–194. [DOI] [PubMed] [Google Scholar]
- 26.Umar A., Boyer,J.C., Thomas,D.C., Nguyen,D.C., Risinger,J.I., Boyd,J., Ionov,Y., Perucho,M. and Kunkel,T.A. (1994) Defective mismatch repair in extracts of colorectal and endometrial cancer cell-lines exhibiting microsatellite instability. J. Biol. Chem., 269, 14367–14370. [PubMed] [Google Scholar]
- 27.Nakabeppu Y. (2001) Regulation of intracellular localization of human MTH1, OGG1, and MYH proteins for repair of oxidative DNA damage. Prog. Nucleic Acid Res. Mol. Biol., 68, 75–94. [DOI] [PubMed] [Google Scholar]
- 28.Stierum R.H., Croteau,D.L. and Bohr,V.A. (1999) Purification and characterization of a mitochondrial thymine glycol endonuclease from rat liver. J. Biol. Chem., 274, 7128–7136. [DOI] [PubMed] [Google Scholar]
- 29.Nilsen H., Otterlei,M., Haug,T., Solum,K., Nagelhus,T.A., Skorpen,F. and Krokan,H.E. (1997) Nuclear and mitochondrial uracil-DNA glycosylases are generated by alternative splicing and transcription from different positions in the UNG gene. Nucleic Acids Res., 25, 750–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cullinane C. and Bohr,V.A. (1998) DNA interstrand cross-links induced by psoralen are not repaired in mammalian mitochondria. Cancer Res., 58, 1400–1404. [PubMed] [Google Scholar]
- 31.Drummond J.T., Genschel,J., Wolf,E. and Modrich,P. (1997) DHFR/MSH3 amplification in methotrexate-resistant cells alters the hMutS alpha/hMutS beta ratio and reduces the efficiency of base-base mismatch repair. Proc. Natl Acad. Sci. USA, 94, 10144–10149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herrnstadt C., Preston,G., Andrews,R., Chinnery,P., Lightowlers,R.N., Turnbull,D.M., Kubacka,I. and Howell,N. (2002) A high frequency of mtDNA polymorphisms in HeLa cell sublines. Mutat. Res., 501, 19–28. [DOI] [PubMed] [Google Scholar]
- 33.Taghian D.G., Hough,H. and Nickoloff,J.A. (1998) Biased short tract repair of palindromic loop mismatches in mammalian cells. Genetics, 148, 1257–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]