

Abstract
Pseudogenes are copies of genes that cannot produce a protein. They can be detected from disruptions to their apparent coding sequence, caused by frameshifts and premature stop codons. They are classed as either processed pseudogenes (made by reverse transcription from an mRNA) or duplicated pseudogenes, arising from duplication in the genomic DNA and subsequent disablement. Historically, there is anecdotal evidence that the fruit fly (Drosophila melanogaster) has few pseudogenes. Investigators have linked this to a high deletion rate of genomic DNA, for which there is evidence from genetic experiments on genome size. Here, we apply a homology-based pipeline that was developed previously to identify pseudogenes in other eukaryotic genomes, to the fruit fly, so as to derive the first complete survey of its pseudogene population. We find approximately 100 pseudogenes, with at least a sixth of these as candidate processed pseudogenes. This gives a much lower proportion of pseudogenes (compared with the size of the proteome) than in the genomes of other eukaryotes for which data are available (human, nematode and budding yeast). Closest matching proteins to Drosophila pseudogenes are significantly longer than the average protein in its proteome (up to ∼60% more than the average protein’s length), in contrast to the situation in the three other eukaryotic genomes. This may be due to the persistence of fragments of longer genes. In the fly pseudogene population, we found most pseudogenes for serine proteases (which are more abundant in the Drosophila lineage compared with the other eukaryotes), immunoglobulin-motif-containing proteins and cytochromes P450. Data on the sequences and positions of the putative pseudogenes are available at: http://www.pseudogene.org/fly. The detection of a small number of pseudogenes in the Drosophila genome and the higher mean length for the closest matching proteins to pseudogenes (possibly because remnants of genes encoding longer proteins are more likely to persist) are further evidence for a high deletion rate of genomic DNA in the fruit fly. The data are useful for molecular evolution study in Drosophila.
INTRODUCTION
Pseudogenes are copies of genes that do not produce a full-length functional protein chain. Their apparent protein-coding sequences are disrupted by frameshifts and premature stop codons as evolution progresses (1–3). They occur in two forms: first, duplicated pseudogenes arise from duplications of a gene (or an exon) that become disabled and subsequently are degraded; secondly, processed pseudogenes arise from reverse transcription of a messenger RNA and reintegration of the resultant cDNA into genomic DNA (1–3). The latter type of pseudogene can arise as a by-product of LINE-1 retrotransposition in humans (4). Surveys have recently been performed on the pseudogene populations of budding yeast, nematode and chromosomes 21 and 22 for human, with a further analysis of over 2000 ribosomal-protein pseudogenes in the whole human genome (5–8). The procedures derived in these papers have been applied to the Drosophila genome in the present study to derive an initial overview of the pseudogene population of this fly. Here, we report the detection of about 100 putative pseudogenes in the Drosophila genome, and present analysis of some of their characteristics, such as the length of their matching proteins and their most common functional groupings.
MATERIALS AND METHODS
Searching for putative pseudogenes in Drosophila melanogaster
We applied procedures for detection of pseudogenes based on the identification of protein homology in the genomic DNA that is disabled by frameshifts or premature stop codons; these procedures have been described in detail previously (7). As for our study of human chromosomes 21 and 22, we ensured that we minimized the number of disabled extensions like those observed for known genes [see methods of ref. (7) for the complete procedure; an extension length minimum of 24 residues was found to be suitable]. We used Releases 1 and 2 of the Drosophila genome and the accompanying annotations (9). We disregarded any sequences that may have arisen from disabled copies of transposable elements (10). As before, we assigned as candidate processed pseudogenes, any sequences that (i) are of substantial length (>70% of the length of the closest matching protein sequence) and that have no obvious introns, or (ii) have evidence of polyadenylation and no obvious introns (7). Evidence of polyadenylation is defined as a discernible canonical AATAAA polyadenylation signal followed within 50 nucleotides by a region of elevated polyadenine content (≥30 adenines in a 50 nucleotide stretch), within 1000 nucleotides from the end of the detected homology (7). Drosophila transcripts have a greater tendency than transcripts of the other eukaryotes to use the canonical AATAAA polyadenylation signal (11). We have re-mapped the pseudogene annotations onto the recent Release 3 of the fly genome.
Comparison with existing pseudogene annotation
In addition, we examined existing annotations for fly pseudogenes downloaded from the FLYBASE website (http://www.flybase.org). We found 10 previously reported pseudogenes that are in euchromatic DNA, that are not obviously associated with a transposable element and whose sequences were available. However, once we set aside those that do not occur in the sequenced fly strain or that are truncations (and would not be detected by our procedures), we are left with only three existing annotations [two cytochrome P450 pseudogenes and one α-esterase pseudogene (9,12)], each of which are recovered in our study.
Assignment of features in pseudogenes
InterPro motif families (13) were assigned to pseudogenes by transferring annotations from the closest matching Drosophila protein. Lists of matches for Drosophila proteins were downloaded from the InterPro proteome analysis website (http://www.ebi.ac.uk/proteome). Similarly, Gene Ontology (GO) annotations for function (downloaded from http://www.geneontology.org) were also transferred (14).
RESULTS AND DISCUSSION
Numbers and distribution of pseudogenes
We found 110 pseudogenes in the Drosophila genome, which is about one for every 130 proteins encoded in the genome. This proportion is much lower than in the other eukaryotic genomes for which studies on pseudogene populations have been completed (Table 1). For example, in the single-celled budding yeast (Saccharomyces cerevisiae) there are over 220 pseudogenic ORFs, which is about one for every 30 encoded proteins (5). In human, our surveys have shown that there may be one duplicated and one processed pseudogene for every four genes (7). A recent paper detailing comparative analysis of the genomes of Anopheles gambiae and D.melanogaster describes detection of 176 pseudogenes in Drosophila by searching for disabled protein homology; however, our methods our more conservative, as we disregard any disabled homology fragments that look like disabled extensions to known genes (such as might arise in the last exon of a gene) (see Materials and Methods) (15); also, we disregard any pseudogenic copies of proteins from transposable elements (10). On a related note, we recently found that the fly has more decayed remnants of genes than other sequenced eukaryotes that are undetectable by standard gene prediction and sequence alignment procedures (16).
Table 1. Numbers and mean lengths for proteins and pseudogenes in four eukaryotes.
| Organism | Number of proteins | Number of pseudogenesa | Number of processed pseudogenes | Mean length of proteinb | Mean length of matching protein for pseudogenes |
|---|---|---|---|---|---|
| Humanc | 927 | 384 (2.4) | 189 | 317 (±43) | 342 |
| Nematode worm | 20732 | 1100 (18.9) | 104 | 435 (±15) | 450 |
| Budding yeast | 6340 | 221 (28.7) | 0 | 467 (±29) | 424d |
| Fruit fly | 14332 | 110 (130.3) | 34e | 500 (±50) | 808f |
aThe proportion of proteins to pseudogenes is given in brackets.
bStandard deviation of the sample mean is given in brackets.
cThese data are for chromosomes 21 and 22 only (7).
dThe difference between mean lengths of yeast proteins in general and those that are closest matches to pseudogenes is marginally significant (P < 0.06) using normal statistics.
eThis value is for pseudogenes (i) that are of substantial length (>70% the length of the closest matching organismal protein) and have no introns (where a matching protein does have introns) or (ii) that have some evidence of polyadenylation. See Materials and Methods for more detail. These procedures are described in (7).
fThe difference between mean lengths of fruit fly proteins in general and those that are closest matches to pseudogenes is very significant (P < 0.0001) using normal statistics. Removing the outlying matchers of seven fragments whose lengths exceed 3000 amino acids, reduces the mean to 610 residues (P < 0.02).
Processed pseudogenes do not have introns (as they are derived from messenger RNA transcripts), and, if recently integrated into the genome, have detectable characteristic features such as a polyadenine tail with an upstream polyadenylation signal (3,7). We examined the fly pseudogenes for evidence of being processed (Table 1). About one-sixth (19/110) of the Drosophila pseudogenes have no obvious introns and both a polyadenylation signal and a downstream polyadenine-rich stretch in the genomic DNA, and up to a third of the pseudogenes (34/110) have some evidence of processing (see Materials and Methods and Table 1 for details). There are six pseudogenic copies of single-exon genes that could be either processed or duplicated pseudogenes. The only previously well-documented evidence of processing in Drosophila is an alcohol dehydrogenase retrosequence, which is part of the gene jing-wei in many Drosophila species (but not melanogaster) (17), and was originally identified as an anomalously conserved processed pseudogene (18,19). Our data show that processed pseudogenes are comparatively rare in the fruit fly genome (Table 1), indicating either a low rate of generation, or a high rate of deletion from the genome. Indeed, our procedures could be over-assigning pseudogenes as processed, particularly in situations where the pseudogene fragment is too small to discern the original intron–exon boundaries, so the figure of 34/110 pseudogenes as processed should be considered an upper bound.
The pseudogene population and its subpopulation of candidate processed pseudogenes appear to be dispersed randomly along the chromosomes (Fig. 1) [as for genes, there are no notable large-scale gradients or clusterings in their positioning (9), although we must emphasize that we only have a small population]. However, there appears to be clustering of pseudogenes within 2 Mb of either side of the 16 Mb of pericentromeric heterochromatin on chromosome 2 (see 2L and 2R in Fig. 1). Such large blocks of heterochromatin are also seen around the X- and third-chromosome centromeres. These clusterings comprise 16 pseudogenes, of which eight were judged to be candidate processed pseudogenes [two of these are homologous to parts of the protein Osa (20)]; of the others, two are homologous to a retroviral reverse transcriptase [InterPro motif IPR000477 (13)]. This pericentromeric area may be a ‘cold-spot’ for genomic DNA deletion.
Figure 1.
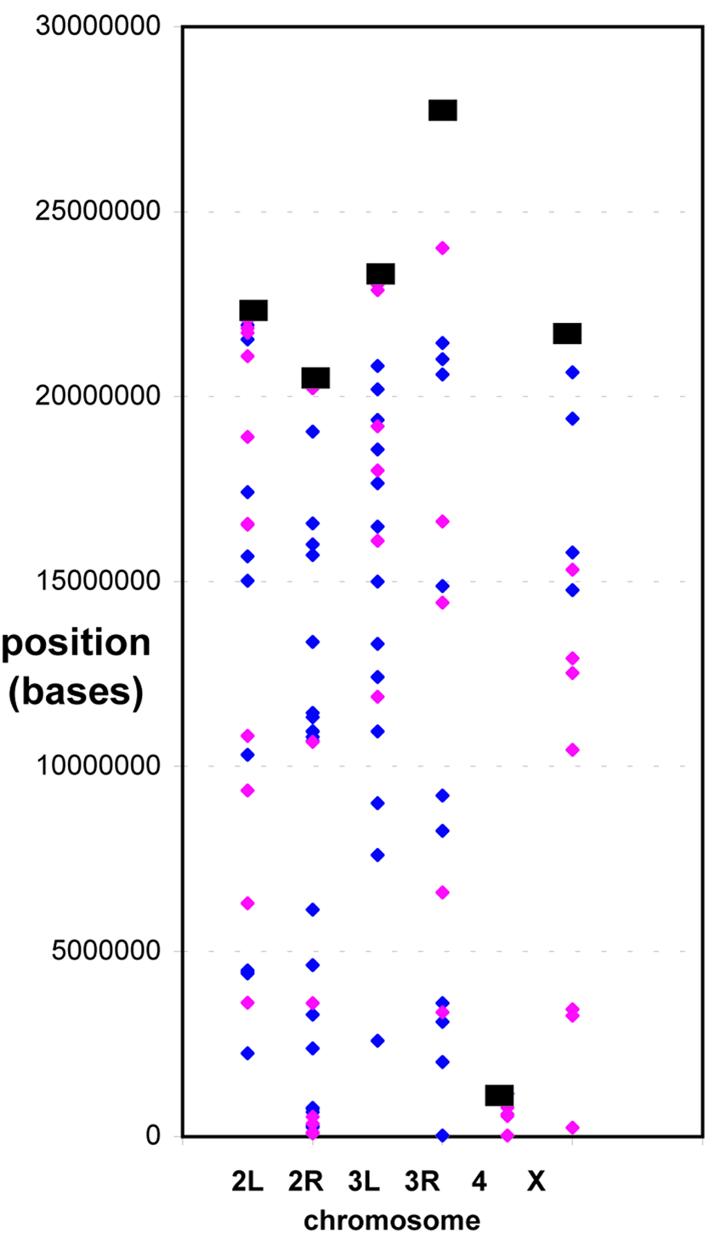
Distribution of putative pseudogenes in the fly genome. The position along each chromosome of each candidate processed pseudogene is indicated by a purple spot. Positions of other pseudogenes are indicated by blue spots. The y-axis is the position in megabases along the chromosome or chromosome arm, with the chromosomes or chromosome arms arrayed along the x-axis.
Length of closest matching proteins to pseudogenes
We calculated the mean length of the closest matching proteins for pseudogenes of the genomes of budding yeast, nematode worm and human (chromosomes 21 and 22 only). We compared this with the same data for the Drosophila genome pseudogenes (Table 1). In Drosophila, coding sequences that give rise to pseudogenes tend to be rather longer than the average coding sequence, in contrast to the situation in other organisms. Specifically, we found that closest matching proteins for pseudogenes tend to be ∼60% longer than the average Drosophila protein (Table 1). [Their mean length reduces to ∼20% longer than average when seven outlying matching proteins of >3000 residues are deleted (Table 1 footnote).] This length observation may arise because remnants of longer genes can persist for longer in the genomic DNA than shorter genes, and withstand very high deletion rates of genomic DNA in Drosophila (21).
There is evidence from experiments investigating genome size that Drosophila has a very high genomic DNA deletion rate (21,22). This has traditionally been thought to be the reason that few Drosophila pseudogenes have been discovered in the past (23). There is some evidence that the underlying deletion rate of genomic DNA is also high in nematodes (24,25); however, some gene families, particularly types of G-protein-coupled receptor (GPCR), appear to acquire and ‘use up’ more novel duplications, resulting in a lowered net rate of deletion of pseudogenes in the nematode genome. There is also a marginally significant difference in the same comparison for the budding yeast pseudogene data (Table 1 footnote); however, in this case, the proteins that are closest matches to pseudogenes tend to be somewhat shorter than the average protein encoded by the genome. This finding may be related to the high concentration of pseudogenes and homologs of pseudogenes near the telomeres of the budding yeast genome (5).
Most common families and functions
InterPro motifs (13) and GO function categories (14) were mapped onto the fruit fly pseudogenes via annotations for their closest matching protein sequences. The top-ranking motifs and functions are listed in Tables 2 and 3.
Table 2. Prevalent InterPro motif families for the fruit fly pseudogenes.
| Number of pseudogenes with motif | Number of proteins with motif | Description of InterPro motif family |
|---|---|---|
| 7 [2] | 187 (4) | Chymotrypsin serine protease, family S1 (IPR001314) |
| 7 [2] | 206 (3) | Serine protease, trypsin family (IPR001254) |
| 6 [2] | 132 (10) | Immunoglobulin/major histocompatibility complex (IPR003006) |
| 5 [2] | 100 (14) | Immunoglobulin C-2 type (IPR003598) |
| 5 [0] | 162 (7) | Proline-rich extensin (IPR002965) |
| 5 [0] | 80 (28) | Chitin-binding peritrophin A (IPR002557) |
| 5 [0] | 347 (1) | C2H2-type zinc finger (IPR000822) |
| 4 [0] | 44 (53) | Protein of unknown function DUF227 (IPR004119) |
| 4 [0] | 88 (20) | EGF-like domain (IPR000561) |
The numbers of pseudogene sequences that have each type of domain (regardless of how many domains are detected in each sequence) are listed in decreasing order. Only the top 10 counts are listed. In square brackets are the counts for candidate processed pseudogenes. In round brackets in the second column are the rankings for the count of genes with motifs in the whole proteome.
Table 3. Prevalent GO categories for the fruit fly pseudogenes.
| Number of pseudogenes with GO category | Description of GO category |
|---|---|
| 4 | Cytochrome P450 (GO:0015034, function) |
| 3 | Serine type endopeptidase (GO:0004252, function) |
| 2 | Myosin ATPase (GO:0008570, function) |
| 2 | RAN protein binding (GO:0008536, function) |
| 2 | Structural constituent of larval cuticle (GO:0008010, function) |
| 2 | DNA binding (GO:0003677, function) [from homology to the gene Osa (16)] |
The counts of GO function categories for the pseudogene sequences in decreasing order of occurrence; only those function categories with multiple occurrences are listed.
The most common InterPro motifs are for serine proteases (there are multiple GO function category designations for these enzymes as well, as ‘serine-type endopeptidase’), and immunoglobulin-like domain motifs. The serine proteases are types of proteins that are very abundant in the fly, but are very rare in the nematode worm (Caenorhabditis elegans), budding yeast (S.cerevisiae) and the weed Arabidopsis thaliana, and of intermediate abundance in the human proteome (see InterPro website: http://www.ebi.ac.uk/interpro). The S1 class of proteases (Table 2) is thought to have roles in digestion, the complement cascade and in various signaling pathways in the fly (9). This finding continues the theme of pseudogenes tending to occur for lineage-specific or lineage-expanded classes of proteins, observed previously for other eukaryotes (1). Interestingly, there are also multiple pseudogenes for the cytochromes P450 (Table 3), which are proteins that have a ‘broad’ substrate specificity. In other organisms, classes of proteins that have a ‘breadth’ of substrate specificity, or ‘binding diversity’, have many pseudogenes, such as the chemoreceptors in the nematode (6), and the immunoglobulins and olfactory receptors in human (7,26). The fact that the cytochromes P450 were not ‘counted’ in the InterPro motif listings demonstrates the utility of combining different methods (here both GO function categories and InterPro motifs) to characterize the functional role of sequences.
Notably, we find only one GPCR pseudogene, which contrasts to the situation in the nematode worm and in the human genome, where several hundred such pseudogenes are found (6,26). This may be because of a fundamental difference in the organization of GPCR genes in the fly; they are distributed among many loci in small clusters of one, two or three genes (9), whereas in the nematode and in human, there are large arrays of dozens of genes with interspersed pseudogenes (6,26). Also, we detect no ribosomal-protein pseudogenes; in contrast, processed pseudogenes from transcripts for these proteins are abundant and ubiquitous in the human genome, suggesting that appropriate reverse transcriptase specificity is not as available or as potent in the fly (7,8). There are two assignments of candidate processed pseudogenes each for serine proteases and for immunoglobulin-like domains; removing them from the counts does not change the identity of the 10 most common domains (Table 2).
CONCLUSIONS
We have completed an initial survey of the pseudogene population in the Drosophila genome. We find about 100 pseudogenes, with at least one-sixth of these as candidate processed pseudogenes. Two features of the fly pseudogene population arguably arise from a comparatively high genomic DNA deletion rate in the fly, relative to the rate of duplication of genes and gene parts: (i) there is a comparatively small number of putative pseudogenes (Table 1), relative to the genomes of other eukaryotes; (ii) closest matching proteins to pseudogenes appear to be rather longer than the average protein sequence in the proteome. Finally, the most pseudogenes occur for serine proteases (which are relatively abundant in the Drosophila lineage, compared with the other eukaryotes), immunoglobulin motif-containing proteins and cytochromes P450. Data relating to this paper are available at http://www.pseudogene.org, including chromosomal positions and protein sequences with disablements. We have re-mapped our annotations onto Release 3 of the genome, and are currently honing our methods for pseudogene detection in Drosophila with consideration of underlying substitution rates in the DNA, and other concepts. Our fruit fly data further add to the picture of evolution of the size and diversity of eukaryotic proteomes. In the human genome, there seems to be a clear correlation between the numbers of processed pseudogenes and the amount of non-coding DNA on a chromosome [(8); Z.Zhang, unpublished data]. However, as can be seen in Table 1 [and refs (1,7,27)], no obvious relationship has yet emerged between the size of a pseudogene population, the size of a proteome, and the amount of coding and non-coding DNA in genomes as whole entities. Detailed analysis of many more genomes will further help in deconvoluting the forces that shape these populations of sequences.
REFERENCES
- 1.Harrison P.M. and Gerstein,M. (2002) Studying genomes through the aeons: protein families, pseudogenes and proteome evolution. J. Mol. Biol., 318, 1155–1174. [DOI] [PubMed] [Google Scholar]
- 2.Mighell A.J., Smith,N.R., Robinson,P.A. and Markham,A.F. (2000) Vertebrate pseudogenes. FEBS Lett., 468, 109–114. [DOI] [PubMed] [Google Scholar]
- 3.Vanin E.F. (1985) Processed pseudogenes: characteristics and evolution. Annu. Rev. Genet., 19, 253–272. [DOI] [PubMed] [Google Scholar]
- 4.Esnault C., Maestre,J. and Heidmann,T. (2000) Human LINE retrotransposons generate processed pseudogenes. Nature Genet., 24, 363–367. [DOI] [PubMed] [Google Scholar]
- 5.Harrison P., Kumar,A., Lan,N., Echols,N., Snyder,M. and Gerstein,M. (2002) A small reservoir of disabled ORFs in the yeast genome and its implications for the dynamics of proteome evolution. J. Mol. Biol., 316, 409–419. [DOI] [PubMed] [Google Scholar]
- 6.Harrison P.M., Echols,N. and Gerstein,M.B. (2001) Digging for dead genes: an analysis of the characteristics of the pseudogene population in the Caenorhabditis elegans genome. Nucleic Acids Res., 29, 818–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harrison P.M., Hegyi,H., Balasubramanian,S., Luscombe,N.M., Bertone,P., Echols,N., Johnson,T. and Gerstein,M. (2002) Molecular fossils in the human genome: identification and analysis of the pseudogenes in chromosomes 21 and 22. Genome Res., 12, 272–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Z., Harrison,P. and Gerstein,M. (2002) Identification and analysis of over 2000 ribosomal protein pseudogenes in the human genome. Genome Res., 12, 1466–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adams M.D., Celniker,S.E., Holt,R.A., Evans,C.A., Gocayne,J.D., Amanatides,P.G., Scherer,S.E., Li,P.W., Hoskins,R.A., Galle,R.F. and Venter,J.C. (2000) The genome sequence of Drosophila melanogaster. Science, 287, 2185–2195. [DOI] [PubMed] [Google Scholar]
- 10.Robertson H.M. (2002) In Craig,N.L. (ed.), Mobile DNA II. ASM Press, Washington DC, pp. 1093–1110.
- 11.Graber J.H., Cantor,C.R., Mohr,S.C. and Smith,T.F. (1999) In silico detection of control signals: mRNA 3′-end-processing sequences in diverse species. Proc. Natl Acad. Sci. USA, 96, 14055–14060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robin G.C., Russell,R.J., Cutler,D.J. and Oakeshott,J.G. (2000) The evolution of an alpha-esterase pseudogene inactivated in the Drosophila melanogaster lineage. Mol. Biol. Evol., 17, 563–575. [DOI] [PubMed] [Google Scholar]
- 13.Apweiler R., Attwood,T.K., Bairoch,A., Bateman,A., Birney,E., Biswas,M., Bucher,P., Cerutti,L., Corpet,F., Croning,M.D., Durbin,R., Falquet,L., Fleischmann,W., Gouzy,J., Hermjakob,H., Hulo,N., Jonassen,I., Kahn,D., Kanapin,A., Karavidopoulou,Y., Lopez,R., Marx,B., Mulder,N.J., Oinn,T.M., Pagni,M. and Servant,F. (2001) The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic Acids Res., 29, 37–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ashburner M., Ball,C.A., Blake,J.A., Botstein,D., Butler,H., Cherry,J.M., Davis,A.P., Dolinski,K., Dwight,S.S., Eppig,J.T., Harris,M.A., Hill,D.P., Issel-Tarver,L., Kasarskis,A., Lewis,S., Matese,J.C., Richardson,J.E., Ringwald,M., Rubin,G.M. and Sherlock,G. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature Genet., 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zdobnov E.M., von Mering,C., Letunic,I., Torrents,D., Suyama,M., Copley,R.R., Christophides,G.K., Thomasova,D., Holt,R.A., Subramanian,G.M., Mueller,H.M., Dimopoulos,G., Law,J.H., Wells,M.A., Birney,E., Charlab,R., Halpern,A.L., Kokoza,E., Kraft,C.L., Lai,Z., Lewis,S., Louis,C., Barillas-Mury,C., Nusskern,D., Rubin,G.M., Salzberg,S.L., Sutton,G.G., Topalis,P., Wides,R., Wincker,P., Yandell,M., Collins,F.H., Ribeiro,J., Gelbart,W.M., Kafatos,F.C. and Bork,P. (2002) Comparative genome and proteome analysis of Anopheles gambiae and Drosophila melanogaster. Science, 298, 149–159. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Z.L., Harrison,P.M. and Gerstein,M. (2002) Digging deep for ancient relics: a survey of protein motifs in the intergenic sequences of four eukaryotic genomes. J. Mol. Biol., 323, 811–822. [DOI] [PubMed] [Google Scholar]
- 17.Long M., Wang,W. and Zhang,J. (1999) Origin of new genes and source for N-terminal domain of the chimerical gene, jingwei, in Drosophila. Gene, 238, 135–141. [DOI] [PubMed] [Google Scholar]
- 18.Jeffs P.S., Holmes,E.C. and Ashburner,M. (1994) The molecular evolution of the alcohol dehydrogenase and alcohol dehydrogenase-related genes in the Drosophila melanogaster species subgroup. Mol. Biol. Evol., 11, 287–304. [DOI] [PubMed] [Google Scholar]
- 19.Jeffs P. and Ashburner,M. (1991) Processed pseudogenes in Drosophila. Proc. R. Soc. Lond. B Biol. Sci., 244, 151–159. [DOI] [PubMed] [Google Scholar]
- 20.Collins R.T., Furukawa,T., Tanese,N. and Treisman,J.E. (1999) Osa associates with the Brahma chromatin remodeling complex and promotes the activation of some target genes. EMBO J., 18, 7029–7040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petrov D.A. and Hartl,D.L. (2000) Pseudogene evolution and natural selection for a compact genome. J. Hered., 91, 221–227. [DOI] [PubMed] [Google Scholar]
- 22.Petrov D.A. (2001) Evolution of genome size: new approaches to an old problem. Trends Genet., 17, 23–28. [DOI] [PubMed] [Google Scholar]
- 23.Petrov D.A., Lozovskaya,E.R. and Hartl,D.L. (1996) High intrinsic rate of DNA loss in Drosophila. Nature, 384, 346–349. [DOI] [PubMed] [Google Scholar]
- 24.Robertson H.M. (1998) Two large families of chemoreceptor genes in the nematodes Caenorhabditis elegans and Caenorhabditis briggsae reveal extensive gene duplication, diversification, movement and intron loss. Genome Res., 8, 449–463. [DOI] [PubMed] [Google Scholar]
- 25.Robertson H.M. (2000) The large srh family of chemoreceptor genes in Caenorhabditis nematodes reveals processes of genome evolution involving large duplications and deletions and intron gains and losses. Genome Res., 10, 192–203. [DOI] [PubMed] [Google Scholar]
- 26.Glusman G., Yanai,I., Rubin,I. and Lancet,D. (2001) The complete human olfactory subgenome. Genome Res., 11, 685–702. [DOI] [PubMed] [Google Scholar]
- 27.Harrison P.M., Kumar,A., Lang,N., Snyder,M. and Gerstein,M. (2002) A question of size: the eukaryotic proteome and the problems in defining it. Nucleic Acids Res., 30, 1083–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]