

Abstract
To test how cell–cell contacts regulate microtubule (MT) and actin cytoskeletal dynamics, we examined dynamics in cells that were contacted on all sides with neighboring cells in an epithelial cell sheet that was undergoing migration as a wound-healing response. Dynamics were recorded using time-lapse digital fluorescence microscopy of microinjected, labeled tubulin and actin. In fully contacted cells, most MT plus ends were quiescent; exhibiting only brief excursions of growth and shortening and spending 87.4% of their time in pause. This contrasts MTs in the lamella of migrating cells at the noncontacted leading edge of the sheet in which MTs exhibit dynamic instability. In the contacted rear and side edges of these migrating cells, a majority of MTs were also quiescent, indicating that cell–cell contacts may locally regulate MT dynamics. Using photoactivation of fluorescence techniques to mark MTs, we found that MTs in fully contacted cells did not undergo retrograde flow toward the cell center, such as occurs at the leading edge of motile cells. Time-lapse fluorescent speckle microscopy of fluorescently labeled actin in fully contacted cells revealed that actin did not flow rearward as occurs in the leading edge lamella of migrating cells. To determine if MTs were required for the maintenance of cell–cell contacts, cells were treated with nocodazole to inhibit MTs. After 1–2 h in either 10 μM or 100 nM nocodazole, breakage of cell–cell contacts occurred, indicating that MT growth is required for maintenance of cell–cell contacts. Analysis of fixed cells indicated that during nocodazole treatment, actin became reduced in adherens junctions, and junction proteins α- and β-catenin were lost from adherens junctions as cell–cell contacts were broken. These results indicate that a MT plus end capping protein is regulated by cell–cell contact, and in turn, that MT growth regulates the maintenance of adherens junctions contacts in epithelia.
INTRODUCTION
Microtubules (MTs) are ubiquitous cytoskeletal polymers in eukaryotic cells that consist of α/β tubulin heterodimers assembled head-to-tail in the 13 protofilaments making up the 25-nm-radius cylindrical MT wall. Both α- and β-tubulin bind GTP, and the relationship between tubulin GTP hydrolysis, MT assembly, and MT stability results in a behavior known as “dynamic instability,” in which growing and shrinking MTs coexist in a population when MTs are in equilibrium with tubulin dimer. In such a population, individual MTs constantly make stochastic transitions between persistent phases of growth and shortening (reviewed in Desai and Mitchison, 1997). The kinetic parameters describing dynamic instability include the velocities of MT growth and shortening and the frequencies of transition between growth and shortening (catastrophe frequency) and between shortening and growth (rescue frequency) (Walker et al., 1988). In addition, the intrinsic polarity of tubulin heterodimers and their unidirectional orientation during association results in a MT polymer with structural polarity, such that dimers add more quickly to the “plus” end and more slowly to the “minus” end (Walker et al., 1988). In vivo, this MT polarity is thought to lend overall polarity and organization to living cells. For example, in tissue cells in culture, MTs are organized with their minus ends either bound to the centrosome adjacent to the nucleus or free and facing toward the cell center and their plus ends radiating out toward the cell periphery (Euteneuer and McIntosh, 1981).
Major questions in MT cell biology include the physiological role of MT plus end dynamic instability and its regulation. Cycles of growth and shortening of MT plus ends during dynamic instability may be a means for MTs to “search” cellular space, as in the case of chromosome kinetochore capture by dynamically unstable MTs during the establishment of the mitotic spindle (reviewed in Rieder and Salmon, 1998) or in the targeting of MTs to focal contacts and promotion of focal contact disassembly in migrating cells (Kaverina et al., 1998, 1999). Alternatively, recent evidence suggests that certain proteins can bind to MT ends only during specific phases of dynamic instability (Perez et al., 1999). Further, MT plus end growth and shortening may activate different signal transduction cascades to produce differential regulation of the actin cytoskeleton (Ren et al., 1999; Waterman-Storer et al., 1999; reviewed in Waterman-Storer and Salmon, 1999). In either case, the glorious advantage of the dynamically unstable MT plus end is its exquisite spatial resolution, making MT dynamic instability a prime candidate for precise control of spatial regulatory processes in the cell. For example, in polarized migrating cells with a free leading edge exhibiting actomyosin-ruffling activity, MTs align along the axis of migration, with their plus ends facing the direction of cell movement (Gotlieb et al., 1981; Kupfer et al., 1982). These polar MTs could then precisely direct the delivery of signaling molecules to drive ruffling, structural components of the motile machinery, or regulators of focal contacts that are required at specific sites at or near the leading edge.
In addition, it has been proposed that selective stabilization of the dynamic instability of individual MT plus ends in specific regions of the cell may promote the establishment of such cellular asymmetries (Kirschner and Mitchison, 1986). Thus, in order to understand the spatial organization of cells, it is of prime importance to understand the regulation of MT plus end dynamic instability in vivo. It is well established that the parameters of MT dynamic instability differ for pure tubulin in vitro and MTs in living cells. Indeed, several protein factors that bind to either tubulin dimers or MT polymer (MAPs) have been identified that regulate specific phases of dynamic instability, such as promotion of catastrophe, enhancing the rates of growth/shortening, or suppressing rescue (reviewed in Cassimeris, 1999). However, the cellular events and/or cellular contexts that regulate these proteins are unclear.
One possible cellular context that may modulate plus end MT dynamic instability is whether cells exist as part of a tissue or are free in culture. To approach this question, we were interested to know whether the dynamic instability of individual MT plus ends was altered by cell–cell contact. In tissues and in culture, contacts between cells are mediated by morphologically distinct structures, including tight junctions, adherens junctions, and desmosomes. They all consist, in some manner, of trans-membrane receptors mediating cell–cell interaction on the outside of the cell, whereas on the inside of the cell, they mediate connections to the cortical cytoskeleton. Tight junctions (TJs) form around the apical domain between polarized epithelial cells and seal the cells' apical surface from their basolateral side. TJs are made up of trans-membrane proteins occludin and claudin, which bind in the cytoplasm to membrane-associated guanylate cyclase kinase homologues, including ZO-1 and ZO-2, which may link to cortical actin filaments (reviewed in Tsukita and Furuse, 1999). Adherens junctions, which do not form a seal, but only anchor neighboring cells to one another, consist of trans-membrane cadherins (E-, N-, and VE-cadherin) that bind to intracellular catenins (α-catenin, β-catenin, plakoglobin), which link to cortical actin via either direct (through β-catenin) or indirect (through vinculin or α-actinin) interactions (Provost and Rimm, 1999; reviewed in Steinberg and McNutt, 1999). Desmosomes consist of trans-membrane desmogleins that interact with desmoplakins, which link to intermediate filaments (reviewed in Troyanovsky, 1999).
In the present study, we were interested to know if the dynamic instability of MT plus ends in cells in the center of the sheet that were contacted on all sides by neighboring cells differed from the dynamic behavior of MT plus ends at the leading free edge of a sheet of squamous epithelial cells migrating during a wound healing reaction in culture. Our results show that plus end dynamic instability is suppressed in fully contacted cells, with individual MTs exhibiting an extended state of pause, suggesting that they become capped. We also find that depolymerization of MTs in fully contacted cells induces disruption of cell–cell adherens junctions. This suggests that a feedback relationship exists in which MT dynamics are modulated by cell–cell contact, and the maintenance of contacts require MTs.
MATERIALS AND METHODS
Cell Culture, Fluorescent Proteins, and Microinjection
Primary cultures of newt lung epithelial cells were established on 22 × 22-mm 1.5 coverslips (Corning) from Taricha granulosa lung tissue and maintained in Rose Chambers at ∼20°C in ½ strength L-15 media containing 5% fetal bovine serum, antibiotics, and antimyocotics as previously described (Reider and Hard, 1990; Waterman-Storer and Salmon, 1997).
Porcine brain tubulin was purified by rounds of temperature-dependent polymerization and depolymerization, followed by phosphocellulose chromatography, and was covalently linked at high pH to succinimidyl ester of X-rhodamine (Molecular Probes, Eugene, OR) as described (Walker et al., 1988; Hyman et al. 1991; Waterman-Storer et al., 1997). C2CF (Mitchison, 1989) was the kind gift of Tim Mitchison and Arshad Desai, and was covalently bound to tubulin as described (Desai and Mitchison, 1998).
Chicken pectoral muscle acetone powder was prepared by the method of Pardee and Spudich (1982). X-rhodamine–labeled globular actin (g-actin) was prepared from acetone powder as described in Turnacioglu et al. (1998). Briefly, g-actin was extracted from acetone powder with water and polymerized by the addition of KCl and MgCl to 100 and 2 mM, respectively. For labeling, the pH was raised to 9 by the addition of sodium bicarbonate, and succinimidyl ester of X-rhodamine was added at a dye:protein ratio of 4:1 and stirred for 1.5 h at 20°C. The labeling reaction was quenched by addition of NH4Cl to 50 mM, and f-actin was pelleted for 1 h at 4°C at 100,000 × g in a 50.2 Ti rotor (Beckman Instruments, Fullerton, CA). F-actin was resuspended in G-Buffer (2 mM Tris, 0.2 mM CaCl2, 0.2 mM MgATP, 0.5 mM β-mercaptoethanol, 0.005% NaN3, pH 8.0) and was depolymerized by dialysis against G-buffer at 4°C for 3 days, clarified by centrifugation at 100,000 × g, and repolymerized by addition of KCl, MgCl, and MgATP to 100, 2, and 1 mM, respectively. F-actin was again pelleted, resuspended in G-buffer, and depolymerized by dialysis for 3 days at 4°C against G-buffer lacking NaN3. Labeled g-actin was clarified by centrifugation, and the concentration adjusted to 4 mg/ml; it was then drop-frozen in liquid nitrogen until use for microinjection.
Coverslips of cells were microinjected with X-rhodamine-labeled tubulin or C2CF-labeled tubulin in injection buffer (50 mM K-glutamate, 0.5 mM MgCl2) at 2 and 5 mg/ml, respectively. For fluorescent speckle imaging of f-actin (Waterman-Storer et al., 1998), cells were injected with 1 mg/ml X-rhodamine–labeled G-actin in G-buffer lacking NaN3. All microinjections were performed on the apparatus described in Waters et al. (1996). After microinjection, cells were allowed to recover for 1–2 h in the dark before being mounted on slides on two strips of double-stick tape in culture media containing 0.3–0.6 U/ml Oxyrase (Oxyrase, Mansfield, OH) to inhibit photobleaching during imaging.
Indirect Immunofluorescence Localization of Cellular Proteins
Coverslips of newt lung cells were permeabilized and prefixed for 5 min in 1% formaldehyde, 0.5% Triton X-100, freshly prepared in PHEM buffer (60 mM Na PIPES, 25 mM Na HEPES, 10 mM EGTA, 4 mM MgSO4, pH 7.2). Cells were then fixed for 15 min in 1% formaldehyde, 0.5% glutaraldehyde, freshly prepared in PHEM, and rinsed three times in PHEM. Free aldehydes were blocked for three 5-min incubations with sodium borohydride, and coverslips were rinsed three times in PBST (15 mM Na2HPO4, 1.6 mM KH2PO4, 2.5 mM KCl, 140 mM NaCl, 0.1% Triton X-100, pH 7.2). To block nonspecific antibody binding, coverslips of cells were incubated 40 min in donkey block (5% boiled donkey serum in PBS [15 mM Na2HPO4, 1.6 mM KH2PO4, 2.5 mM KCl, 140 mM NaCl, pH 7.2]). Cells were then incubated in a humid chamber for 1 h at 37°C with primary antibodies at the proper dilution in donkey block, rinsed four times in PBST, and incubated similarly with fluorescently labeled secondary antibodies (1:50 in donkey block; Jackson ImmunoResearch, West Grove, PA). If localizing f-actin, 0.5 U/ml Texas red phalloidin (Molecular Probes) were included with the secondary antibody. Coverslips were then rinsed four times in PBST and one time in PBS, mounted on slides in 50% glycerol, 50% PBS containing n-propyl-gallate, and sealed with nail polish. For localizing tubulin, monoclonal mouse anti–α-tubulin clone DM 1A (Blose et al., 1984; Sigma Chemical, St. Louis, MO) was used at 1:500; for localizing α- or β-catenin, rabbit α- or β-catenin polyclonal sera (Sigma Chemical) were used at 1:1000.
Time-Lapse Digital Fluorescence Microscopy
Digital images were obtained with a 12-bit Hamamatsu C-4880 camera containing a Texas Instruments TC-215 charge-coupled device (CCD) with 12-μm2 pixels cooled to −40°C on the multimode microscope described in Salmon et al. (1998). Images were collected on a Nikon Microphot FXA with a 60× 1.4 NA objective, a 1.25× body tube magnifier, and a 1.5× optavar. For epi-fluorescence imaging, illumination was provided by a 100-W mercury arc lamp and passed through a narrow band pass 570-nm excitation filter (Chroma, Brattleboro, VT) in an electronically controlled dual filter wheel/shutter device (Metaltek, Raleigh, NC), reflected off of a triple band pass dichromatic mirror (Chroma), and focused onto the specimen. Emission from the specimen of 590 nm was collected by the objective, passed through the dichromatic mirror and a triple band-pass emission filter (Chroma) and collected by the camera. For photoactivation of C2CF fluorescence, a 360-nm excitation filter was used, and a 25 μm × 1-mm slit (Melles Griot, Rochester, NY) was placed in the field diaphragm plane of the epi-illumination pathway to allow exposure of UV light to a bar-shaped region of the cell as described in Waters et al. (1996). Filter wheel, shutter, and camera image acquisition timing were computer controlled using the MetaMorph Digital imaging system software (Universal Imaging, Brandywine, PA) and the Mutech MV-1000 frame grabber board in a PC computer.
Time-Lapse Phase Contrast Microscopy
Cells were observed in Rose Chambers on a Zeiss Universal microscope (Thornwood, NY) equipped with a 25× objective lens, illumination from a quartz-halogen lamp, and components for phase contrast image formation. Images were collected with an 8-bit video-rate CCD camera (Hamamatsu C2400, Bridgewater, NJ), contrast was enhanced with a real-time image processor (Hamamatsu Argus-10), and images were recorded onto SVHS videotape at 120× real time on a time-lapse VCR (Panasonic AG-6750-A, Seacacus, NJ). Nocodazole-containing media were exchanged with drug-free media via syringes with 16-gauge needles inserted into the rubber gasket in the Rose Chamber.
Data Analysis
All position, length, and intensity measurements were made using the analysis functions in MetaMorph software, values were exported to Microsoft Excel for file formatting, and determination of instantaneous velocity was performed using the custom-written RTM software (Walker et al., 1988). Parameters of individual MT dynamic instability were obtained exactly as described previously (Waterman-Storer et al., 1997). Dynamicity for individual MTs was calculated as the sum of the absolute value of all the detectable growth and shortening velocities measured for that MT × 1624 tubulin dimers/μm and divided by the total time observed (Toso et al., 1993). Comparison of intensity of f-actin in adherens junctions was performed on measurements of cells from the same experiment stained equally with Texas red phalloidin, and measurements were corrected for camera exposure time. All values are expressed as means ± SD, and significant differences were determined with a two-tailed Student's t test.
RESULTS
MT Plus Ends Are Quiescent in Fully Contacted Cells
To test the hypothesis that MT plus end dynamic instability was modulated by cell–cell contact, we microinjected X-rhodamine–labeled tubulin into newt lung epithelial cells that were contacted on all sides by neighboring cells and situated in the center of an epithelial sheet. This sheet migrates from the cut edges of an explant of lung tissue during a wound healing response. In these cultures, the cells on the edge of the sheet are specialized for motility with polarized f-actin–based ruffling and adhesive contacts with the substrate at the non-contacted “leading edge,” whereas cells in the center of the sheet lack adhesive contacts with the substrate and do not contribute to motility (Waterman-Storer and Salmon, 1997). Incorporation of X-rhodamine tubulin into all cellular MTs was complete by 2 h post-injection, as assayed by comparing X-rhodamine MTs to immunolocalization of MTs in fixed cells (our unpublished results). After incorporation of the labeled tubulin into MTs (∼2–4 h), we imaged MT dynamics by time-lapse digital epi-fluorescence microscopy, collecting images at 5- to 7-s intervals. In these “fully contacted cells,” MTs were organized with many ends facing the cell periphery and terminating within a few micrometers from the cell–cell junctions (Figure 1a), whereas in the cell center, there was a concentration of bent and sinuous MTs of random orientation (not shown). We concentrated our studies on the MT ends that were easily visible in the periphery of the fully contacted cells. In stark contrast to MT plus ends in the lamella of motile cells, which exhibit dynamic instability (Cassimeris et al., 1988; Waterman-Storer and Salmon, 1997), time-lapse movies of MTs in the periphery of fully contacted cells revealed that plus end MT assembly/disassembly was greatly suppressed. MTs in these fully contacted cells exhibited Brownian vibrations in the cytoplasm, but their plus ends were surprisingly quiescent, exhibiting only very short, infrequent excursions of growth or shortening (Figure 1b) during observation periods of up to ∼20 min.
Figure 1.

(A) MT plus ends are quiescent, and parallel MTs do not flow rearward in contacted cells. A cell in the center of the epithelial sheet that was contacted on all sides by neighboring cells was microinjected with X-rhodamine–labeled tubulin, and images of fluorescent MTs were captured at 7-s intervals. Neighboring cells cannot be seen because they were not injected with fluorescent protein. Selected images from the time-lapse sequence are shown. MT plus ends (arrows) near the cell edge remain quiescent over the ∼10-min observation period. An MT that is parallel to the leading edge (large arrow) maintains a constant distance from the edge of the cell. Time is in min:s; bar, 5 μm. (B and C) Comparison of dynamic life history plots of MT plus ends near the edge of contacted cells and MT plus ends near the leading edge of migrating cells. Cells were microinjected with X-rhodamine–labeled tubulin, and images of fluorescent MTs were captured at 7-s intervals for a total of 7 min. In (B), three MT plus ends (MT 1–3) were tracked in the periphery of different cells, all of which were contacting neighboring cells on all sides. MTs in these contacted cells exhibited only brief excursions of growth and shortening, spending most of their time in pause. In (C), three MT plus ends (MT 1–3) were tracked near the contact-free leading edges in different cells at the periphery of the epithelial cell sheet. MTs 1 and 2 were oriented parallel to the leading edge of the cell, whereas MT 3 was oriented perpendicular to the leading edge. These MTs underwent dynamic instability similar to that reported in this and other cell types for cells with free edges. (D–G) Comparison of the distributions of dynamicity (total dimer exchange per second) of individual MT plus ends in newt lung epithelial cells). (D) and (E) are calculated from the results of Waterman-Storer and Salmon (1997).
The dynamic behavior of MTs in fully contacted cells was quantified and compared with MT plus ends in the lamella of motile cells as observed in our previous study (Waterman-Storer and Salmon, 1997) (Table 1). In that analysis, we noted significant differences in the assembly/disassembly dynamics between lamella MTs that were oriented parallel to the leading cell edge (parallel MTs) and those oriented perpendicular to the leading edge (perpendicular MTs). In particular, perpendicular MTs exhibited slower growth rates, more frequent catastrophes, and spent more time in a “paused” state, neither growing nor shortening, than parallel MTs. We have now compared the total tubulin dimer exchange per second per MT plus end (“dynamicity”) (Toso et al., 1993) and confirmed that in motile cells, perpendicular MT plus ends (average dynamicity = 63.9 ± 33.0 dimers/s, n = 22 MTs in 13 cells) are much less dynamic than parallel MT plus ends (average dynamicity = 171.5 ± 79.3 dimers/s, n = 22 MTs in 13 cells). In contrast, analysis of MT plus ends in the periphery of fully contacted cells shows that these MTs are even less dynamic than perpendicular MTs in migrating cells, exhibiting an average dynamicity of 15.3 ± 29.5 dimers/s (n = 45 MTs in 8 cells). This difference is attributed to the MT plus ends in fully contacted cells spending 87.4% of their time in pause (n = 45 MTs in 8 cells) versus 40.1% for perpendicular MTs (n = 22 MTs in 13 cells). Otherwise, rates and durations of growth and shortening and frequencies of catastrophe and rescue were not significantly different between plus ends of perpendicular MTs in migrating cells and MT plus ends in the periphery of fully contacted cells. We noted no qualitative differences in the behavior of MTs of different orientations with respect to the cell edge in fully contacted cells.
Table 1.
Quantification of MT plus end dynamic behavior in contacted and noncontacted edges of newt lung epithelial cells
| Parameter | Fully contacted cells, +MT enda (n = 45 MTs, 8 cells) | Contacted sides of migrating cellsa
|
Leading edge of migrating cells, +MT end perpendicularb (n = 22 MTs, 13 cells) | Leading edge of migrating cells, +MT end parallelb (n = 22 MTs, 13 cells) | ||
|---|---|---|---|---|---|---|
| Dynamic & Quiescent Combined (n = 39 MTs, 5 cells) | Dynamic MTs (n = 18 MTs, 3 cells) | Quiescent MTs (n = 21 MTs, 5 cells) | ||||
| % Time in Growth/Shortening/Pause | 7.1/5.5/87.4 | 14.5/11.7/73.7 | 46.2/38.7/15.05 | 3.4/4.7/94.8 | 35.7/24.2/40.1 | 75.6/17.7/6.7 |
| Growth rate (μm/min) | 4.3 ± 3.6 | 9.1 ± 5.1 | 8.9 ± 4.4 | 10.5 ± 8.7 | 4.5 ± 2.7 | 6.8 ± 3.9 |
| Growth duration (min) | 0.5 ± 0.3 | 0.4 ± 0.2 | 0.4 ± 0.2 | 0.4 ± 0.3 | 0.6 ± 0.4 | 0.9 ± 0.7 |
| Shortening rate (μm/min) | 3.7 ± 2.4 | 8.0 ± 5.0 | 8.5 ± 5.0 | 6.3 ± 4.7 | 5.2 ± 4.3 | 7.6 ± 5.4 |
| Shortening duration (μm/min) | 0.5 ± 0.3 | 0.4 ± 0.2 | 0.4 ± 0.2 | 0.3 ± 0.1 | 0.5 ± 0.5 | 0.4 ± 0.2 |
| Catastrophe frequency (min−1) | 1.30 | 1.65 | 1.43 | 1.12 | 1.57 | 0.61 |
| Rescue frequency (min−1) | 1.90 | 1.80 | 1.48 | 1.34 | 2.31 | 2.59 |
| Dynamicity (dimers/s) | 15.3 ± 29.5 | 125.4 ± 142.1c | 253.1 ± 113.1c | 13.4 ± 29.2 | 63.9 ± 33.0c | 171.5 ± 79.3c |
Values are means ± SD.
No qualitative differences between the dynamic behavior for MTs of different orientations with respect to the cell edge were noted.
Data taken from Waterman-Storer and Salmon (1997). For comparison of assembly dynamics of parallel and perpendicular MTs in migrating cells, 22 MTs that grew from 0 to 30° from perpendicular to the cell's edge and then underwent a bend and grew from 0–30° from parallel to the cell edge and were followed. Assembly parameters were determined for the periods during which the MT was within these angular ranges. The period when the MT was bending was discounted in the averages.
A statistically significant difference from fully contacted cells (p < 0.01).
These results indicate that compared with the free noncontacted leading edge of migrating cells of the same type, cells that are contacted with neighboring cells on all sides have a decrease in MT dynamic turnover because of an increase in the percent time that MT plus ends spend in pause.
Cell–Cell Contacts Locally Regulate MT Dynamics
The marked difference that we observed between the assembly/disassembly behavior of MTs in the leading lamella of migrating cells and MTs in the periphery of fully contacted cells led us to question whether the free noncontacted leading edge of migrating cells globally increases MT dynamics throughout the cell or if cell–cell contacts locally modulate MT dynamics. To approach this question, we examined and quantitated the assembly/disassembly behavior of MT ends in the contacted sides and rear of migrating cells at the edge of the epithelial sheet. We analyzed MT plus ends in regions of these cells away from the free leading edge and adjacent to contacts with neighboring cells. Time-lapse movies of X-rhodamine–labeled MTs in contacted regions in the rear or sides of migrating cells revealed that in contrast to MT plus ends in the lamella of the same cell (not shown), many MT plus ends remained in an extended state of pause. However, there was also a subset of MT plus ends in contacted regions that were very dynamic, undergoing long excursions of very rapid growth or shortening (Table 1). The two populations of MTs seen in contacted regions of migrating cells are easily differentiated in a histogram of the dynamicity of individual MTs (Figure 1g), which exhibits a bimodal distribution with peaks at 0–49 and 150–299 dimers/s. This is in contrast to the unimodal distributions of MT dynamicity in fully contacted cells (Figure 1f) and in parallel (Figure 1d) and perpendicular MTs (Figure 1e) in the noncontacted leading lamellae. We analyzed these two populations separately, considering those microtubules in the contacted sides and rear with a dynamicity of <100 to be “quiescent” and those with a dynamicity of >100 to be “dynamic.” This revealed that dynamic MT plus ends in contacted regions of migrating cells have the greatest dynamicity of all microtubules thus far analyzed in newt lung cells. Average values for MT plus ends in the contacted regions of migrating cells indicate that as a population, they spend 73.7% of their time in pause and that when they are exhibiting growth or shortening, these phases are significantly more rapid than growth or shortening of any other type of MT measured in newt lung epithelial cells (average growth velocity = 9.0 ± 5.1 μm/min, average shortening velocity = 8.0 ± 5.0 μm/min, n = 39 MTs in 5 cells).
These results suggest that MT dynamics are regulated differently in regions of migrating cells adjacent to cell–cell contacts compared with MTs at the leading edge. Near cell–cell contacts, many MT plus ends are quiescent, similar to those in the periphery of fully contacted cells, whereas there is also a population of highly dynamic MTs.
MTs and f-Actin Do Not Undergo Retrograde Flow in Fully Contacted Cells
In our previous study of MT dynamics in migrating cells, we found that MTs in the lamella moved away from the leading edge toward the cell center at 0.4 μm/min in an actomyosin-dependent manner (Waterman-Storer and Salmon, 1997). In contrast to this, when we examined MTs that were parallel to the cell edge in fully contacted cells, no retrograde movement was observed (Figure 1a). To determine if either parallel or perpendicular MTs were moving toward the cell center of fully contacted cells, we performed photoactivation of fluorescence marking of the MTs. Cells were coinjected with X-rhodamine–labeled tubulin to allow visualization of MTs before photoactivation and caged-fluorescein tubulin (C2CF tubulin), to mark the MTs (Mitchison, 1989). Following incorporation of the tubulins into MTs, the cells were exposed at ∼1/3 the distance from the cell edge to the nucleus to a narrow bar of 360 nm light to activate the fluorescence of C2CF (Figure 2). Monitoring the position of the bar of activated green fluorescence on MTs over time confirmed that there was no retrograde MT movement in fully contacted cells. Furthermore, most of the microtubules marked by photoactivated fluorescein were present at the end of the ∼20- to 30-min observation sequence. The majority of the decay in photoactivated signal in the marked region is due to diffusion of unpolymerized tubulin (Salmon et al., 1984) and photobleaching. This confirmed the slow rate of MT turnover in fully contacted cells. Similarly, we found that MTs near contacted regions of migrating cells did not flow from the edge toward the cell center (not shown).
Figure 2.

In contacted cells, MTs do not exhibit retrograde flow toward the cell center. A cell in the center of the epithelial sheet that was contacted on all sides by neighboring cells was microinjected with a mixture of X-rhodamine (red) and caged fluorescein (yellow-green) labeled tubulins. Neighboring cells cannot be seen because they were not injected with fluorescent protein. A narrow bar-shaped region of the cell between the cell edge and the nucleus was exposed to UV (360 nm) light to activate the fluorescein label on MTs spanning this region. Images of X-rhodamine– and fluorescein-labeled tubulin were captured in succession at ∼2-min intervals, color encoded, and overlaid to show the position of the fluorescein marks relative to the MT lattice. The marks remained stationary (0.00 μm/min velocity) with respect to the cell edge over the ∼15-min observation period. Time is in min:sec; bar, 5 μm.
This lack of retrograde MT movement in contacted cells could be explained by one of two hypotheses: first, that there is no retrograde actin flow in contacted cells, or second, that MTs are uncoupled from retrograde actin flow. To differentiate between these two possibilities, we imaged actin retrograde flow directly in migrating and contacted cells using fluorescent speckle microscopy (FSM) (Waterman-Storer et al., 1998). In this technique, cells are microinjected with a low level of X-rhodamine–labeled actin (amounting to ∼ 0.5% of the total cellular actin pool). Thus, the f-actin meshwork in the lamella appears speckled in diffraction-limited digital fluorescence images (Figure 3) due to random variation in incorporation of the few fluorescent actin monomers into the f-actin meshwork (Waterman-Storer et al., 1998). This speckle pattern acts as a fiduciary pattern on the f-actin meshwork and in time-lapse FSM images allows for the observation of movements of and turnover within the meshwork (Waterman-Storer et al., 1998). Kymograph analysis of a time-lapse series of actin FSM images recorded at 15-s intervals of the leading edge of migrating cells revealed two distinct rates of retrograde f-actin flow: 1.61 ± 0.42 μm/min in the lamellipodia region within 3–5 μm of the leading edge and more slowly at 0.40 ± 0.22 μm/min in the lamella at distances >3–5 μm from the edge (Figure 3, a and c) (Waterman-Storer et al., 1998). In contrast, similar analysis of time-lapse actin FSM in fully contacted cells gave no indication of retrograde f-actin movement in the region between the cell edge and the nucleus (Figure 3, b and d). However, this analysis revealed that the f-actin concentrated in the cell–cell adherens junctions is highly dynamic, as indicated by the change in speckle pattern of the junctions (Figure 3d, the top of the kymograph) compared with the relatively constant speckle pattern in the region between the cell edge and the nucleus (Figure 3d, the center of the kymograph).
Figure 3.

Actin flows rearward in the lamella of migrating cells but not in contacted cells. Cells were microinjected with low levels of X-rhodamine–labeled actin to label actin filaments with fluorescent speckles, and images of the fluorescent actin meshwork were captured at 15-s intervals for 15 min. (A and B) Individual images from the time-lapse series. The low level of fluorescent actin in the cells results in a fluorescence image that exhibits a very fine speckled labeling of the actin meshwork due to stochastic incorporation of labeled and unlabeled actin subunits into actin polymer. The cell in (A) is at the edge of the epithelial cell sheet, with the free leading edge facing the top of the page. Note the concentration of actin polymer in the lamellipodia. The cell in (B) is surrounded on all sides by contacting neighboring cells, which are invisible because they were not injected with fluorescent protein. The cell–cell adhesions are brightly labeled with fluorescent actin. Images of the regions covered by lines in (A) and (B) from each image in the time lapse series were used to construct the kymographs in (C) and (D), respectively. Inhomogeneities in fluorescent labeling of the actin meshwork are seen as streaks across the kymograph. Slopes of the streaks represent velocities of actin meshwork movement. In the free-edged cell in (C), actin moves toward the cell center at 1.7 μm/min in the lamellipodia and at 0.4 μm/min in the lamella. In the cell that is contacted, the actin meshwork remains stationary. Scale bar in (A), 10 μm (equal for all frames).
These results indicate that neither MTs nor f-actin exhibit retrograde flow in contacted cells.
Suppression of MT Growth Induces Breakage of Cell–Cell Contacts
Knowing that the presence of oriented, dynamic MTs are required for cell motility of migrating cells (Vasiliev et al., 1970; Goldman, 1971; Gotleib et al., 1981; Kupfer et al., 1982; Liao et al., 1995), we wanted to determine what role(s) MTs play in contacted cells. To approach this question, we treated newt lung epithelial cells with nocodazole to block plus end growth and promote MT disassembly. We then imaged the cells by low-magnification, time-lapse, phase-contrast video microscopy to observe both the migrating cells at the edge of the cell sheet and the fully contacted cells in the center of the sheet. Within minutes after application of 100 nM nocodazole, advancement of the migrating cells ceased, as expected from previous studies (Liao et al., 1995) (Figure 4, times:12:03–02:35). However, surprisingly, after ∼60–90 min (Figure 4, time: 66:27), cells that had been in contact with neighboring cells began to locally lose cell–cell adhesion at sites along their periphery (Figure 4, arrows). When these cells retracted from one another, the edges that had been quiescent and contacted began to undergo ruffling activity. The dissolution of cell–cell contact was not synchronous throughout the cell sheet, but continued at different sites around the sheet for several hours (arrows, 66:27–115:34). We tested both a high concentration of nocodazole that instantly inhibits microtubule growth and promotes rapid depolymerization of MTs (10 μM, not shown), and a low concentration that rapidly suppresses both MT growth and shortening but does not induce immediate disassembly in newt lung cells (100 nM, Figure 4) (Vasquez et al., 1997) and found similar results on a similar time-scale with both treatments. However, unlike other studies in different cell types, which found that 100 nM nocodazole did not decrease MT polymer levels (Liao et al., 1995), we found that after 60–90 min, newt lung cells treated with 100 nM nocodazole showed a substantially reduced complement of cellular MTs (Figure 5). In contrast, nearly all microtubules were depolymerized after 60–120 min in 10 μM nocodazole (not shown). Because high and low concentrations of nocodazole affect microtubule depolymerization in newt lung cells with such different kinetics, this suggests that the similar time course of the two treatments on cell–cell junction disruption is not due to their effects on MT depolymerization, but their similar inhibition of microtubule growth. Thus, these results indicate that MT growth is required to maintain the integrity of epithelial cell–cell junctions.
Figure 4.
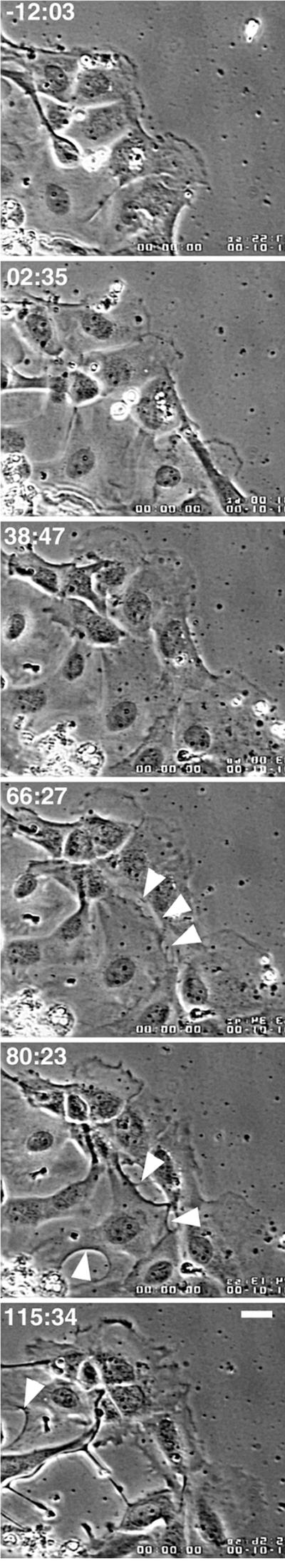
Treatment of newt lung epithelial cells with nocodazole results in breakage of cell–cell contacts. Phase contrast images taken from a time-lapse series. Time (in min:sec) is relative to time of addition of 100 nM nocodazole. During the first 16 h of filming (not shown), the epithelial sheet advanced ∼50 μm. After application of 100 nM nocodazole (at time 00:00), advancement of the cells ceased. By 66:27, cells that had previously been in contact with neighboring cells began to locally lose cell–cell adhesion at sites along their periphery (arrows). Loss of contact with neighboring cell continued for several hours (arrows, 80:23–115:34). Bar, 50 μm.
Figure 5.

Actin and MT localization in contacted newt lung cells after treatment with 100 nM nocodazole. After application of 100 nM nocodazole (time in min in lower left), cells were fixed. F-actin was localized with Texas red phalloidin (top panels), and MTs were visualized in the same cells by indirect immunofluorescence (bottom panels). Before nocodazole treatment (time 0), f-actin is concentrated in the cell–cell junctions. At 15 min after nocodazole application, f-actin is recruited into stress fibers, and the density of MTs is unaffected. By 60–90 min, f-actin stress fibers have disassembled and f-actin is reduced in cell–cell junctions, MTs are reduced in the cytoplasm, and contacts between cells begin to break (arrowheads). Bar, 10 μm.
Suppression of MT Growth Induces Loss of F-actin from Adherens Junctions
Suppression of MT growth in fibroblasts has been shown to result in the activation of the Rho small GTPase, which increases cell contractility by inducing the formation of actomyosin stress fibers and focal adhesions to the extra cellular matrix (Danowski, 1989; Bershadsky et al., 1996; Ren et al., 1999). One hypothesis for why MT depolymerization induced the disruption of cell–cell contacts in the present study is that an increase in contractility caused adjacent cells to rip apart from their neighbors as they contracted. To test this hypothesis, we fixed cells at various times after application of nocodazole (either 10 μM [not shown] or 100 nM [Figure 6]), processed them for immunofluorescent localization of tubulin to visualize MTs, stained them with Texas red phalloidin to visualize f-actin, and used the assembly state of f-actin stress fibers as an indirect indication of the contractile state of the cell.
Figure 6.

During nocodazole treatment, β- and α-catenins are lost at cell–cell junctions as cell–cell contacts are broken. Newt lung epithelial cells were treated with 100 nM nocodazole then fixed at the time (in min top left) indicated after application of nocodazole. F-actin and α-catenin or MTs and β-catenin were then double-localized in the cells as indicated. F-actin and α-catenin are concentrated in cell–cell junctions before treatment with nocodazole (time 0). After 120 min in 100 nM nocodazole, f-actin is reduced, and α-catenin becomes punctate and is lost at sites where cell–cell contacts have recently been broken (arrowheads). After 120 min in 100 nm nocodazole, MTs are reduced in the cell, whereas β-catenin is punctate in cell–cell contacts and is lost at sites of recent contact disruption. Bar, 10 μm.
Before treatment of fully contacted cells with 100 nM nocodazole, MTs were distributed throughout the cytoplasm, whereas f-actin was localized to a fine meshwork in the cytoplasm, concentrated in cell–cell junctions and in a few brightly labeled foci in the cytoplasm. F-actin was infrequently organized into stress fibers in fully contacted cells, but when present, they tended to align around the periphery of the cell a few micrometers from the cell–cell junctions. After 15 min in 100 nM nocodazole, MT polymer did not noticeably decrease in the cell; however, f-actin stress fibers dramatically increased and spanned across the central regions of the cell. Other f-actin–containing structures appeared to be unchanged after 15 min in nocodazole. Stress fibers remained prominent for 30–60 min after nocodazole application; however, by 90–120 min after addition of nocodazole, MT polymer levels were markedly decreased, and the f-actin stress fibers had disassembled. Although cytoplasmic foci of f-actin still remained, the level of f-actin in cell–cell junctions decreased substantially. At 60 min after the application of nocodazole, the fluorescence intensity of f-actin staining per pixel along adherens junctions was reduced to 24.87% of control values (578 measurements for control, 372 measurement for experimental, means significantly different, p = 2.3 × 10−6). By 120 min, f-actin was nearly absent from cell–cell junctions, and this coincided with approximately the same time that cell–cell contacts began to disrupt. A similar time-course of stress fiber assembly, disassembly and loss of f-actin from cell–cell junctions was obtained upon treatment of cells with 10 μM nocodazole, although MTs depolymerized more quickly (our unpublished results).
These results suggest that stress fiber-mediated contractility is not responsible for the breakage of cell–cell junctions that is induced by nocodazole treatment, but instead, that f-actin within cell–cell junctions may be regulated by MT depolymerization.
α- and β-Catenin Are Lost from Adherens Junctions as Cell–Cell Contacts Are Broken
The loss of f-actin from cell–cell junctions after MT depolymerization suggested that other molecular components of cellular junctions may be disassembling from these sites in response to MT depolymerization. To test this hypothesis, newt lung epithelial cells were treated with 10 μM or 100 nM nocodazole and then fixed and processed for indirect immunolocalization of α- and β-catenin (Figure 6). In Figure 6, α-catenin and f-actin were localized in untreated cells and in cells treated with 100 nM nocodazole for 120 min. In untreated cells, both f-actin and α-catenin were highly concentrated in a uniform distribution along the cell margin in cell–cell adherens junctions. After 120 min in 100 nM nocodazole, actin in all adherens junctions maintained the same even distribution, but was not as abundant compared with controls. In contrast, α-catenin became more punctate along junctions and was lost completely from sites where contacts appeared to have recently been broken (Figure 6, arrows).
In similar experiments, β-catenin and MTs were localized in cells after 10 μM and 100 nM nocodazole treatment. Before nocodazole treatment, like α-catenin, β-catenin was concentrated evenly along cell edges in adherens junctions (our unpublished results). One hundred twenty minutes after treatment with 100 nM nocodazole, β-catenin also became punctate along the contacted edges of the cell margin and was gone from cell edges that apparently had detached recently from their neighbors. The shift from a uniform to punctate distribution of α/β-catenin localization with nocodazole treatment represent substantial changes in the molecular composition of the adherens junctions, because punctae could only be detected by the light microscope if the space between them were >240 nm. Thus, loss of up to hundreds of α/β-catenin molecules occurs while the cells are still in contact, and the contacts break as larger sections of the junctions lose α/β-catenins. Similar results were obtained for localization of both α- and β-catenin after treatment of cells with either 10 μM or 100 nM nocodazole. These results demonstrate that inhibition of MT growth causes rearrangement of adherens junction proteins into punctae before their disappearance from junctions during breakage of cell–cell contacts.
DISCUSSION
We sought to determine if plus end MT dynamic behavior is modulated by cell–cell contact. Our results demonstrate that MT plus end dynamic instability is greatly suppressed in cells that are fully contacted around their periphery. Furthermore, regulation of plus end MT dynamics by cell–cell contacts may occur regionally within a single cell, such that many MT plus ends adjacent to cell–cell contacts exhibit suppressed dynamics whereas MT plus ends near free noncontacted cell edges are very dynamic. In addition, cell–cell contact also regulates actin dynamics, inhibiting the retrograde f-actin flow that is characteristic of migrating cells with free leading edges. Surprisingly, by treating contacted cells with nocodazole, we also found that MT growth is required for the maintenance of adherens junctions, and this may occur via regulation of f-actin in adherens junctions. These results thus identify a novel feedback loop in newt lung epithelial cells in which MT and f-actin dynamics are regulated by cell–cell contact, and cell–cell contacts and f-actin within adherens junctions, in turn, require MT growth for their maintenance.
Modulation of Cytoskeletal Dynamics by Cell–Cell Contacts
The striking suppression of plus end MT dynamic instability that we have observed in contacted cells raises the question of how MT dynamics are regulated by cell–cell contact. We will consider three possible mechanisms. One possibility is a difference in the tubulin pool in fully contacted versus partially contacted cells, which results in the suppression of dynamic instability. However, this is very unlikely, because we found that cell–cell contacts locally affected MT dynamics in different regions of the cell. Indeed, tubulin dimer diffuses freely in the cytoplasm, making intracellular gradients in tubulin concentration an unlikely explanation (Salmon et al., 1984; Saxton et al., 1984).
A second possibility for how plus end MT dynamics could become suppressed in contacted cells is by differential regulation of MAPs that bind along the MT lattice. However, because there were not substantial differences in velocities of growth and shortening or frequencies of catastrophe and rescue between contacted and noncontacted cells, but instead there were major differences in the time spent in pause, this possibility is also unlikely. Indeed, all MAPs thus far characterized in nonneuronal cells, including MAP4, XMAP 230, XMAP 310, or XMAP 215, bind to the MT lattice and stabilize MTs by inhibiting catastrophe or promoting rescue (reviewed in Cassimeris, 1999). However, the activity of XMAP 215, which induces very rapid MT plus end growth and shortening (Vasquez et al., 1994), could be responsible for the highly dynamic subset of MTs that we observed near the contacted sides and rear of migrating cells.
On the basis of our observation that MT plus ends in fully contacted cells and most MT plus ends adjacent to contacts in partially contacted cells were in an extended state of pause, we think it is more likely that cell–cell contact promotes the activity of a plus end capping protein. The only well-characterized protein known to specifically bind MT ends and inhibit their growth is γ-tubulin. However, γ-tubulin only binds minus ends and is not active at MT plus ends (reviewed in Jeng and Stearns, 1999). Several proteins recently have been found to localize specifically to MT plus ends in cells, including CLIP-170, EB-1, components of the dynein/dynactin motor complex, and the adenomatous polyposis coli protein APC (Pierre et al., 1992; Nathke et al., 1996; Vaughan et al., 1999). However, so far none of these proteins have been shown to inhibit growth and shortening at MT plus ends. E-MAP-115/ensconsin (Masson and Kreis, 1993; Bulinski and Bossler, 1994), which promotes the stabilization of MTs (Masson and Kreis, 1995), recently has been shown to be upregulated and redistributed from MT shafts to MT ends after the formation of cell–cell contacts in human keratinocyte epithelial cells (Fabre-Jonca et al., 1999). It would be interesting to know the effects of E-MAP-115/ensconsin on MT dynamics in vitro and whether it is localized to MT plus ends in contacted newt lung epithelial cells.
Previous studies have shown that cells possess a population of MTs that are stable to nocodazole-induced depolymerization and that can be recognized by their content of detyrosinated tubulin (Glu MTs). Because Glu MTs tend to be coiled around the nucleus of contacted cells and rarely extend to the cell periphery (Nagasaki et al., 1992) and because the microtubules incorporated labeled tubelin, we think it is unlikely that the MTs with the suppressed dynamic instability that we observe in peripheral regions of contacted newt lung cells are Glu MTs.
The question of how cell–cell contact affects MT dynamics and organization has been touched on in previous studies, in which MTs were examined in fixed Madin–Darby kidney cells (MDCK) epithelial cells as they established contacts and underwent differentiation into a polarized monolayer (Bre et al., 1987, 1990; Bacallao et al., 1989; Buendia et al., 1990). However, these studies primarily concentrated on the changes in MT behavior as the cells underwent polarized differentiation, with less focus on the initial establishment of cell–cell contacts. Because newt lung cells are squamous and do not undergo polarization, the differences in MT dynamics are likely to be due primarily to the differences in the cell–cell contacts. One aspect of this question was addressed more directly in MDCK cells by Pepperkok et al., (1990). MT fluorescence recovery after photobleaching was used to demonstrate that the MT half-life for turnover doubles as these cells form contacts with their neighbors. This finding supports our results that MT dynamics are regulated by cell–cell contact; however, in that study, the behavior of individual MTs was not observed.
We have also found that cell–cell contact has profound effects on the dynamics of f-actin in cells. It is well established that cell–cell contact inhibits retrograde membrane surface structures and ruffling activity at the contacting edges of motile cells (e.g., Trinkaus et al., 1971). Our experiments using f-actin FSM provide the first direct demonstration that the continuous polymerization and retrograde movement of f-actin that occurs at the free edge of motile cells is shut down in contacted cells. This observation suggests that cell–cell contact is a major regulator of proteins that control the assembly and structural dynamics of f-actin. Thus, the proteins involved in lamellipodial activity and retrograde flow in motile cells including the f-actin pointed end capper/nucleator/cross-linker Arp2/3 complex, the f-actin depolymerizing factor ADF/cofilin (Cramer, 1997; reviewed in Carlier, 1998; Loisel et al., 1999) as well as the f-actin cross-linker α-actinin (Loisel et al., 1999), and f-actin–based myosin motors (Lin et al., 1997) must somehow be dramatically modulated or inhibited after establishment of cell–cell contact. How these proteins are regulated by cell–cell contact is completely unknown, but likely involves the activity of small GTPases of the Rho family (Braga et al., 1997, 1999; Takaishi et al., 1997; Jou and Nelson, 1998; see below). Concomitant with the downregulation of f-actin–associated proteins involved in generation of lamellipodial activity, the proteins mediating contact and establishment of cellular junctions, including cadherins, catenins, and α-actinin and vinculin, must be assembled and establish their respective associations with the cortical actin cytoskeleton after formation of cell–cell contacts (Adams et al., 1998). The dynamics of this process has just begun to be analyzed in living cells (Vasioukhin et al., 2000; Adams et al., 1998; Krendel et al., 1999; Krendel and Bonder, 1999), but the changes in regulation at the biochemical level are completely unknown.
Modulation of Cell–Cell Contacts by MTs
We have found that cell–cell contacts became disrupted that after several hours in nocodazole, which inhibits the growth and promotes the subsequent depolymerization of MTs. We will consider three possible molecular mechanisms for why depolymerization of MTs or suppression of MT growth leads to disruption of cell–cell adherens junctions. One possibility is that depolymerization of MTs results in the disruption of MT-based delivery to the cell periphery of material that is required for the maintenance of adherens junctions. In this case, plus end–directed kinesin or kinesin-related motor proteins would likely be involved. However, because disruption of cell contacts occurred in 100 nM nocodazole when microtubules still were extended to the cell periphery (see Figures 5 and 6), this is unlikely. In addition, thus far no kinesin-related protein has been identified that is thought to play a role in maintenance or formation of cell–cell junctions, although there are many kinesins whose function is unknown (reviewed in Goldstein and Philp, 1999).
In the second scenario, adherens junction stability may be regulated by the balance between soluble and MT-bound APC. APC is a protein that binds MTs in vitro (Munemitsu et al., 1994; Smith et al., 1994) and localizes to MT ends in vivo (Nathke et al., 1996). APC also competes with α-catenin for binding to β-catenin (Hulsken et al., 1994; Rubinfeld et al., 1995), and binding of β-catenin to APC targets β-catenin for destruction via the ubiquitination/proteasome pathway (Munemitsu et al., 1995; Aberle et al., 1997). Thus, in our experiments it is possible that depolymerization of MTs or suppression of MT growth released APC from the MT ends into the cytoplasm, this free APC then could compete with β-catenin away from interaction with α-catenin and thus induce the breakdown of adherens junctions. We attempted to test this hypothesis by immunolocalizing APC before and during nocodazole-induced breakdown of adherens junctions; however, antibodies to APC that we obtained (the kind gift of Inke Nathke) did not cross-react with newt tissue (our unpublished results). Although regulation of β-catenin in adherens junctions via MT release of APC seems plausible, our data show that the reduction of f-actin in adherens junctions was the first indication of their breakdown. Only at later times was there rearrangement of catenins into punctae and then finally a loss from junctions concurrent with breakdown of cell–cell adhesions.
Recent evidence has shown that members of the Rho family of small GTPases regulate cell–cell adhesion (reviewed in Kaibuchi et al., 1999), and MTs may modulate Rho-family signaling (reviewed in Waterman-Storer and Salmon, 1999). Thus, it is also possible that cell–cell adherens junctions are disrupted by MT mediation of a Rho-family small GTPase signal transduction pathway that regulates f-actin dynamics in adherens junctions. Indeed, there is recent mounting evidence that specific phases of plus end MT dynamic instability may influence the activity of Rho GTPases in fibroblasts, such that MT shortening results in the activation of RhoA and the recruitment of f-actin into stress fibers (Ren et al., 1999) and that plus end MT growth activates Rac1 and induces the polymerization of f-actin in lamellipodia (Waterman-Storer et al., 1999; reviewed in Waterman-Storer and Salmon, 1999). Thus, this is a likely mechanism for the disruption of cell–cell junctions by suppression of microtubule growth.
Why has the disruption of cell–cell contacts by MT perturbing drugs not been observed previously? Indeed, there have been multiple studies in which various types of confluent cultured epithelial cell lines have been treated with nocodazole, and this phenomenon has not been reported (e.g., Middleton et al., 1989; Hunziker et al., 1990). However, there is one report that primary cultures of thyroid epithelia lose transepithelial electrical resistance across the cell monolayer 3–6 h after treatment with colchicine and show disruption of the distribution of cell junctional marker proteins (Yap et al., 1995). This suggests that regulation of cell adhesion by MTs may be specific to primary cultures, and this regulation may be lost in transformed cells. In any case, our observation has important implications for the stability and integrity of cell–cell contacts in tissues during the use of antimicrotubule agents in cancer therapy.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Arshad Desai and Tim Mitchison for the kind gift of C2CF, Bertolt Kreft and Keith Burridge for anti–α- and β-catenin antibodies, Inke Nathke for anti-APC antibodies, Kerry Bloom for support of W.C.S., and Albert Harris and Mark Peifer for interesting discussions. During this work, C.M.W.S. was supported by a fellowship from the Jane Coffin Childs Fund for Cancer Research. C.M.W.S and W.C.S. are currently supported by The Scripps Research Institute and the Institute for Childhood and Neglected Diseases. This work was supported by National Institutes of Health Grant GM 24364 to E.D.S.
Footnotes
REFERENCES
- Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. Beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997;16:3797–3804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams CL, Chen YT, Smith SJ, Nelson WJ. Mechanisms of epithelial cell-cell adhesion and cell compaction revealed by high-resolution tracking of E-cadherin-green fluorescent protein. J Cell Biol. 1998;142:1105–1119. doi: 10.1083/jcb.142.4.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacallao R, Antony C, Dotti C, Karsenti E, Stelzer EH, Simons K. The subcellular organization of Madin-Darby canine kidney cells during the formation of a polarized epithelium. J Cell Biol. 1989;109:2817–2832. doi: 10.1083/jcb.109.6.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bershadsky A, Chausovsky A, Becker E, Lyubimova A, Geiger B. Involvement of microtubules in the control of adhesion-dependent signal transduction. Curr Biol. 1996;6:1279–1289. doi: 10.1016/s0960-9822(02)70714-8. [DOI] [PubMed] [Google Scholar]
- Blose SH, Meltzer DI, Feramisco JR. 10-nm filaments are induced to collapse in living cells microinjected with monoclonal and polyclonal antibodies against tubulin. J Cell Biol. 1984;98:847–858. doi: 10.1083/jcb.98.3.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braga VM, Machesky LM, Hall A, Hotchin NA. The small GTPases Rho and Rac are required for the establishment of cadherin-dependent cell-cell contacts. J Cell Biol. 1997;137:1421–1431. doi: 10.1083/jcb.137.6.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braga VM, Del Maschio A, Machesky L, Dejana E. Regulation of cadherin function by Rho and Rac: modulation by junction maturation and cellular context. Mol Biol Cell. 1999;10:9–22. doi: 10.1091/mbc.10.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bre MH, Kreis TE, Karsenti E. Control of microtubule nucleation and stability in Madin-Darby canine kidney cells: the occurrence of noncentrosomal, stable detyrosinated microtubules. J Cell Biol. 1987;105:1283–1296. doi: 10.1083/jcb.105.3.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bre MH, Pepperkok R, Hill AM, Levilliers N, Ansorge W, Stelzer EH, Karsenti E. Regulation of microtubule dynamics and nucleation during polarization in MDCK II cells. J Cell Biol. 1990;111:3013–3021. doi: 10.1083/jcb.111.6.3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buendia B, Bre MH, Griffiths G, Karsenti E. Cytoskeletal control of centrioles movement during the establishment of polarity in Madin-Darby canine kidney cells. J Cell Biol. 1990;110:1123–1135. doi: 10.1083/jcb.110.4.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulinski JC, Bossler A. Purification and characterization of ensconsin, a novel microtubule stabilizing protein. J Cell Sci. 1994;107:2839–2849. doi: 10.1242/jcs.107.10.2839. [DOI] [PubMed] [Google Scholar]
- Carlier MF. Control of actin dynamics. Curr Opin Cell Biol. 1998;10:45–51. doi: 10.1016/s0955-0674(98)80085-9. [DOI] [PubMed] [Google Scholar]
- Cassimeris L, Pryer NK, Salmon ED. Real-time observations of microtubule dynamic instability in living cells. J Cell Biol. 1988;107:2223–2231. doi: 10.1083/jcb.107.6.2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassimeris L. Accessory protein regulation of microtubule dynamics throughout the cell cycle. Curr Opin Cell Biol. 1999;11:134–141. doi: 10.1016/s0955-0674(99)80017-9. [DOI] [PubMed] [Google Scholar]
- Cramer LP. Molecular mechanism of actin-dependent retograde flow in lamellipodia of motile cells. Front Biosci. 1997;2:d260–d270. doi: 10.2741/a189. [DOI] [PubMed] [Google Scholar]
- Danowski BA. Fibroblast contractility and actin organization are stimulated by microtubule inhibitors. J Cell Sci. 1989;93:255–266. doi: 10.1242/jcs.93.2.255. [DOI] [PubMed] [Google Scholar]
- Desai A, Mitchison TJ. Microtubule polymerization dynamics. Annu Rev Cell Dev Biol. 1997;13:83–117. doi: 10.1146/annurev.cellbio.13.1.83. [DOI] [PubMed] [Google Scholar]
- Desai A, Mitchison TJ. Preparation and characterization of caged fluorescein tubulin. Methods Enzymol. 1998;298:125–132. doi: 10.1016/s0076-6879(98)98014-4. [DOI] [PubMed] [Google Scholar]
- Euteneuer U, McIntosh JR. Polarity of some motility-related microtubules. Proc Natl Acad Sci USA. 1981;78:372–376. doi: 10.1073/pnas.78.1.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre-Jonca N, Viard I, French LE, Masson D. Upregulation and redistribution of E-MAP-115 (epithelial microtubule-associated protein of 115 kDa) in terminally differentiating keratinocytes is coincident with the formation of intercellular contacts. J Invest Dermatol. 1999;112:216–225. doi: 10.1046/j.1523-1747.1999.00500.x. [DOI] [PubMed] [Google Scholar]
- Goldman RD. The role of three cytoplasmic fibers in BHK-21 cell motility. I. Microtubules and the effects of colchicine. J Cell Biol. 1971;51:752–762. doi: 10.1083/jcb.51.3.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein LS, Philp AV. The road less traveled: emerging principles of kinesin motor utilization. Annu Rev Cell Dev Biol. 1999;15:141–183. doi: 10.1146/annurev.cellbio.15.1.141. [DOI] [PubMed] [Google Scholar]
- Gotlieb AI, May LM, Subrahmanyan L, Kalnins VI. Distribution of microtubule organizing centers in migrating sheets of endothelial cells. J Cell Biol. 1981;91:589–594. doi: 10.1083/jcb.91.2.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulsken J, Birchmeier W, Behrens J. E-cadherin and APC compete for the interaction with beta-catenin and the cytoskeleton. J Cell Biol. 1994;127:2061–2069. doi: 10.1083/jcb.127.6.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunziker W, Male P, Mellman I. Differential microtubule requirements for transcytosis in MDCK cells. EMBO J. 1990;9:3515–3525. doi: 10.1002/j.1460-2075.1990.tb07560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman A, Drechsel D, Kellogg D, Salser S, Sawin K, Steffen P, Wordeman L, Mitchison T. Preparation of modified tubulins. Methods Enzymol. 1991;196:478–485. doi: 10.1016/0076-6879(91)96041-o. [DOI] [PubMed] [Google Scholar]
- Jeng R, Stearns T. Gamma-tubulin complexes: size does matter. Trends Cell Biol. 1999;9:339–342. doi: 10.1016/s0962-8924(99)01621-9. [DOI] [PubMed] [Google Scholar]
- Jou TS, Nelson WJ. Effects of regulated expression of mutant RhoA and Rac1 small GTPases on the development of epithelial (MDCK) cell polarity. J Cell Biol. 1998;142:85–100. doi: 10.1083/jcb.142.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaibuchi K, Kuroda S, Fukata M, Nakagawa M. Regulation of cadherin-mediated cell-cell adhesion by the Rho family GTPases. Curr Opin Cell Biol. 1999;11:591–596. doi: 10.1016/s0955-0674(99)00014-9. [DOI] [PubMed] [Google Scholar]
- Kaverina I, Rottner K, Small JV. Targeting, capture, and stabilization of microtubules at early focal adhesions. J Cell Biol. 1998;142:181–190. doi: 10.1083/jcb.142.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaverina I, Krylyshkina O, Small JV. Microtubule targeting of substrate contacts promotes their relaxation and dissociation. J Cell Biol. 1999;146:1033–1044. doi: 10.1083/jcb.146.5.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner M, Mitchison T. Beyond self-assembly: from microtubules to morphogenesis. Cell. 1986;45:329–342. doi: 10.1016/0092-8674(86)90318-1. [DOI] [PubMed] [Google Scholar]
- Krendel M, Gloushankova NA, Bonder EM, Feder HH, Vasiliev JM, Gelfand IM. Myosin-dependent contractile activity of the actin cytoskeleton modulates the spatial organization of cell-cell contacts in cultured epitheliocytes. Proc Natl Acad Sci USA. 1999;96:9666–9670. doi: 10.1073/pnas.96.17.9666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krendel MF, Bonder EM. Analysis of actin filament bundle dynamics during contact formation in live epithelial cells. Cell Motil Cytoskeleton. 1999;43:296–309. doi: 10.1002/(SICI)1097-0169(1999)43:4<296::AID-CM3>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Kupfer A, Louvard D, Singer SJ. Polarization of the Golgi apparatus and the microtubule-organizing center in cultured fibroblasts at the edge of an experimental wound. Proc Natl Acad Sci USA. 1982;79:2603–2607. doi: 10.1073/pnas.79.8.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao G, Nagasaki T, Gundersen GG. Low concentrations of nocodazole interfere with fibroblast locomotion without significantly affecting microtubule level: implications for the role of dynamic microtubules in cell locomotion. J Cell Sci. 1995;108:3473–3483. doi: 10.1242/jcs.108.11.3473. [DOI] [PubMed] [Google Scholar]
- Lin CH, Espreafico EM, Mooseker MS, Forscher P. Myosin drives retrograde F-actin flow in neuronal growth cones. Biol Bull. 1997;192:183–185. doi: 10.2307/1542600. [DOI] [PubMed] [Google Scholar]
- Loisel TP, Boujemaa R, Pantaloni D, Carlier MF. Reconstitution of actin-based motility of Listeria and Shigella using pure proteins [see comments] Nature. 1999;401:613–616. doi: 10.1038/44183. [DOI] [PubMed] [Google Scholar]
- Masson D, Kreis TE. Identification and molecular characterization of E-MAP-115, a novel microtubule-associated protein predominantly expressed in epithelial cells. J Cell Biol. 1993;123:357–371. doi: 10.1083/jcb.123.2.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson D, Kreis TE. Binding of E-MAP-115 to microtubules is regulated by cell cycle-dependent phosphorylation. J Cell Biol. 1995;131:1015–1024. doi: 10.1083/jcb.131.4.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton CA, Brown AF, Brown RM, Karavanova ID, Roberts DJ, Vasiliev JM. The polarization of fibroblasts in early primary cultures is independent of microtubule integrity. J Cell Sci. 1989;94:25–32. doi: 10.1242/jcs.94.1.25. [DOI] [PubMed] [Google Scholar]
- Mitchison TJ. Polewards microtubule flux in the mitotic spindle: evidence from photoactivation of fluorescence. J Cell Biol. 1989;109:637–652. doi: 10.1083/jcb.109.2.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munemitsu S, Souza B, Muller O, Albert I, Rubinfeld B, Polakis P. The APC gene product associates with microtubules in vivo and promotes their assembly in vitro. Cancer Res. 1994;54:3676–3681. [PubMed] [Google Scholar]
- Munemitsu S, Albert I, Souza B, Rubinfeld B, Polakis P. Regulation of intracellular beta-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc Natl Acad Sci USA. 1995;92:3046–3050. doi: 10.1073/pnas.92.7.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasaki T, Chapin CJ, Gundersen GG. Distribution of detyrosinated microtubules in motile NRK fibroblasts is rapidly altered upon cell-cell contact: Implications for contact inhibition of locomotion. Cell Motil Cytoskel. 1992;23:45–65. doi: 10.1002/cm.970230106. [DOI] [PubMed] [Google Scholar]
- Nathke IS, Adams CL, Polakis P, Sellin JH, Nelson WJ. The adenomatous polyposis coli tumor suppressor protein localizes to plasma membrane sites involved in active cell migration. J Cell Biol. 1996;134:165–179. doi: 10.1083/jcb.134.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardee JD, Spudich JA. Purification of muscle actin. Methods Enzymol. 1982;85, Pt B:164–181. doi: 10.1016/0076-6879(82)85020-9. [DOI] [PubMed] [Google Scholar]
- Pepperkok R, Bre MH, Davoust J, Kreis TE. Microtubules are stabilized in confluent epithelial cells but not in fibroblasts. J Cell Biol. 1990;111:3003–3012. doi: 10.1083/jcb.111.6.3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez F, Diamantopoulos GS, Stalder R, Kreis TE. CLIP-170 highlights growing microtubule ends in vivo. Cell. 1999;96:517–527. doi: 10.1016/s0092-8674(00)80656-x. [DOI] [PubMed] [Google Scholar]
- Pierre P, Scheel J, Rickard JE, Kreis TE. CLIP-170 links endocytic vesicles to microtubules. Cell. 1992;70:887–900. doi: 10.1016/0092-8674(92)90240-d. [DOI] [PubMed] [Google Scholar]
- Provost E, Rimm DL. Controversies at the cytoplasmic face of the cadherin-based adhesion complex. Curr Opin Cell Biol. 1999;11:567–572. doi: 10.1016/s0955-0674(99)00015-0. [DOI] [PubMed] [Google Scholar]
- Ren XD, Kiosses WB, Schwartz MA. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 1999;18:578–585. doi: 10.1093/emboj/18.3.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder CL, Salmon ED. The vertebrate cell kinetochore and its roles during mitosis. Trends Cell Biol. 1998;8:310–318. doi: 10.1016/s0962-8924(98)01299-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reider CL, Hard RH. Newt lung epithelial cells: cultivation, use and advantages for biomedical science. Int Rev Cytol. 1990;122:153–220. doi: 10.1016/s0074-7696(08)61208-5. [DOI] [PubMed] [Google Scholar]
- Rubinfeld B, Souza B, Albert I, Munemitsu S, Polakis P. The APC protein and E-cadherin form similar but independent complexes with alpha-catenin, beta-catenin, and plakoglobin. J Biol Chem. 1995;270:5549–5555. doi: 10.1074/jbc.270.10.5549. [DOI] [PubMed] [Google Scholar]
- Salmon ED, Saxton WM, Leslie RJ, Karow ML, McIntosh JR. Diffusion coefficient of fluorescein-labeled tubulin in the cytoplasm of embryonic cells of a sea urchin: video image analysis of fluorescence redistribution after photobleaching. J Cell Biol. 1984;99:2157–2164. doi: 10.1083/jcb.99.6.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon ED, Shaw SL, Waters J, Waterman-Storer CM, Maddox PS, Yeh E, Bloom K. A high-resolution multimode digital microscope system. Methods Cell Biol. 1998;56:185–215. [PMC free article] [PubMed] [Google Scholar]
- Saxton WM, Stemple DL, Leslie RJ, Salmon ED, Zavortink M, McIntosh JR. Tubulin dynamics in cultured mammalian cells. J Cell Biol. 1984;99:2175–2186. doi: 10.1083/jcb.99.6.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KJ, Levy DB, Maupin P, Pollard TD, Vogelstein B, Kinzler KW. Wild-type but not mutant APC associates with the microtubule cytoskeleton. Cancer Res. 1994;54:3672–3675. [PubMed] [Google Scholar]
- Steinberg MS, McNutt PM. Cadherins and their connections: adhesion junctions have broader functions. Curr Opin Cell Biol. 1999;11:554–560. doi: 10.1016/s0955-0674(99)00027-7. [DOI] [PubMed] [Google Scholar]
- Takaishi K, Sasaki T, Kotani H, Nishioka H, Takai Y. Regulation of cell-cell adhesion by rac and rho small G proteins in MDCK cells. J Cell Biol. 1997;139:1047–1059. doi: 10.1083/jcb.139.4.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toso RJ, Jordan MA, Farrell KW, Matsumoto B, Wilson L. Kinetic stabilization of microtubule dynamic instability in vitro by vinblastine. Biochemistry. 1993;32:1285–1293. doi: 10.1021/bi00056a013. [DOI] [PubMed] [Google Scholar]
- Trinkaus JP, Betchaku T, Krulikowski LS. Local inhibition of ruffling during contact inhibition of cell movement. Exp Cell Res. 1971;64:291–300. doi: 10.1016/0014-4827(71)90079-6. [DOI] [PubMed] [Google Scholar]
- Troyanovsky SM. Mechanism of cell-cell adhesion complex assembly. Curr Opin Cell Biol. 1999;11:561–566. doi: 10.1016/s0955-0674(99)00021-6. [DOI] [PubMed] [Google Scholar]
- Tsukita S, Furuse M. Occludin and claudins in tight-junction strands: leading or supporting players? Trends Cell Biol. 1999;9:268–273. doi: 10.1016/s0962-8924(99)01578-0. [DOI] [PubMed] [Google Scholar]
- Turnacioglu KK, Sanger JW, Sanger JM. Sites of monomeric actin incorporation in living PtK2 and REF-52 cells. Cell Motil Cytoskeleton. 1998;40:59–70. doi: 10.1002/(SICI)1097-0169(1998)40:1<59::AID-CM6>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Vasiliev JM, Gelfand IM, Domnina LV, Ivanova OY, Komm SG, Olshevskaja LV. Effect of colcemid on the locomotory behavior of fibroblasts. J Embryol Exp Morphol. 1970;24:625–640. [PubMed] [Google Scholar]
- Vasioukhin V, Bauer C, Yin M, Fuchs E. Directed actin polymerization is the driving force for epithelial cell-cell adhesion. Cell. 2000;100:209–219. doi: 10.1016/s0092-8674(00)81559-7. [DOI] [PubMed] [Google Scholar]
- Vasquez RJ, Gard DL, Cassimeris L. XMAP from Xenopus eggs promotes rapid plus end assembly of microtubules and rapid microtubule polymer turnover. J Cell Biol. 1994;127:985–993. doi: 10.1083/jcb.127.4.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez RJ, Howell B, Yvon AM, Wadsworth P, Cassimeris L. Nanomolar concentrations of nocodazole alter microtubule dynamic instability in vivo and in vitro. Mol Biol Cell. 1997;8:973–985. doi: 10.1091/mbc.8.6.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan KT, Tynan SH, Faulkner NE, Echeverri CJ, Vallee RB. Colocalization of cytoplasmic dynein with dynactin and CLIP-170 at microtubule distal ends. J Cell Sci. 1999;112:1437–1447. doi: 10.1242/jcs.112.10.1437. [DOI] [PubMed] [Google Scholar]
- Walker RA, O'Brien ET, Pryer NK, Soboeiro MF, Voter WA, Erickson HP, Salmon ED. Dynamic instability of individual microtubules analyzed by video light microscopy: rate constants and transition frequencies. J Cell Biol. 1988;107:1437–1448. doi: 10.1083/jcb.107.4.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman-Storer CM, Salmon ED. Actomyosin-based retrograde flow of microtubules in the lamella of migrating epithelial cells influences microtubule dynamic instability and turnover and is associated with microtubule breakage and treadmilling. J Cell Biol. 1997;139:417–434. doi: 10.1083/jcb.139.2.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman-Storer CM, Desai A, Bulinski JC, Salmon ED. Fluorescent speckle microscopy, a method to visualize the dynamics of protein assemblies in living cells. Curr Biol. 1998;8:1227–1230. doi: 10.1016/s0960-9822(07)00515-5. [DOI] [PubMed] [Google Scholar]
- Waterman-Storer CM, Worthylake RA, Liu BP, Burridge K, Salmon ED. Microtubule growth activates Rac1 to promote lamellipodial protrusion in fibroblasts [see comments] Nat Cell Biol. 1999;1:45–50. doi: 10.1038/9018. [DOI] [PubMed] [Google Scholar]
- Waterman-Storer CM, Salmon E. Positive feedback interactions between microtubule and actin dynamics during cell motility. Curr Opin Cell Biol. 1999;11:61–67. doi: 10.1016/s0955-0674(99)80008-8. [DOI] [PubMed] [Google Scholar]
- Waters JC, Mitchison TJ, Rieder CL, Salmon ED. The kinetochore microtubule minus-end disassembly associated with poleward flux produces a force that can do work. Mol Biol Cell. 1996;7:1547–1558. doi: 10.1091/mbc.7.10.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap AS, Stevenson BR, Abel KC, Cragoe EJ, Jr, Manley SW. Microtubule integrity is necessary for the epithelial barrier function of cultured thyroid cell monolayers. Exp Cell Res. 1995;218:540–550. doi: 10.1006/excr.1995.1189. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.