

Abstract
Background
Doherty and Zinkernagel, who discovered that antigen presentation is restricted by the major histocompatibility complex (MHC, called HLA in humans), hypothesized that individuals heterozygous at particular MHC loci might be more resistant to particular infectious diseases than the corresponding homozygotes because heterozygotes could present a wider repertoire of antigens. The superiority of heterozygotes over either corresponding homozygote, which we term allele-specific overdominance, is of direct biological interest for understanding the mechanisms of immune response; it is also a leading explanation for the observation that MHC loci are extremely polymorphic and that these polymorphisms have been maintained through extremely long evolutionary periods. Recent studies have shown that in particular viral infections, heterozygosity at HLA loci was associated with a favorable disease outcome, and such findings have been interpreted as supporting the allele-specific overdominance hypothesis in humans.
Methods
An algebraic model is used to define the expected population-wide findings of an epidemiologic study of HLA heterozygosity and disease outcome as a function of allele-specific effects and population genetic parameters of the study population.
Results
We show that overrepresentation of HLA heterozygotes among individuals with favorable disease outcomes (which we term population heterozygote advantage) need not indicate allele-specific overdominance. On the contrary, partly due to a form of confounding by allele frequencies, population heterozygote advantage can occur under a very wide range of assumptions about the relationship between homozygote risk and heterozygote risk. In certain extreme cases, population heterozygote advantage can occur even when every heterozygote is at greater risk of being a case than either corresponding homozygote.
Conclusion
To demonstrate allele-specific overdominance for specific infections in human populations, improved analytic tools and/or larger studies (or studies in populations with limited HLA diversity) are necessary.
Background
The role of genetics in modulating the immune response to infectious diseases is a topic of longstanding interest among epidemiologists, clinicians, population geneticists, and immunologists [1,2]. Zinkernagel and Doherty's Nobel Prize-winning discovery that cellular immunity to viral infections was "restricted" by the highly variable proteins of the Major Histocompatibility Complex (MHC) brought the field of immunogenetics into sharper focus [3]. Their demonstration that T lymphocytes recognize virus antigens displayed on a host cell "in the context of" MHC proteins immediately suggested the idea that genetic differences at the loci encoding the MHC might modulate the intensity and effectiveness of host response to infection.
The relationship between MHC genotype and infectious disease resistance can take several forms. Most simply, individual MHC alleles may be especially effective, or especially ineffective, at presenting antigens from particular infections, so that carrying one or two copies of a given MHC allele might predispose an infected individual to a more or less favorable disease outcome. A second, distinct but compatible hypothesis was suggested by Doherty and Zinkernagel soon after their discovery of MHC restriction: since each MHC allele provides an infected individual with the ability to present a particular set of antigens, individuals who are heterozygous (say, genotype XY) at a particular MHC locus may mount a more vigorous immune response to a given infection, resulting in a better outcome, than individuals who are homozygous for either of the corresponding alleles (XX or YY) [4].
Determining whether either or both of these mechanisms operates for a particular disease is of interest for a variety of basic and applied purposes. Epidemiologically, HLA genotype partially accounts for inter-patient variation in disease severity or progression rates for such long-term viral infections as HIV, human T-cell lymphotropic virus, type 1 (HTLV-1), and hepatitis B and C [5-9]. Such associations can lead to mechanistic studies to test hypotheses about the immunologic basis of these associations [10,11] and could, in principle, be used to help predict individual prognoses [12,13]. At least one clinical study has shown that HLA genotype affects the immune response to a candidate HIV vaccine [14]. Vaccine designers have begun to take account of the HLA genotypes of potential recipients in choosing antigens for inclusion in a vaccine [15], and it has been suggested that HLA genotype be considered in designing samples for inclusion in epidemiologic studies of anti-HIV immune response [12]. In evolutionary biology, the possibility that HLA heterozygotes are more resistant to infectious diseases is the basis for a leading hypothesis to explain the unparalleled diversity of HLA genes (and MHC genes in other vertebrates) and the maintenance of this diversity over long periods of evolutionary time [16].
Of the two ways described above in which MHC genotype may affect disease outcome, the first – association between a particular allele and disease outcome – has been repeatedly documented in human populations by various methods of genetic epidemiology [17]. The second kind of effect, more vigorous immune responses to a pathogen by a heterozygote as compared to homozygotes for the same alleles, which we term "allele-specific overdominance," has been suggested by several animal studies [18-21], although none of these studies is unambiguous (see DISCUSSION). There have been no studies (to our knowledge) in humans that directly compare the infectious disease outcomes of heterozygotes to those of homozygotes for the same alleles (interestingly, the situation is very different for autoimmune diseases; see DISCUSSION for further consideration).
As an alternative to directly examining the hypothesis of allele-specific overdominance, several investigators have compared the infectious disease outcomes of heterozygotes at a given HLA locus, as a group, to the outcomes of homozygotes at the same locus, as a group. In many cases, heterozygotes as a group have shown better infectious disease outcomes (slower disease progression or more rapid clearance of viral infection) than homozygotes as a group [5,6,8,9,14,22], a phenomenon we call "population heterozygote advantage." Because these studies group all homozygotes and group all heterozygotes, they do not, in fact, test the hypothesis of allele-specific overdominance, which is conditional on the alleles involved. Although population heterozygote advantage and allele-specific overdominance are different [10], reports of population heterozygote advantage have frequently been interpreted as confirmations of the Doherty-Zinkernagel hypothesis of allele-specific overdominance.
In this report, we show that although population heterozygote advantage is compatible with allele-specific overdominance, it is also compatible with the opposite; i.e., population heterozygote advantage could arise in a population in which every heterozygote had a worse disease outcome than either of the corresponding homozygotes ("allele-specific underdominance"). The reason for this disconnect is an unusual form of confounding in which particular (protective or detrimental) alleles are over- or under-represented among heterozygotes, as we discuss below; an additional factor that may be involved results from the effects of dominance in creating an asymmetry between homozygotes and heterozygotes. As we have suggested above, it is biologically and epidemiologically useful to determine (a) the beneficial or harmful effects of specific alleles, and (b) the effects of heterozygosity, such as allele-specific overdominance; we show here that unconditional comparisons of all heterozygotes to all homozygotes can reflect either or both of these, and cannot distinguish them. To measure the quantitites of direct interest, analyses that compare disease outcomes conditional on the alleles involved, rather than grouped comparisons of heterozygotes to homozygotes, will be more informative.
In the first section below, we outline a general model for the relationship between genotype at a particular HLA locus and infectious disease outcome (which we dichotomize into favorable and unfavorable), and we use the model to define the conditions under which population heterozygote advantage is expected. We show that, depending on allele frequencies at the HLA locus of interest, population heterozygote advantage may occur when there is allele-specific overdominance, but may also occur under other conditions, including allele-specific underdominance. Two examples are given to illustrate the reasons for the lack of concordance between population and allele-specific effects. In the Discussion, we suggest some possible approaches for estimating allele-specific effects.
General Model
To determine the precise conditions under which population heterozygote advantage will be observed, we consider a general model that predicts the expected outcome of a comparison between heterozygotes and homozygotes in an epidemiological study as a function of (i) the frequencies of resistant and susceptible alleles at a particular locus and (ii) the relationship between genotype at that locus and phenotype. Note that this is not a model for the evolution of genotype frequencies or for the maintenance of MHC heterozygosity, but simply an algebraic framework for predicting the outcome of an epidemiological study of the type cited above, given current allele frequencies and genotype-phenotype mappings.
The model is summarized in Table 1. Suppose that individual alleles of a particular locus confer either susceptibility or resistance to a given disease, and that there are m resistance alleles, R1,R2,.,Rm with frequencies p1,p2,.,pm in the population, and n susceptibility alleles S1,S2,.,Sn with frequencies q1,q2,.,qn. Let 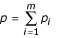 be the total frequency of resistant alleles, and
be the total frequency of resistant alleles, and 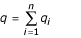 be the total frequency of susceptible alleles, with p + q = 1. Further, define
be the total frequency of susceptible alleles, with p + q = 1. Further, define 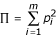 and
and 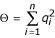 are the sums of squared frequencies of the resistant and susceptible alleles, respectively. We assume Hardy-Weinberg genotype frequencies [23] throughout. Thus, Π is the frequency of RR homozygotes and is an inverse measure of the diversity of resistant alleles, while Θ, the frequency of SS homozygotes, is an inverse measure of the diversity of susceptible alleles.
are the sums of squared frequencies of the resistant and susceptible alleles, respectively. We assume Hardy-Weinberg genotype frequencies [23] throughout. Thus, Π is the frequency of RR homozygotes and is an inverse measure of the diversity of resistant alleles, while Θ, the frequency of SS homozygotes, is an inverse measure of the diversity of susceptible alleles.
Table 1.
Frequency and disease risk of 5 classes of genotypes under the model.
| Class | Susceptible homozygotes | Heterozygote of susceptible alleles | Heterozygote with one R and one S allele | Heterozygote of resistant alleles | Resistant homozygotes |
| Genotype | SiSi | SiSi, i ≠ j | RiSj | RiRj, i ≠ j | RiRi |
| Frequency | Θ | q2 - Θ | 2 pq | p2 - Π | Π |
| Probability of Favorable Outcome | x | ax | bx | cx | dx |
Notation: Frequency of allele Si is qi; frequency of allele Ri is pi. p = ![]() pi; q =
pi; q = ![]() qi = 1 - p;
qi = 1 - p; ![]() ;
; ![]() . The subscripts of R alleles and the subscripts of the S alleles are unrelated.
. The subscripts of R alleles and the subscripts of the S alleles are unrelated.
We assume that SS homozygotes have a probability x of a favorable disease course, and that SS heterozygotes (carrying two different susceptible alleles), SR heterozygotes, RR heterozygotes, and RR homozygotes have probabilities ax, bx, cx, and dx respectively (see Table 1). To give meaning to the notions of resistant and susceptible alleles, we assume d > 1 (RR homozygotes do better than SS homozygotes) and a ≤ b ≤ c (given that one is heterozygous, more R alleles are better). This model can accommodate dominance (a = 1; b = c = d), additivity (a = 1;c = d; b = (1 + d)/2), or recessiveness (a = b = 1 and c = d) of the resistant alleles. It can also accommodate overdominance of the resistant alleles (a > 1; b >d; c >d), in which each heterozygote does better than either corresponding homozygote, and underdominance, in which each heterozygote does worse than either corresponding homozygote (a < 1; b < 1; c <d). In this model, susceptibility and resistance are relative and simply refer respectively to lower and higher probabilities of favorable disease course, given infection. For simplicity, we assume that all S alleles are equivalent and all R alleles are equivalent; our conclusions could obviously be generalized to cases where there is a whole range of effects for different alleles.
Under these assumptions, we can calculate fhom and fhet, the probability of a favorable disease course for homozygotes (the first and fifth classes in Table 1) and for heterozygotes (the 2nd, 3rd, and 4th classes in Table 1).
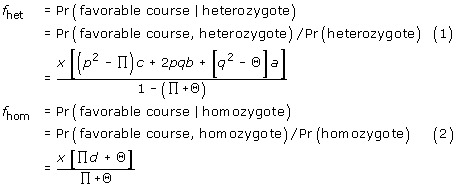
The relative risk (RR) of a favorable outcome for a heterozygote compared to a homozygote is defined as:
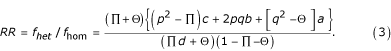
Population heterozygote advantage corresponds to RR > 1. Various formulations for relative risk (or odds ratio, used in case-control studies, such as [6]) are used in studies of HLA heterozygosity and disease outcome, but in all cases the cutoff is one and a value larger or smaller than one (depending on the precise definition used) corresponds to population heterozygote advantage. Using the relative risk equation above, it can be shown by taking partial derivatives with respect to the parameters p, Π and Θ, that (under the assumptions stated above) the relative risk increases with p and Θ and decreases with Π. Thus, population heterozygote advantage is most likely to be observed when resistant alleles are common (p large) but highly diverse (Π is small), and when susceptible alleles are not diverse (large Θ). These trends can be understood intuitively. Homozygotes will be predominantly resistant if R alleles are common and have little diversity and sensitive when S alleles are common and have little diversity. Heterozygotes will be predominantly resistant if R alleles are highly diverse and sensitive if S alleles are highly diverse.
Figure 1 shows the parameter regions in which population heterozygote advantage is expected and those in which the contrary is expected: homozygotes on average are more likely to have a favorable disease course. Each panel reflects a different assumption about the true genetic basis of resistance – assuming that resistant alleles are overdominant; dominant; additive; recessive; or underdominant. In each case, population heterozygote advantage, shown as the black region and corresponding to RR > 1, is most likely when resistant alleles are highly diverse and susceptible alleles have low diversity (bottom right of each panel).
Figure 1.

Population heterozygote advantage as a function of allele-specific effects and allele frequencies. Parameter regions in which heterozygotes will on average have a higher probability of a favorable disease outcome than homozygotes (regions of population heterozygote advantage) are shown in black. Population heterozygote advantage occurs when diversity of resistant alleles is sufficiently high and diversity of susceptible alleles is sufficiently low i.e., toward the bottom right of the parameter space in each panel of the figure. Different panels indicate various assumptions about the genotype-specific relative risks a-d (defined in Table 1). Parameters: Overdominant (a = 1.1, b = 1.6, c = 2, d = 1.5); dominant (a = 1, b = c = d); additive (a = 1, b = (1 + c)/2, c = d); recessive (a = b = 1, c = d); underdominant (a = 0.5, b = 0.9, c = 1.4, d = 1.5). These curves are drawn for p = 0.5. Dominant, additive and recessive curves are valid for all possible values of the free parameters, while underdominant and overdominant curves are examples whose positions depend on the particular values of the parameters a, b, c and d.
Figure 1 shows that allele-specific overdominance (the biological phenomenon of interest) and population heterozygote advantage (the finding of epidemiological studies such as those cited above) are two different things. Population heterozygote advantage (black) may be observed even when resistance is not overdominant, but only dominant, additive, recessive or only underdominant, as long as allele frequencies are sufficiently far toward the lower right of the parameter space (high diversity of resistant alleles and low diversity of susceptible alleles). Figure 1 shows that this is possible under a fairly broad range of parameter combinations (i.e., there are substantial black areas in the dominant, additive and recessive panels).
The converse is also true, though only in what seem to be very special circumstances. That is, even when allele-specific overdominance holds, it is possible that heterozygotes on average will do worse than homozygotes, so population heterozygote advantage will not be observed. This occurs with genotype frequencies sufficiently far toward the top left of Figure 1, with a high diversity of susceptible alleles and low diversity of resistant ones. This occurs only rarely for the parameter values we have chosen (most of the leftmost panel is black), and this seems to be the case for a broad range of parameter values.
Although neither population heterozygote advantage nor allele-specific overdominance implies the other, the two phenomena are of course related. Specifically, the conditions to observe population heterozygote advantage are broadest when allele-specific overdominance holds and become narrower as the underlying genetics becomes more "different" from overdominance of resistance (dominance -> additivity -> recessiveness -> underdominance).
Two Examples
The intuition behind our result that population heterozygote advantage need not reflect allele-specific overdominance can be seen in two simple examples.
Example 1: Suppose that there are two alleles, each at 50% frequency in the population, one (R) conferring resistance and one (S) conferring susceptibility in the homozygous state. Population heterozygote advantage will be observed if the probability of a favorable outcome for heterozygotes is greater than the arithmetic mean of the probabilities of favorable outcomes for RR homozygotes and for SS homozygotes. In this situation, population heterozygote advantage does not require overdominance, but merely allele effects that are more than additive (partial dominance). Dominance of particular HLA alleles conferring resistance, and/or recessiveness of susceptibility (poor outcome) alleles, have been documented for schistosomiasis [24], leprosy [25], acute lymphoblastic leukemia (for which an infectious cause is hypothesized) [26], and hepatitis B (in this case the outcome was vaccine responsiveness) [27].
When allele frequencies are equal, as in Example 1, partial dominance is sufficient to create population heterozygote advantage [10,11]. When allele frequencies differ, the conditions for population heterozygote advantage may become broader or narrower. This can be seen in a second deliberately extreme example.
Example 2: Suppose there are 10 alleles, each with frequency 5%, each conferring resistance to a particular disease, and one allele, with frequency 50%, conferring susceptibility. In the notation of Table 1, this corresponds to p = q = 0.5; Π = (10)(.05)2 = 0.025; Θ = 0.25. In this case, >90% of homozygotes in the population will be homozygous for a susceptible allele, since the frequency of SS homozygotes is Θ = 0.25, while the frequency of RR homozygotes is Π = 0.025. In contrast, 100% of heterozygotes will have at least one resistant allele. In epidemiological terms, this is a form of confounding, in which possession of a resistant allele is positively associated with heterozygosity (the exposure of interest) and positively associated with having a favorable disease course (the outcome of interest). Because of this confounding, under some parameter values, population heterozygote advantage can occur even when heterozygotes are not at an advantage relative to their corresponding homozygotes. Specifically, population heterozygote advantage may be observed when resistance is additive (heterozygotes have risks equal to the average of the risks of the corresponding homozygotes), when resistance is recessive, or even when it is underdominant (heterozygotes have higher risk of disease than either corresponding homozygote).
Continuing this example, suppose that the resistant alleles are recessive to the susceptible one, so that individuals with one or two copies of the susceptible allele have favorable outcomes with probability .3 and individuals with two resistant alleles have favorable outcomes with probability .7; these assumptions correspond to x = 0.3; a = b = 1; c = d = 2.33. In this example, there are no SS heterozygotes since there is only one S allele; SR heterozygotes make up 50% of the population (2pq = 0.5), while RR heterozygotes (carrying two different R alleles) make up p2 - Π = 0.225 = 22.5% of the population. The probability of a favorable outcome for heterozygotes will be the weighted average of the probabilities of a favorable outcome for SR and RR heterozygotes (using equation 1):
![]()
For homozygotes (using equation 2), the probability of a favorable outcome fhom will be the weighted average of the probabilities for SS homozygotes (25% of the population) and for RR homozygotes (2.5% of the population):
![]()
Thus, heterozygotes on average will be 1.26 times more likely to have a favorable outcome, even though each heterozygote has the same outcome as if s/he were homozygous for the worse of the two alleles s/he carries. In epidemiological terms, heterozygotes would have a relative risk of 0.87 of an unfavorable outcome compared to homozygotes.
Both of these examples were chosen for the purposes of clarity, rather than for precise reflection of the allele frequencies in real populations; in particular, few if any populations have a single HLA allele with a frequency of 50%. Moreover, we have simplified the effects of alleles into two categories, resistant and susceptible (R and S), which are simply relative notions. In fact, real alleles will likely have a spectrum of effects, ranging from highly resistant, to highly susceptible, with some alleles having "no effect." Note, however, that "no effect" is also a relative term, and refers to an allele whose effect on disease outcome is close to the population average.
Discussion
The foregoing examples show that the finding of population heterozygote advantage, as in the infectious disease studies cited, does not support an inference of allele-specific overdominance, the condition of primary interest as an immunological hypothesis and a mechanism for the maintenance of MHC diversity. Put another way, population heterozygote advantage may appear due to a combination of the two distinct mechanisms we defined in the Introduction: the protective or detrimental effects of particular alleles (R and S alleles in our model), and the effects of heterozygosity itself. The effects of R and S alleles appear as effects of heterozygosity vs. homozygosity because heterozygotes and homozygotes will in general carry different distributions of S and R alleles; thus, in an analysis that fails to condition on the alleles carried, heterozygosity is confounded with the alleles carried.
One advantage of correctly separating the effects of individual alleles from the effects of heterozygosity conditional on those alleles, is that each of these measures is a characteristic of an individual, rather than a population. For example, if genotypes XX, XY and YY have relative risks 0.6, 2.1, and 1 for clearance of a viral infection, then this should hold true regardless of who else is in the population. In contrast, we have shown that population heterozygote advantage depends on not only the effects of individual genotypes on disease outcome, but also on allele frequencies. Therefore, even if biological and epidemiologic mechanisms were identical in two populations, but allele frequencies differed in those two populations, it would be perfectly reasonable to find population heterozygote advantage in one but not the other.
The problem we have described with measuring population heterozygote advantage is not in principle limited to susceptibility/resistance studies of infectious diseases. In principle, the same problem could occur in any study of HLA associations with disease outcome. Interestingly, however, in our review of the literature on HLA-chronic disease associations, we have found no examples of the problem. Moreover, direct estimates of the allele-specific effects of heterozygosity (relative to the corresponding homozygotes), nearly absent in the infectious disease-HLA literature, are frequently found in studies of HLA genotype and chronic (mostly autoimmune) diseases [28-30]. We do not know the reason for this difference in approach, but suspect that some infectious disease investigators, informed by the evolutionary hypothesis of overdominance for infectious disease resistance and by animal studies, may have a special interest in detecting effects of heterozygosity per se. Many studies of HLA-autoimmune disease associations, on the other hand, seem to focus more on the effects of individual alleles and, having established these, move on to investigate the dependence of these effects on genetic modifiers, including the identity of the individual's other allele at the same locus.
We have shown that when allele-specific overdominance exists, it will often be manifest as population heterozygote advantage, but that a finding of population heterozygote advantage may be consistent with other patterns of allele-specific effects; for example, when resistant and susceptible alleles are equally common and equally diverse, population heterozygote advantage will occur if allele-specific effects are additive, dominant or overdominant. It is difficult to generalize, without doing a specific epidemiologic study, about how the prevalence and diversity of R and S alleles would be likely to occur in a given population. O'Brien et al. [12] found a spectrum of estimated effects of Class I HLA alleles on HIV-1 disease progression, and this spectrum seems visually to be roughly symmetric, but further information would be required to determine plausible values of p, Θ or Π from these data. One might expect the distribution to be skewed toward resistant alleles for diseases that have exerted long-term selection on a population, but it is difficult to make confident predictions about such patterns without detailed knowledge of host-pathogen coevolution.
Our results do not deny that allele-specific overdominance at HLA exists in human populations with respect to infectious disease resistance, but simply raise doubts about the reliability of the major evidence that has been adduced in support of this phenomenon. Existing data suggest that allele-specific overdominance may exist in some animal infectious diseases, but also that simple dominance and other outcomes are commonly observed [11]. Several experimental studies have examined this question using single-strain infections in animals, and while suggestions of allele-specific overdominance have been made, none of the studies has been entirely convincing. Doherty and Zinkernagel [20] demonstrated using congenic mice that MHC homozygotes had more vigorous immune responses to lymphocytic choriomeningitis virus (LCMV) than the corresponding homozygotes, but since the pathology of the infection they used is due to the immune response, the enhanced immune response actually resulted in reduced survival. A study in mice of resistance to Toxoplasma gondii showed that F1 offspring of two genetically divergent parent strains were more resistant than either parent strain, but there seems to be no demonstration that heterozygosity at an MHC locus is responsible [18], while another reference [19] that has been cited [10] as showing overdominance at MHC in the same host-pathogen system does not appear to address the question directly. A study of Marek's disease virus in chickens seems to demonstrate allele-specific overdominance at the MHC using defined genetic backgrounds, but the report does not clearly specify how homogeneous the genetic background was in these experiments [21].
From the perspective of the population genetics debate concerning the role of overdominance in maintaining polymorphism at the MHC, we should note that this mechanism requires allele-specific overdominance for total fitness, not for resistance to individual diseases. As noted by Doherty and Zinkernagel [4], simple dominance for resistance to each of several diseases can create allele-specific overdominance for total fitness, if different alleles confer resistance to different diseases [10,31]. For these reasons, our results, while relevant to the longstanding debate over the relative importance of various kinds of balancing selection in maintaining MHC diversity [32], raise doubts about only one of several lines of evidence for the overdominance hypothesis.
Simple and accurate methods exist to determine for a single pair of alleles how the three possible genotypes (2 homozygotes and one heterozygote) affect disease outcome, and these methods have been used frequently in the literature on autoimmunity and HLA [28-30] and once (to our knowledge) in the HLA-infectious disease literature [33]. It is of additional interest for epidemiologists, immunologists, vaccine designers and evaluators, and population geneticists to know whether HLA heterozygosity in general improves the immune response to and outcome of infectious diseases. To answer this question in a meaningful way, it will be necessary to develop improved methods that estimate the effect of heterozygosity while conditioning on the alleles involved. Meanwhile, findings of population heterozygote advantage should not be interpreted as confirming the mechanism of allele-specific overdominance.
Competing interests
None declared.
Authors' contributions
The methodological principles in this report grew out of discussions held by the three authors. ML formulated the problem and drafted the manuscript, which was edited and revised by CB and RA. All authors read and approved the final manuscript.
Appendix
In this appendix we prove that the relative risk defined in Equation (3) is increasing in p and Θ, and decreasing in Π.
As a preliminary result that will be useful later, note that 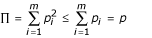 and
and 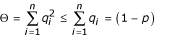 .
.
Proof that the relative risk is increasing in p. We must show that 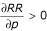 . To do so, we calculate:
. To do so, we calculate:
![]()
By definition, p and Π + Θ are each between zero and one, and all of the parameters Π, Θ, a, b, and c are positive; and by assumption, a ≤ b ≤ c. Therefore; therefore, all terms in the partial derivative are positive, so the relative risk is increasing in p. QED.
Proof that relative risk is decreasing in Π. Since RR = fhet/fhom, it is sufficient to show that the numerator, fhet, is decreasing in Π and the denominator, fhom, is increasing in Π. (This is intuitively clear: fhet is the weighted average of the risk of individuals in the 2nd, 3rd, and 4th columns of Table 1; increasing Π reduces the weight on the highest-risk category, the 4th column with risk cx, thereby reducing the weighted average. Similarly, increasing Π increases the representation of RR homozygotes among homozygotes, thereby increasing the average risk of homozygotes.) Formally: first, we show that fhet, is decreasing in Π, namely that 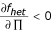 . Taking the partial derivative and rearranging we find:
. Taking the partial derivative and rearranging we find:
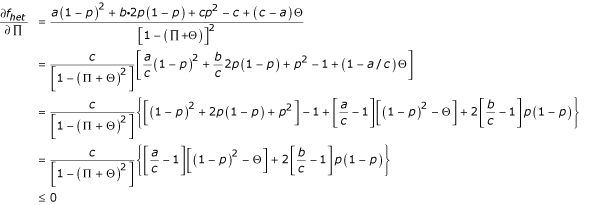
This is justified as follows: The first term is positive by defnition. Inside the curly brackets in the penultimate line above, we have the sum of two terms, each of which itself is the product of a nonpositive term and a nonnegative term; thus, the curly brackets are nonpositive. Specifically, 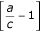 and
and 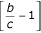 are both nonpositive because we assumed a ≤ b ≤ c. [(1 - p)2 - Θ] is nonnegative as noted at the beginning of the appendix. The term in curly brackets is nonpositive, and the term outside is positive, so the whole expression is nonpositive, as stated.
are both nonpositive because we assumed a ≤ b ≤ c. [(1 - p)2 - Θ] is nonnegative as noted at the beginning of the appendix. The term in curly brackets is nonpositive, and the term outside is positive, so the whole expression is nonpositive, as stated.
It is apparent by inspection that the denominator of the relative risk, fhom, is increasing in Π. Since the numerator of the relative risk is nonincreasing in Π, and the denominator of the relative risk is increasing, the relative risk is decreasing in Π.
The argument that the relative risk is increasing in Θ is exactly symmetric to the argument for Π.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Acknowledgments
Acknowledgements
This work was supported in part by National Insitutes of Health grants R01AI48935 to ML and R29GM54268 to RA. The authors thank the Helen Riaboff Whiteley Center at Friday Harbor for providing a beautiful and conducive environment for beginning this work. Dr. Brian Greenwood, Megan McCloskey, and Dr. John Mittler are thanked for comments on early versions of the MS, and Dr. Wayne Potts is thanked for a particularly helpful referee's report. R.H. Lipsitch is thanked for comments during the final revision.
Contributor Information
Marc Lipsitch, Email: mlipsitc@hsph.harvard.edu.
Carl T Bergstrom, Email: cbergst@u.washington.edu.
Rustom Antia, Email: rantia@biology.emory.edu.
References
- Kallman FJ, Reisner D. Twin studies on the significance of genetic factors in tuberculosis. American Review of Tuberculosis. 1943;47:549–574. [Google Scholar]
- Hill AV. The genomics and genetics of human infectious disease susceptibility. Annu Rev Genomics Hum Genet. 2001;2:373–400. doi: 10.1146/annurev.genom.2.1.373. [DOI] [PubMed] [Google Scholar]
- Zinkernagel RM, Doherty PC. Immunological surveillance against altered self components by sensitised T lymphocytes in lymphocytic choriomeningitis. Nature. 1974;251:547–548. doi: 10.1038/251547a0. [DOI] [PubMed] [Google Scholar]
- Doherty PC, Zinkernagel RM. A biological role for the major histocompatibility antigens. Lancet. 1975;1:1406–1409. doi: 10.1016/S0140-6736(75)92610-0. [DOI] [PubMed] [Google Scholar]
- Tang J, Costello C, Keet IP, Rivers C, Leblanc S, Karita E, Allen S, Kaslow RA. HLA class I homozygosity accelerates disease progression in human immunodeficiency virus type 1 infection. AIDS Res Hum Retroviruses. 1999;15:317–324. doi: 10.1089/088922299311277. [DOI] [PubMed] [Google Scholar]
- Thursz MR, Thomas HC, Greenwood BM, Hill AV. Heterozygote advantage for HLA class-II type in hepatitis B virus infection. Nat Genet. 1997;17:11–12. doi: 10.1038/ng0997-11. [DOI] [PubMed] [Google Scholar]
- Hill AV. The immunogenetics of human infectious diseases. Annu Rev Immunol. 1998;16:593–617. doi: 10.1146/annurev.immunol.16.1.593. [DOI] [PubMed] [Google Scholar]
- Asti M, Martinetti M, Zavaglia C, Cuccia MC, Gusberti L, Tinelli C, Cividini A, Bruno S, Salvaneschi L, Ideo G, Mondelli MU, Silini EM. Human leukocyte antigen class II and III alleles and severity of hepatitis C virus-related chronic liver disease. Hepatology. 1999;29:1272–1279. doi: 10.1002/hep.510290445. [DOI] [PubMed] [Google Scholar]
- Jeffery KJ, Siddiqui AA, Bunce M, Lloyd AL, Vine AM, Witkover AD, Izumo S, Usuku K, Welsh KI, Osame M, Bangham CR. The influence of HLA class I alleles and heterozygosity on the outcome of human T cell lymphotropic virus type I infection. J Immunol. 2000;165:7278–7284. doi: 10.4049/jimmunol.165.12.7278. [DOI] [PubMed] [Google Scholar]
- Penn DJ. The scent of genetic compatibility: Sexual selection and the major histocompatibility complex. Ethology. 2002;108:1–21. doi: 10.1046/j.1439-0310.2002.00768.x. [DOI] [Google Scholar]
- Penn DJ, Damjanovich K, Potts WK. MHC heterozygosity confers a selective advantage against multiple-strain infections. Proc Natl Acad Sci U S A. 2002;99:11260–11264. doi: 10.1073/pnas.162006499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrington M, Nelson G, O'Brien SJ. Considering genetic profiles in functional studies of immune responsiveness to HIV-1. Immunol Lett. 2001;79:131–140. doi: 10.1016/S0165-2478(01)00275-9. [DOI] [PubMed] [Google Scholar]
- Kaslow RA, Carrington M, Apple R, Park L, Munoz A, Saah AJ, Goedert JJ, Winkler C, O'Brien SJ, Rinaldo C, Detels R, Blattner W, Phair J, Erlich H, Mann DL. Influence of combinations of human major histocompatibility complex genes on the course of HIV-1 infection. Nat Med. 1996;2:405–411. doi: 10.1038/nm0496-405. [DOI] [PubMed] [Google Scholar]
- Kaslow RA, Rivers C, Tang J, Bender TJ, Goepfert PA, El Habib R, Weinhold K, Mulligan MJ. Polymorphisms in HLA class I genes associated with both favorable prognosis of human immunodeficiency virus (HIV) type 1 infection and positive cytotoxic T-lymphocyte responses to ALVAC-HIV recombinant canarypox vaccines. J Virol. 2001;75:8681–8689. doi: 10.1128/JVI.75.18.8681-8689.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aidoo M, Lalvani A, Gilbert SC, Hu JT, Daubersies P, Hurt N, Whittle HC, Druihle P, Hill AV. Cytotoxic T-lymphocyte epitopes for HLA-B53 and other HLA types in the malaria vaccine candidate liver-stage antigen 3. Infect Immun. 2000;68:227–232. doi: 10.1128/iai.68.1.227-232.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahata N, Nei M. Allelic genealogy under overdominant and frequency-dependent selection and polymorphism of major histocompatibility complex loci. Genetics. 1990;124:967–978. doi: 10.1093/genetics/124.4.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill AVS, Motulsky AG. Natural selection for disease susceptibility and resistance genes: examples and prospects. In: Stearns SC, editor. Evolution in Health and Disease. Oxford: Oxford University Press; 1999. pp. 50–61. [Google Scholar]
- Williams DM, Grumet FC, Remington JS. Genetic control of murine resistance to Toxoplasma gondii. Infect Immun. 1978;19:416–420. doi: 10.1128/iai.19.2.416-420.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLeod R, Skamene E, Brown CR, Eisenhauer PB, Mack DG. Genetic regulation of early survival and cyst number after peroral Toxoplasma gondii infection of A × B/B × A recombinant inbred and B10 congenic mice. J Immunol. 1989;143:3031–3034. [PubMed] [Google Scholar]
- Doherty PC, Zinkernagel RM. Enhanced immunological surveillance in mice heterozygous at the H-2 gene complex. Nature. 1975;256:50–52. doi: 10.1038/256050a0. [DOI] [PubMed] [Google Scholar]
- Martin A, Dunnington EA, Briles WE, Briles RW, Siegel PB. Marek's disease and major histocompatibility complex haplotypes in chickens selected for high or low antibody response. Anim Genet. 1989;20:407–414. doi: 10.1111/j.1365-2052.1989.tb00896.x. [DOI] [PubMed] [Google Scholar]
- Carrington M, Nelson GW, Martin MP, Kissner T, Vlahov D, Goedert JJ, Kaslow R, Buchbinder S, Hoots K, O'Brien SJ. HLA and HIV-1: heterozygote advantage and B*35-Cw*04 disadvantage. Science. 1999;283:1748–1752. doi: 10.1126/science.283.5408.1748. [DOI] [PubMed] [Google Scholar]
- Hartl DL, Clark AG. Principles of Population Genetics. 3. Sunderland, MA: Sinauer Associates; 1997. [Google Scholar]
- McManus DP, Ross AG, Williams GM, Sleigh AC, Wiest P, Erlich H, Trachtenberg E, Guanling W, McGarvey ST, Li YS, Waine GJ. HLA class II antigens positively and negatively associated with hepatosplenic schistosomiasis in a Chinese population. Int J Parasitol. 2001;31:674–680. doi: 10.1016/S0020-7519(01)00132-1. [DOI] [PubMed] [Google Scholar]
- van Eden W, de Vries RR, Mehra NK, Vaidya MC, D'Amaro J, van Rood JJ. HLA segregation of tuberculoid leprosy: confirmation of the DR2 marker. J Infect Dis. 1980;141:693–701. doi: 10.1093/infdis/141.6.693. [DOI] [PubMed] [Google Scholar]
- Dorak MT, Lawson T, Machulla HK, Darke C, Mills KI, Burnett AK. Unravelling an HLA-DR association in childhood acute lymphoblastic leukemia. Blood. 1999;94:694–700. [PubMed] [Google Scholar]
- Kruskall MS, Alper CA, Awdeh Z, Yunis EJ, Marcus-Bagley D. The immune response to hepatitis B vaccine in humans: inheritance patterns in families. J Exp Med. 1992;175:495–502. doi: 10.1084/jem.175.2.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonagh JE, Dunn A, Ollier WE, Walker DJ. Compound heterozygosity for the shared epitope and the risk and severity of rheumatoid arthritis in extended pedigrees. Br J Rheumatol. 1997;36:322–327. doi: 10.1093/rheumatology/36.3.322. [DOI] [PubMed] [Google Scholar]
- Wordsworth P, Pile KD, Buckely JD, Lanchbury JS, Ollier B, Lathrop M, Bell JI. HLA heterozygosity contributes to susceptibility to rheumatoid arthritis. Am J Hum Genet. 1992;51:585–591. [PMC free article] [PubMed] [Google Scholar]
- Jean S, Quelvennec E, Alizadeh M, Guggenbuhl P, Birebent B, Perdriger A, Grosbois B, Pawlotsky PY, Semana G. DRB1*15 and DRB1*03 extended haplotype interaction in primary Sjogren's syndrome genetic susceptibility. Clin Exp Rheumatol. 1998;16:725–728. [PubMed] [Google Scholar]
- Hedrick PW. Pathogen resistance and genetic variation at MHC loci. Evolution Int J Org Evolution. 2002;56:1902–1908. doi: 10.1111/j.0014-3820.2002.tb00116.x. [DOI] [PubMed] [Google Scholar]
- Hedrick PW. Balancing selection and MHC. Genetica. 1998;104:207–214. doi: 10.1023/A:1026494212540. [DOI] [PubMed] [Google Scholar]
- Flores-Villanueva PO, Yunis EJ, Delgado JC, Vittinghoff E, Buchbinder S, Leung JY, Uglialoro AM, Clavijo OP, Rosenberg ES, Kalams SA, Braun JD, Boswell SL, Walker BD, Goldfeld AE. Control of HIV-1 viremia and protection from AIDS are associated with HLA-Bw4 homozygosity. Proc Natl Acad Sci U S A. 2001;98:5140–5145. doi: 10.1073/pnas.071548198. [DOI] [PMC free article] [PubMed] [Google Scholar]