


Abstract
The transcription factor Ets2 has a role in cancer development and represents an attractive therapeutic target. In this study, we designed a triplex-forming oligonucleotide (TFO) directed to a homopurine:homopyrimidine sequence in the Ets2 promoter. Transcription factors of the Sp family bound to this sequence and mutation of the Sp1 site reduced Ets2 promoter activity. The Ets2-TFO had high binding affinity for the target sequence and inhibited binding of Sp1/Sp3 to the overlapping site. This effect occurred with a high degree of sequence specificity. Mismatched oligonucleotides did not inhibit Sp1/Sp3 binding and mutations in the target sequence that abolished triplex formation prevented inhibition of Sp1/Sp3 binding by the TFO. The Ets2-TFO inhibited Ets2 promoter activity and expression of the endogenous gene in prostate cancer cells at nanomolar concentrations. The TFO did not affect reporter constructs with mutations in the TFO binding site and promoters of non-targeted genes. Expression of non-targeted genes was also not affected in TFO-treated cells. Collectively, these data demonstrated that the anti-transcriptional activity of the Ets2-TFO was sequence- and target-specific, and ruled out alternative, non-triplex mediated mechanisms of action. This anti-transcriptional approach may be useful to examine the effects of selective downregulation of Ets2 expression and may have therapeutic applications.
INTRODUCTION
The human Ets family includes 25 genes that code for positively and negatively acting transcription factors involved in various aspects of cell proliferation and differentiation. Ets factors share a highly conserved DNA binding domain (Ets domain), which binds to DNA elements (Ets binding sites) characterized by the purine-rich core sequence GGAA/T (1). A large number of genes, including genes for transcription factors, matrix metalloproteinases, cell cycle regulators, extracellular matrix receptors and growth factors, are known to contain Ets binding sites (1). Ets factors are downstream effectors of the Ras-signaling pathway, which undergoes oncogenic activation in many cancers (2–4). The oncogenic potential of various Ets factors, including Ets2, has been demonstrated in several experimental systems (5–9). Recent studies indicate that Ets2 contributes to neoplastic transformation and maintenance of the malignant phenotype in various cancer types, including prostate, breast and thyroid cancers (10–14). The oncogenic effects of Ets2 are likely related to its ability to activate expression of multiple genes that promote cell proliferation and invasion, or prevent apoptotic cell death. Therefore, targeting this transcription factor may be a valid therapeutic strategy for various forms of cancer.
Oligonucleotide-directed triple helix formation offers a means to target specific sequences in DNA and interfere with gene expression at the transcriptional level (15–17). Antigene or triplex-forming oligonucleotides (TFOs) bind to homopurine:homopyrimidine sequences forming a stable, sequence-specific complex with duplex DNA. Recent studies have provided convincing evidence of triplex formation at extrachromosomal and chromosomal sites in cells by analyzing site-specific mutagenesis and covalent crosslinking of target DNA induced by TFOs (18–22). Purine-rich sequences are frequent in gene regulatory regions and TFOs directed to promoter sequences have been shown to prevent binding of transcription activators and inhibit transcription initiation in vitro (15,16). TFOs have also been shown to downregulate expression of targeted genes in cells by blocking transcription initiation or elongation (16,17). This anti-transcriptional approach could complement other gene-targeted strategies, such as antisense, small-interfering RNA, and dominant negative constructs, which interfere with gene expression at post-transcriptional level. However, a clear demonstration of triplex-mediated and sequence-specific inhibition of transcription initiation by TFOs has generally been lacking in most cell culture studies. Oligonucleotides are known to elicit non-specific effects by interacting with proteins or non-target nucleic acids both in sequence and non-sequence specific manners (23–25). These effects may depend on nucleotide sequence, chemical composition, and tendency of oligonucleotides to form particular secondary structures (24,26–28). Therefore, one cannot always rule out that the effects of TFOs on transcription initiation observed both in cells and cell-free systems might be due to non-specific and non-triplex mediated effects.
Our goal in the present study was to design a TFO directed to a homopurine:homopyrimidine sequence in the Ets2 promoter with the intent to selectively inhibit transcription of this gene. The sequence selected for triplex-mediated gene targeting was located ∼40 bp upstream of the transcription initiation sites in the Ets2 promoter (29–31). Our data show that transcription factors of the Sp family bound to this region and that mutation of the Sp1 site significantly reduced promoter activity. The Ets2-targeting TFO had high affinity and specificity for the target sequence, inhibited binding of Sp1/Sp3 transcription factors to the target site in vitro and was effective as repressor of Ets2 transcription in cells. Experiments with mutated oligonucleotide targets and mutated promoter reporter constructs provided evidence that the effects of the TFO both in vitro and in cells were sequence- and target-specific. These results demonstrated that the anti-transcriptional activity of the Ets2-TFO was due to triplex formation, and ruled out alternative, non-triplex mediated mechanisms of action. The activity of the TFO combined with a high degree of specificity suggest that this anti-transcriptional approach may be useful to analyze the effects of selective downregulation of Ets2 expression in experimental settings and may have potential therapeutic applications.
MATERIALS AND METHODS
Oligonucleotides
Phosphodiester (PO) and phosphorothioate (PS) oligonucleotides were purchased from Genset (La Jolla, CA). PO and PS oligonucleotides were purified by polyacrylamide gel electrophoresis (PAGE) or high-performance liquid chromatography. Stock solutions of oligonucleotides were made in sterile water. Oligonucleotide concentrations were determined with a spectrophotometer using appropriate nucleotide extinction coefficients (32). Immediately before each experiment, oligonucleotide solutions were heated at 65°C for 10 min and then chilled on ice. This heating step was performed to eliminate self-aggregates (e.g. tetraplex and homoduplex) that might have formed during storage of the oligonucleotides at low temperatures. Sequences of the TFO, control oligonucleotides and oligonucleotides corresponding to purine and pyrimidine strands of the target site are shown in Figure 1.
Figure 1.


Position of the target site in the Ets2 promoter and triplex DNA formation by the Ets2-TFO. (A) The 25-bp homopurine:homopyrimidine target sequence is located ∼40 bp upstream of the transcription start site in the Ets2 promoter. Putative binding sites for Sp1 and AP2 (black boxes), inverted repeat sequence (small arrows) and nuclease hypersensitive site (vertical arrow) adjacent to the target site are indicated. Sequences of the target site, TFO and control oligonucleotides are shown below the promoter map. The Sp1 consensus sequence within the TFO target site is boxed. (B) The oligonucleotide corresponding to the pyrimidine strand of the target sequence was 5′-end labeled with [γ-32P]ATP and annealed to the complementary purine-rich oligonucleotide. Duplex DNA (1 nM) was incubated with the indicated concentrations of TFO and control oligonucleotides M1 and M2 for 24 h at 37°C. Samples were resolved on a 12% polyacrylamide gel under non-denaturating conditions. Positions of double- and triple-stranded DNA are indicated.
Gel mobility shift assays
Oligonucleotides corresponding to the pyrimidine-rich strand of the wild type or mutated targets were 5′-end labeled with [γ-32P]ATP and T4 polynucleotide kinase and annealed to complementary oligonucleotides as previously described (33). Samples containing 1 nM duplex DNA and increasing concentrations of either TFO or control oligonucleotides were incubated for 24 h at 37°C in a buffer containing 90 mM Tris, 90 mM borate (pH 8) and 10 mM MgCl2 (TBM buffer). To examine triplex DNA formation, samples were resolved by PAGE under non-denaturing conditions (33). Apparent dissociation constants (Kd) of TFOs were estimated as previously described (33). To examine nuclear protein binding to the Ets2 promoter site, nuclear extracts were prepared from breast cancer cells as described previously (32). Nuclear extracts were incubated with 1 nM of duplex DNA, which had been pre-incubated with or without TFOs overnight in TBM buffer. Binding reactions were incubated for 20 min at 10°C and then were resolved on 4% polyacrylamide gels (32). Sources of antibodies for supershift assays and double-stranded oligonucleotide competitors and related experimental conditions have been reported previously (32).
Promoter reporter constructs
To generate pGL3-Ets2, a pBluescript plasmid containing a 3.6 kb fragment of the human Ets2 promoter was digested with SacII (29). After filling-in with T4 DNA polymerase (Promega), the SacII fragment was subcloned into the SmaI site of the pGEM-3Z vector (Promega). A pGEM-3Z-Ets2 clone with the Ets2 promoter insert in the 5′–3′ orientation was expanded, digested with KpnI and HindIII, and the resulting fragment was subcloned into the pGL3-basic (Promega, Madison, WI). Promoter reporter constructs containing either three or five mutations in the TFO target sequence were produced from pGL3-Ets2 using the QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). Ets2-Mut3 oligonucleotides were used as primers in the PCR to generate the pGL3-Ets2-Mut3. The pGL3-Ets2-Mut5 was then produced using pGL3-Ets2-Mut3 as template and Ets2-Mut5 oligonucleotides as primers. The pGL3-Ets2-mSp1, which contained a mutated Sp1 site in the TFO target sequence, was produced by changing the wild type sequence 5′-TCCCTCCT-3′ to 5′-AGACTCCT-3′ using the QuickChange Site-Directed Mutagenesis kit. Incorporation of the desired mutations in the isolated clones was confirmed by sequencing. The pGL3-Myc-P1 plasmid was generated by subcloning a KpnI–XhoI fragment of 1126 bp of the c-myc promoter into pGL3-basic. The pGL3-Src was obtained by sucloning a KpnI–XbaI fragment of the c-src promoter into pGL3-basic (34).
Transfection with reporter plasmid and luciferase assay
Human prostate cancer cells (DU145 and PC3) were grown in RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen, Carlsbad, CA). Cells were transfected with reporter vectors in the absence or presence of oligonucleotides using either DOTAP (Roche, Indianapolis, IN) or Oligofectamine (Invitrogen) as transfection reagent. For transfection with DOTAP, cells were plated in 96-well plates at a density of 8 × 103 cells/well. After 24 h, 100 ng of pGL3-Ets2 was mixed with TFO or control oligonucleotide in 20 mM HEPES and incubated with DOTAP at a mass ratio of 5:1 (DOTAP:DNA) for 15 min at room temperature. Either pRL-SV40 (5 ng) or pRL-TK (50 ng) control vector was added to each sample to monitor transfection efficiency. The mixture was then diluted in 100 µl of serum-containing medium and added to the cells. After 6 h, medium was removed and replaced with fresh medium. When Oligofectamine was used, cells were plated in 48-well plates at a density of 1 × 104 cells/well. Reporter vectors (100 ng), control vector (5 ng), oligonucleotides and Oligofectamine (0.8 µl) were diluted in 20 µl of Opti-MEM, incubated for 15 min at room temperature and then added to the cells seeded in 80 µl of Opti-MEM. After 4 h, growth medium containing 10% serum was added. Cells were lysed 24 h later and luciferase activity was measured using dual-luciferase assay system (Promega). Each experiment was repeated at least three times to ensure reproducibility of the data. In the co-transfection experiments, oligonucleotides represented a significant amount (100–200 ng) of the total DNA to be transfected and preliminary experiments indicated that the efficiency of transfection depended greatly on the amount of DNA and transfection reagent used. Thus, reporter activity of TFO-treated cells was compared with the reporter activity of cells transfected with an equal amount of control oligonucleotide rather than reporter plasmid alone, therefore keeping total amount of DNA (plasmid plus oligonucleotide) and transfection reagent constant. Hence, similar transfection efficiencies were achieved in the different experimental groups.
RNA analysis
DU145 cells were seeded at 7 × 104 cells/well in 6-well plates and transfected 24 h later with TFO or control oligonucleotide using Oligofectamine as described above. Total RNA was extracted from control and TFO-treated cells using Trizol (Invitrogen). RNA concentrations were determined by spectrophotometry. RT–PCR was performed using the SuperScript One Step RT–PCR system (Invitrogen) and gene-specific primers. Forward and reverse primers for Ets2 were 5′-TCAGCTCTGAGCAGGAGTTTCAGA-3′ and 5′-GGTTGGCTTATTGAGGCAGAGAGA-3′, respectively, which amplified a fragment of 296 bp. Sequences of the GAPDH primers have been published previously (33). Sequences of forward and reverse primers for Ets1 were 5′-TGCTATCAAACAAGAAGTCGTCAC-3′ and 5′-GACAGGAGATGGCTGGGAATTCA. RT–PCR was performed using 100 ng of total RNA. Each reaction contained 0.2 µM forward and reverse gene-specific primers in addition to reagents present in the SuperScript PCR buffer. RNA was reverse-transcribed for 30 min at 50°C and then subjected to 26 cycles of PCR (94°C, 15 s; 55°C, 30 s; 72°C, 15 s). Samples were analyzed on 2% agarose gels. Following staining with ethidium bromide, PCR products were visualized using ChemiImager 4400 AlphaInnotech (San Leandro, CA). Densitometric analysis was performed using AlphaEase software (AlphaInnotech). The amount of RNA (100 ng) and number of PCR cycles (26 cycles) were optimized in preliminary experiments to ensure that PCR was performed in the exponential phase of amplification and there was a linear relationship between the amount of RNA template and yield of amplified products. Three individual RNA preparations were used with identical results and RT–PCR was performed at least two times with each individual preparation to ensure data reproducibility. Northern blot analysis was done as previously described using 3 µg of total RNA and a radio-labeled Ets2 cDNA fragment as hybridization probe (11,33).
RESULTS
Selection of the target site and design of the TFO
The Ets2 gene is transcribed from multiple initiation sites directed by a promoter lacking typical TATA and CAAT elements (29,30). To target this gene using the triplex DNA-based approach, we selected a 25-bp homopurine:homopyrimidine sequence located ∼40 bp upstream of the cluster of transcription initiation sites and overlapping a putative Sp1 binding site (Fig. 1). The selected sequence is located within a 160-bp region that has been previously shown to be essential for maximal promoter activity (29,30). This region includes multiple putative transcription factor binding sites, an inverted repeat and a long polypyrimidine (CT) tract, which is able to stimulate transcriptional activity of heterologous promoters (29,30). Nuclease hypersensitive sites, which are generally found in the promoter of actively transcribed genes and indicate the presence of an open chromatin structure, have been mapped in this region of the promoter in Ets2-expressing cells (29). In particular, a nuclease hypersensitive site co-localizes with the inverted repeat within the target sequence.
We designed a GT-rich oligonucleotide to bind to this sequence. As shown in Figure 1, the Ets2-targeting TFO had G residues opposite G:C base pairs, and T residues opposite A:T base pairs. The TFO was not identical or complementary to either strand of the target duplex and was expected to bind to the purine-rich strand of the duplex in antiparallel orientation. Similar antiparallel GT-rich TFOs have been shown to bind with high affinity to target DNA at physiological pH (33,35–38). Two mismatched control oligonucleotides, designed as M1 and M2, which had nucleotide composition similar to the Ets2-TFO but scrambled sequences, were used in the study (Fig. 1A). Both control oligonucleotides conserved potentially critical sequence elements (e.g. short G2 and G3 runs) present in the TFO. M2 was identical to the TFO at the 5′ and 3′ ends and had extensive sequence homology to the TFO in the remaining part. TFO and control oligonucleotides were synthesized as both PO and PS oligonucleotides. All the data presented herein were obtained with oligonucleotides synthesized with a fully modified PS backbone to increase resistance to nuclease degradation.
Triplex DNA formation by the Ets2-targeting TFO
To examine binding of the Ets2-TFO, a radio-labeled duplex DNA corresponding to the target sequence was incubated with increasing concentrations of Ets2-TFO or control oligonucleotides. Binding was then assessed by gel mobility shift assay. In these experiments, samples were incubated for 24 h to ensure that binding was evaluated under equilibrium conditions as done in previous studies (38–40). In addition, potassium ions, which have been often shown to reduce the ability of PO and PS oligonucleotides to form triplex DNA at physiological intracellular concentration (15,16), were not included in the binding reaction buffer. As shown in Figure 1B, the PS-modified Ets2-TFO exhibited high affinity for the target sequence under these conditions. Its apparent Kd, estimated by gel mobility shift assay, was ∼5 nM. Complete formation of triplex DNA was observed at a concentration of 50–100 nM of TFO, which corresponded to a 50- to 100-fold molar excess of TFO compared with the target DNA. PS-modified TFOs have shown reduced binding affinity compared with the PO counterpart in previous studies (38–40). However, the PS modification did not appear to affect binding of the Ets2-TFO to its target, since similar results were observed with both PS and PO TFO (data not shown). As shown in Figure 1B, EMSA was performed also with the control oligonucleotides M1 and M2. Both control oligonucleotides did not form triplex DNA at concentrations as high as 1 µM, confirming the sequence specificity of the interaction of the Ets2-TFO with its target sequence.
Inhibition of Sp1/Sp3 protein binding to the Ets2 promoter by the Ets2-TFO
The TFO target site directly overlaps a Sp1 consensus sequence (Fig. 1). Sp1 sites are thought to be involved in determining the position of the initiation site in absence of a classical TATA box and be critical for activity of genes with TATA-less promoters, like Ets2 (41). However, the identity of the proteins binding to this region and their role in the activation of Ets2 transcription were not known. To examine nuclear protein binding, a double-stranded oligonucleotide probe, Ets2-WT, which included the TFO target sequence and the putative Sp1 consensus site was prepared. Gel mobility shift assay with nuclear extracts and the Ets2-WT probe showed formation of three major protein/DNA complexes, which were identified as Sp1 and Sp3 complexes (Fig. 2). Formation of these complexes was inhibited by the addition of a competitor oligonucleotide containing a Sp1 consensus sequence and antibodies against Sp1 and Sp3 (Fig. 2, and data not shown). Next, we determined whether formation of the Sp1/Sp3 complexes with the duplex DNA could be blocked by the Ets2-TFO. Incubation of the Ets2-WT probe with the TFO under conditions that allowed triplex formation resulted in a clear reduction of Sp1/Sp3 complex formation (Fig. 3). A >50% reduction of Sp1/Sp3 binding was already observed at 10 nM of TFO, i.e. only 10-fold molar excess compared with target DNA. These data were consistent with the high binding affinity of the Ets2-TFO estimated by gel mobility shift assay. Both control oligonucleotides M1 and M2 did not affect Sp1/Sp3 binding, indicating sequence specificity of the TFO effects. These data suggested that the TFO formed a stable complex that was able to block binding of Sp factors to the Ets2 promoter. This could lead to transcription inhibition by preventing assembly of an active initiation complex.
Figure 2.
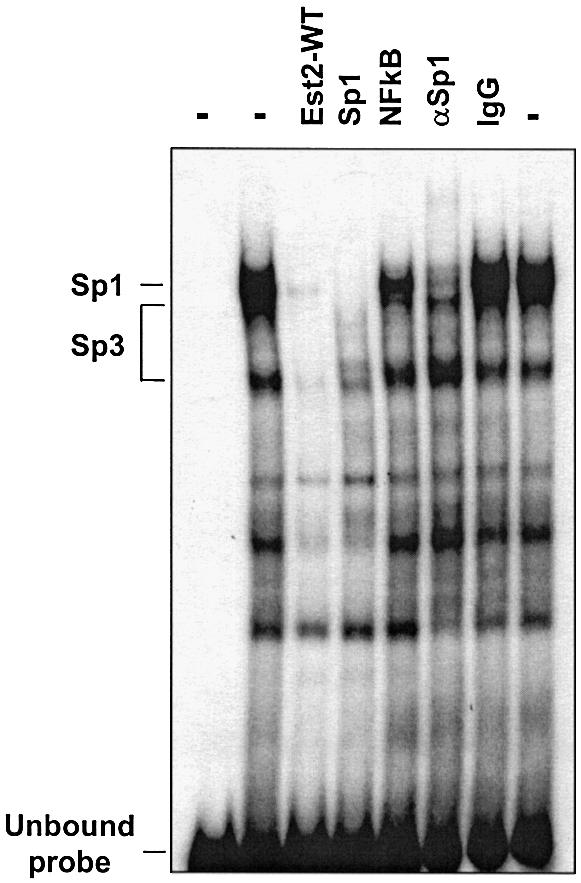
Nuclear protein binding to the Ets2 promoter site. Radiolabeled double-stranded oligonucleotide corresponding to the Ets2 target sequence (Ets2-WT) was incubated with nuclear extracts, with the exception of the first lane. To identify proteins binding to the site in the Ets2 promoter, binding reactions contained either unlabeled Ets2 target (Ets2-WT), Sp1 and NFkB consensus oligonucleotides (Sp1 and NFkB), anti-Sp1 antibody (αSp1), and normal rabbit IgG (IgG). Samples were electrophoresed on a polyacrylamide gel under non-denaturing conditions. Positions of unbound probe and Sp1/Sp3 complexes are indicated.
Figure 3.

TFO-induced inhibition of Sp1/Sp3 protein binding to the Ets2 promoter. Double-stranded oligonucleotide Ets2-WT was radiolabeled and incubated without or with the indicated concentrations of Ets2-TFO or control oligonucleotides for 24 h at 37°C. Nuclear extracts were added to the binding reactions, with the exception of the first lane, incubated for 20 min and then samples were analyzed by gel electrophoresis as described in the legend to Figure 2.
Inhibition of Sp1/Sp3 binding by the Ets2-TFO is mediated by triplex formation
The TFO-induced inhibition of Sp1/Sp3 binding to the Ets2 promoter could be due to a sequence-specific but non-triplex mediated effect (e.g. a protein decoy mechanism). To rule out this possibility, we used double-stranded oligonucleotide probes, Ets2-Mut3 and Ets2-Mut5, which contained mutations in the TFO target sequence (Fig. 4A). These mutations were specifically designed to disrupt the ability of the TFO to form triplex DNA but not binding of Sp1/Sp3 factors. Gel mobility shift assays showed that the Ets2-TFO was unable to bind to the mutated probes, while it was clearly able to form triplex DNA with the probe containing the wild type sequence (Fig. 4B). Next, the mutated probes were incubated with nuclear extract to examine nuclear protein binding. Gel mobility shift assays with the Ets2-Mut3 probes showed formation of three major protein/DNA complexes similar to those formed with the wild type probe (Fig. 4C). These complexes were identified as Sp1/Sp3 complexes by oligonucleotide competition and antibody supershift assays (Fig. 4C). When the Ets2-Mut3 probe was incubated with the Ets2-TFO or control oligonucleotide, the TFO was unable to prevent binding of Sp1 and Sp3 under conditions that inhibited binding of these proteins to the wild type target (Fig. 4C). Formation of Sp1/Sp3 complexes on the Ets2-Mut5 probe was also not affected by pre-incubation with the Ets2-TFO (data not shown). Taken together, these data confirmed the sequence specificity of the interaction between the TFO and the Ets2 target site. Moreover, these results demonstrated that inhibition of Sp1/Sp3 binding by the Ets2-TFO required triplex formation with the target DNA and was not due to a direct interaction of the TFO with the transcription factors or other proteins in the nuclear extract.
Figure 4.


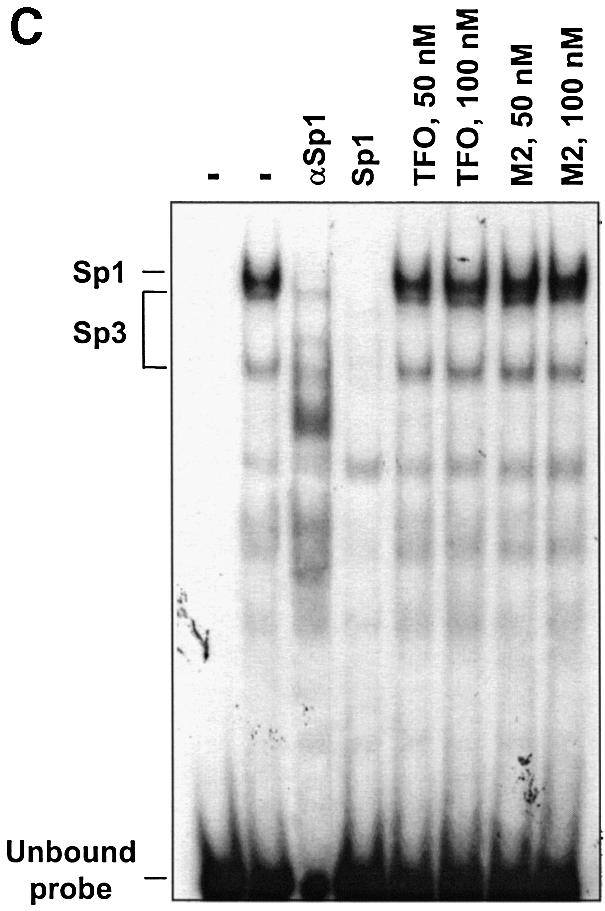
TFO-induced inhibition of nuclear protein binding to the Ets2 promoter is sequence specific and triplex mediated. (A) Sequence of wild type and mutated oligonucleotides. Sp1 sites in the Ets2-WT, Ets2-Mut3 and Ets2-Mut5 oligonucleotides are boxed. TFO target sequence is underlined. Mutated bases in the Ets2-Mut3 and Ets2-Mut5 oligonucleotides are shown in small bold letters. (B) Binding of the Ets2-TFO to wild type and mutated targets. Ets2-WT, Ets2-Mut3 and Ets2-Mut5 oligonucleotides were radiolabeled and incubated with the indicated concentrations of Ets2-TFO. Binding and electrophoresis were performed as described in the legend of Figure 1. (C) Nuclear protein binding to the mutated Ets2 target and effects of the TFO. Ets2-Mut3 was radiolabeled and incubated either in the absence or presence of the indicated concentrations of Ets2-TFO or control oligonucleotide M2. Nuclear extracts were added to the binding reactions with the exception of the first lane. Where indicated, anti-Sp1 antibody (αSp1) and a Sp1 consensus oligonucleotide (Sp1) were added to the samples to confirm presence of Sp1 factors in the protein/DNA complexes. Samples were separated on a polyacrylamide gel as described in the legend to Figure 2.
Inhibition of Ets2 promoter activity by the Ets2-TFO in prostate cancer cells
To evaluate the biological activity of the Ets2-TFO, we determined whether it had any effect on the Ets2 promoter in prostate cancer cells known to express high levels of this gene. The pGL3-Ets2 plasmid contained an 880-bp fragment of the Ets2 promoter, including the transcription start site and TFO target sequence, cloned upstream of the firefly luciferase gene. Hence, the level of luciferase activity in transfected cells was a direct measurement of transcription initiation within the Ets2 promoter. DU145 and PC3 cells were transfected with the pGL3-Ets2 plasmid along with either pRL-TK or pRL-SV40 control vectors. After 24 h, the activity of both firefly and Renilla luciferase were measured in cell extracts. Upon transfection of the Ets2 promoter construct, luciferase activity was induced ∼100-fold in DU145 and PC3 cells compared with cells transfected with pGL3 basic vector, indicating that the Ets2 promoter effectively activated luciferase expression in these cells. To determine the functional relevance of the Sp1 site overlapping the TFO target sequence, we mutated the corresponding sequence in the pGL3-Ets2 reporter construct. As shown in Figure 5A, Ets2 promoter activity was significantly reduced in the pGL3-Ets2-mSp1 compared with wild type reporter.
Figure 5.


Inhibition of Ets2 promoter activity by the Ets2-TFO. (A) pGL3-Ets2 and pGL3-Ets2mSp1 were transfected into DU145 cells along with a pRL-SV40 control vector using DOTAP. Luciferase activity was measured after 24 h from transfection. Renilla luciferase activity was used to control for transfection efficiency. Asterisk, P < 0.005 compared with wild type reporter. (B) The pGL3-Ets2 plasmid was transfected into DU145 cells along with Ets2-TFO or control oligonucleotide M2 and pRL-SV40 control vector using DOTAP. Firefly and Renilla luciferase activities were measured after 24 h from transfection. Firefly luciferase activity was normalized to the Renilla luciferase activity and data are presented as percent of luciferase activity compared with control transfected cells. Asterisks, P < 0.05 and P < 0.0005 at 125 and 250 nM of Ets2-TFO compared with mismatched- treated cells.
Next, prostate cancer cells were co-transfected with pGL3-Ets2 and either Ets2-TFO or control oligonucleotide at concentrations of 125 and 250 nM. When DU145 cells were incubated with the Ets2-TFO, the activity of the reporter gene was significantly reduced compared with cells incubated with identical concentrations of control oligonucleotide (Fig. 5B). Inhibition of Ets2 promoter activity by the TFO was dose dependent with ∼50 and 75% reduction at 125 and 250 nM, respectively. A similar effect of the TFO on Ets2 promoter activity was observed in PC3 cells (data not shown). Significantly, the extent of inhibition of Ets2 promoter activity by the Ets2-TFO was similar to that observed by mutating the Sp1 site overlapping the TFO target sequence. This finding suggested that inhibition of Sp1/Sp3 binding at the target site was a likely mechanism of transcription inhibition by the TFO. Furthermore, the significant inhibition of promoter activity by the Ets2-TFO and the lack of effect of the non-triplex forming oligonucleotide suggested that the inhibition was likely due to triplex DNA formation at the TFO target site. Since the short incubation prior to transfection should not permit triplex formation, these data suggested that the TFO was able to interact with the target DNA once inside the cells and to form a stable complex that inhibited promoter activity.
Triplex formation is required for inhibition of promoter activity by the Ets2-TFO
To examine the mechanism of inhibition of Ets2 promoter activity by the TFO, we generated promoter reporter constructs that contained mutations in the TFO target sequence identical to those tested in gel mobility shift assays. The pGL3-Ets2-Mut3 and pGL3-Ets2-Mut5 constructs were derived from the pGL3-Ets2 plasmid and differed only in the presence of either three or five mutations in the TFO target sequence (Fig. 4A). In both constructs, the Sp1 site overlapping the target sequence was not affected by the mutations, and gel shift mobility assays showed binding of Sp1/Sp3 proteins to oligonucleotide probes containing identical base changes (Fig. 4B). In luciferase reporter assays, activity of the mutated promoters was decreased ∼25% compared with the wild type reporter (Fig. 6A). This reduction in promoter activity was probably due to the partial disruption of the inverted repeated adjacent to the Sp1 site. When DU145 cells were transfected with the TFO or control oligonucleotide along with reporter constructs, the Ets2-TFO was unable to affect the activity of mutated promoters under conditions that resulted in significant inhibition of the wild type promoter (Fig. 6B). Thus, the presence of the intact target sequence, which was required for triplex formation and inhibition of transcription factor binding in vitro, was also essential for inhibition of promoter activity by the TFO in cells. These data strongly support the conclusion that inhibition of transcription initiation by the Ets2-TFO was due to triplex formation and rule out that alternative mechanisms, such as a decoy-like mechanism, were responsible for these effects.
Figure 6.


TFO-induced inhibition of Ets2 promoter activity requires triplex formation. (A) DU145 cells were transfected with pGL3-Ets2, pGL3-Ets2-Mut3 or pGL3-Ets2-Mut5 along with pRL-SV40. Firefly and Renilla luciferase activities were measured 24 h later. (B) DU145 cells were transfected with pGL3-Ets2, pGL3-Ets2-Mut3 or pGL3-Ets2-Mut5 along with 200 nM of Ets2-TFO or control oligonucleotide M2 using Oligofectamine. The pRL-SV40 control vector was included to monitor transfection efficiency. Luciferase activity was measured after 24 h. Data are presented as described in the legend to Figure 5. Asterisk, P < 0.0005 compared with control treated cells.
Inhibition of promoter activity by the Ets2-TFO is target specific
To further prove the selectivity of the Ets2-TFO for the targeted gene promoter, we evaluated its effects on promoter reporter constructs with significant structural and functional similarity to the Ets2 promoter. Both the c-myc and c-src promoter contained purine-rich sequences similar to that targeted by the Ets2-TFO and multiple Sp1 binding sites (32–34,42). The c-src promoter contained a long polypyrimidine region located upstream of the transcription initiation site known to be critical for promoter activity (42). A 25-bp sequence within this polypyrimidine region had only four mismatches compared with the Ets2 target sequence. The c-myc P1 promoter construct used in this study also contained a polypyrimidine tract (CT element) upstream of the P1 promoter (33). To prevent potential secondary effects due to Ets2 downregulation by the TFO, however, the Ets binding site near the c-myc P2 promoter had been deleted (43). Gel mobility shift assays confirmed that Sp1/Sp3 proteins bound to sites proximal to the purine-rich sequences in both c-myc and c-src promoters (32,42). We also determined that binding of Sp1/Sp3 to oligonucleotides containing the myc P1 promoter sequence was not affected by the Ets2-TFO (data not shown). As shown in Figure 7, the Ets2-TFO did not inhibit the activity of the c-myc and c-src promoter constructs at concentrations that induced significant inhibition of Ets2 promoter. Thus, these data indicated that the effects of the TFO depended strictly on the presence of the complementary target sequence and were specific for the targeted gene promoter. The TFO was unable to affect activity of promoters with similar purine-rich sequences and Sp1 sites, suggesting that the inhibition of Ets2 promoter by the TFO was not due to non-specific interactions of the TFO with transcription factors or other components of the transcription machinery.
Figure 7.

Target specificity of the effects of the Ets2-TFO on promoter reporter activity. DU-145 cells were transfected using DOTAP with the reporter constructs pGL3-c-myc-P1 and pGL3-c-src along with 250 nM of TFO or control oligonucleotide M2 and the pRL-SV40 control vector. Luciferase activity was measured as described in the legend to Figure 5.
TFO-mediated inhibition of expression of the endogenous Ets2 gene
The results described above indicated that the Ets2-TFO was able to bind to the target sequence in vitro and inhibit Ets2 promoter activity in cells in a sequence- and target-specific manner. These data suggested that the Ets2-TFO could be used to selectively downregulate expression of the endogenous Ets2 gene. To determine the effect of the TFO on Ets2 expression, DU145 cells were transfected with 200 nM of TFO or control oligonucleotide and total RNA was isolated after 24 and 48 h. The level of Ets2 RNA was determined by RT–PCR under conditions that ensured that the amount of amplified product was proportional to the initial amount of RNA template as described in Materials and Methods (Fig. 8A). Ets2 RNA was reduced by ∼40 and 50% at 24 and 48 h, respectively, in TFO-treated cells compared with untreated and control oligonucleotide-treated cells (Fig. 8B and C). The level of GAPDH RNA was similar in TFO- and control-treated cells. In addition, we measured RNA levels of another transcription factor, Ets1, which is highly homologous to Ets2 and is expressed in prostate cancer cells. Ets1 expression was unaffected by the Ets2-targeting TFO (Fig. 8B). We also investigated whether increasing the concentration of TFO would result in greater inhibition of Ets2 transcription. As shown in Figure 8D and E, Ets2 RNA was reduced by ∼50 and 65% in cells incubated for 48 h with 200 and 400 nM of TFO compared with untreated control cells, indicating a dose-dependent effect of the TFO on Ets2 gene expression. The control oligonucleotide did not affect Ets2 RNA level at these concentrations. We did not detect a significant effect of the TFO on Ets2 gene expression at concentrations <200 nM (data not shown). Analysis of total RNA from cells incubated with 400 nM TFO or control oligonucleotide by northern blot analysis also showed a reduced level of Ets2 RNA in TFO-treated cells compared with control-treated cells, confirming the data obtained by RT–PCR (Fig. 8F). Similar amounts of ribosomal RNA were detected in the ethidium bromide stained gel indicating equal RNA loading. Collectively, these data indicated that the TFO was able to inhibit transcription of the endogenous Ets2 gene and this effect was specific for the targeted gene.
Figure 8.

Downregulation of endogenous Ets2 gene expression by the Ets2-TFO. (A) Increased amounts of total RNA from DU145 cells were reverse- transcribed and amplified with Ets2-specific primers. PCR products were separated on 2% agarose gels, stained with ethidium bromide, visualized and quantified using a ChemiImager. (B) DU145 cells were transfected with 200 nM Ets2-TFO or control oligonucleotide M2 using Oligofectamine. After 24 and 48 h total RNA from untreated control (Co), TFO-treated (TFO) and mismatched oligonucleotide-treated (M2) cells was extracted and RT–PCR performed. (C) Densitometric analysis of Ets2 RNA levels at 24 and 48 h after transfection. Data are mean ± SD from three separate experiments. Asterisks, P < 0.005 at 24 h and P < 0.0005 at 48 h compared with untreated control and M2-treated cells. (D) DU145 cells were transfected with 200 and 400 nM of TFO or control oligonucleotide M2. RNA was extracted after 48 h and analyzed by RT–PCR. (E) Densitometric analysis of Ets2 RNA levels in cells incubated with increasing concentrations of TFO and control oligonucleotide. Data are mean ± SD from two experiments. Asterisks, P <0.005 compared with untreated control and M2-treated cells. (F) RNA from cells transfected with 400 nM of TFO and control oligonucleotide and incubated for 48 h after transfection was subjected to northern blot analysis using an Ets2 cDNA probe. Ethidium bromide-stained gel is shown to confirm integrity and equal loading of RNA.
DISCUSSION
Several studies have established a link between various members of the Ets family of transcription factors and neoplastic transformation (1,9). Increased expression of Ets2 has been associated with initiation and progression of various cancer types (11–14). Inhibition of Ets2 by antisense or dominant negative constructs has been shown to reduce anchorage-independent growth of prostate, breast and thyroid cancer cells (11,12,14). Even partial reduction of Ets2 expression, such as that obtained by disruption of a single Ets2 allele, limited growth of breast tumors in transgenic mice (13). Thus, a selective inhibitor of this transcription factor would be useful to study its role in cancer development and might have therapeutic applications as an anticancer agent. Our goal in this study was to design an inhibitor of Ets2 using the triplex DNA-based approach. Recent studies have shown successful applications of this approach to induce site-specific mutagenesis, recombination and transcriptional repression in various experimental systems (17). Convincing evidence of triplex formation on chromosomal targets has also been presented (18–22). However, whether the anti-transcriptional effects of TFOs, particularly of those targeting regulatory elements in gene promoters and blocking transcription initiation, are indeed due to a triplex DNA-based mechanism is still controversial. Oligonucleotides could interact in a sequence or non-sequence-specific manner with transcription factors and other components of the transcription apparatus, and inhibit transcription initiation regardless of triplex formation (44–46). In this study, we selected a 25-bp homopurine:homopyrimidine sequence in the Ets2 promoter that appeared optimal for triplex-mediated gene targeting. The sequence was immediately upstream of the transcription start site and overlapped a putative Sp1 binding site. We demonstrated binding of Sp1/Sp3 transcription factors to this sequence by gel mobility shift assay and its relevance for Ets2 transcription by promoter reporter assays. A PS-modified GT-TFO, directed to this sequence, was able to inhibit nuclear protein binding, promoter activity and expression of the endogenous Ets2 gene. Several lines of evidence support the conclusion that the effects of the TFO were sequence- and target-specific, and that formation of a triplex DNA structure at the target site was required for the anti-transcriptional activity of the TFO. Our results indicate that this TFO can act as a selective transcriptional repressor and suggest that it can be used to antagonize the effects of Ets2 over-expression in cancer cells.
Analysis of the binding properties of the Ets2-TFO showed that it bound to the target DNA with very high affinity. This was consistent with previous reports showing that antiparallel GT-rich oligonucleotides were able to form stable triplex DNA at low concentrations and physiological pH and temperature (33,35–38). The high binding affinity of the Ets2-TFO was probably due to the perfect homopurine: homopyrimidine composition of the target sequence. Any pyrimidine interruption in the purine strand would decrease stability of the triple helix and lower TFO binding affinity (15). Furthermore, the PS modification, which was required to increase nuclease resistance, did not affect binding of the Ets2-TFO. In fact, the Kd value of the PS Ets2-TFO, estimated by gel mobility shift assays, was very similar to that of the corresponding PO TFO. Despite its high affinity, binding of the Ets2-TFO required exact pairing with the target sequence according to the triplex DNA code. Control oligonucleotides with similar length and base composition but a number of mismatched bases relative to the target sequence did not form triplex DNA. Furthermore, the Ets2-TFO did not recognize targets containing even a limited number of mutations compared with the wild type target. It is important to note that we measured binding of the Ets2-TFO in the absence of physiological concentrations of potassium ions, which have been shown to inhibit triplex DNA formation in vitro (15,16). However, the relevance of potassium-mediated inhibition of triplex DNA formation by PO and PS oligonucleotides in cells is unclear. Indeed, a number of studies have now provided both direct and indirect evidence of oligonucleotide-mediated triplex DNA formation in cells (18–22). Our present data showing triplex-mediated effects of a PS TFO on promoter activity and endogenous gene expression also support the conclusion that the intracellular concentration of potassium ions may not be a major limiting factor for activity of TFOs in cells.
The sequence targeted by the Ets2-TFO was immediately upstream to the transcription initiation site in the Ets2 gene. The region comprising the target sequence contained multiple putative transcription factor binding sites (29,30). However, the identity of proteins binding to this region and their role in the transcriptional activation was not known. Using gel mobility shift assays, we determined that transcription factors of the Sp1 family bound to the site overlapping the TFO target sequence. The Ets2-TFO inhibited binding of these proteins to the Ets2 promoter in a dose- and triplex-dependent manner. Control oligonucleotides with similar length and base composition that did not bind to the target DNA did not inhibit Sp1/Sp3 binding. Moreover, the introduction of mutations in the target sequence adjacent to the Sp1 site abolished triplex formation and any effect of the TFO on Sp1/Sp3 binding. These data confirmed that the activity of the TFO depended strictly on the formation of a triple helical complex with the target DNA. These findings also ruled out that the inhibition of Sp1/Sp3 binding was mediated by a direct interaction of the TFO with transcription factors or other proteins in the nuclear extract. Inhibition of Sp1/Sp3 binding, demonstrated in the gel mobility shift assays, could be an important mechanism of TFO-induced inhibition of transcription initiation in cells. In this regard, it is significant that the TFO inhibited Ets2 promoter activity to a degree similar to that achieved by mutating the Sp1 site overlapping the TFO target sequence. It is also relevant that mutations adjacent to the Sp1 site that did not affect Sp1/Sp3 binding induced only a minimal reduction of the Ets2 promoter activity. Sp1 sites may be involved in positioning the transcription start site and may be critical for promoter activity of genes, like Ets2, which lack a TATA box (41). Sp factors can also enhance transcription by facilitating DNA loop formation via protein–protein interaction (41). Thus, by binding to its target sequence, the Ets2-TFO may prevent binding of these trans-activating factors and inhibit formation of an active transcription complex.
In addition to its activity in the cell-free system, the Ets2-TFO was able to inhibit transcription in promoter reporter assays and expression of the endogenous gene in cells at similarly low concentrations. In these experiments a mismatched oligonucleotide, which was unable to form triplex DNA at the Ets2 site, did not inhibit promoter activity and Ets2 expression. The lack of effects of the Ets2-TFO on mutated reporters was also consistent with a triplex-mediated mechanism and indicated that alternative mechanisms of inhibition of transcription initiation by the Ets2-TFO were unlikely. Furthermore, the Ets2-TFO did not affect the activity of promoters of non-targeted genes, although they had significant structural similarity to the Ets2 promoter. Expression of non-targeted genes, such as Ets1 and GAPDH, was also not affected by the Ets2-TFO. Together, these data suggest that binding of the TFO to non-targeted DNA sites might be an unlikely event in the absence of a perfectly matching sequence. The nuclease resistance and high binding affinity of the PS-modified TFO was certainly important in these cellular studies. Intracellular accumulation of PS oligonucleotides is greatly enhanced compared with PO oligonucleotides. As a result of the increased stability, intact TFO would be present in cells for a sustained period, thus favoring triplex formation. However, the Ets2-TFO appeared to be less active on the endogenous gene than on the promoter reporter construct when tested at similar concentrations and under similar conditions. This difference is not surprising and may be due to various factors. Accessibility of a chromosomal target site may be restricted and depend on the transcriptional state of the gene. Faria et al. (22) reported significant variability in gene expression levels in single cells over time and markedly different temporal responses to a TFO among individual cells in an unsynchronized cell population. Indeed, the endogenous Ets2 promoter may be accessible to both regulatory proteins and TFO only in cells that are actively transcribing the gene. Thus, restricted accessibility of the endogenous gene promoter combined with heterogeneity of cellular and nuclear uptake of the TFO among cells may have contributed to the apparently reduced efficacy of the Ets2-TFO in inhibiting the endogenous gene.
In the present study, we have shown that transcription inhibition by a TFO targeting the Ets2 promoter was sequence specific and triplex mediated. A concern that we had carrying out these studies was that PS-modified oligonucleotides were known to cause non-specific effects, particularly when used at high concentration (28). An additional concern was the tendency of G-rich oligonucleotides to form self-aggregates (15). Formation of homoduplex and tetraplex structures has been suggested as a possible source of non-antisense or non-triplex mediated effects of G-rich oligonucleotides (24,26,27). Therefore, in this study we used multiple experimental controls to demonstrate the specificity of the TFO effects. These included: (i) mismatched oligonucleotides that conserved nucleotide content and critical sequence elements of the TFO; (ii) oligonucleotide targets and promoter reporter constructs with mutations that prevented binding of the TFO; and (iii) promoter reporter constructs from non-targeted genes that had similar purine-rich tracts and transcription factor binding sites. Control mismatched oligonucleotides ruled out non-sequence-dependent effects that might be due to chemical or nucleotide composition of the TFO. The other experimental controls were important to exclude sequence-dependent, but non triplex-mediated effects of the TFO. Using this combined approach we have shown triplex-mediated inhibition of transcription initiation by a PS-modified GT-rich TFO both in a cell-free system and in cells. Our data suggest that high-affinity and nuclease-resistant TFOs can be used as sequence-specific DNA ligands and gene-specific transcriptional repressors in cells.
Acknowledgments
ACKNOWLEDGEMENTS
We acknowledge the assistance of the Biomolecular Computing Resource Facility and DNA sequencing Facility of the Medical University of South Carolina. This work was supported by NIH grant CA-70735 (to C.V.C.) and a postdoctoral award from US Army Breast Cancer Research Program (to E.M.M.).
REFERENCES
- 1.Sementchenko V.I. and Watson,D.K. (2000) Ets target genes: past, present and future. Oncogene, 19, 6533–6548. [DOI] [PubMed] [Google Scholar]
- 2.Yang B.S., Hauser,C.A., Henkel,G., Colman,M.S., Van Beveren,C., Stacey,K.J., Hume,D.A., Maki,R.A. and Ostrowski,M.C. (1996) Ras-mediated phosphorylation of a conserved threonine residue enhances the transactivation activities of c-Ets1 and c-Ets2. Mol. Cell. Biol., 16, 538–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McCarthy S.A., Chen,D., Yang,B.S., Garcia Ramirez,J.J., Cherwinski,H., Chen,X.R., Klagsbrun,M., Hauser,C.A., Ostrowski,M.C. and McMahon,M. (1997) Rapid phosphorylation of Ets-2 accompanies mitogen-activated protein kinase activation and the induction of heparin-binding epidermal growth factor gene expression by oncogenic Raf-1. Mol. Cell. Biol., 17, 2401–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gioeli D., Mandell,J.W., Petroni,G.R., Frierson,H.F.,Jr and Weber,M.J. (1999) Activation of mitogen-activated protein kinase associated with prostate cancer progression. Cancer Res., 59, 279–284. [PubMed] [Google Scholar]
- 5.Seth A., Watson,D.K., Blair,D.G. and Papas,T.S. (1989) c-ets-2 protooncogene has mitogenic and oncogenic activity. Proc. Natl Acad. Sci. USA, 86, 7833–7837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seth A. and Papas,T.S. (1990) The c-ets-1 proto-oncogene has oncogenic activity and is positively autoregulated. Oncogene, 5, 1761–1767. [PubMed] [Google Scholar]
- 7.Wasylyk C., Maira,S.M., Sobieszczuk,P. and Wasylyk,B. (1994) Reversion of Ras transformed cells by Ets transdominant mutants. Oncogene, 9, 3665–3673. [PubMed] [Google Scholar]
- 8.Galang C.K., Der,C.J. and Hauser,C.A. (1994) Oncogenic Ras can induce transcriptional activation through a variety of promoter elements, including tandem c-Ets-2 binding sites. Oncogene, 9, 2913–2921. [PubMed] [Google Scholar]
- 9.Gilliland D.G. (2001) The diverse role of the ETS family of transcription factors in cancer. Clin. Cancer Res., 7, 451–453. [PubMed] [Google Scholar]
- 10.Liu A.Y., Corey,E., Vessella,R.L., Lange,P.H., True,L.D., Huang,G.M., Nelson,P.S. and Hood,L. (1997) Identification of differentially expressed prostate genes: increased expression of transcription factor ETS-2 in prostate cancer. Prostate, 30, 145–153. [DOI] [PubMed] [Google Scholar]
- 11.Sementchenko V.I., Schweinfest,C.W., Papas,T.S. and Watson,D.K. (1998) ETS2 function is required to maintain the transformed state of human prostate cancer cells. Oncogene, 17, 2883–2888. [DOI] [PubMed] [Google Scholar]
- 12.Sapi E., Flick,M.B., Rodov,S. and Kacinski,B.M. (1998) Ets-2 transdominant mutant abolishes anchorage-independent growth and macrophage colony-stimulating factor-stimulated invasion by BT20 breast carcinoma cells. Cancer Res., 58, 1027–1033. [PubMed] [Google Scholar]
- 13.Neznanov N., Man,A.K., Yamamoto,H., Hauser,C.A., Cardiff,R.D. and Oshima,R.G. (1999) A single targeted Ets2 allele restricts development of mammary tumors in transgenic mice. Cancer Res., 59, 4242–4246. [PubMed] [Google Scholar]
- 14.de Nigris F., Mega,T., Berger,N., Barone,M.V., Santoro,M., Viglietto,G., Verde,P. and Fusco,A. (2001) Induction of ETS-1 and ETS-2 transcription factors is required for thyroid cell transformation. Cancer Res., 61, 2267–2275. [PubMed] [Google Scholar]
- 15.Chan P.P. and Glazer,P.M. (1997) Triplex DNA: fundamentals, advances, and potential applications for gene therapy. J. Mol. Med., 75, 267–282. [DOI] [PubMed] [Google Scholar]
- 16.Praseuth D., Guieysse,A.L. and Helene,C. (1999) Triple helix formation and the antigene strategy for sequence-specific control of gene expression. Biochim. Biophys. Acta, 1489, 181–206. [DOI] [PubMed] [Google Scholar]
- 17.Winters T.A. (2000) Gene targeted agents: new opportunities for rational drug development. Curr. Opin. Mol. Ther., 2, 670–681. [PubMed] [Google Scholar]
- 18.Wang G., Seidman,M.M. and Glazer,P.M. (1996) Mutagenesis in mammalian cells induced by triple helix formation and transcription-coupled repair. Science, 271, 802–805. [DOI] [PubMed] [Google Scholar]
- 19.Giovannangeli C., Diviacco,S., Labrousse,V., Gryaznov,S., Charneau,P. and Helene,C. (1997) Accessibility of nuclear DNA to triplex-forming oligonucleotides: the integrated HIV-1 provirus as a target. Proc. Natl Acad. Sci. USA, 94, 79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oh D.H. and Hanawalt,P.C. (1999) Triple helix-forming oligonucleotides target psoralen adducts to specific chromosomal sequences in human cells. Nucleic Acids Res., 27, 4734–4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vasquez K.M., Narayanan,L. and Glazer,P.M. (2000) Specific mutations induced by triplex-forming oligonucleotides in mice. Science, 290, 530–533. [DOI] [PubMed] [Google Scholar]
- 22.Faria M., Wood,C.D., Perrouault,L., Nelson,J.S., Winter,A., White,M.R., Helene,C. and Giovannangeli,C. (2000) Targeted inhibition of transcription elongation in cells mediated by triplex-forming oligonucleotides. Proc. Natl Acad. Sci. USA, 97, 3862–3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lebedeva I. and Stein,C.A. (2001) Antisense oligonucleotides: promise and reality. Annu. Rev. Pharmacol. Toxicol., 41, 403–419. [DOI] [PubMed] [Google Scholar]
- 24.Bates P.J., Kahlon,J.B., Thomas,S.D., Trent,J.O. and Miller,D.M. (1999) Antiproliferative activity of G-rich oligonucleotides correlates with protein binding. J. Biol. Chem., 274, 26369–26377. [DOI] [PubMed] [Google Scholar]
- 25.Scaggiante B., Morassutti,C., Dapas,B., Tolazzi,G., Ustulin,F. and Quadrifoglio,F. (1998) Human cancer cell lines growth inhibition by GTn oligodeoxyribonucleotides recognizing single-stranded DNA-binding proteins. Eur. J. Biochem., 252, 207–215. [DOI] [PubMed] [Google Scholar]
- 26.Benimetskaya L., Berton,M., Kolbanovsky,A., Benimetsky,S. and Stein,C.A. (1997) Formation of a G-tetrad and higher order structures correlates with biological activity of the RelA (NF-kappaB p65) ‘antisense’ oligodeoxynucleotide. Nucleic Acids Res., 25, 2648–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burgess T.L., Fisher,E.F., Ross,S.L., Bready,J.V., Qian,Y.X., Bayewitch,L.A., Cohen,A.M., Herrera,C.J., Hu,S.S., Kramer,T.B. et al. (1995) The antiproliferative activity of c-myb and c-myc antisense oligonucleotides in smooth muscle cells is caused by a nonantisense mechanism. Proc. Natl Acad. Sci. USA, 92, 4051–4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crooke S.T. and Bennett,C.F. (1996) Progress in antisense oligonucleotide therapeutics. Annu. Rev. Pharmacol. Toxicol., 36, 107–129. [DOI] [PubMed] [Google Scholar]
- 29.Mavrothalassitis G.J., Watson,D.K. and Papas,T.S. (1990) The human ETS-2 gene promoter: molecular dissection and nuclease hypersensitivity. Oncogene, 5, 1337–1342. [PubMed] [Google Scholar]
- 30.Mavrothalassitis G.J., Watson,D.K. and Papas,T.S. (1990) Molecular and functional characterization of the promoter of ETS2, the human c-ets-2 gene. Proc. Natl Acad. Sci. USA, 87, 1047–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mavrothalassitis G.J. and Papas,T.S. (1991) Positive and negative factors regulate the transcription of the ETS2 gene via an oncogene-responsive-like unit within the ETS2 promoter region. Cell Growth Differ., 2, 215–224. [PubMed] [Google Scholar]
- 32.McGuffie E.M. and Catapano,C.V. (2002) Design of a novel triple helix-forming oligodeoxyribonucleotide directed to the major promoter of the c-myc gene. Nucleic Acids Res., 30, 2701–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Catapano C.V., McGuffie,E.M., Pacheco,D. and Carbone,G.M. (2000) Inhibition of gene expression and cell proliferation by triple helix-forming oligonucleotides directed to the c-myc gene. Biochemistry, 39, 5126–5138. [DOI] [PubMed] [Google Scholar]
- 34.Bonham K. and Fujita,D.J. (1993) Organization and analysis of the promoter region and 5′ non-coding exons of the human c-src proto-oncogene. Oncogene, 8, 1973–1981. [PubMed] [Google Scholar]
- 35.Durland R.H., Kessler,D.J., Gunnell,S., Duvic,M., Pettitt,B.M. and Hogan,M.E. (1991) Binding of triple helix forming oligonucleotides to sites in gene promoters. Biochemistry, 30, 9246–9255. [DOI] [PubMed] [Google Scholar]
- 36.Tu G.C., Cao,Q.N. and Israel,Y. (1995) Inhibition of gene expression by triple helix formation in hepatoma cells. J. Biol. Chem., 270, 28402–28407. [DOI] [PubMed] [Google Scholar]
- 37.Aggarwal B.B., Schwarz,L., Hogan,M.E. and Rando,R.F. (1996) Triple helix-forming oligodeoxyribonucleotides targeted to the human tumor necrosis factor (TNF) gene inhibit TNF production and block the TNF-dependent growth of human glioblastoma tumor cells. Cancer Res., 56, 5156–5164. [PubMed] [Google Scholar]
- 38.McGuffie E.M., Pacheco,D., Carbone,G.M. and Catapano,C.V. (2000) Antigene and antiproliferative effects of a c-myc-targeting phosphorothioate triple helix-forming oligonucleotide in human leukemia cells. Cancer Res., 60, 3790–3799. [PubMed] [Google Scholar]
- 39.Hacia J.G., Wold,B.J. and Dervan,P.B. (1994) Phosphorothioate oligonucleotide-directed triple helix formation. Biochemistry, 33, 5367–5369. [DOI] [PubMed] [Google Scholar]
- 40.Kim H.G., Reddoch,J.F., Mayfield,C., Ebbinghaus,S., Vigneswaran,N., Thomas,S., Jones,D.E.,Jr and Miller,D.M. (1998) Inhibition of transcription of the human c-myc protooncogene by intermolecular triplex. Biochemistry, 37, 2299–2304. [DOI] [PubMed] [Google Scholar]
- 41.Black A.R., Black,J.D. and Azizkhan-Clifford,J. (2001) Sp1 and kruppel-like factor family of transcription factors in cell growth regulation and cancer. J. Cell Physiol., 188, 143–160. [DOI] [PubMed] [Google Scholar]
- 42.Ritchie S., Boyd,F.M., Wong,J. and Bonham,K. (2000) Transcription of the human c-Src promoter is dependent on Sp1, a novel pyrimidine binding factor SPy, and can be inhibited by triplex-forming oligonucleotides. J. Biol. Chem., 275, 847–854. [DOI] [PubMed] [Google Scholar]
- 43.Roussel M.F., Davis,J.N., Cleveland,J.L., Ghysdael,J. and Hiebert,S.W. (1994) Dual control of myc expression through a single DNA binding site targeted by ets family proteins and E2F-1. Oncogene, 9, 405–415. [PubMed] [Google Scholar]
- 44.Michelotti E.F., Tomonaga,T., Krutzsch,H. and Levens,D. (1995) Cellular nucleic acid binding protein regulates the CT element of the human c-myc protooncogene. J. Biol. Chem., 270, 9494–9499. [DOI] [PubMed] [Google Scholar]
- 45.Kinniburgh A.J., Firulli,A.B. and Kolluri,R. (1994) DNA triplexes and regulation of the c-myc gene. Gene, 149, 93–100. [DOI] [PubMed] [Google Scholar]
- 46.Simonsson T., Pecinka,P. and Kubista,M. (1998) DNA tetraplex formation in the control region of c-myc. Nucleic Acids Res., 26, 1167–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]