

Abstract
RNA interference has emerged as a powerful tool for the silencing of gene expression in animals and plants. It was reported recently that 21 nt synthetic small interfering RNAs (siRNAs) specifically suppressed the expression of endogenous genes in several lines of mammalian cells. However, the efficacy of siRNAs is dependent on the presence of a specific target site within the target mRNA and it remains very difficult to predict the best or most effective target site. In this study, we demonstrate that siRNAs that have been generated in vitro by recombinant human Dicer (re-hDicer) significantly suppress not only the exogenous expression of a puromycin-resistance gene but also the endogenous expression of H-ras, c-jun and c-fos. In our system, selection of a target site is not necessary in the design of siRNAs. However, it is important to avoid homologous sequences within a target mRNA in a given protein family. Our diced siRNA system should be a powerful tool for the inactivation of genes in mammalian cells.
INTRODUCTION
RNA interference (RNAi) is a phenomenon whereby double-stranded RNA (dsRNA) induces the sequence-dependent degradation of a cognate mRNA in animal or plant cells (1–4). The mechanism responsible for dsRNA-induced gene silencing, which proceeds via a two-step mechanism, appears to have been strongly conserved during evolution (5–8). In the first step, long dsRNAs are recognized by a nuclease in the RNase III family known as Dicer, which cleaves the dsRNA into small interfering RNAs (siRNAs) (7) of 21–23 nt. These siRNAs are incorporated into a multicomponent nuclease complex, known as RISC, that is then responsible for the destruction of cognate mRNAs (9–11).
Since dsRNAs act as inactivating agents of specific genes, they have been utilized as tools for the functional analysis of genes in a nematode, the fruit fly and plants (12–14). It has been reported that, in mammalian cells, long dsRNAs induce the sequence-specific silencing of genes in mouse embryonal carcinoma cells and embryonic stem cells (15,16). However, long dsRNAs (of >30 nt in length) activate a dsRNA-dependent protein kinase (PKR) and 2′,5′-oligoadenylate synthetase (17) in mammalian somatic cells and the activities of these enzymes lead to a non-specific reduction in levels of mRNAs.
It was reported recently that synthetic 21 nt siRNAs specifically suppressed the expression of endogenous genes in several lines of mammalian cells (18). Use of these 21 nt siRNA duplexes circumvented the activation of PKR and 2′,5′-oligoadenylate synthetase and suggested that siRNAs might be useful as gene-inactivating agents in mammals. However, the efficacy of siRNAs is dependent on identification of a specific target site within a target mRNA (19). To obtain effective siRNAs, it is necessary, although both costly and time-consuming, to design and synthesize many different siRNAs.
In this report, we demonstrate that siRNAs generated in vitro by recombinant human Dicer (re-hDicer) significantly suppressed not only the exogenous expression of a puromycin-resistance gene but also the endogenous expression of H-ras, c-jun and c-fos. As reported recently, it is possible to produce short dsRNAs using RNase III from Escherichia coli (20). However, since dsRNAs of 12–15 nt in length, on average, are generated by this RNase III (21), it is better to use re-hDicer, which generates a more uniform population of 21–23 nt siRNAs. Use of re-hDicer to generate siRNAs might provide a powerful tool for studies of the mechanism of RNAi and the functions of various genes in mammalian cells, with potential utility in a clinical setting.
MATERIALS AND METHODS
Cloning of the human gene for Dicer and purification of re-hDicer
Partial cDNAs for human dicer/HERNA (nucleotides 379–1657 and 1390–7037) in the pBluescript vector (Stratagene, CA) were a gift from Dr S. Matsuda of the University of Nagoya (22). We amplified the Dicer 5′ coding region (nucleotides 183–902) from a HeLa cDNA library by PCR with specific primers (forward primer, 5′-ATG AAA AGC CCT GCT TTG CAA CCC CT-3′; reverse primer, 5′-AGT TGC AGT TTC AGC ATT ACT CTT-3′) and then we cloned the amplified DNA into the TA cloning vector (Invitrogen, CA). Then we cloned the full-length human gene for Dicer from these partial cDNAs for human Dicer (hDicer). We digested full-length cDNA for hDicer and subcloned the blunt-ended fragment into the PinPoint™-Xa vector (Promega, Madison, WI), which contained the coding sequence for a biotin-binding region, using an EcoRV site. The hDicer expression plasmid was introduced into E.coli with 2 µM biotin and expressed upon induction with 100 µM IPTG. Then, cells were collected and pelleted by centrifugation at 5000 r.p.m. for 10 min. Cells were resuspended in lysis buffer [100 mM NaCl, 0.5 mM EDTA, 1% Triton X-100, 1 mM dithiothreitol, 2 mM phenylmethylsulfonyl fluoride, 50 mM Tris–HCl (pH 8.0)]. Then re-hDicer was purified on SoftLink™ resin (Promega) that included bound streptavidin, according to the manufacturer’s protocol. For detection of re-hDicer, an aliquot of the resultant preparation of re-hDicer (0.5 µg) was fractionated by SDS–PAGE (10% polyacrylamide) and transferred to a PVDF membrane (Funakoshi Co., Tokyo, Japan) by electroblotting. Then re-hDicer was visualized with an ECL kit (Amersham Co., Little Chalfont, UK) and streptavidin-conjugated alkaline phosphatase.
Preparation of long dsRNAs and siRNAs
To generate the long dsRNA, a puromycin-resistance gene (nucleotides 1–300), the H-ras gene (nucleotides 370–570), the c-jun gene (nucleotides 1–200) and the c-fos gene (nucleotides 1–200) were amplified by PCR with a specific forward primer that contained a T7 promoter and a specific reverse primer that contained an SP6 promoter. Then, sense strand RNAs were generated by T7 RNA polymerase and antisense strand RNAs were generated by SP6 RNA polymerase. All siRNAs directed against puromycin-resistance mRNA and H-ras mRNA were synthesized by Japan Bio Service Co. Ltd (Saitama, Japan). Ten target sites (sites 1–10) were chosen for the synthetic siRNAs directed against the puromycin-resistance mRNA (site 1, 5′-ATG ACC GAG TAC AAG CCC A-3′; site 2, 5′-CTC GCC ACC CGC GAC GAC G-3′; site 3, 5′-CAC CGT CGA CCC GGA CCG C-3′; site 4, 5′-GAA CTC TTC CTC ACG CGC G-3′; site 5, 5′-GGT GTG GGT CGC GGA CGA C-3′; site 6, 5′-CAG ATG GAA GGC CTC CTG G-3′; site 7, 5′-GGA GCC CGC GTG GTT CCT G-3′; site 8, 5′-GGG TCT GGG CAG CGC CGT C-3′; site 9, 5′-CCT CCC CTT CTA CGA GCG G-3′; site 10, 5′-GCC CGG TGC CTG ACG CCC G-3′). Ten target sites (sites 1–10) were chosen for the synthetic siRNAs against H-ras mRNA (site 1, 5′-ATG ACG GAA TAT AAG CTT G-3′; site 2, 5′-GTT GGC GCC GGC GGT GTG G-3′; site 3, 5′-TAC GAC CCC ACT ATA GAG G-3′; site 4, 5′-AGG AGG AGT ACA GCG CCA T-3′; site 5, 5′-CAA CAC CAA GTC TTT TGA G-3′; site 6, 5′-GGA CTC GGA TGA CGT GCC C-3′; site 7, 5′-TCT CGG CAG GCT CAG GAC C-3′; site 8, 5′-GAC CCG GCA GGG AGT GGA G-3′; site 9, 5′-GCT GCGGAA GCT GAA CCC T-3′; site 10, 5′-GTG TGT GCT CTC CTG AGG A-3′). Then, to generate siRNAs, all RNAs were annealed by the standard method (6).
Processing in vitro by re-hDicer and purification of diced siRNAs
To examine the activity of hDicer, we mixed 10 µg of dsRNA with 1 µg of re-hDicer in 200 µl of reaction buffer [100 mM NaCl, 20 mM HEPES, 1 mM ATP, 5 mM MgCl2, 50 mM Tris–HCl (pH 7.0)]. The mixture was incubated for 30 min at 37°C. Then, 20 µl of the reaction mixture were fractionated by electrophoresis on a non-denaturing 12% polyacrylamide gel. Bands of RNA were detected with Syber™ green II reagent (Nippon Gene, Toyama, Japan). We recovered siRNAs of 21–23 nt in length from the reaction mixture using a QIAquick™ nucleotide-removal kit (Qiagen, Hilden, Germany). The siRNAs were precipitated in ethanol and then dissolved in TE buffer. The concentration of diced siRNAs was determined by monitoring absorbance at 260 nm.
Transfection of cells and assay of cell viability
HeLa cells were cultured in DMEM supplemented with 10% fetal bovine serum. Transfections with either 20 nM siRNAs or diced siRNAs were performed using the Oligofectamine™ reagent (Invitrogen) in accordance with the manufacturer’s instructions. Cell viability was determined with trypan blue as described.
Western blotting
HeLa cells that had been transfected with individual siRNAs were harvested. Proteins were resolved by SDS–PAGE (10% polyacrylamide) and transferred to a PVDF membrane (Funakoshi Co.) by electroblotting. Immune complexes were visualized with an ECL™ kit, using specific polyclonal antibodies against H-Ras (Oncogene Research Products, San Diego, CA), N-Ras (Oncogene Research Products), K-Ras (Oncogene Research Procducts), c-Jun (Santa Cruz Biotechnology, Santa Cruz, CA), c-Fos (Santa Cruz Biotechnology) and actin (Oncogene Research Products), as an endogenous control. Quantitation was performed by densitometry and NIH Image Analysis.
RESULTS
Cloning of a human gene for Dicer and purification and characterization of re-hDicer
To prepare heterogeneous siRNAs that could target various sites in a specific target mRNA, we used hDicer, which participates in RNAi in human cells. Initially, to generate re-hDicer using a bacterial expression system, we cloned the full-length human gene for Dicer using partial cDNAs for hDicer (see Materials and Methods; 7,22). The full-length cDNA was then subcloned into the PinPoint™-Xa vector that contained the coding region for the biotin-binding region of a biotin ligase for subsequence purification of the recombinant protein.
We introduced the hDicer expression plasmid (phDicer) into E.coli and, with biotin in the culture medium, re-hDicer was expressed upon induction with IPTG. The re-hDicer with bound biotin was purified by a ‘pull-down’ method with beads to which streptavidin had been bound (see Materials and Methods) and analyzed by SDS–PAGE, after which re-hDicer was detected by western blotting with visualization using alkaline phosphatase-conjugated streptavidin. As shown in Figure 1A, we detected re-hDicer with a biotin tag by western blotting. The putative molecular mass of the biotin-labeled re-hDicer was ∼220 kDa. Mock transfection with a vector that did not include the cDNA for hDicer did not yield the corresponding protein, confirming the production of re-hDicer in this expression system in E.coli.
Figure 1.
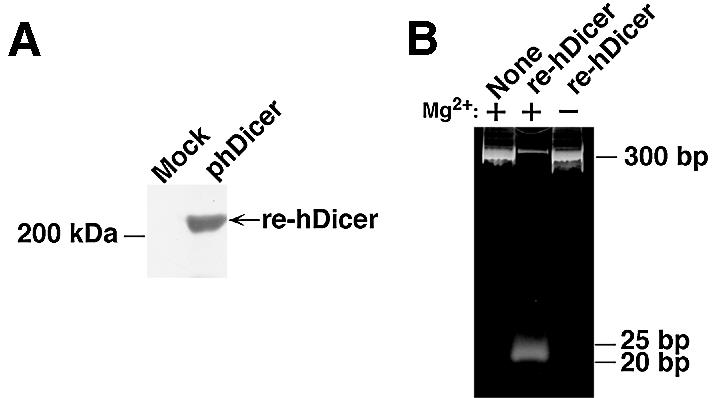
(A) Detection of the hDicer-mediated generation of diced siRNAs. Re-hDicer was synthesized in E.coli and detected by western blotting analysis, as described in the text. The preparation of re-hDicer (0.5 µg) was fractionated by SDS–PAGE (10% polyacrylamide) and transferred to a PVDF membrane by electroblotting. Then re-hDicer was visualized with an ECL kit and streptavidin-conjugated alkaline phosphatase. Mock, preparation from cells transfected with the empty plasmid; phDicer, preparation from cells infected with an expression plasmid that encoded hDicer. (B) Generation of siRNAs by re-hDicer. Long dsRNA (10 µg) was mixed with 1 µg of re-hDicer in 200 µl of reaction buffer, with or without 5 mM MgCl2. The reaction mixtures were incubated for 30 min at 37°C. Then 20 µl of each reaction mixture were fractionated by electrophoresis in a 12% non-denaturing polyacrylamide gel. siRNAs of 20–25 nt in length were detected with Syber™ green II.
Next, to determine whether the re-hDicer had RNase III activity, we performed a processing assay in vitro using long dsRNAs that had been generated by transcription in vitro. To construct long dsRNAs for use as substrates, we amplified a puromycin-resistance gene (nucleotides 1–300) by PCR using a specific forward primer that contained a T7 promoter and a specific reverse primer that contained an SP6 promoter. Then sense strand RNAs were generated by T7 RNA polymerase and antisense strand RNAs were generated by an SP6 RNA polymerase. These transcribed RNAs were annealed by the standard method (6). Then, the long dsRNAs and re-hDicer were combined and incubated in an appropriate reaction mixture (see Materials and Methods) for 30 min at 37°C. As shown in Figure 1B, siRNAs of 20–25 nt in length were generated in the presence of re-hDicer. In contrast, in the absence of either re-hDicer or Mg2+ ions, no such siRNAs were detected (lanes 1 and 3). Thus, re-hDicer exhibited Mg2+-dependent dsRNA processing activity. It was reported recently that recombinant hDicer in insect cells also exhibits RNase III activity (23,24).
Effects of diced siRNAs on expression of an exogenous puromycin-resistance gene
In mammalian cells, synthetic 21 nt siRNAs suppress the expression of both reporter genes and endogenous genes to a significant extent (18,19,25). While long dsRNAs (>30 nt) activate PKR and 2′,5′-oligoadenylate synthetase, use of these shorter siRNAs circumvents the activation of these enzymes and activates the RNAi pathway. Thus, these siRNAs might be expected to be useful for the sequence-specific silencing of gene expression. However, the dependence of the activity of siRNAs on the target site has been reported (19), and it remains very difficult to predict the best and most effective target site. Since the dependence of the activity of siRNAs on the target site was examined only in the case of the human gene for tissue factor (19), we further investigated the dependence on target site using siRNAs targeted to a puromycin-resistance gene and directed against 10 target sites within this gene (Fig. 2A and B). We used siRNA targeted to GL3-luciferase as a control.
Figure 2.


Suppression of the expression of an exogenous puromycin-resistance gene. (A) Ten sites (sites 1–10) were chosen as targets for synthetic siRNAs, as detailed in the text. For preparation of diced siRNAs, long dsRNAs corresponding to the 5′ region of the puromycin-resistance gene (nucleotides 1–300) were generated and treated with re-hDicer. (B) The secondary structure of the puromycin-resistance mRNA as predicted by the mfold program (24). (C) Diced siRNAs were the most effective suppressors of the expression of the exogenous puromycin-resistance gene (as indicated by the bar on the far right of the histogram). The efficiency of transfections with siRNAs was monitored with a reporter gene for luciferase from Renilla.
To prepare diced siRNAs, we transcribed both sense and antisense strands of a partial mRNA for puromycin resistance (nucleotides 1–300) in vitro, using T7 and SP6 RNA polymerases (Fig. 1B). After the dicing of dsRNAs in vitro, we purified the diced siRNAs using a QIAquick™ nucleotide-removal kit. Next, we introduced each siRNA (20 nM) and the expression plasmid for a puromycin-resistance gene into HeLa cells using the Oligofectamine™ reagent. After 36 h, we treated the HeLa cells with puromycin and determined cell viability using trypan blue. The efficiency of transfection of each siRNA was measured with the luciferase gene from Renilla as a reporter gene.
As shown in Figure 2C, in the presence of siRNA targeted to GL3-luciferase, cell viability remained the same as that of wild-type HeLa cells. The viability of cells that had been treated with site 2-, site 3-, site 4-, site 5- and site 10-specific siRNAs was significantly lower than that of wild-type cells that harbored the puromycin-resistance gene. In contrast, site 1-, site 6-, site 7-, site 8- and site 9-specific siRNAs had lower growth-inhibitory activity. These results indicated that the efficiency of siRNAs that were specific for the puromycin-resistance gene depended on the target site within the target mRNA.
As we had anticipated, diced siRNAs that corresponded to unlimited target sites within the specific mRNA were more effective than any specific siRNA or a mixture of site 1-, site 2-, site 3-, site 4- and site 5-specific siRNAs. When we analyzed the GC content of target sites and the secondary structure of the target gene, as predicted with the mfold program (Fig. 2B) (26), we failed to recognize a clear correlation between either secondary structure or GC content (Table 1) and effective and less effective target sites. In the case of the target gene, siRNAs targeted to regions from position 20 to position 280 and near the 3′ end of the target gene were more effective than others that we tested.
Table 1. GC contents (%) of the target site of each siRNA and levels of inhibition of gene expression.
| Target site | Puromycin-resistance mRNA | H-ras mRNA | ||
|---|---|---|---|---|
| GC (%) | Inhibition (%) | GC (%) | Inhibition (%) | |
| 1 | 52.6 | 37.2 | 36.0 | 63.2 |
| 2 | 78.9 | 88.6 | 78.9 | 81.7 |
| 3 | 78.9 | 86.9 | 52.6 | 78.6 |
| 4 | 63.1 | 79.5 | 57.8 | 87.8 |
| 5 | 73.6 | 84.6 | 42.0 | 70.5 |
| 6 | 73.6 | 24.8 | 68.4 | 45.3 |
| 7 | 73.6 | 45.1 | 68.4 | 23.6 |
| 8 | 78.9 | 40.5 | 73.6 | 36.8 |
| 9 | 68.4 | 36.8 | 63.1 | 74.2 |
| 10 | 84.0 | 78.2 | 57.8 | 82.1 |
Effects of diced siRNAs on expression of an endogenous H-ras gene
To examine the effects of diced siRNAs on expression of an endogenous gene, we selected the H-ras gene as a target. The H-ras gene is a member of the ras family, which also includes K-ras and N-ras (27). Again, to check the dependence on target site of the effects of siRNAs, we selected 10 target sites in the H-ras gene (Fig. 3A and B). For construction of diced siRNAs, we searched for a region of low homology to other members of the ras family in the H-ras gene. We selected such a region of the H-ras gene (nucleotides 370–570) as the long dsRNA substrate. Diced siRNAs targeted to the H-ras gene were generated by the same method as described above. Each individual siRNA (20 nM) was introduced into HeLa cells using the Oligofectamine™ reagent. After 72 h, cells were collected and total proteins were extracted. Levels of H-Ras protein were monitored by western blotting with specific antibodies. We determined levels of actin similarly as an endogenous control. Quantitation of proteins was performed by densitometry and NIH Image Analysis.
Figure 3.


Suppression of the expression of the endogenous H-ras gene. (A) Ten sites (sites 1–10) were chosen as targets for synthetic siRNAs, as detailed in the text. For the preparation of diced siRNAs, long dsRNAs corresponding to the 3′ region of the H-ras gene (nucleotides 370–570), which exhibited weak homology to the sequences of related ras genes, were generated and treated with re-hDicer. (B) The secondary structure of H-ras mRNA, as predicted by the mfold program (24). (C) Diced siRNAs were the most effective suppressors of the expression of the endogenous H-ras gene (as indicated by a bar on the far right of the histogram). The results were normalized by reference to levels of actin, as a control. (D) Effects of diced siRNAs targeted to the H-ras gene on the expression of the related K-ras and N-ras genes. K-Ras and N-Ras were detected by western blotting analysis with specific antibodies. Levels were quantitated by densitometry and NIH Image Analysis.
As shown in Figure 3C, the levels of H-Ras in cells that had been treated with site 2-, site 3-, site 4-, site 5- and site 9-specific siRNAs were significantly lower than that in wild-type HeLa cells. In contrast, site 1-, site 6-, site 7- and site 8-specific siRNAs had lower inhibitory activity. We normalized the results by reference to levels of actin and the results indicated that the efficiency of the siRNAs depended on the target site in the endogenous H-ras gene. In the case of both the puromycin-resistance gene and the H-ras gene, synthetic siRNAs targeted to the central regions of the genes were less effective. As for the diced siRNAs, they were significantly more effective than all the other siRNAs tested and than a mixture of siRNAs (site 7-, site 8-, site 9- and site 10-specific siRNAs) targeted to the H-ras gene. The levels of K-Ras and N-Ras proteins in cells that had been treated with diced siRNAs were, as anticipated, similar to those in wild-type HeLa cells (Fig. 3D). Thus, the diced siRNAs did not affect the expression of either the K-ras or the N-ras gene.
A structural analysis (Fig. 3B) similar to that shown in Figure 2B again failed to indicate any correlation between GC content (Table 1) and the extent of inhibition by siRNAs. Since results obtained in a previous study suggested that the effects of siRNAs, oligonucleotides and ribozymes might share some common features (28), it seems likely that more experimental data related to the accessibility of target mRNAs in vitro and in vivo (rather than computer-predicted secondary structures, as shown, for example, in Fig. 3B) are needed for a more precise examination of such putative relationships. In the case of H-ras, again, siRNAs targeted to certain regions (from position 20 to 280 and near the 3′ end) in the target gene were more effective than others, and diced siRNAs were again much more effective than individual siRNAs or a mixture of such siRNAs (Fig. 3C). These results resembled those obtained with the puromycin-resistance gene. It is possible that the higher activity of the diced siRNAs than that of individual siRNAs might simply have been due to an additive effect of targeting multiple sites along a message. However, the results obtained with the mixture of siRNAs (site 7-, site 8-, site 9- and site 10-specific siRNAs) failed to support this interpretation. It is more likely that the effects of siRNAs are changed by even a small change in the target site (of even just a few nucleotides) and, thus, diced siRNAs appeared to include some populations that were significantly more effective than the combined individual siRNAs. The observations suggest the clear advantages of diced siRNAs over individual synthetic siRNAs, at least in the experiments carried out in this study.
Diced siRNAs directed against c-jun and c-fos mRNAs
Finally, to confirm the effects of diced siRNA, we generated diced siRNAs against c-jun mRNA and c-fos mRNA, respectively. We introduced these diced siRNAs into HeLa cells and, 72 h later, we collected the cells and extracted the total proteins. The levels of the c-Jun and c-Fos proteins were examined by western blotting with specific antibodies. As shown in Figure 4, the level of c-Jun in cells that had been transfected with diced siRNAs was significantly lower than that in wild-type HeLa cells. The levels of actin were similar in both transfected and wild-type cells. We obtained analogous results using diced siRNAs directed against c-fos mRNA. Our results confirmed the high potential utility of diced siRNAs in the silencing of specific genes in human cells.
Figure 4.
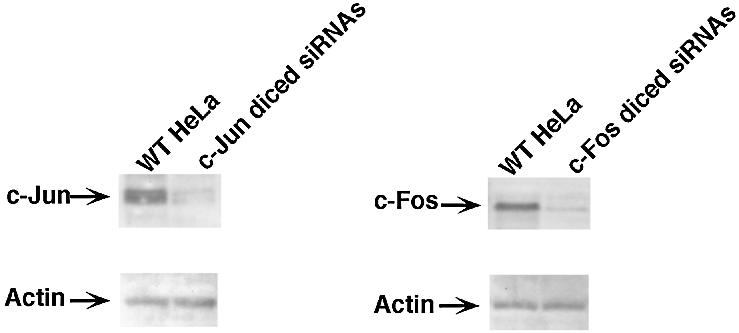
Suppression of the expression of the endogenous c-jun and c-fos genes by diced siRNAs. For the preparation of diced siRNAs, long dsRNAs corresponding to the 5′ region of the c-jun gene (nucleotides 1–200) and the c-fos gene (nucleotides 1–200) were generated and treated with re-hDicer. Both c-Jun and c-Fos were detected by western blotting analysis with specific antibodies. Actin was used as an endogenous control. WT-HeLa, wild-type HeLa cells; c-Jun (c-Fos) diced siRNs, siRNAs generated from c-jun-specific (c-fos-specific) dsRNA.
DISCUSSION
In this study, we developed an effective gene-silencing method, which we refer to as diced siRNA technology, using re-hDicer. RNAi has been shown to be a powerful tool for studies of gene function in a nematode, the fruit fly and plants (12–14). Effective sequence-specific gene silencing in several animals requires dsRNA of >150 bp (8,9,29,30). However, in mammals, long dsRNAs (>30 nt) induce the non-specific reduction of gene expression via the activation of PKR. Synthetic siRNAs that can effectively suppress the expression of endogenous genes have been reported by several groups (18,19,25). However, the efficiency of siRNAs depends on a specific target site within the target gene (Figs 2 and 3).
In RNAi, siRNAs become associated with RISC and function as guide RNAs in the search for target sites (3). However, we do not yet know how the siRNA–RISC complex can gain access to its target site and how cleavage at the target site occurs. In the nematode Caenorhabditis elegans, a RNA-directed RNA polymerase (RdRP) chain reaction with siRNA amplifies the interference that is caused by a small amount of ‘trigger’ dsRNA (31). Thus, siRNA acts not only as a guide but also as a primer and long dsRNAs are generated by RdRP. These long dsRNAs are then cleaved into fragments of ∼21 nt in length by Dicer.
Since accessibility of the siRNA might depend on the secondary structure of the target mRNA, we have to design and synthesize many different siRNAs to obtain effective siRNAs, and, at present, success depends on trial and error. To overcome these problems, we utilized re-hDicer for dicing of long dsRNAs in vitro and no longer needed to select a target site. However, it remained important to avoid the selection of sequences homologous to those of other members of the family to which the target protein belonged. Using our method, we succeeded in suppressing the expression of several endogenous genes that included H-ras, c-jun and c-fos, as shown in Figures 3 and 4. It appears, therefore, that the diced siRNA technology might be a powerful tool for the functional analysis of genes of interest, as well as for inactivation of specific genes in a clinical setting.
Acknowledgments
ACKNOWLEDGEMENTS
The authors thank Dr S. Matsuda of Nagoya University for gifts of partial cDNAs for hDicer and Dr Laura Nelson of AIST for critical reading of and helpful comments on the original manuscript. This research was supported by grants from the Ministry of Economy, Trade and Industry (METI) of Japan and by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Culture (MEXT) of Japan.
REFERENCES
- 1.Fire A., Xu,S., Montgomery,M.K., Kostas,S.A., Driver,S.E. and Mello,C.C. (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature, 391, 806–811. [DOI] [PubMed] [Google Scholar]
- 2.Sharp P.A. (2001) RNA interference-2001. Genes Dev., 15, 485–490. [DOI] [PubMed] [Google Scholar]
- 3.Hutvagner G. and Zamore,P.D. (2002) RNAi: nature abhors a double-strand. Curr. Opin. Genet. Dev., 12, 225–232. [DOI] [PubMed] [Google Scholar]
- 4.Hannon G.J. (2002) RNA interference. Nature, 418, 244–251. [DOI] [PubMed] [Google Scholar]
- 5.Zamore P., Tuschl,T., Sharp,P. and Bartel,D. (2000) RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21- to 23-nucleotide intervals. Cell, 101, 25–33. [DOI] [PubMed] [Google Scholar]
- 6.Elbashir S.M., Lendeckel,W. and Tuschl,T. (2001) RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev., 15, 188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bernstein E., Caudy,A.A., Hammond,S.M. and Hannon,G.J. (2001) Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature, 409, 363–366. [DOI] [PubMed] [Google Scholar]
- 8.Tuschl T., Zamore,P.D., Lehmann,R., Bartel,D.P. and Sharp,P.A. (1999) Targeted mRNA degradation by double-stranded RNA in vitro. Genes Dev., 13, 3191–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hammond S.M., Bernstein,E., Beach,D. and Hannon,G.J. (2000) An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature, 404, 293–296. [DOI] [PubMed] [Google Scholar]
- 10.Hammond S.M., Boettcher,S., Caudy,A.A., Kobayashi,R. and Hannon,G.J. (2001) Argonaute 2, a link between genetic and biochemical analysis of RNAi. Science, 293, 1146–1150. [DOI] [PubMed] [Google Scholar]
- 11.Bernstein E., Denli,A.M. and Hannon,G.J. (2001) The rest is silence. RNA, 7, 1509–1521. [PMC free article] [PubMed] [Google Scholar]
- 12.Vaucheret H., Beclin,C. and Fagard,M. (2001) Post-transcriptional gene silencing in plants. J. Cell Sci., 114, 3083–3091. [DOI] [PubMed] [Google Scholar]
- 13.Hammond S.M., Caudy,A.A. and Hannon,G.J. (2001) Post-transcriptional gene silencing by double-stranded RNA. Nature Rev. Genet., 2, 110–119. [DOI] [PubMed] [Google Scholar]
- 14.Grishok A. and Mello,C.C. (2002) RNAi (nematodes: Caenorhabditis elegans). Adv. Genet., 46, 339–360. [DOI] [PubMed] [Google Scholar]
- 15.Billy E., Brondani,V., Zhang,H., Müller,U. and Filipowicz,W. (2001) Specific interference with gene expression induced by long, double-stranded RNA in mouse embryonal teratocarcinoma cell lines. Proc. Natl Acad. Sci. USA, 98, 14428–14433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paddison P.J., Caudy,A.A. and Hannon,G.J. (2002) Stable suppression of gene expression by RNAi in mammalian cells. Proc. Natl Acad. Sci. USA, 99, 1443–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stark G.R., Kerr,I.M., Williams,B.R., Silverman,R.H. and Schreiber,R.D. (1998) How cells respond to interferons. Annu. Rev. Biochem., 67, 227–264. [DOI] [PubMed] [Google Scholar]
- 18.Elbashir S.M., Harborth,J., Lendeckel,W., Yalcin,A., Weber,K. and Tuschl,T. (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in mammalian cell culture. Nature, 411, 494–498. [DOI] [PubMed] [Google Scholar]
- 19.Holen T., Amarzguioui,M., Wiiger,M.T., Babaie,E. and Prydz,H. (2002) Positional effects of short interfering RNAs targeting the human coagulation trigger Tissue Factor. Nucleic Acids Res., 30, 1757–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang D., Buchholz,F., Huang,Z., Goga,A., Chen,C.Y., Brodsky,F.M. and Bishop,J.M. (2002) Short RNA duplexes produced by hydrolysis with Escherichia coli RNase III mediate effective RNA interference in mammalian cells. Proc. Natl Acad. Sci. USA, 99, 9942–9947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amarasinghe A.K., Calin-Jageman,I., Harmouch,A., Sun,W. and Nicholson,A.W. (2001) Escherichia coli ribonuclease III: affinity purification of hexahistidine-tagged enzyme and assays for substrate binding and cleavage. Methods Enzymol., 342, 143–158. [DOI] [PubMed] [Google Scholar]
- 22.Matsuda S., Ichigotani,Y., Okuda,T., Irimura,T., Nakatsugawa,S. and Hamaguchi,M. (2000) Molecular cloning and characterization of a novel human gene (HERNA) which encodes a putative RNA-helicase. Biochim. Biophys. Acta, 1490, 163–169. [DOI] [PubMed] [Google Scholar]
- 23.Harborth J., Elbashir,S.M., Bechert,K., Tuschl,T. and Weber,K. (2001) Identification of essential genes in cultured mammalian cells using small interfering RNAs. J. Cell Sci., 114, 4557–4565. [DOI] [PubMed] [Google Scholar]
- 24.Provost P., Dishart,D., Doucet,J., Frendewey,D., Samuelsson,B. and Radmark,O. (2002) Ribonuclease activity and RNA binding of recombinant human Dicer. EMBO J., 21, 5864–5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang H., Kolb,F.A., Brondani,V., Billy,E. and Filipowicz,W. (2002) Human Dicer preferentially cleaves dsRNAs at their termini without a requirement for ATP. EMBO J., 21, 5875–5885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zuker M., Mathews,D.H. and Turner,D.H. (1999) Algorithms and thermodynamics for RNA secondary structure prediction: a practical guide. In Barciszewski,J. and Clark,B.F.C. (eds), RNA Biochemistry and Biotechnology. Kluwer Academic, Hingham, MA.
- 27.Adjei A.A. (2001) Blocking oncogenic Ras signaling for cancer therapy. J. Natl Cancer Inst., 93, 1062–1074. [DOI] [PubMed] [Google Scholar]
- 28.Miyagishi M., Hayashi,M. and Taira,K. (2003) Comparison of the suppressive effects of antisense oligonucleotides and siRNAs directed against the same targets in mammalian cells. Antisense Nucleic Acid Drug Dev., in press. [DOI] [PubMed] [Google Scholar]
- 29.Caplen N.J., Fleenor,J., Fire,A. and Morgan,R.A. (2000) dsRNA-mediated gene silencing in cultured Drosophila cells: a tissue culture model for the analysis of RNA interference. Gene, 252, 95–105. [DOI] [PubMed] [Google Scholar]
- 30.Ngo H., Tschudi,C., Gull,K. and Ullu,E. (1998) Double-stranded RNA induces mRNA degradation in Trypanosoma brucei. Proc. Natl Acad. Sci. USA, 95, 14687–14692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sijen T., Fleenor,J., Simmer,F., Thijssen,K.L., Parrish,S., Timmons,L., Plasterk,R.H. and Fire,A. (2001) On the role of RNA amplification in dsRNA-triggered gene silencing. Cell, 107, 465–476. [DOI] [PubMed] [Google Scholar]