


Abstract
The human ABCA2 transporter gene encodes a member of a large family of ATP-binding proteins that transport a variety of macromolecules across biological membranes. We have performed luciferase reporter gene assays with promoter constructs comprising the 5′-flanking region to identify cis-regulatory DNA elements and have mapped the minimal promoter region to 321 bp upstream of the translation start site. We have discovered a functional role for two GC-boxes located in the proximal promoter of the ABCA2 gene that contain overlapping sites for the EGR-1 and Sp1 transcription factors. We observed that oligonucleotides containing overlapping EGR-1/Sp1 sites bind the Sp1, Sp3 and Sp4 transcription factors. When BE(2)-M17 cells were treated with phorbol 12-myristate 13-acetate, we observed inducible expression and binding of the EGR-1 transcription factor to the two GC-boxes. Transfection of Sp1, Sp3 or Sp4 expression constructs into Drosophila S2 induced a dose-dependent increase in transcriptional activation of the ABCA2 promoter, but transfection of EGR-1 alone failed to activate transcription. When increasing amounts of EGR-1 were transfected into the BE(2)-M17 neuroblastoma cells we observed a dose-dependent decrease in expression of the ABCA2 promoter, although expression of the endogenous ABCA2 gene increased following transfection of EGR-1.
INTRODUCTION
ATP-binding-cassette (ABC) transporters utilize the energy of ATP hydrolysis to pump substrates unidirectionally across membranes (1). ABC transporters consist minimally of a membrane-spanning domain (MSB), usually containing six transmembrane segments, which function in substrate recognition, and a nucleotide-binding domain (NBD) that binds ATP on the cytosolic face of the membrane. The ABC proteins are synthesized either as half transporters with a single MSB and NBD that function as homodimers or as a single polypeptide with two MSBs and NBDs (2). The characteristic ABC consensus sequence is comprised of conserved Walker A and Walker B motifs, separated by 90–100 amino acids containing a characteristic ‘ABC signature’ motif (3).
ABC transporters have been identified that are specific for a variety of substrates, including amino acids, sugars, inorganic ions, polysaccharides, lipids, peptides and proteins (4). Several ABC transporters have been implicated in human diseases, including the cystic fibrosis transmembrane receptor protein (CTFR or ABCC7) and cystic fibrosis (5), the mitochondrial half-transporter (ABCB7) and X-linked sideroblastic anemia and ataxia (6), the rod photoreceptor ABC transporter (ABCA4) involved in the etiology of Stargardt’s disease and other retinal disorders (7), and the ABCA1 transporter and Tangier’s disease, affecting cholesterol transport and susceptibility to atherosclerosis (8). The multidrug resistance protein (MDR) and the multidrug resistance-related protein subfamilies are also ABC transporters with demonstrated roles in acquired resistance to chemotherapeutic drugs (9).
This laboratory, investigating the mechanism of acquired resistance of ovarian carcinoma cells to the anticancer drug estramustine, detected a heterogeneously staining region in chromosomal spreads, indicative of gene amplification at position 9q34 (10). Chromosomal mapping has assigned the human ABCA2 gene to position 9q34 (11). Southern blot analysis of genomic DNA isolated from estramustine-resistant human ovarian carcinoma cells revealed that the ABCA2 gene was indeed amplified (10). We cloned the human ABCA2 cDNA and subsequent northern blot analysis of expression patterns revealed that transcripts were dramatically elevated in the brain relative to other tissues (12). We further determined the subcellular localization of ABCA2 by immunofluorescence and observed co-localization with the late-endosomal/lysosomal marker lysosomal-associated membrane protein 1 (12).
In order to understand the mechanisms responsible for regulating transcription of the ABCA2 gene we have performed studies to identify the basal promoter and DNA regulatory elements. We mapped the basal promoter to 321 bp upstream of the ATG start codon and demonstrated a functional role for two GC-boxes containing overlapping early growth response protein-1 (EGR-1) and Sp1 binding sites that bind several of the Sp-family factors and the EGR-1 transcription factor and regulate transcription of the promoter. This work represents the first functional characterization of a mammalian ABCA2 transporter promoter.
MATERIALS AND METHODS
Isolation of the 5′ flanking region of human ABCA2 gene
A human cosmid library in pWE15 (Clontech, Palo Alto, CA) was screened using a 1015 bp template generated by RT–PCR on a human brain cDNA template (Clontech) and the probe radiolabeled with [α-32P]dCTP using the Radprime kit (Invitrogen, Carlsbad, CA). Approximately 0.5 × 106 colonies were screened. The colonies were lysed and the DNA denatured on the filters by sequential incubations for 15 min in denaturing solution (0.5 M NaOH, 1.5 M NaCl), neutralizing solution (1 M Tris–HCl pH 7.5) and wash solution (1 M Tris–HCl pH 7.5, 1.5 M NaCl). The DNA was fixed to the filters by UV crosslinking (Stratalinker; Stratagene, La Jolla, CA). Filters were hybridized with the radiolabeled probe overnight at 65°C in buffer containing 0.5 M NaH2PO4 pH 7.2, 1 mM EDTA pH 8.0, 7% SDS, and 1% BSA, 2 mg/ml denatured salmon sperm DNA. Filters were washed twice for 15 min each at room temperature in 40 mM NaH2PO4 pH 7.2, 1 mM EDTA pH 8.0, 5% SDS, and 0.5% BSA, twice for 30 min each in NaH2PO4 pH 7.2, 1 mM EDTA pH 8.0, 1% SDS and once for 30 min in 1× SSC, 0.1% SDS. Filters were exposed to X-ray film and autoradiographed. Positive colonies were identified and the cosmid DNA sequenced using a Perkin-Elmer ABI 377 automated sequencer.
Transcription start site mapping by S1 nuclease protection
S1 nuclease mapping. S1 nuclease mapping of the ABCA2 transcription start site was performed as described (13). Briefly, a sense primer 5′-CCGGATGCCTCTGGGAGA AGAG-3′, located in the 5′-flanking DNA, 422 bp upstream of the ATG translation start codon, was 5′ phosphorylated with T4 polynucleotide kinase (Invitrogen) and PCR performed on 479 bp template of cloned human genomic DNA spanning the putative transcription start site with the above phosphorylated primer and an antisense primer 5′-GCGTTTGAGCG TCACGTTCTTCCA-3′, located 57 bp downstream of the ATG start codon, to generate a phosphorylated 479 bp double-stranded PCR product. Two micrograms of the PCR product was digested with λ exonuclease (Invitrogen) in 67 mM glycine–KOH pH 9.4, 2.5 mM MgCl2, 50 µg/ml BSA at 37°C for 30 min to generate the antisense single-stranded DNA. The antisense single-stranded DNA was end labeled with [γ-32P]ATP (10 mCi/ml; Amersham) and T4 polynucleotide kinase (Invitrogen). Total RNA from BE(2)-M17 neuroblastoma cells (100 µg) was hybridized with the radiolabeled antisense probe (1 × 105 c.p.m.) in 80% formamide, 0.4 M NaCl, 0.2 M PIPES, pH 6.5 at 55°C overnight. S1 nuclease digestion buffer, 30 mM sodium acetate pH 4.6, 50 mM NaCl, 1 mM zinc acetate, 5% (v/v) glycerol and S1 nuclease (100 U; Invitrogen) were added to the hybridization mixture and digestion performed at 37°C for 30 min. The protected fragment was precipitated, resuspended in 90% formamide, 5 mM EDTA, 0.05% bromophenol blue, 0.05% xylene cyanol, boiled for 5 min and loaded on a 6% polyacrylamide–7 M urea sequencing gel. The φX174 Hinf DNA ladder was radiolabeled with [γ-32P]ATP (10 mCi/ml; Amersham) and T4 polynucleotide kinase and 1 × 105 c.p.m. loaded on the gel. The 479 bp cloned template DNA was sequenced using the antisense primer radiolabeled with [γ-32P]ATP (10 mCi/ml; Amersham) and T4 polynucleotide kinase according to the manufacturer’s instructions (Epicentre Technologies, Madison, WI). Following electrophoresis the gel was dried and autoradiographed using a Fuji phosphorimager.
Cell culture
Neuroblastoma cells. Human BE(2)-M17 neuroblastoma cells were obtained from the American Type Culture Collection (ATCC, Rockville, MD) and maintained in 1:1 modified Eagle’s medium/Hams’ F12 supplemented with 10% fetal bovine serum and 100 mg/ml streptomycin/penicillin at 37°C, 5% CO2. For electrophoretic mobility shift analysis experiments, where indicated, cells were serum starved for 16 h and then were treated with 50 ng/ml of phorbol 12-myristate 13-acetate (PMA; Sigma, St Louis, MO) for 1 h before the preparation of nuclear extracts.
Drosophila S2 cells. Drosophila Schneider 2 cells were obtained from Invitrogen and maintained in Drosophila Expression System (DES) medium supplemented with 10% fetal bovine serum and 100 mg/ml streptomycin/penicillin at 25°C.
Plasmid constructions
Cloning of ABCA2 5′-flanking DNA and construction of 5′ nested reporter deletions. The ABCA2 cosmid clone was digested with XhoI and MluI restriction enzymes and the fragments were subcloned into the pGL3 basic luciferase reporter vector (Promega, Madison, WI) to generate plasmid p-5780, extending from 5780 bp upstream to 396 bp downstream of the ATG start codon of exon 1B. The ATG start codon was mutated to ATT to ensure that translation initiated at the ATG start codon present in the luciferase reporter gene using the QuickChange™ XL site-directed mutagenesis system (Stratagene). The primers for the mutagenesis were upper, 5′-GCCATTGGCTTCCTGCACCAG-3′, lower, 5′-GAAGC CAATGGCGGGGCCACG-3′, and mutagenesis was performed according to the manufacturer’s instructions. The 5′ nested reporter deletion constructs were generated by sequential enzymatic activities of exonuclease III, S1 nuclease, Klenow and T4 DNA ligase (14). Briefly, the reporter plasmid p-5780 was digested sequentially with MluI and SacI to linearize the plasmid and generate a 5′-overhanging end that is susceptible to ExoIII digestion and a 3′-overhanging end that is resistant to ExoIII digestion. Timed samples were removed from the ExoIII digestion buffer, blunt ended with S1 nuclease and Klenow, recircularized by ligation with T4 DNA ligase and the plasmids were cloned into DH5α Escherichia coli competent cells (Invitrogen) to generate the 5′ nested reporter deletion library.
Mutagenesis of EGR-1 and Sp1 sites in GC-boxes of ABCA2 promoter. For mutagenesis of EGR-1 and Sp1 transcription factor binding sites in GC-boxes 1 and 2 of the ABCA2 promoter, we used the QuickChange™ XL site-directed mutagenesis kit. The template for mutagenesis was the p-321 luciferase reporter construct that contained the ABCA2 promoter 5′-flanking sequence extending from 321 bp upstream of the ATG translation start site to 396 bp downstream of the ATG start site and that contained both GC-boxes with overlapping EGR-1 and Sp1 sites. The primers used for mutation of the Sp1 site on GC-box 1 were upper primer, 5′-GGCGTTCGGGGCGCGGGCAGAG-3′, lower primer, 5′-CCCGAACGCCCCCGCTTAAAGG-3′; for the Sp1 site on GC-box 2, upper primer, 5′-CCGAGCGCAACGCCCC CGCGCG-3′, lower primer, 5′-GGGCGTTGCGCTCGGG CGTCCG-3′; for the EGR-1 site on box 1, upper primer, 5′-TTAAGCTAGGGCGGGCGGGGC-3′, lower primer, 5′-CGCCCTAGCTTAAAGGCGCCG-3′; for the EGR-1 site on box 2, upper primer, 5′-CCCATGCGCGGGCGATGCC-3′, lower primer, 5′-GCGCATGGGCGGGGCGCTC-3′. The 50 µl PCR mixture contained 10 ng of the p-321 plasmid DNA and 10 pmol of the upper and lower primers. The PCR cycling conditions were 95°C for 1 min, 18 cycles of denaturation at 95°C for 50 s, annealing at 66°C (Sp1) or 62°C (EGR-1) for 50 s and extension at 68°C for 20 min, and a final extension at 68°C for 7 min. The PCR products were digested with 2 µl of DpnI for 2 h at 37°C to eliminate the parental plasmid DNA. The DpnI digested plasmid DNA was transformed into XL10-Gold ultracompetent cells and isolated plasmid DNA was isolated (Qiagen Maxiprep, Qiagen) and DNA sequenced to identify clones containing the desired mutation.
Synthesis and cloning of transcription factor expression constructs. The Sp1 expression construct was prepared by PCR on a reverse transcribed template derived from neuroblastoma BE(2)-M17 cell total RNA using sense primer 5′-ATGGTACCGCCACCATGAGCGACCAAGAT-3′ and antisense primer 5′-ATGAATTCTGGCTCTCTCTTCCAT GTCT-3′. The 2475 bp PCR product was gel purified, digested with KpnI and EcoRI and subcloned into the pAc5.1/V5-His A vector (Invitrogen). The EGR-1 expression construct was similarly prepared as above by PCR using sense primer 5′-ATGGTACCCCCGACACCAGCTCTCCAGC-3′ and antisense primer 5′-TATCTAGAGAACCCTCCTCTCCTA TGGC-3′. The 1773 bp PCR product was gel purified, digested with KpnI and XbaI and subcloned into pAc5.1/V5-His A vector or the mammalian expression vector pcDNA 3.1(+) (Invitrogen) to generate pCMV-EGR-1. The pPac Sp3 expression construct was a gift from Dr Kathleen Scotto (Fox Chase Cancer Center). The Sp4 expression construct was generated by PCR using sense primer 5′-ATGGTACCC TCTATCCCAGTGTCTCCGTCT-3′ and antisense primer 5′-ATTCTAGACAGCCTGCAAACTACTAAGGTC-3′. The 2.7 kb PCR product was gel purified, digested with KpnI and XbaI and subcloned into the pAc5.1/V5-His A vector (Invitrogen). Mammalian expression constructs were generated by subcloning the Sp1, Sp3, Sp4 and EGR-1 cDNAs into the pcDNA 3.1 vector (Invitrogen). DNA sequencing as described above verified the nucleotide sequence of all the expression constructs.
Transient transfection and reporter gene assays
Neuroblastoma BE(2)-M17 cells. The day before transfection, 8 × 105 neuroblastoma BE(2)-M17 cells were grown in triplicate in six-well plates for each construct in complete medium without antibiotic selection. The medium was replaced with 1 ml OptiMEM I medium (Invitrogen) and the cells were transfected with 1 µg of each reporter plasmid construct expressing firefly luciferase and 0.1 µg of the control plasmid expressing renilla luciferase under the control of the cytomegalovirus (CMV) promoter and 8 µl of Lipo fectamine 2000 transfection reagent (Invitrogen). The medium was replaced with complete medium 5 h after transfection and the cells cultured for an additional 43 h. Cell lysates were prepared with 1× passive lysis buffer (Promega) and 20 µl of cell lysate was analyzed for luciferase activity using the Dual Luciferase Assay System (Promega) on a Zylux Sirus FB-15-2 luminometer. To control for transfection efficiency, the activity of each reporter plasmid construct was normalized to the activity of the control plasmid expressing renilla luciferase. For analysis of the ABCA2 5′-flanking DNA, transfection of reporter constructs into BE(2)-M17 neuroblastoma cells was performed in triplicate and each experiment was performed a minimum of three times. For experiments to evaluate a functional role for the EGR-1/Sp1 transcription factor binding sites, transfection of reporter constructs into BE(2)-M17 neuroblastoma cells was performed in triplicate and each experiment was performed a minimum of two times. The data are expressed as the mean luciferase activity (± SEM). For evaluation of the role of transient expression of Sp-factors and Egr-1 on ABCA2 expression, 5 µg per 1 × 106 cells of the Sp1, Sp3, Sp4 or Egr-1 expression plasmids were transfected using Lipofectamine 2000™ reagent (Invitrogen), according to manufacturer’s instructions and 72 h post transfection total RNA was isolated for northern blot analysis.
Drosophila S2 cells. The cells were plated the night before transfection at a density of 6 × 105 cells in DES medium. Transfection of plasmid constructs was performed according to the manufacturer’s instructions, using CellFectin (4 µl/100 ml) transfection reagent (Invitrogen) with the addition of 1 µg of p-321 reporter construct and the indicated amounts of co-transfected Sp1, Sp3, Sp4 or EGR-1 expression constructs as described in the figure legends. Equivalent amounts of total DNA were transfected in each experiment by the addition of Bluescript KS II+ plasmid (Stratagene). Cell lysates were obtained as described above and reporter gene activity measured using the Luciferase Assay System. Luciferase activity was corrected to total protein in cell lysates as determined by the DC protein assay (Bio-Rad). Results are expressed as the mean ± the standard deviation (SD) of three independent transfection experiments.
Electrophoretic mobility shift assays (EMSA)
Nuclear extracts were prepared from 1 × 106 neuroblastoma BE(2)-M17 cells as described (15). Protein concentrations in the nuclear extracts were 1–2 mg/ml, as determined by the DC protein assay (Bio-Rad). Oligonucleotides for EMSA analysis were GC-box 1, EGRwt/Sp1wt 5′-GGATCCAGCGGGG GCGGGCGGACG-3′, EGRm/Sp1wt 5′-GGATCCAGCTA GGGCGGGCGGACG-3′, EGRwt/Sp1m 5′-GGATCCAGC GGGGGCGTTCGGACG-3′, EGRm/Sp1m 5′-GGATCCA GCTAGTTCGGGCGGACG-3′; GC-box 2, Sp1wt/EGRwt 5′-CGTCGCCCCGCCCCCGCTGGATCC-3′, Sp1m/EGRwt 5′-CGTCGCAACGCCCCCGCTGGATCC-3′, Sp1wt/EGRm 5′-CGTCGCCCCGCCCATGCTGGATCC-3′, Sp1m/EGRm 5′-CGTCGCCCCGAACATGCTGGATCC-3′. The binding sites for the designated transcription factors are underlined and the sites for mutagenesis are in bold. The consensus Sp1 and EGR double-stranded competitor oligonucleotides were prepared as those described above from the consensus sequences suggested from Santa Cruz Biotechnology. The sense oligonucleotides (20 pmol) were radiolabeled with [γ-32P]ATP and polynucleotide kinase and annealed to a molar excess (30 pmol) of cold antisense oligonucleotides by heating to 90°C for 10 min in oligo annealing buffer (10 mM Tris–HCl pH 8.0, 4 mM MgCl2, 0.1 M NaCl, 2 mM EDTA pH 8.0) and cooled slowly to room temperature (∼3 h) to generate the radiolabeled double-stranded oligonucleotide probes. The Sp1, Sp3, Sp4 and EGR-1 antibodies used for super-shift experiments were obtained from Santa Cruz Biotechnology. Gel-shift assays were performed as described (16) with slight modifications. Briefly, 4 µg of nuclear extract were incubated with radiolabeled probes (4 ng) for 30 min at room temperature in a 20 µl binding reaction containing 20 mM HEPES pH 7.9, 50 mM KCl, 0.5 mM EDTA pH 8.0, 5% glycerol, 1 mM dithiothreitol, 1 mg/ml bovine serum albumin, 0.1% Nonidet P-40 and 12.5 µg/ml poly(dI-dC). For competition studies, 50-fold molar excesses of unlabeled double-stranded competitor oligonucleotides were added to the binding reactions as indicated and incubated for 30 min at room temperature before addition of the radiolabeled oligonucleotide probes. For antibody super-shift experiments, 4 µg of rabbit anti-peptide antibodies to Sp1, Sp3, Sp4 or EGR-1 were added as indicated to binding reactions and incubated for 1 h on ice before addition of the radiolabeled oligonucleotide probes and incubation for an additional 30 min at room temperature. DNA–protein complexes and free DNA probe were separated on a 6% non-denaturing polyacrylamide gel in 0.5× TBE at 200 V for 3.5 h at 4°C. Gels were dried and autoradiographed. Imaging was performed using a BAS 2000 image analyzer (Fuji Film, Japan).
Northern blot
Total RNA was isolated using the Nucleospin™ RNA purification kit (Clontech) according to the manufacturer’s instructions and 5 µg of total RNA was separated on a 1.2% agarose gel, transferred to nitrocellulose and probed with a 32P radiolabeled ABCA2-specific probe using NorthernMax™ (Ambion) reagents according to the manufacturer’s instructions. Following autoradiography, imaging was performed as described above.
RESULTS
Organization of the 5′-flanking region of the human ABCA2 gene
In order to identify elements in the ABCA2 gene that regulate its expression we isolated 5′-flanking DNA by screening a human genomic cosmid DNA library. Using a cDNA probe generated by PCR comprising ∼1 kb of the 5′ end of the cDNA we isolated several positive clones that were characterized further. One clone, containing an insert of ∼30 kb, was partially sequenced and comparison with the ABCA2 cDNA sequence suggested that it contained the 5′-flanking DNA including the putative ABCA2 promoter. Potential transcription factor binding sites were evaluated by employing the Transcription Factor Element Search Software (17) that utilizes transcription factor binding site consensus sequences included in the TRANSFAC database. In a region comprising –549 to +397 relative to the ATG start codon, we detected putative binding sites for multiple transcription factors, including hepatic nuclear factor-3 (HNF-3), c-myc, activating protein 1 (AP1), and two GC-rich boxes at –195 and –108 bp, respectively, containing overlapping binding sites for the EGR-1 and Sp1 families of transcription factors (Fig. 1). Analysis of the sequence in a region comprising 1 kb, from –604 to + 397 relative to the ATG start codon, reveals that the putative ABCA2 promoter lacks a TATA-box and is contained within a CpG island. This region is extraordinarily GC rich, and has a G+C content of nearly 80% (0.791) and the CpG/GpC ratio is 0.882 (18), which exceeds the 0.6 threshold for designation as a CpG island (19).
Figure 1.

Human ABCA2 5′-flanking DNA from –549 to +397 bp relative to ATG translation start site. The putative ABCA2 promoter region DNA sequence is shown with upper case letters comprising the 5′-flanking DNA and first exon and the lower case letters comprising the beginning of the first intron. The numbering of the DNA sequence is in base pairs relative to the ATG start site, depicted in bold. The large arrow depicts the major transcription start site and the small arrow depicts a minor transcription start site. Binding sites that match the consensus DNA sequence as listed in the TRANSFAC database are lined, including HNF-3, EGR-1, Sp1, c-myc and AP1. Of particular note are two GC-boxes at positions –195 and –108 bp that contain overlapping 13 bp recognition sites for the transcription factors EGR-1 and Sp1.
Transcription start site mapping
To determine the ABCA2 transcription start site, we employed S1 nuclease mapping by hybridization of a 479 bp radiolabeled antisense DNA probe that spanned the putative start site to total RNA and subsequent digestion of the single-stranded non-overlapping regions with S1 nuclease. A 152 bp protected fragment was evident following polyacrylamide gel electrophoresis and autoradiography (Fig. 2) that maps the major start site to a region 95 bp upstream of the ATG start codon, although several weakly protected fragments were also evident, suggestive of multiple transcription start sites that are common in the promoters of many GC-rich TATA-less housekeeping genes.
Figure 2.
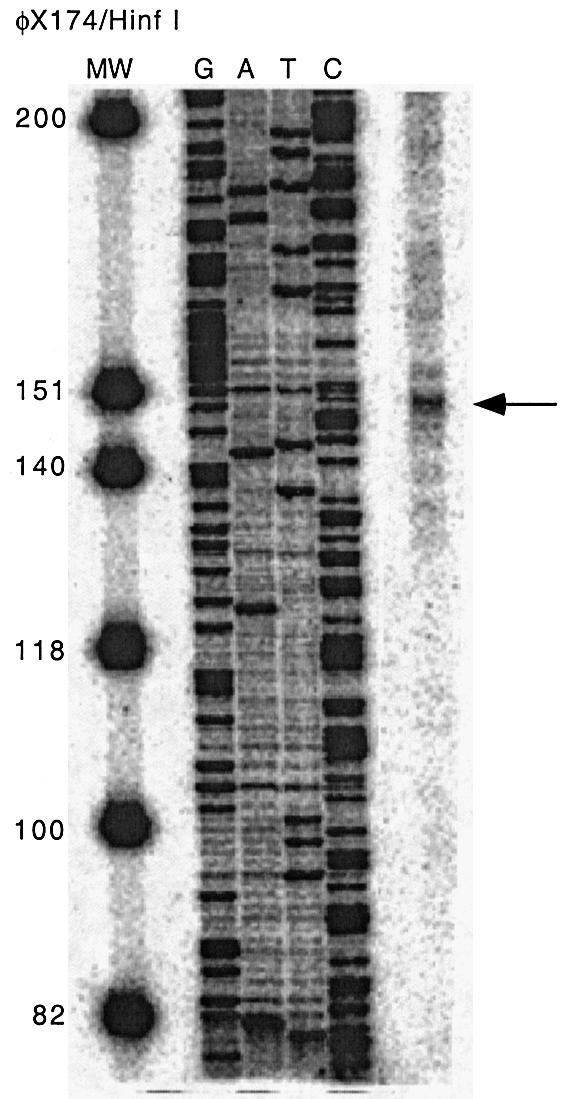
S1 nuclease protection mapping of the ABCA2 transcription start site. A 5′-end phosphorylated 479 bp double-stranded DNA template for the S1 probe was generated by PCR and digested with λ exonuclease to generate an antisense single-stranded DNA probe that was end labeled with [γ-32P]ATP and hybridized to total RNA from BE(2)-M17 neuroblastoma cells. Following digestion with S1 nuclease, the protected fragment was separated by electrophoresis in a 6% polyacrylamide–7 M urea gel followed by autoradiography. The antisense strand of the 479 bp DNA template was manually sequenced and the φX174 Hinf DNA ladder was radiolabeled with [γ-32P]ATP to serve as sequence and size markers respectively. An arrow indicates a major protected fragment of 152 bp, located 95 bp upstream of the ATG translation start site that represents the major transcription start site.
Transcriptional activity of the human ABCA2 promoter
We investigated the regulation of the ABCA2 promoter by generation of a series of progressive 5′ deletion reporter gene constructs that extended from over 5 kb upstream to 396 bp downstream of the ATG translation start site in the pGL3 luciferase expression vector and subsequent transfection of these promoter constructs into the neuroblastoma cell line BE(2)-M17 (Fig. 3). We selected the BE(2)-M17 neuroblastoma cell line for ABCA2 expression studies because we have previously demonstrated elevated expression of ABCA2 transcripts in the human brain by northern blot (12), the mouse neuroblastoma cell line, Neuro2A line exhibited elevated expression (11) and by RT–PCR analysis we measured elevated ABCA2 transcript levels in the BE(2)-M17, among the human neuroblastoma cell lines evaluated (data not shown). Analysis of these reporter constructs revealed that a region 321 bp upstream and 397 bp downstream of the ATG start codon was sufficient to provide >90% of the transcriptional activity of the ABCA2 promoter, i.e., relative to the maximal expression observed in these studies for the p-403 reporter construct. Further deletion to –103 bp reduced expression by over 60% from the level of the p-321 basal promoter and deletion beyond –103 bp virtually abolished expression (∼10% residual activity). Inspection of the DNA sequence in the basal promoter revealed two GC-boxes with overlapping EGR-1/Sp1 transcription factor binding sites at –195 bp (GC-box 1) and –108 bp (GC-box 2), respectively. We should note that deletion to –103 and subsequent religation of the deletion construct regenerates the Sp1 site of the GC-box at position –108 (see Fig. 1), and this accounts for similar expression levels observed for the p-112 and p-103 constructs.
Figure 3.

Reporter gene analysis of promoter activity of progressive deletions of ABCA2 5′-flanking DNA identifies the basal promoter. Shown are the results of reporter gene analysis following transfection of luciferase reporter gene constructs of ABCA2 5′-flanking DNA into human BE(2)-M17 neuroblastoma cells. Cell lysates were analyzed for luciferase activity using the Dual Luciferase Assay kit (Promega). The data are represented as the fold increase in expression of each construct relative to the promoter-less pGL3 vector (± SEM) following correction for transfection efficiency using the pRL-CMV renilla expression vector. For each experiment, transfections were performed in triplicate and each experiment was performed a minimum of three times.
Sp1, Sp3, Sp4 and EGR-1 factors bind to GC-box 1 and GC-box 2
In order to evaluate whether the overlapping Sp1/EGR-1 sites present in the two GC-boxes of the basal promoter bind these transcription factors, we performed gel mobility shift assays (EMSA) using GC-box 1 and GC-box 2 [γ-32P]ATP radiolabeled oligonucleotide probes and nuclear extracts from neuroblastoma BE(2)-M17 cells. We observed several prominent retarded complexes using GC-box 1 or GC-box 2 radiolabeled probes (Fig. 4). Virtually identical results of binding assays were observed for both GC-box probes and the results are presented for GC-box 2. Previous reports have demonstrated the appearance of an EGR-1 activity following cell culture in the presence of the phorbol ester PMA (16,20,21). When we cultured the neuroblastoma cells for 1 h in the presence of 50 ng/ml PMA before preparation of the nuclear extracts, we detect the appearance of a putative EGR-1-containing complex following EMSA (Fig. 4). To confirm the specificity of protein binding to the Sp1/EGR-1 sites, we performed oligonucleotide competition experiments. In these experiments an unlabeled oligonucleotide is incubated with the nuclear extract prior to the addition of the radiolabled GC-box probe. When extracts were incubated with unlabeled Sp1 consensus oligonucleotides before addition of radiolabeled GC-box 2 probe, putative Sp-family transcription factor binding was dramatically reduced. Similarly, when unlabeled EGR-1 consensus oligonucleotides were added, the putative EGR-1 transcription factor binding was abrogated. A faster migrating complex that was competed with all unlabeled oligonucleotide probes was detected that we believe to be a non-specific complex. Competition studies using the unlabeled GC-box 2 probe decreased binding of both putative Sp-family and EGR-1 factors to the radiolabeled GC-box 2 probe. Mutation of the EGR-1 binding site of the box 2 competitor probe while maintaining the wild-type Sp1 site reduced putative Sp-family binding and failed to compete putative EGR-1 protein binding. Alternatively, mutation of the Sp1 site of the box 1 competitor probe while maintaining the wild-type EGR-1 site specifically reduced putative EGR-1 binding while only moderately affecting putative Sp-family binding. Mutation of both EGR-1 and Sp1 sites of the GC-box 2 competitor probe failed to compete with the radiolabeled probe for binding to either EGR-1 or Sp-family transcription factors. These findings suggest that Sp-family and EGR-1 transcription factors bind to both GC-box elements in the ABCA2 basal promoter. Additionally, the results of these gel-shift experiments indicate that the binding of Sp-family and EGR-1 proteins to the overlapping EGR-1/Sp1 sites was mutually exclusive, i.e., double occupancy of the GC-box probes simultaneously by these factors was not observed. We base this conclusion on the observation that there was no shift in the mobility of the individual complexes that remain after addition of the competitor oligonucleotides. To confirm the identity of the specific Sp-family and EGR-1 transcription factors that bind to GC-boxes 1 and 2, we employed antibody super-shift experiments. When the nuclear extract was incubated with an EGR-1-specific antibody before addition of the radiolabeled GC-box 2 probe, the EGR-1 protein complex was specifically super-shifted to a slower migrating species (Fig. 5). Experiments using antibodies to other family members, EGR-2, EGR-3, EGR-4 and Wilms’ Tumor protein-1, failed to shift any of the complexes (data not shown). Antibodies to Sp1 specifically shifted the Sp1-containing complex and antibodies to Sp3 resulted in the specific loss of complexes at ∼110 kDa and two faster migrating complexes of ∼60–70 kDa that migrate as a duplex. It appears that the antibodies specifically block the binding of the Sp3 proteins to GC-box 1 probes. The mobility of the Sp-family member complexes that we observe in our experiments is consistent with the results reported in numerous studies evaluating these factors (22). Antibodies to the Sp4 transcription factor also resulted in the specific loss of the Sp4-containing complex, which may be due to antibody blockade of Sp4 binding to GC-box 2 probes. When Sp1 and Sp3 antibodies are used together, the Sp1 and Sp3 complexes are specifically targeted. Similarly, when Sp1 and Sp4 or Sp3 and Sp4 are used together, their respective complexes are specifically targeted. Antibodies to the Sp2 transcription factor failed to super-shift or reduce protein binding in any of the complexes (data not shown). Virtually identical results were observed in antibody super-shift experiments with GC-box 1 probes. These experiments indicate that the Sp-family transcription factors Sp1, Sp3 and Sp4 bind GC-boxes 1 and 2 and that following PMA stimulation of the neuroblastoma cells, EGR-1 also binds these elements present in the ABCA2 basal promoter.
Figure 4.

EMSA analysis of nuclear protein binding to GC-box 2 using binding site oligonucleotide competitors. Displayed are the consensus DNA sequences for the EGR-1 and Sp1 binding sites and the 13 bp overlapping EGR-1 and Sp1 binding sites of GC-box 2. Also depicted is the DNA sequence of the wild-type GC-box 2 probe and the mutated nucleotide residues in bold that abrogate EGR-1 or Sp-factor binding. The results of EMSA analysis of binding of nuclear proteins from nuclear extracts of BE(2)-M17 neuroblastoma cells are shown, and demonstrate the appearance of an EGR-1 binding activity with PMA treatment of cells. The results of EMSA are shown following the addition of 50-fold molar excesses of individual competitor oligonucleotides, Sp1 consensus, EGR-1 consensus, wild-type GC-box 2, or GC-box 2 nucleotides containing mutations in the binding sites for EGR-1, Sp1 or both EGR-1 and Sp1 to nuclear extracts prior to addition of radiolabeled GC-box 2 probe.
Figure 5.

EMSA super-shift analysis of nuclear protein binding to GC-box 2 using antibodies to specific transcription factors. The results of EMSA super-shift experiments are shown following the addition of antibodies that are specific for the EGR-1, Sp1, Sp3, Sp4 factors or pair-wise combinations of Sp1, Sp3 and Sp4 to nuclear extracts prior to the addition of radiolabeled GC-box 2 probe. Arrows mark the positions of super-shifted EGR-1 and Sp1 complexes, respectively.
Overlapping Sp1/EGR-1 elements regulate ABCA2 promoter activity
In order to demonstrate a functional role for the overlapping EGR-1/Sp1 binding sites of the two GC-boxes in regulating ABCA2 promoter activity we mutated the EGR-1 and Sp1 sites to abrogate transcription factor binding and evaluated the effects on expression of the p-321 basal reporter construct in BE(2)-M17 neuroblastoma cells. Mutation of both EGR-1 sites in GC-boxes 1 and 2 did not significantly reduce expression of the promoter compared to the wild-type p-321 basal promoter construct (Fig. 6). The mutation of both Sp1 sites in GC-boxes 1 and 2 reduced expression by ∼60%, which was additive to the effect observed when the individual Sp1 sites were mutated. Mutation of EGR-1 and Sp1 elements in GC-box 1 resulted in a ∼20% reduction in expression; however, when both EGR-1 and Sp1 sites of GC-box 2 were mutated, expression was reduced by ∼90%. The 90% reduction in expression is similar to that observed in reporter deletion experiments for the construct deleted beyond GC-box 2 (p+102). The sum of these findings suggest that both GC-boxes are functional in regulating expression of the basal ABCA2 promoter, that the GC-box 2 element is responsible for the majority of transcriptional activity, and that the Sp1 sites play a greater role under basal conditions.
Figure 6.

Analysis of the effect of mutation of EGR-1 and Sp1 elements in GC-boxes 1 and 2 on ABCA2 promoter activity. Shown are the results of reporter gene analysis following transfection of wild-type and mutant GC-box 1 and 2 luciferase constructs into human BE(2)-M17 neuroblastoma cells. The p-321 reporter construct contains the elements required for basal expression of the ABCA2 promoter. The EGR-1 dm construct contains the mutated EGR-1 elements and wild-type Sp1 elements in GC-boxes 1 and 2. The GC-box 1 or GC-box 2 Sp1 constructs contain individual Sp1 site mutations in the respective GC-boxes. The Sp1 dm construct contains mutated Sp-1 elements and wild-type EGR-1 elements in GC-boxes 1 and 2. The mBox 1 construct contains mutated EGR-1 and Sp1 elements in GC-box 1 and wild-type elements in GC-box 2. The mBox 2 construct contains mutated Sp1 and EGR-1 elements in GC-box 2 and wild-type elements in GC-box 1. The data are represented as the mean luciferase expression (± SD) relative to the activity of the wild-type p-321 construct (set at 100%).
Sp-family transcription factors activate the ABCA2 promoter in Drosophila S2 cells
In order to evaluate the role of Sp-family transcription factors and EGR-1 in regulating transcription of the ABCA2 promoter, we co-transfected increasing amounts of Sp1, Sp3, Sp4 and EGR-1 expression constructs together with the p-321 basal promoter reporter construct into Drosophila S2 cells. These insect cells do not manifest an endogenous Sp-family activity (23). We observed a dose-dependent increase in the transcriptional activity of the ABCA2 promoter with each of the Sp-family transcription factors tested, but transfection of the EGR-1 factor alone failed to activate transcription (Fig. 7).
Figure 7.

The effect of expression of Sp-family and EGR-1 transcription factors on ABCA2 promoter activity in Drosophila S2 cells. Shown are the results of co-transfection of the p-321 reporter construct containing the basal ABCA2 promoter and increasing amounts of Sp1, Sp3, Sp4 and EGR-1 expression constructs into Drosophila S2 cells. The data are represented as the fold-increase in luciferase expression (± SD) relative to the activity of the p-321 reporter construct.
EGR-1 factor represses expression of the ABCA2 promoter in neuroblastoma cells
Since EGR-1 failed to activate transcription of the ABCA2 promoter in Drosophila S2 cells we evaluated the consequences of transient transfection of an EGR-1 expression construct into BE(2)-M17 neuroblastoma cells on ABCA2 promoter activity. When increasing amounts of the EGR-1 expression construct were introduced into the neuroblastoma cells, we measured a dose-dependent decrease in the expression of the p-321 basal promoter reporter construct (Fig. 8). Interestingly, when both EGR-1 binding sites of the p-321 basal promoter construct were mutated to abrogate EGR-1 binding to its cognate site, we measured a greater dose-dependent repression of expression.
Figure 8.

The effect of EGR-1 on expression of the ABCA2 promoter in neuroblastoma cells. Shown are the results of reporter gene analysis following transfection of the p-321 luciferase reporter gene or p-dmEGR-1, where both EGR-1 binding sites were mutated, as described in Materials and Methods, and increasing amounts of the pCMV-EGR-1 expression construct into human BE(2)-M17 neuroblastoma cells. Cell lysates were analyzed for luciferase activity using the Dual Luciferase Assay kit (Promega). The data are represented as the mean luciferase expression (± SD) following correction for transfection efficiency using the pRL-CMV renilla expression vector, relative to the activity of p-321 reporter construct in the absence of pCMV-EGR-1 (set at 100%).
Transient overexpression of Sp-factors and EGR-1 elevates expression of the ABCA2 gene
To provide physical evidence of transcriptional regulation of the ABCA2 gene by the Sp-family and EGR-1 transcription factors we transiently transfected BE(2)-M17 cells with expression constructs for the individual factors. We did not detect a major elevation of ABCA2 transcripts, as measured by northern blot, following forced expression of Sp1 or Sp3, but observed a significant increase mediated by Sp4 (Fig. 9). However, forced expression of EGR-1 resulted in a significant increase in ABCA2 transcripts, which is in contrast to the results observed for the basal reporter gene construct or those observed in the Drosophila S2 cells.
Figure 9.
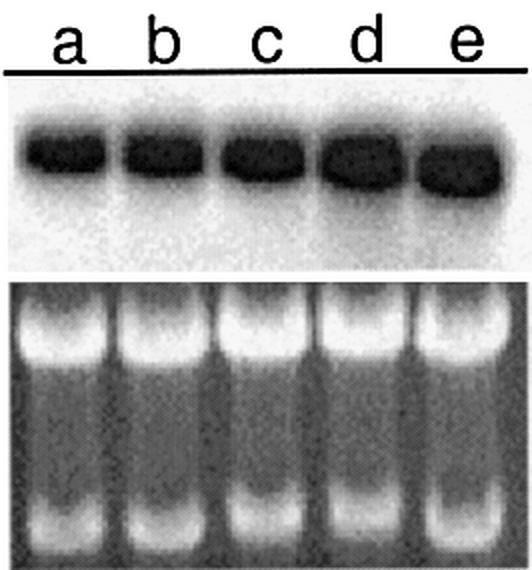
Northern blot analysis of ABCA2 expression following transient transfection of Sp-factors or EGR-1. The effects of forced expression of Sp1, Sp3, Sp4 or EGR-1 mammalian expression constructs on the expression of the ABCA2 endogenous gene is measured by northern blot as described in Materials and Methods. a, Mock-transfected; b, Sp1; c, Sp3; d, Sp4; e, EGR-1. Shown below is the ethidium bromide stained gel to demonstrate equivalent RNA loading.
DISCUSSION
In this report we have identified the human ABCA2 basal promoter region and characterized a functional role for two GC-rich regions that contain overlapping binding sites for the EGR-1 and Sp1 transcription factors in regulating ABCA2 transcription. The promoter region is GC rich and lacks a TATA-box element and is contained within a CpG island that is common for many housekeeping genes (18). Transcription start site mapping by S1 nuclease protection identifies a major transcription start site 95 bp upstream of the translation start site.
We show that the transcription factors Sp1, Sp3 and Sp4 can bind to the Sp1 binding site of both GC-boxes by oligonucleotide competition and antibody super-shift experiments. Further, we show that when neuroblastoma cells are cultured in the presence of the phorbol ester PMA, an additional binding activity to GC-boxes 1 and 2 is detected that we identify to be the EGR-1 transcription factor. The results of mutation of Sp1, EGR-1 or both sites in GC-box 1 and 2 reveal that both GC-boxes are functional in regulating expression of the ABCA2 promoter and that the Sp1 sites play a greater role under basal conditions. Additionally, we show that both the EGR-1 and Sp1 sites in GC-box 2 are required for the majority of promoter activity because mutation of both sites reduced basal expression of the promoter by 90%. These findings suggest that mutation of both elements in GC-box 2 essentially abrogate any role for Sp factors in mediating basal expression. Alternatively, under basal conditions, cooperative interactions between Sp factors and additional proteins that bind these elements in GC-box 2 may be required for transcription from the human ABCA2 promoter.
A growing number of genes have been identified that contain GC-boxes with overlapping EGR-1 and Sp1 factor binding sites, including the human tissue factor gene (16,24), the human EGR-1 gene (25), the human synapsin I gene (26), the murine adenosine deaminase gene (27), the rat tyrosine hydroxylase gene (20) and the receptor tyrosine kinase human Flt-1 gene (21).
The Sp-family of transcription factors includes Sp1, Sp2, Sp3 and Sp4 that are all structurally similar (22). The DNA-binding domain is comprised of three highly conserved zinc fingers near the C-terminus and glutamine-rich activation domains at their N-terminus. They also contain serine/threonine residues that are the targets of phosphorylation that affect DNA binding. The Sp-factors bind to the DNA sequence 5′-GGGGCGGGG-3′ with equal affinity (with the exception Sp2) and although most show ubiquitous distribution in cell lines and tissues (28,29) the Sp4 factors exhibit expression predominantly in neuronal cells (29,30) and we observe that forced expression of Sp4 elevates ABCA2 expression in the BE(2)-M17 neuroblastoma cell line. The Sp1 factor functions as an activator of transcription (23) although it can also function as a ‘superactivator’ by the synergistic activity of Sp1 proteins in promoters that contain multiple GC-boxes (31). Our results also suggest the synergistic activation of the ABCA2 basal promoter by the cooperative interaction of proteins that bind to the Sp1 sites of GC-boxes 1 and 2 as evidenced by the additive effect of repression of promoter expression when the Sp1 sites of both GC-boxes 1 and 2 are mutated to abrogate Sp-family factor binding. The Sp3 factor exists as three isoforms of 110–115 kDa, and two 60–70 kDa that arise by translational initiation at internal AUG codons resulting in N-terminal truncations of the full-length protein (32). We detect the binding of all three Sp3 isoforms to both of the Sp1 sites of GC-boxes 1 and 2 by gel-shift analysis. The role of Sp3 as an activator or repressor of transcription of genes is currently unresolved, as there are many reports for both activities. Evidence suggests that in vivo its function may be due to the structure and arrangement of Sp-recognition sites as well as by cell type-specific differences (22,33). Our experiments in Drosophila S2 cells suggest that Sp3 is a strong activator of the ABCA2 basal promoter. The EGR-1 transcription factor [also known as NGFI-A, Zif268, krox24 and TIS8 (34–38)] is a member of a transcription factor family that contain three zinc fingers and binds the DNA sequence 5′-GCGGGGGCG-3′, each finger contacts three bases within this sequence (39). These genes were initially characterized by their rapid synthesis in response to treatment of PC12 cells with nerve growth factor, which induces their subsequent differentiation (40). As a result these proteins were termed ‘early growth response’ analogous to the immediate-early genes, which include transcription factors that are rapidly induced following viral infection. A role for EGR-1-regulated transcription has been identified in EGR-1-deficient mice that have a deficiency in the neuropeptide, luteinizing hormone (LH) whose transcription is mediated by an EGR-1 binding site present in the LHβ promoter (41). Additionally, EGR-family members have been implicated in the transcription of genes whose expression is critical for the establishment and maintenance of neuronal plasticity, e.g. long term potentiation (40). Whether EGR-1 functions as a transcriptional activator or repressor appears also to be gene dependent and examples of its role as an activator of transcription, multidrug resistance gene 1 (MDR1) (42), human tissue factor gene (16,24), Flt-1 gene (21) or as a repressor of transcription, murine adenosine deaminase (27), Pgp2/mdr1b (43), β1-adrenergic receptor (44) are both well documented. Our evidence suggests that for the human ABCA2 promoter, EGR-1 functions as a transcriptional repressor, as forced expression of the factor resulted in a dose-dependent reduction of Sp-mediated transcription of the promoter in the neuroblastoma cell line. Interestingly, when the binding site of EGR-1 was mutated to abrogate binding we observed a further repression of the promoter expression. This suggests that EGR-1 binding to its cognate site is not required for transcriptional repression when the factor is overexpressed. Evidence for reciprocal modulation between Sp1 and EGR-1 through binding of EGR-1 to its own site or the Sp1 site, displacing Sp1 binding (45) is consistent with our observations for its role in repression of Sp-mediated transcriptional activation of the ABCA2 promoter. Paradoxically, forced expression of EGR-1 in neuroblastoma cells results in an elevation in expression of the endogenous ABCA2 gene, as measured by northern blot. There are several possible explanations for these apparently contradictory findings. The ABCA2 gene may contain EGR-1 sites that lie outside the promoter region characterized that function to activate ABCA2 expression. Alternatively, the forced expression of EGR-1 may elevate the levels of additional transcription factors that act on sites outside of the basal promoter to increase ABCA2 expression. We also observed an increase in expression of the ABCA2 gene following treatment of cells with PMA (data not shown), which is consistent with the effect of PMA on elevation of EGR-1 binding activity to the GC-boxes that we report in the gel-shift experiments. Whereas the evidence presented here suggests that effect of EGR-1 on the basal promoter as characterized is repressive to ABCA2 expression, the net effect of EGR-1 on expression of the ABCA2 gene will be a function of the inducing stimulus and the sum of effects of transcription factors acting on elements that regulate its expression.
In summary we have performed the first functional characterization of a mammalian ABCA2 transporter gene promoter and have mapped the basal promoter necessary for transcription and demonstrated a functional role for two GC-boxes containing overlapping EGR-1/Sp1 binding sites. We have also shown that PMA can induce binding of the EGR-1 transcription factor to its cognate site, which suggests that PMA-responsive signaling pathways may regulate the endogenous activation of this gene. The Sp-family and EGR-1 transcription factors have been shown to activate numerous genes required for cell proliferation and differentiation and those stimuli that elevate the levels of these factors are also expected to be correlated with elevated ABCA2 levels. Inasmuch as the ABCA2 gene shares significant sequence and putative structural homology to the ABCA1 gene and given the dramatic elevation of ABCA2 expression in the brain relative to ABCA1 or other cloned ABC transporters, the ABCA2 gene may have significant involvement in brain homeostasis or degenerative disease states as well as in mediating response to drugs used in chemotherapy. Future work will focus on the identification of endogenous ABCA2 transport substrates that are required for normal cell functions.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported in part by National Institutes of Health grants CA06927, CA75266 and CA883778 to K.D.T., by an appropriation from the Commonweath of Pennsylvania, and a grant from the Active Pass Pharmaceuticals Company, Vancouver, BC. It contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Cancer Institute.
REFERENCES
- 1.Higgins C.F. (1992) ABC transporters: from microorganisms to man. Annu. Rev. Cell. Biol., 8, 67–113. [DOI] [PubMed] [Google Scholar]
- 2.Dean M. and Allikmets,R. (1995) Evolution of ATP-binding cassette transporter genes. Curr. Opin. Genet. Dev., 5, 779–785. [DOI] [PubMed] [Google Scholar]
- 3.Croop J.M. (1998) Evolutionary relationships among ABC transporters. Methods Enzymol., 292, 101–116. [DOI] [PubMed] [Google Scholar]
- 4.Holland I.B. and Blight,M.A. (1999) ABC-ATPases, adaptable energy generators fuelling transmembrane movement of a variety of molecules in organisms from bacteria to humans. J. Mol. Biol., 293, 381–389. [DOI] [PubMed] [Google Scholar]
- 5.Kunzelmann K. (1999) The cystic fibrosis transmembrane conductance regulator and its function in epithelial transport. Rev. Physiol. Biochem. Pharmacol., 137, 1–70. [DOI] [PubMed] [Google Scholar]
- 6.Allikmets R., Rashkind,W.H., Hutchinson,A., Schueck,N.D., Dean,M. and Koeller,D.M. (1999) Mutation of a putative mitochondrial iron transporter gene (ABC7) in X-linked sideroblastic anemia and ataxia (XLSA/A). Hum. Mol. Genet., 8, 743–749. [DOI] [PubMed] [Google Scholar]
- 7.Shroyer N.F., Lewis,R.A., Allikmets,R., Singh.N., Dean,M., Leppert,M. and Lupski,J.R. (1999) The rod photoreceptor ATP-binding cassette transporter gene, ABCR and retinal disease: from monogenic to multifactorial. Vision Res., 39, 2537–2544. [DOI] [PubMed] [Google Scholar]
- 8.Bodzioch M., Orso,E., Klucken,J., Langmann,T., Bottcher,A., Diederich,W., Drobnik,W., Barlage,S., Buchler,C., Porsch-Ozcurumez,M., Kaminski,W.E., Hahmann,H.W., Oette,K., Roethe,G., Aslandis,C., Lackner,K.J. and Schmitz,G. (1999) The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier Disease. Nature Genet., 22, 347–351. [DOI] [PubMed] [Google Scholar]
- 9.Gottesman M.M. (1993) Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu. Rev. Biochem., 62, 385–427. [DOI] [PubMed] [Google Scholar]
- 10.Laing N.M., Belinsky,M.G., Kruh,G.D., Bell,D.W., Boyd,J.T., Barone,L., Testa,J.R. and Tew,K.D. (1998) Amplification of the ATP-binding cassette transporter 2 gene is functionally linked with enhanced efflux of estramustine in ovarian carcinoma cells. Cancer Res., 58, 1332–1337. [PubMed] [Google Scholar]
- 11.Luciani M.F., Denizot,F., Savary,S., Mattei,M.G. and Chimini,G. (1994) Cloning of two novel ABC transporters mapping on human chromosome 9. Genomics, 21, 150–159. [DOI] [PubMed] [Google Scholar]
- 12.Vulevic B., Chen,Z., Boyd,J.T., Davis,W.,Jr, Walsh,E.S., Belinsky,M.G. and Tew,K.D. (2001) Cloning and characterization of human adenosine 5′-triphosphate binding cassette, sub-family A, transporter 2 (ABCA2). Cancer Res., 61, 3339–3347. [PubMed] [Google Scholar]
- 13.Quan T. and Fisher,G.J. (1999) Cloning and characterization of the human protein kinase C-eta promoter. J. Biol. Chem., 274, 28566–28574. [DOI] [PubMed] [Google Scholar]
- 14.Henikoff S. (1984) Unidirectional digestion with exonuclease III creates targeted breakpoints for DNA sequencing. Gene, 28, 351. [DOI] [PubMed] [Google Scholar]
- 15.Schreiber E., Matthias,P., Muller,M.M. and Schaffner,W. (1989) Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res., 17, 6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cui M.Z., Parry,G.C., Oeth,P., Larson,H., Smith,M., Huang,R.P., Adamson,E.D. and Mackman,N. (1996) Transcriptional regulation of the tissue factor gene in human epithelial cells is mediated by Sp1 and Egr-1. J. Biol. Chem., 271, 2731–2739. [DOI] [PubMed] [Google Scholar]
- 17.Shug J. and Overton,G.C. (1997) TESS: Transcription Element Search Software on the WWW. Technical Report CBIL-TR-1997-1001-v0.0, of the Computational Biology and Informatics Laboratory, School of Medicine, University of Pennsylvania.
- 18.Anbazhagan R., Herman,J.G., Enika,K. and Gabrielson,E. (2001) Spreadsheet-based program for the analysis of DNA methylation. Biotechniques, 30, 110–114. [DOI] [PubMed] [Google Scholar]
- 19.Gardiner-Garden M. and Frommer,M. (1987) CpG islands in vertebrate genomes. J. Mol. Biol., 196, 261–282. [DOI] [PubMed] [Google Scholar]
- 20.Papanikolaou N.A. and Sabban,E.L. (2000) Ability of Egr-1 to activate tyrosine hydroxylase transcription in PC12 cells. J. Biol. Chem., 275, 26683–26689. [DOI] [PubMed] [Google Scholar]
- 21.Akuzawa N., Kurabayashi,M. and Ohyama,Y. (2000) Zinc finger transcription factor Egr-1 activates Flt-1 gene expression in THP-1 cells on induction for macrophage differentiation. Arteriosclero. Thromb. Vasc. Biol., 20, 377–384. [DOI] [PubMed] [Google Scholar]
- 22.Suske G. (1999) The Sp-Family of trancription factors. Gene, 238, 291–300. [DOI] [PubMed] [Google Scholar]
- 23.Courey A.J. and Tjian,R. (1988) Analysis of Sp1 in vivo reveals multiple transcriptional domains, including a novel glutamine-rich activation motif. Cell, 55, 887–898. [DOI] [PubMed] [Google Scholar]
- 24.Cui M.Z., Penn,M.S. and Chisolm,G.M. (1999) Native and oxidized low density lipoprotein induction of tissue factor gene expression in smooth muscle cells is mediated by both Egr-1 and Sp1. J. Biol. Chem., 274, 32795–32802. [DOI] [PubMed] [Google Scholar]
- 25.Cao X., Mahendran,R., Guy,G.R. and Tan,Y.H. (1993) Detection and characterization of cellular Egr-1 binding to its recognition site. J. Biol. Chem., 268, 16949–16957. [PubMed] [Google Scholar]
- 26.Thiel G., Schoch,S. and Petersohn,D. (1994) Regulation of synapsin I gene expression by the zinc finger trancription factor zif 268/Egr-1. J. Biol. Chem., 269, 15294–15301. [PubMed] [Google Scholar]
- 27.Ackerman S.L., Minden,A.G., Williams,G.T., Bobonis,C. and Yeung,C.-Y. (1991) Functional significance of an overlapping consensus binding motif for Sp1 and Zif268 in the murine adenosine deaminase gene promoter. Proc. Natl Acad. Sci. USA, 88, 7523–7527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saffer J.D., Jackson,S.P. and Annarellla,M.B. (1991) Developmental expression of Sp1 in the mouse. Mol. Cell. Biol., 11, 2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hagen G., Muller,S., Beato,M. and Suske,G. (1992) Cloning by recognition site screening of two novel GT box binding proteins: a family of Sp1 related genes. Nucleic Acids Res., 20, 5519–5525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Supp D.M., Witte,D.P., Branford,W.W., Smith,E.P. and Potter,S.S. (1996) Sp4, a member of the Sp-1 family of zinc finger transcription factors, is required for normal murine growth, viability and male fertility. Dev. Biol., 176, 284–299. [DOI] [PubMed] [Google Scholar]
- 31.Pascal E. and Tjian,R. (1991) Different activation domains of Sp1 govern formation of multimers and mediate transcriptional synergism. Genes Dev., 5, 1646–1656. [DOI] [PubMed] [Google Scholar]
- 32.Kennett S.B., Udvadia,A.J. and Horowitz,J.M. (1997) Sp3 encodes multiple proteins that differ in their capacity to stimulate or repress transcription. Nucleic Acids Res., 25, 3110–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Majello B., De,L.P. and Lania,L. (1997) Sp3 is a bifunctional transcription regulator with modular independent activation and repression domains. J. Biol. Chem., 272, 4021–4026. [DOI] [PubMed] [Google Scholar]
- 34.Sukhatme V.P., Cao,X., Chang,L.C., Tsai-Morris,C.-H., Stamenkovich,D., Ferreira,P.C.P., Cohen,D.R., Edwards,S.A., Shows,T.B., Curran,T., Le Beau,M.M. and Adamson,E.D. (1988) A zinc finger-encoding gene coregulated with c-fos during growth and differentiation and after cellular polarization. Cell, 53, 37–43. [DOI] [PubMed] [Google Scholar]
- 35.Chisty B. and Nathans,D. (1989) DNA binding site of the growth factor-inducible protein Zif268. Proc. Natl Acad. Sci. USA, 86, 8737–8741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Milbrandt J. (1987) A nerve growth factor-induced gene encodes a possible trancriptional regulatory factor. Science, 238, 797–799. [DOI] [PubMed] [Google Scholar]
- 37.LeMaire P., Revelant,O., Bravo,R. and Charnay,P. (1988) Two mouse genes encoding potential transcription factors with identical DNA-binding domains are activated by growth factors in cultured cells. Proc. Natl Acad. Sci. USA, 85, 4691–4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lim R.W., Varnum,B.C., O’Brien,T.G. and Herschman,H.R. (1989) Induction of tumor promoter-inducible genes in murine 3T3 cell lines and tetradecanoyl phorbol acetate-nonproliferative 3T3 variants can occur through protein kinase C-dependent and independent pathways. Mol. Cell. Biol., 9, 1790–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Latchman D. (1998) Eukaryotic Transcription Factors, 3rd Edn. Academic Press, London, UK, pp. 240–244.
- 40.O’Donovan K.J., Tourtellotte,W.G., Milbrandt,J. and Baraban,J.M. (1999) The EGR family of transcription-regulator factors: progress at the interface of molecular and systems neuroscience. Trends Neurosci., 22, 167–173. [DOI] [PubMed] [Google Scholar]
- 41.Lee S.L., Sadovsky,Y., Swirnoff,A.H., Polish,J.A., Goda,P., Gavrilina,G. and Milbrandt,J. (1996) Luteinizing hormone deficiency and female infertility in mice lacking the transcription factor NGFI-A (Egr-1). Science, 273, 1219–1221. [DOI] [PubMed] [Google Scholar]
- 42.McCoy C., Smith,D.E. and Cornwell,M.M. (1995) 12-O-tetradecanoylphorbol-13-acetate activation of the MDR1 promoter is mediated by EGR-1. Mol. Cell. Biol., 15, 6100–6108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thottassery J.V., Sun,D., Zambetti,G.P., Troutman,A., Sukhatme,V.P., Schuetz,E.G. and Schuetz,J.D. (1999) Sp1 and egr-1 have opposing effects on the regulation of the rat Pgp2/mdr1b gene. J. Biol. Chem., 274, 3199–3206. [DOI] [PubMed] [Google Scholar]
- 44.Bahouth S.W., Beauchamp,M.J. and Vu,K.N. (2002) Reciprocal regulation of beta(1)-adrenergic receptor gene transcription by Sp1 and early growth response gene 1: induction of EGR-1 inhibits the expression of the beta(1)-adrenergic receptor gene. Mol. Pharmacol., 61, 379–390. [DOI] [PubMed] [Google Scholar]
- 45.Huang R.-P., Fan,Y., Ni,Z., Mercola,D. and Adamson,E.D. (1997) Reciprocal modulation between Sp1 and Egr-1. J. Cell. Biochem., 66, 489–499. [PubMed] [Google Scholar]