

Abstract
Over the past two decades, the incidence of infections due to Candida glabrata, a yeast with intrinsic low susceptibility to azole antifungals, has increased markedly. Respiratory deficiency due to mutations in mitochondrial DNA (mtDNA) associated with resistance to azoles frequently occurs in vitro in this species. In order to specify the relationships between respiration and azole susceptibility, the effects of respiratory chain inhibitors on a wild-type isolate of C. glabrata were evaluated. Respiration of blastoconidia was immediately blocked after extemporaneous addition of potassium cyanide, whereas a 4-h preincubation was required for sodium azide. Antifungal susceptibility determined by a disk diffusion method on Casitone agar containing sodium azide showed a significant decrease in the susceptibility to azoles. Biweekly subculturing on Casitone agar supplemented with sodium azide was therefore performed. This resulted after 40 passages in the isolation of a respiration-deficient mutant, as suggested by its lack of growth on glycerol-containing agar. This respiratory deficiency was confirmed by flow cytometric analysis of blastoconidia stained with rhodamine 123 and by oxygraphy. Moreover, transmission electron microscopy and restriction endonuclease analysis of the mtDNA of mutant cells demonstrated the mitochondrial origin of the respiratory deficiency. Finally, this mutant exhibited cross-resistance to all the azoles tested. In conclusion, blockage of respiration in C. glabrata induces decreased susceptibility to azoles, culminating in azole resistance due to the deletion of mtDNA. This mechanism could explain the induction of petite mutations by azole antifungals which have been demonstrated to act directly on the mitochondrial respiratory chain.
With the development of antibiotic treatments, the widespread use of immunosuppressive therapy, and the emergence of the AIDS epidemic, the incidence of life-threatening fungal infections has increased significantly worldwide over the past two decades (3). The major agents of fungal infections are Candida species, and among them, Candida albicans is the most frequent, representing about 50% to 60% of overall yeast isolates (19). However, as a consequence of the resulting extensive use of antifungal agents, a shift in the nature of the infecting organisms has been reported (17). C. albicans remains the most frequent causative agent, but infections due to other yeast species such as Candida glabrata and Candida krusei are increasingly reported (20).
Azole antifungals are widely used in current therapies against these infections. Fluconazole, a water-soluble triazole with greater than 90% bioavailability after oral administration, has been used extensively in prophylaxis and therapy of candidosis in organ and bone marrow transplant recipients, patients undergoing chemotherapy, and AIDS patients (4, 23). However, fluconazole-resistant isolates of C. albicans have been reported increasingly since 1990 in many immunocompromised and immunosuppressed patients (17). Moreover, it is now well established that the prolonged use of ketoconazole or fluconazole may give rise to the emergence of C. glabrata infections (4, 17, 20). Thus, C. glabrata, which was for a long time the second most frequent yeast in candiduria and vulvovaginal candidosis, has also become the second most frequently isolated yeast species in candidemia and systemic mycoses (10). For instance, its frequency in candidemia increased from 2% in 1987 to 26% in 1992 in the United States (22) and from 6% in 1987 to 17% in 1995 in the Netherlands (36).
In a recent study of the Sentry Program realized in the United States, Canada, Latin America, and Europe, 55% of the yeast bloodstream infections were due to C. albicans, followed by C. glabrata and C. parapsilosis, each accounting for 15% of these mycoses (19). Likewise, similar frequencies in candidemia have been reported in Alabama over a 6-year period (1995 to 2000) for C. albicans and C. glabrata (50% and 24%, respectively) with 10% of fluconazole-resistant C. glabrata isolates (2).
Different mechanisms have been suggested to be involved in resistance of clinical yeast isolates to azole antifungal agents. Azole resistance may result from an increased cellular content of the azole target Erg11p, a hemoprotein supporting lanosterol 14α-demethylase activity (15, 27, 38), or from a reduced affinity of azoles for Erg11p because of point mutations in the corresponding ERG11 gene (14, 25, 35, 39). More frequently, resistance has been mediated by overexpression of genes encoding efflux pumps that reduce intracellular drug accumulation (16, 21, 26, 27, 38). In addition, alterations of the membrane sterol composition have been described in some resistant isolates (12).
Concerning C. glabrata, we showed previously that mutations in mitochondrial DNA (mtDNA) frequently occur in vitro, leading to a respiratory deficiency associated with resistance to all the azole compounds tested except tioconazole (6). Moreover, petite mutants were obtained from a wild-type strain by exposure to ethidium bromide, and these mutants were shown to be resistant to azoles. The present study was therefore conducted to explore the relationships between respiration and azole susceptibility.
MATERIALS AND METHODS
Yeast isolates and culture conditions.
The clinical isolate C. glabrata 90.1085, obtained from a urine sample and cloned by limiting dilution, was used throughout. This isolate was maintained by biweekly passages at 37°C on yeast extract-peptone-glucose (YEPD) agar, containing, in grams per liter, yeast extract, 5; peptone, 10; glucose, 20; chloramphenicol, 1; and agar, 20.
The isolate was also subcultured twice a week on Casitone agar plates (Bacto-Casitone, 9 g/liter; glucose, 20 g/liter; chloramphenicol, 0.5 g/liter; yeast extract, 5 g/liter; and agar, 18 g/liter [pH 7.2]) supplemented with 1 mM sodium azide (Sigma Chemical Co., St. Louis, Mo.). Every five passages, growth on yeast extract-peptone agar containing glycerol 2% as the sole carbon source and susceptibility to azole antifungals were evaluated.
Antifungal susceptibility testing. (i) Disk diffusion method.
Antifungal susceptibility was determined by a disk diffusion method on Casitone agar with antifungal Neosensitab tablets from Rosco Diagnostica (Taastrup, Denmark) as described previously (6). Briefly, inoculum in sterile distilled water was prepared from fresh cultures on YEPD agar, and 10 ml of the fungal suspensions was deposited onto Casitone agar plates. Excess fluid was removed immediately, and the plates were dried for 15 min at 37°C. Antifungal Neosensitab tablets (containing 8 μg of drug for itraconazole; 10 μg for amphotericin B, clotrimazole, econazole, isoconazole, and miconazole; 15 μg for ketoconazole; and 50 μg for nystatin) were then applied to the surface. Following a preincubation period of 30 min at room temperature, plates were incubated for 48 h at 37°C, and the diameter of the inhibition zones was measured.
(ii) Etest procedure.
Susceptibility to fluconazole, ketoconazole, and amphotericin B was also determined by measuring the MICs of the antifungal agents by the Etest procedure, performed as recommended by the manufacturer (AB Biodisk, Solna, Sweden). In this assay, one or two colonies from fresh cultures on YEPD agar plates were suspended in 2 ml of 0.15 M phosphate-buffered saline (PBS, pH 7.2) to achieve a turbidity comparable to a 0.5 McFarland standard. The inoculum was swabbed onto Casitone agar plates, which were allowed to dry for 15 min before the Etest strips were applied. Plates were incubated at 37°C, and MICs were read after 48 h as the drug concentration at which the inhibition ellipse intercepted the scale on the antifungal strips.
Respiration measurements.
Exogenous respiration of the cells was determined with stationary-growth-phase blastoconidia (48 h at 37°C) from YEPD agar plates. After washing in sterile distilled water and enumeration by hemacytometer counts, cells were suspended in 10 mM potassium phosphate buffer (pH 7.4) containing 20 mM glucose to give a cell density of 109 cells/ml. Oxygen consumption was measured at 37°C with an Oxytherm oxygraph (Hansatech Instruments Ltd., Norfolk, England). Initially, the respiration of approximately 5 × 107 cells in a 1-ml final volume was measured, and the effect of the addition of potassium cyanide or sodium azide at a final concentration of 1 mM was evaluated. Respiration rates are expressed as nanomoles of oxygen consumed per milliliter per minute.
Flow cytometry.
The respiratory status of the cells was also investigated by flow cytometry after staining with rhodamine 123 (30). Blastoconidia grown to stationary phase in YEPD broth were harvested by centrifugation and washed in PBS. Cells (2 × 106/ml) were then incubated with rhodamine 123 (Sigma) at a final concentration of 10 μg/ml for 30 min at 37°C under constant shaking. To inhibit the electron flow of the respiratory chain, cell suspensions were first incubated for 2 h at 37°C with 1 mM sodium azide before the addition of rhodamine 123. After washing in PBS, the cell fluorescence was quantified with a FACScan flow cytometer (BDIS Europe, Erembodegem, Belgium) equipped with an air-cooled 15-mW argon-ion laser operating at 488 nm. Cell debris were excluded by gating the blastoconidia on the basis of their rectilinear forward and side light-scattering properties. The fluorescence intensity was quantified at 535 nm. Each sample was collected in a list mode file of 10,000 events, and data were analyzed with the CellQuest software from BDIS. The data presented correspond to fluorescence frequency distribution histograms (relative number of blastoconidia versus relative fluorescence intensity expressed in arbitrary units on a logarithmic scale).
Transmission electron microscopy.
Samples for electron microscopy were prepared by the method of Aoki et al. (1) with minor modifications. Blastoconidia grown on YEPD agar plates were washed with PBS and fixed for 1 h at room temperature with 2.5% glutaraldehyde (Merck, Darmstadt, Germany) buffered at pH 7.2 with 0.1 M sodium cacodylate (Sigma). Cells were then washed with cacodylate buffer and fixed with1.5% KMnO4 (Sigma) for 18 h at 4°C. After washing, the cells were postfixed with osmium tetroxide (SPI Supplies, West Chester, Pa.) 1% for 2 h at room temperature. Samples were dehydrated with a graded series of ethanol and embedded in Epon (SPI). Ultrathin sections were stained with uranyl acetate and examined with a 100 CX JEOL microscope (Jeol, Paris, France).
mtDNA extraction and analysis.
mtDNA was extracted according to a previously described technique (6), slightly modified. Blastoconidia from 48-h-old cultures in YEPD broth were pelleted at 500 × g for 5 min. The pellets were washed twice in sterile distilled water and resuspended in 10 ml of buffer A (0.5 M sorbitol, 10 mM EDTA, 50 mM Tris-HCl [pH 7.6]) containing 1 mg of Zymolyase 100 T (Seikagaku Corporation, Tokyo, Japan) per ml and 0.2% β-mercaptoethanol. After incubation at 37°C for 1.5 h under constant shaking, the cellular lysate was transferred to sterile microtubes and centrifuged at 1,000 × g for 10 min. The supernatant containing the mtDNA was then centrifuged at 10,000 × g for 10 min, and the mitochondrial pellet was resuspended in 500 μl of buffer A and deposited on a 0 to 40% discontinuous Percoll gradient. After centrifugation at 3,000 × g for 10 min, mitochondria found at the 0 to 20% interface of the Percoll gradient were then washed four times with buffer A. The resulting pellet was lysed for 30 min at room temperature by the addition of 600 μl of a 50 mM Tris-HCl buffer, pH 7.6, containing 100 mM NaCl, 10 mM EDTA, and 1% Sarkosyl. Nucleic acids were extracted by phenol-chloroform, precipitated by isopropanol, and then resuspended in sterile distilled water. RNAs were eliminated by the addition of DNase-free RNase.
mtDNA was digested with EcoRV (Roche Diagnostics GmbH, Mannheim, Germany) at 37°C for 2 h in a 20-μl volume with 1 U of the enzyme. Finally, the digestion products were analyzed by 0.8% agarose gel electrophoresis in TAE buffer (40 mM Tris acetate, 1 mM EDTA, pH 8.0), stained with ethidium bromide, and visualized under UV illumination.
RESULTS
Influence of respiratory chain inhibitors. (i) Oxygen consumption.
Initially, exogenous oxygen consumption by blastoconidia of the clinical isolate 90.1085 was measured at 37°C on 5 × 107 cells. Then an inhibitor of the cytochrome oxidase, potassium cyanide or sodium azide, was added at a final concentration of 1 mM. The results are shown in Fig. 1A. Cells without inhibitors of the respiratory chain consumed the oxygen in about 7 min. Oxygen consumption was immediately blocked by the addition of potassium cyanide, whereas sodium azide had a minor effect, with a respiration rate of 27.6 nmol ml−1 min−1 versus 39.2 nmol ml−1 min−1 for the control.
FIG. 1.

Oxygen consumption by parent blastoconidia. (A) Effect of extemporaneous addition (arrow) of 1 mM potassium cyanide (dashed line) or sodium azide (dotted line). (B) Effect of a 2-h (dashed line) or 4-h (dotted line) preincubation of the cells with 1 mM sodium azide. The continuous line represents oxygen consumption by the cells in the absence of an inhibitor.
As extemporaneous addition of sodium azide only slightly modified the respiration, the cells were also preincubated with this inhibitor. Under these conditions, sodium azide led to a reduction in respiration that was even more pronounced when the incubation time was longer (Fig. 1B). Indeed, a marked decrease in the respiration rate was observed after a 2-h incubation of the cells with sodium azide (9.4 nmol ml−1 min−1 versus 20 nmol ml−1 min−1 for untreated cells), and the inhibition of respiration became almost complete when preincubation was prolonged to 4 h.
(ii) Antifungal susceptibility.
The influence of potassium cyanide and sodium azide on antifungal susceptibility was evaluated by an agar disk diffusion method with Neosensitab tablets. Ten colonies of the clinical isolate 90.1085 were tested on Casitone agar plates containing or lacking the respiratory inhibitor (final concentration, 1 mM). The addition of potassium cyanide to the culture medium did not modify the antifungal susceptibility. This may be related to the persistence of respiration, as attested by growth on glycerol-containing agar supplemented with 1 mM potassium cyanide. Conversely, the addition of sodium azide, which completely inhibited growth on glycerol-containing agar, resulted in a significant decrease in the diameters of the inhibition zones for all the azoles tested, with a concomitant increased susceptibility to polyenes (Table 1).
TABLE 1.
Effects of sodium azide on azole susceptibility of the parent isolate
| Antifungal agent | Mean diameter (mm) of inhibition zonea (SD)
|
|
|---|---|---|
| Control | Sodium azide-containing agar | |
| Amphotericin B | 28.6 (1.6) | 36.8 (1.5) |
| Nystatin | 28.2 (1.5) | 37.7 (1.7) |
| Isoconazole | 23.0 (1.4) | 17.8 (2.0) |
| Miconazole | 28.6 (1.0)∗ | 26.8 (1.4)* |
| Clotrimazole | 30.6 (1.3) | 26.4 (1.6) |
| Ketoconazole | 34.8 (1.0) | 29.8 (1.5) |
| Fluconazole | 29.4 (1.3) | 21.6 (1.3) |
| Itraconazole | 21.4 (1.3) | 17.4 (1.3) |
Results correspond to the mean diameter of the inhibition zones obtained for 10 colonies. Data were analyzed with Student's t test. P < 0.001 except * (P = 0.01).
Subcultures on Casitone agar containing sodium azide.
Considering the decreased azole susceptibility of the clinical isolate in the presence of sodium azide, biweekly subcultures were performed on Casitone agar plates containing 1 mM sodium azide. After 40 passages, a mutant designated P40, which did not grow on glycerol-containing agar, suggesting a respiratory deficiency, was isolated.
Evidence for respiratory deficiency of mutant P40. (i) Flow cytometry.
Flow cytometric analysis of cells stained with rhodamine 123, a lipophilic cationic fluorochrome which is incorporated into mitochondria through a transmembrane potential-dependent mechanism, further supported evidence for the respiratory deficiency of mutant P40. Indeed, incubation of blastoconidia with this mitochondrial marker resulted in a high fluorescence intensity for the parent isolate, and a significant reduction of this fluorescence was observed when cells were pretreated with the respiratory chain inhibitor sodium azide (Fig. 2A). Conversely, cells of the mutant were poorly stained by the fluorochrome, and their fluorescence intensity was not affected by their pretreatment with sodium azide (Fig. 2B). Analysis of the cells incubated without rhodamine 123 attested to the absence of autofluorescence.
FIG. 2.

Flow cytometry of rhodamine 123-stained parent (A) and mutant P40 (B) cells. Yeast cells were preincubated (thick line) or not (grey area) with 1 mM sodium azide before rhodamine 123 staining. The fluorescence of cells incubated without the fluorochrome (black area) is presented as a control.
(ii) Oxygraphy.
Comparison of exogenous respiration of parent and mutant blastoconidia confirmed the respiratory deficiency. Figure 3 shows that mutant cells did not respire, whereas parent blastoconidia consumed the oxygen present in the medium in 10 min.
FIG. 3.

Oxygen consumption by parent (solid line) and mutant P40 (dashed line) blastoconidia. Comparison of the respiration rates showed a marked decrease in oxygen consumption by the mutant cells (1.5 nmol ml−1 min−1 versus 27.3 nmol ml−1 min−1 for the parent cells).
Mitochondrial origin of the respiratory deficiency.
Transmission electron microscopy revealed the mitochondrial origin of the respiratory deficiency of mutant P40. Indeed, numerous mitochondrial sections with obvious cristae were present in the blastoconidia of the parent isolate (Fig. 4A, arrowheads), whereas mutant cells exhibited no mitochondria (Fig. 4B).
FIG. 4.

Transmission electron micrographs of parent (A) and mutant P40 (B) cells. Note the absence of mitochondria in the respiration-deficient blastoconidia compared to the numerous mitochondrial sections with obvious cristae (arrowheads) in the parent cells. N, nucleus. Bars, 1 μm.
The mitochondrial origin of the respiratory deficiency was finally confirmed by mtDNA analysis. Great differences were observed between parent and mutant cells. Indeed, restriction endonuclease analysis of the mtDNA performed with EcoRV resulted in two restriction fragments of about 20 and 5 kbp for the respiration-competent isolate (Fig. 5, lane 1), while no mtDNA was detected for blastoconidia of the respiration-deficient mutant (Fig. 5, lane 2).
FIG. 5.
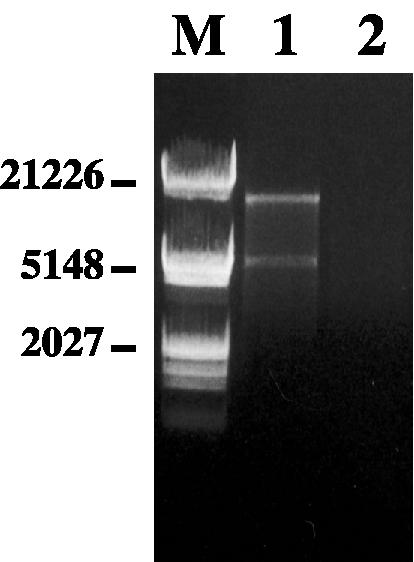
Electrophoretic patterns of mtDNA of parent (lane 1) and mutant P40 (lane 2) cells. mtDNA was analyzed by agarose gel electrophoresis after digestion with EcoRV. Lane M, molecular size markers (Marker III; Roche Molecular Biochemicals, Meylan, France). Sizes are shown in base pairs.
Antifungal susceptibility pattern of mutant P40. (i) Disk diffusion method.
In vitro antifungal susceptibility testing performed by the agar disk diffusion method revealed distinct susceptibility patterns for the parent and mutant isolates. The parent isolate, which showed well-defined inhibition zones, was sensitive to all the antifungals tested (Fig. 6A). However, resistant colonies randomly distributed within the inhibition zones were seen for the azoles, some even growing in direct contact with the antifungal tablet. Conversely, mutant P40 was completely resistant to all the azole compounds tested and strikingly remained sensitive to polyenes, with larger inhibition zones than the parent isolate (Fig. 6B).
FIG. 6.

Antifungal susceptibility of parent (A) and mutant P40 (B) cells. Susceptibility testing was performed by the disk diffusion method on Casitone agar plates with Neosensitab tablets. 1, amphotericin B; 2, nystatin; 3, miconazole; 4, ketoconazole; 5, clotrimazole; 6, fluconazole; and 7, econazole.
(ii) MIC determination.
Determination of MICs by the Etest method confirmed these results. Mutant P40 exhibited complete resistance to the azoles tested (ketoconazole MIC, >32 μg/ml, and fluconazole MIC, >256 μg/ml, versus 0.5 and 32 μg/ml, respectively, for the parent isolate). Likewise, the mutant showed a greater susceptibility to amphotericin B, with a MIC of 0.008 μg/ml, whereas it was 0.032 μg/ml for the parent isolate.
DISCUSSION
Antifungal resistance mechanisms have been extensively studied in the pathogenic yeast C. albicans. However, as a consequence of the wide use of azole antifungals in prophylaxis and therapy of candidosis, other yeast species have emerged in medical pathology, particularly C. glabrata, which is usually less susceptible and sometimes resistant to azoles.
Previous in vitro experiments performed in our laboratory established a close relationship between azole resistance in C. glabrata and mtDNA deficiency (6). All the mutants randomly selected for resistance to fluconazole or ketoconazole proved to be completely resistant to all the azoles tested except tioconazole and exhibited respiratory deficiency due to total or partial deletion of the mtDNA. Likewise, petite mutants induced by ethidium bromide were revealed to be completely resistant to the azoles except tioconazole.
In order to specify the relationships between respiration and azole susceptibility, we evaluated the respiration of a wild-type isolate of C. glabrata in the presence of different inhibitors of the mitochondrial respiratory chain and their effects on antifungal susceptibility. Potassium cyanide immediately blocked oxygen consumption by the blastoconidia but did not modify the antifungal susceptibility pattern. This may be related to its inactivation in agar-based media or to an alternative cyanide-insensitive respiratory pathway induced upon exposure of cells to this inhibitor, as occurs for C. albicans (28). Conversely, sodium azide completely inhibited respiration and reduced the susceptibility to azoles. Likewise, 40 biweekly passages of the wild-type isolate on Casitone agar plates supplemented with sodium azide resulted in a respiration-deficient mutant of mitochondrial origin, presenting resistance to all the azoles tested.
Cytoplasmic petite mutants of Saccharomyces cerevisiae have been known for many years (8), and more recently such mutants of C. glabrata and some petite negative yeasts such as C. albicans have been found. Interestingly, petite mutations are associated with a decreased susceptibility of yeasts to various chemicals, such as tannic acid or the alkaloid lycorine (7, 37). Likewise, Gyurko et al. (11) recently demonstrated that respiration-deficient mutants of C. albicans were resistant to histatin 5, a small cationic salivary antimicrobial peptide, mitochondrial ATP synthesis being required for effective killing activity of this salivary peptide.
Strikingly, petite mutations may also lead to acquired resistance to commercially available azole antifungals. For instance, fluconazole activity in S. cerevisiae requires the combination of an efficient Δ5,6-sterol desaturase (Erg3p) and intact mitochondria. Petite mutants of S. cerevisiae therefore present an acquired resistance to fluconazole involving an active oxidative phosphorylation, according to Kontoyiannis (13). In C. glabrata, Sanglard et al. (24) recently showed that the high frequency of azole resistance acquired in vitro after exposure to fluconazole was linked with mitochondrial loss and with the subsequent upregulation of the ATP-binding cassette transporter genes CDR1 and CDR2. Indeed, it is well known that the functional state of mitochondria influences nuclear gene expression in yeasts (18); for instance, petite mutations induce the overexpression of genes encoding membrane transporters in S. cerevisiae as well as in C. glabrata (24, 32).
In contrast to the cross-resistance to azoles, the respiration-deficient mutant described here exhibited a significant increase in susceptibility to polyenes, the diameter of the inhibition zones being statistically greater for the mutant than for the parent isolate. This greater susceptibility to polyenes, which is a common feature of all petite mutants of C. glabrata that we tested (6), may be related to an increased membrane content of ergosterol, the target of these antifungals. Sterol analysis of petite mutants selected by exposure to fluconazole or induced by ethidium bromide treatment revealed a marked increase in the ergosterol content compared to the parent strains (data not shown). However, one may also speculate on increased synthesis of ergosterol in response to the inhibition of respiration by sodium azide. Indeed, ERG11 is a member of the hypoxic gene family (33), and the synthesis of ergosterol is therefore enhanced under hypoxic conditions. Interestingly, previous studies reported increased expression of the ERG11 gene for azole-resistant isolates of C. glabrata and C. albicans (15, 38).
A wide range of chemical compounds have been described as inducers of the petite phenotype in S. cerevisiae (9). Most of them are effective petite mutagens on growing yeasts, inducing partial or total loss of the mitochondrial genome by intercalating into the mtDNA. Thus, petite mutants are easily obtained with ethidium bromide as well as propidium iodide or other intercalating agents such as acridine derivatives. However, intercalation is not necessary for petite mutagenesis, and some DNA-alkylating agents have also been used. Moreover, the induction of petite mutations may also result from the inhibition of protein synthesis. For example, chloramphenicol and erythromycin inhibit the translation and therefore the synthesis of proteins encoded by the mitochondrial genome by binding to the mitochondrial ribosomes of yeasts (40). This inhibition of protein synthesis by antibiotics, as well as the blockage of the enzyme activity of these proteins by sodium azide, would lead to the accumulation of reactive oxygen species in mitochondria and therefore to oxidant damage of the mtDNA.
Together, these results demonstrate a close relationship between respiration and azole susceptibility in C. glabrata, since the blockage of complex IV of the respiratory chain by sodium azide is sufficient to induce decreased susceptibility to azoles, probably through upregulation of the expression of nuclear genes encoding the efflux proteins. Likewise, exposure of the wild-type isolate to sodium azide leads to deletion of the mtDNA with a correlated acquired resistance to azoles after several passages. This mechanism could also explain petite mutagenesis due to azoles because a direct action of miconazole and ketoconazole on respiration has already been demonstrated in C. albicans with intact blastoconidia or isolated mitochondria (29, 31, 34).
The clinical relevance of these petite mutations, which may occur in vivo in patients undergoing fluconazole therapy or prophylaxis (5), remains to be defined. Works are in progress to specify the mechanisms of azole resistance of these petite mutants and to evaluate their adherence abilities and their virulence.
REFERENCES
- 1.Aoki, S., and S. Ito-Kuwa. 1987. Induction of petite mutation with acriflavine and elevated temperature in Candida albicans. J. Med. Vet. Mycol. 25:269-277. [DOI] [PubMed] [Google Scholar]
- 2.Baddley, J. W., A. M. Smith, S. A. Moser, and P. G. Pappas. 2001. Trends in frequency and susceptibilities of Candida glabrata bloodstream isolates at a university hospital. Diagn. Microbiol. Infect. Dis. 39:199-201. [DOI] [PubMed] [Google Scholar]
- 3.Beck-Sague, C. M., and W. R. Jarvis. 1993. Secular trends in the epidemiology of nosocomial fungal infections in the United States, 1980-1990. National Nosocomial Infections Surveillance System. J. Infect. Dis. 167:1247-1251. [DOI] [PubMed] [Google Scholar]
- 4.Bodey, G. P., M. Mardani, H. A. Hanna, M. Boktour, J. Abbas, E. Girgawy, R. Y. Hachem, D. P. Kontoyiannis, and I. I. Raad. 2002. The epidemiology of Candida glabrata and Candida albicans fungemia in immunocompromised patients with cancer. Am. J. Med. 112:380-385. [DOI] [PubMed] [Google Scholar]
- 5.Bouchara, J. P., R. Zouhair, S. Le Boudouil, G. Renier, R. Filmon, D. Chabasse, J. N. Hallet, and A. Defontaine. 2000. In vivo selection of an azole-resistant petite mutant of Candida glabrata. J. Med. Microbiol. 49:977-984. [DOI] [PubMed] [Google Scholar]
- 6.Defontaine, A., J. P. Bouchara, P. Declerk, C. Planchenault, D. Chabasse, and J. N. Hallet. 1999. In vitro resistance to azoles associated with mitochondrial DNA deficiency in Candida glabrata. J. Med. Microbiol. 48:663-670. [DOI] [PubMed] [Google Scholar]
- 7.Del Guidice, L., D. R. Massardo, F. Manna, A. Evidente, G. Randazzo, and K. Wolf. 1984. Differential effect of the alkaloid lycorine on rho+, mit−, rho− and rho0 strains of Saccharomyces cerevisiae. Curr. Genet. 8:493-497. [DOI] [PubMed] [Google Scholar]
- 8.Ephrussi, B., H. Hottinguer, and A.-M. Chimenes. 1949. Action de l'acriflavine sur les levures. I. La mutation “petite colonie.” Ann. Inst. Pasteur 76:351-367. [Google Scholar]
- 9.Ferguson, L. R., and R. C. von Borstel. 1992. Induction of the cytoplasmic “petite” mutation by chemical and physical agents in Saccharomyces cerevisiae. Mutat. Res. 265:103-148. [DOI] [PubMed] [Google Scholar]
- 10.Fidel, P. L., J. A. Vasquez, and J. D. Sobel. 1999. Candida glabrata: review of epidemiology, pathogenesis, and clinical disease with comparison to C. albicans. Clin. Microbiol. Rev. 12:80-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gyurko, C., U. Lendenmann, R. F. Troxler, and F. G. Oppenheim. 2000. Candida albicans mutants deficient in respiration are resistant to the small cationic salivary antimicrobial peptide histatin 5. Antimicrob. Agents Chemother. 44:348-354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kelly, S. L., D. C. Lamb, D. E. Kelly, N. J. Manning, J. Löffler, H. Hebart, U. Schumacher, and H. Einsele. 1997. Resistance to fluconazole and cross-resistance to amphotericin B in Candida albicans from AIDS patients caused by defective sterol Δ5,6-desaturation. FEBS Lett. 400:80-82. [DOI] [PubMed] [Google Scholar]
- 13.Kontoyiannis, D. P. 2000. Modulation of fluconazole sensitivity by the interaction of mitochondria and Erg3p in Saccharomyces cerevisiae. J. Antimicrob. Chemother. 46:191-197. [DOI] [PubMed] [Google Scholar]
- 14.Lamb, D. C., D. E. Kelly, W.-H. Schunck, A. Z. Shyadehi, M. Akhtar, D. J. Lowe, B. D. Baldwin, and S. L. Kelly. 1997. The mutation T315A in Candida albicans sterol 14α-demethylase causes reduced enzyme activity and fluconazole resistance through reduced affinity. J. Biol. Chem. 272:5682-5688. [DOI] [PubMed] [Google Scholar]
- 15.Marichal, P., H. Vanden Bossche, F. C. Odds, G. Nobel, D. W. Warnock, V. Timmerman, C. van Broeckhoven, Y. Fay, and P. Mose-Larsen. 1997. Molecular biological characterization of an azole-resistant Candida glabrata isolate. Antimicrob. Agents Chemother. 41:2229-2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miyazaki, H., Y. Miyazaki, A. Geber, T. Parkinson, C. Hitchcock, D. J. Falconer, D. J. Ward, K. Marsden, and J. E. Bennett. 1998. Fluconazole resistance associated with drug efflux and increased transcription of a drug transporter gene, PDH1, in Candida glabrata. Antimicrob. Agents Chemother. 42:1695-1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Odds, F. C. 1996. Epidemiological shifts in opportunistic and nosocomial Candida infections: mycological aspects. Int. J. Antimicrob. Agents 6:141-144. [DOI] [PubMed] [Google Scholar]
- 18.Parikh, V. S., M. M. Morgan, R. Scott, L. S. Clements, and R. A. Butow. 1987. The mitochondrial genome can influence nuclear gene expression in yeast. Science 235:576-580. [DOI] [PubMed] [Google Scholar]
- 19.Pfaller, M. A., D. J. Diekema, R. N. Jones, H. S. Sader, A. C. Fluit, R. J. Hollis, S. A. Messer, and the SENTRY Participant Group. 2001. International surveillance of bloodstream infections due to Candida species: frequency of occurrence and in vitro susceptibility to fluconazole, ravuconazole, and voriconazole of isolates collected from 1997 through 1999 in the SENTRY Antimicrobial Surveillance Program. J. Clin. Microbiol. 39:3254-3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pfaller, M. A., R. N. Jones, S. A. Messer, M. B. Edmond, and R. P. Wenzel. 1998. National surveillance of nosocomial blood stream infection due to species of Candida other than Candida albicans: frequency of occurrence and antifungal susceptibility in the SCOPE program. Diagn. Microbiol. Infect. Dis. 31:327-332. [DOI] [PubMed] [Google Scholar]
- 21.Prasad, R., P. De Wergifosse, A. Goffeau, and E. Balzi. 1995. Molecular cloning and characterization of a novel gene of Candida albicans, CDR1, conferring multiple resistance to drugs and antifungals. Curr. Genet. 27:320-329. [DOI] [PubMed] [Google Scholar]
- 22.Price, M. F., M. T. LaRocco, and L. O. Gentry. 1994. Fluconazole susceptibility of Candida species and distribution of species recovered from blood cultures over a 5-year period. Antimicrob. Agents Chemother. 38:1422-1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rex, J. H., M. A. Pfaller, A. L. Barry, P. W. Nelson, and C. D. Webb for the NIAID Mycoses Study Group and the Candidemia Study Group. 1995. Antifungal susceptibility testing of isolates from a randomized, multicenter trial of fluconazole versus amphotericin B as treatment of nonneutropenic patients with candidemia. Antimicrob. Agents Chemother. 39:40-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanglard, D., F. Ischer, and J. Bille. 2001. Role of ATP-binding-cassette transporter genes in high-frequency acquisition of resistance to azole antifungals in Candida glabrata. Antimicrob. Agents Chemother. 45:1174-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanglard, D., F. Ischer, L. Koymans, and J. Bille. 1998. Amino acid substitutions in the cytochrome P-450 lanosterol 14α-demethylase (CYP51A1) from azole-resistant Candida albicans clinical isolates contribute to resistance to azole antifungal agents. Antimicrob. Agents Chemother. 42:241-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanglard, D., F. Ischer, M. Monod, and J. Bille. 1997. Cloning of Candida albicans genes conferring resistance to azole antifungal agents: characterization of CDR2, a new multidrug ABC transporter gene. Microbiology 143:405-416. [DOI] [PubMed] [Google Scholar]
- 27.Sanglard, D., K. Kuchler, F. Ischer, J.-L. Pagani, M. Monod, and J. Bille. 1995. Mechanisms of resistance to azole antifungal agents in Candida albicans isolates from AIDS patients involve specific multidrug transporters. Antimicrob. Agents Chemother. 39:2378-2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shepherd, M. G., C. M. Chin, and P. A. Sullivan. 1978. The alternate respiratory pathway of Candida albicans. Arch. Microbiol. 116:61-67. [DOI] [PubMed] [Google Scholar]
- 29.Shigematsu, M. L., J. Uno, and T. Arai. 1982. Effect of ketoconazole on isolated mitochondria from Candida albicans. Antimicrob. Agents Chemother. 21:919-924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Skowronek, P., G. Krummeck, O. Haferkamp, and G. Rödel. 1990. Flow cytometry as a tool to discriminate respiratory-competent and respiratory-deficient yeast cells. Curr. Genet. 18:265-267. [DOI] [PubMed] [Google Scholar]
- 31.Sreedhara Swamy, K. H., M. Sirsi, and G. Ramanda Rao. 1974. Studies on the mechanism of action of miconazole: effect of miconazole on respiration and cell permeability of Candida albicans. Antimicrob. Agents Chemother. 5:420-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Traven, A., J. M. S. Wong, D. Xu, M. Sopta, and C. J. Ingles. 2001. Interorganellar communication: altered nuclear gene expression profiles in a yeast mitochondrial mutant. J. Biol. Chem. 276:4020-4027. [DOI] [PubMed] [Google Scholar]
- 33.Turi, T. G., and J. C. Loper. 1992. Multiple regulatory elements control expression of the gene encoding the Saccharomyces cerevisiae cytochrome P450, lanosterol 14α-demethylase (ERG11). J. Biol. Chem. 267:2046-2056. [PubMed] [Google Scholar]
- 34.Uno, J., M. L. Shigematsu, and T. Arai. 1982. Primary site of action of ketoconazole on Candida albicans. Antimicrob. Agents Chemother. 21:912-918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vanden Bossche, H., P. Marichal, J. Gorrens, D. Bellens, H. Moereels, and P. A. J. Janssen. 1990. Mutation in cytochrome P450-dependent 14α-demethylase results in decreased affinity for azole antifungals. Biochem. Soc. Trans. 18:56-59. [DOI] [PubMed] [Google Scholar]
- 36.Voss, A., J. A. Kluytmans, J. G. Koeleman, L. Spanjaard, C. M. Vandenbroucke-Grauls, H. A. Verbrugh, M. C. Vos, A. Y. Weersink, J. A. Hoogkamp-Korstanje, and J. F. Meis. 1996. Occurrence of yeast bloodstream infections between 1987 and 1995 in five Dutch university hospitals. Eur. J. Clin. Microbiol. Infect. Dis. 15:909-912. [DOI] [PubMed] [Google Scholar]
- 37.Wauters, T., D. Iserentant, and H. Verachtert. 2001. Impact of mitochondrial activity on the cell wall composition and on the resistance to tannic acid in Saccharomyces cerevisiae. J. Gen. Appl. Microbiol. 47:21-26. [DOI] [PubMed] [Google Scholar]
- 38.White, T. C. 1997. Increased mRNA levels of ERG16, CDR, and MDR1 correlate with increases in azole resistance in Candida albicans isolates from a patient infected with human immunodeficiency virus. Antimicrob. Agents Chemother. 41:1482-1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.White, T. C. 1997. The presence of an R467K amino acid substitution and loss of allelic variation correlate with an azole-resistant lanosterol 14α-demethylase in Candida albicans. Antimicrob. Agents Chemother. 41:1488-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Williamson, D. H., N. G. Maroudas, and D. Wilkie. 1971. Induction of the cytoplasmic petite mutation in Saccharomyces cerevisiae, by the antibacterial antibiotics erythromycin and chloramphenicol. Mol. Gen. Genet. 111:209-223. [DOI] [PubMed] [Google Scholar]