

Abstract
Background
Since lung epithelial cells are constantly being exposed to reactive oxygen intermediates (ROIs), the alveolar surface is a major site of oxidative stress, and each cell type may respond differently to oxidative stress. We compared the extent of oxidative DNA damage with that of mitochondrial injury in lung epithelial cells at the single cell level.
Result
DNA damage and mitochondrial injury were measured after oxidative stress in the SV-40 transformed lung epithelial cell line challenged with hydrogen peroxide (H2O2). Single cell analysis of DNA damage was determined by assessing the number of 8-oxo-2-deoxyguanosine (8-oxo-dG) positive cells, a marker of DNA modification, and the length of a comet tail. Mitochondrial membrane potential, ΔΨm, was determined using JC-1. A 1 h pulse of H2O2 induced small amounts of apoptosis (3%). 8-oxo-dG-positive cells and the length of the comet tail increased within 1 h of exposure to H2O2. The number of cells with reduced ΔΨm increased after the addition of H2O2 in a concentration-dependent manner. In spite of a continual loss of ΔΨm, DNA fragmentation was reduced 2 h after exposure to H2O2.
Conclusion
The data suggest that SV-40 transformed lung epithelial cells are resistant to oxidative stress, showing that DNA damage can be dissociated from mitochondrial injury.
Background
Aerobic cells are constantly exposed to reactive oxygen intermediates (ROIs). Increased intracellular levels of the ROIs, superoxide (O2-), hydroxyl radical (·OH), or hydrogen peroxide (H2O2) are referred to as oxidative stress [1,2]. The alveolar surface of the lung is a major target for oxidative stress. Also, cigarette smoking can cause an acute inflammatory reaction in the lung, characterized by the accumulation and activation of leukocytes in vivo, producing ROIs and nitrogen species in high concentrations [3,4]. These different reactive species might be responsible for much of the tissue injuries and disease states associated with inflammation. Increased oxidative DNA damage, such as 8-hydroxyguanine formation in human lung tissue [5], is found in smokers compared with non-smokers. Oxidative stress may play an important role in the pathogenesis of smoking-associated diseases, such as chronic obstructive pulmonary diseases (COPD) [6,7], asthma [8], and carcinogenesis [9-11].
ROIs damage DNA, resulting in base modifications, e.g. 8-oxo-2'-deoxyguanosine (8-oxo-dG) [12,13], as well as causing DNA strand-breaks [14]. The presence of 8-oxo-dG in DNA is considered as a marker of oxidative stress and DNA damage [13]. We quantified DNA modifications induced by H2O2, as shown by 8-oxo-dG-positive cells at the single cell level, using laser-scanning cytometry (LSC). In general, this method of analysis can be applied to stained cells adherent to slides, eliminating the cell loss that inevitably occurs due to repeated centrifugations during sample preparation for flow cytometry. And also, the alkaline version of the single cell gel electrophoresis (SCGE) or Comet assay represents a sensitive technique for the detection of single-stranded DNA breaks [15]. We used the Comet assay to measure the extent of single-stranded DNA breaks in epithelial cells treated with H2O2, and compared this with the number of base modifications at the single cell level.
An altered mitochondrial membrane potential followed by increased ROI generation, the loss of mitochondrial cardiolipin, and increased intracellular Ca2+ concentrations have recently been described as common features of apoptosis [16]. Apoptosis is a programmed event, which is regulated by genes, and is irreversible in terms of the DNA cleavage.
The aim of the current study was to investigate the susceptibility of DNA damage and mitochondrial injury caused by H2O2 in cultured SV-40 transformed lung epithelial cells, which may prove a useful in vitro model of the lung. The extent of oxidative DNA damage and mitochondrial injury was assessed at the single cell level. We hypothesized that lung epithelial cells would be relatively resistant to oxidative stress and one may therefore be able to distinguish between DNA damage and mitochondrial injury.
Results
Cell cycle analysis
To compare the cell cycle effects of different culture conditions on starved BEAS-2B cell lines treated with H2O2, the number of proliferating cells (defined as BrdU-positive cells) and apoptotic cells were measured after culturing with growth factors (GF) or in GF-free medium. Quiescent cells starved of GF for 48 h were exposed to H2O2 (100–500 μM) for 1 h, after which the cells were cultured in fresh media without GF. After 1 day, cycle analysis indicated that the cells treated with H2O2 were similar to untreated controls (Fig. 1A and 1C). However, when quiescent cells were exposed to H2O2 followed by culturing in fresh medium containing GF to initiate entry into cycle, a higher percentage of G1 phase-cells (83%) and a lower percentage of S phase-cells (6%) were found compared to the controls (70% and 16% respectively; Fig. 1B and 1C). H2O2 inhibited cell proliferation in a concentration-dependent manner in conjunction with GF (Fig. 1C). As a result, few apoptotic cells (3%) were detected, seen as a "pre-G1" phase population (Fig. 1B).
Figure 1.

Distribution of cell cycle phases in BEAS-2B cells exposed to H2O2. A, quiescent cells were exposed to H2O2 for 1 h, followed by culturing in growth factor (GF) free-media for 24 h. B, quiescent cells were exposed to H2O2 for 1 h, followed by culturing in media supplemented with GF for 24 h. All floating and attached cells were collected, incubated with BrdU, and stained with anti-BrdU and PI. C, graphic representation of H2O2-exposed BrdU-positive cells. *, significantly different from control at p < 0.05 (Mann-Whitney U test).
8-oxo-dG for detecting DNA damage
It had previously been shown by high-performance liquid chromatography (HPLC) that H2O2 induces 8-oxo-dG, a DNA damage marker [16]. Exposure of quiescent WI-26VA4 cells, another SV-40 transformed epithelial cell line, to H2O2 had no effect on the number of 8-oxo-dG-positive cells (Fig. 2A). In the present study, we used a direct binding fluorescent probe to detect 8-oxo-dG in the DNA, which was quantified by LSC. The rate of DNA damage measured was that induced by a 1 h exposure of BEAS-2B cells to H2O2. We also assessed the number of 8-oxo-dG-positive cells by LSC (Fig. 2B). H2O2 treatment increased the number of 8-oxo-dG-positive cells increased in a concentration-dependent manner (2.1 versus 19.6%; Fig. 2C).
Figure 2.
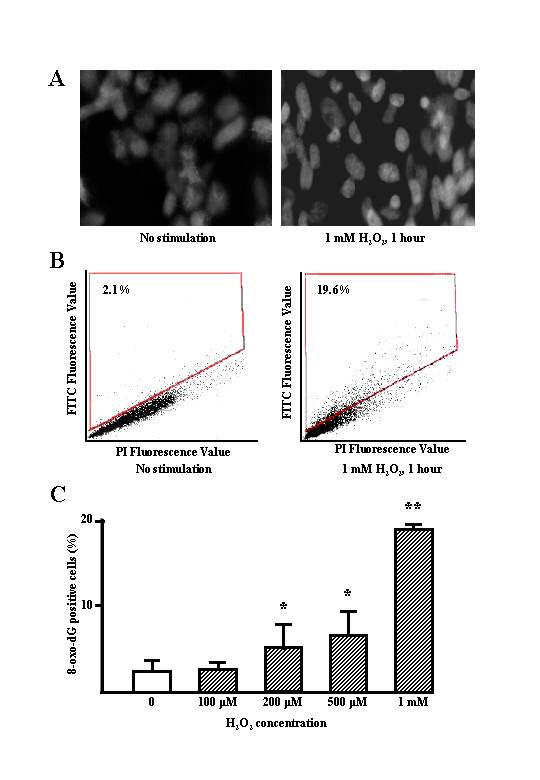
A, green-fluorescent image of intranuclear 8-oxoguanine expression. WI-26VA4 cells were cultured with and without 1 mM H2O2 for 1 h. The cells were fixed with cold methanol in situ, then stained with a protein, which binds specifically and directly to 8-oxo-dG. B, representative dot plot of DNA content vs. 8-oxo-dG of FITC staining in WI-26VA4 cells treated with or without 1 mM H2O2 for 1 h. Cells were counterstained with PI, and analyzed by laser scanning cytometer. The number in each dot-plot indicates the percentage of 8-oxo-dG positive cells. C, graphic representation of the 8-oxo-dG positive cells that increased in an H2O2 concentration-dependent manner. *, **, significantly different from controls at p < 0.05 and p < 0.01, respectively (Mann-Whitney U-test).
Comet assay
To determine the effect of H2O2 on single-strand DNA breaks in BEAS-2B cells, cells were treated with H2O2 (200–1000 μM) and the tail moment (TM) determined. The mean tail moment relative to time of H2O2 treatment is shown in Figure 3B. For the first 1h after H2O2 treatment, the mean TM showed both a time- and concentration-dependent decrease. However, the mean TM increased at every concentration of H2O2 between 60 and 120 min.
Figure 3.
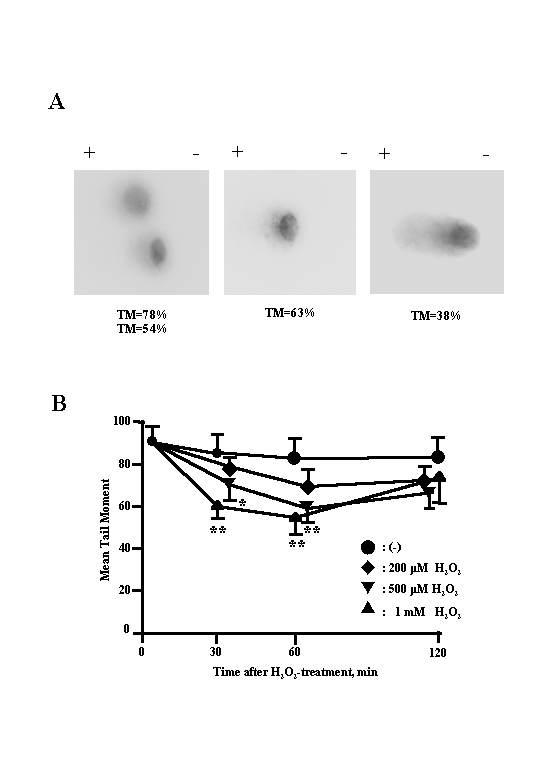
A, typical DNA migration patterns of BEAS-2B cells treated with 500 μM H2O2 for 1 h at 37°C. B, tail moment (TM) representing the DNA breaks induced by H2O2 in BEAS-2B cells. Cells were exposed to H2O2 at 37°C for 1 h and then subjected to alkaline single-cell gel electrophoresis (Comet assay). Values are expressed as the means ± SD of 3 experiments carried out in duplicate (n = 3). *, **, significantly different from controls at p < 0.05 and p < 0.01, respectively (Mann-Whitney U test).
JC-1 staining
Previous studies have suggested that a decline of ΔΨm may be an early event in the process of cell death, and so we determined ΔΨm at various times after H2O2 treatment in BEAS-2B cells, using the membrane potential-sensitive probe, JC-1, which forms monomers (green fluorescence) at a low membrane potential and J-aggregates (red fluorescence) at a higher membrane potential [17]. The ratio between the red and the green signals is indicative of the ΔΨm. There was a dramatic drop of the red fluorescence in cells exposed to H2O2, indicating a loss of ΔΨm. Figure 4A shows a clear increase in the percentage of cells that emitted only green fluorescence after H2O2 treatment, representing cells with a depolarized mitochondrial membrane. H2O2 caused a concentration-dependent reduction or loss in ΔΨm (Fig. 4B).
Figure 4.

A, flow cytometric analysis of the preconditioning-induced changes in the inner mitochondrial membrane potential in BEAS-2B cells. The BEAS-2B cells were exposed to H2O2, and stained with the potentiometric dyes JC-1. Bivariate plots of red (FL2) versus green (FL1) fluorescence were used as an estimate of mitochondrial membrane potential (ΔΨm). B, percent of cells with ΔΨm disruption vs. exposure time. Cells were treated with 200, 500 μM, and 1 mM of H2O2 for 10, 30, 60, and 120 min. Untreated controls and H2O2-treated cells were then stained with JC-1 and analyzed by flow cytometry as described in Methods. n = 10, 000 cells/analysis. *, **, significantly different from controls at p < 0.05 and p < 0.01, respectively (Mann-Whitney U-test).
Immunohistochemistry for nitrotyrosine
An important pathway that enhances NO cytotoxicity is the rapid reaction with superoxide anion radicals (O2·-) to yield peroxynitrite (ONOO-), which has a wide range of biologic activities that includes oxidation of biomolecules and protein tyrosine nitration [18]. It is widely accepted that peroxynitrite is a key mediator of tissue injury in oxidative stress. To determine whether H2O2-induced peroxynitrite formation occurred in BEAS-2B cells, cells were stained with an anti-nitrotyrosine antibody. H2O2 (60 min, 500–1000 μM) induced clear perinuclear nitrotyrosine staining (Fig. 5B and 5C). The peroxynitrite donor, SIN-1, induced a predominantly nuclear membrane distribution of nitrotyrosine staining (Fig. 5D).
Figure 5.
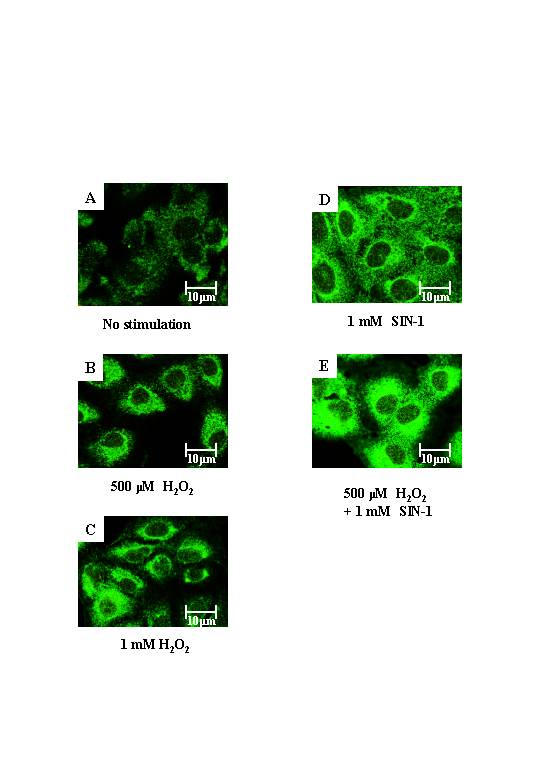
Indirect immunofluorescence of nitrotyrosine expression in BEAS-2B cells. A, cells without stimulation, B, cells exposed to 500 μM H2O2, C, cells exposed to 1 mM H2O2, D, cells exposed to 1 mM SIN-1, and E, cells exposed to 500 μM H2O2 and 1 mM SIN-1 for 1 h were stained with anti-nitrotyrosine antibody. Each image was captured by laser confocal microscope. Results are representative of 3 individual experiments.
Discussion
The alveolar surface of the lung is a major site of oxidant injury. In lung epithelial cells, oxidative damage to DNA is caused not only by air pollutants, but also by neutrophil products [19]. Using high-density DNA microarrays, lung epithelial cells exposed to oxidative injury showed early induction of bcl-2 and mdm-2, genes which are known to be are involved in the regulation of apoptosis [20]. Both DNA and mitochondrial events combine to regulate cell cycle progression and apoptosis after treatment with H2O2. This study has assessed DNA damage and mitochondrial injury at the single cell level.
First, we have confirmed an effect of H2O2 on replicating DNA and apoptosis during the cell cycle. Oxidative stress is believed to induce the cyclin dependent kinase inhibitor, p21, thereby inhibiting DNA replication and hence allowing additional time to repair damaged DNA. Consistent with a previous report [21] demonstrating that H2O2 induced G1 arrest in A549 cells, we have shown that a 1-h pulse of H2O2 induced cell arrest at G0/G1 phase and small amounts of apoptosis (only 3 %) in SV-40 transformed lung epithelial cells in the presence of growth factors.
A variety of endogenous and environmental factors can cause DNA damage [22,23]. Estimates indicate that a human cell sustains ~10, 000 lesions per day due to metabolism-generated free radicals [23]. There are two types of DNA damage induced by the genotoxic substance H2O2, DNA modifications and DNA breaks. Over 100 oxidative stress-induced DNA modifications have been characterized [24,25], of which the 8-oxo-dG lesion is the most common and extensively studied oxidative lesion [26]. To locate of oxidative damage in situ, a number of different investigators have developed antibodies against the oxidative DNA lesion, 8-oxo-dG [27-29]. We used an FITC-conjugated substrate to detect 8-oxo-dG and analyzed this by LSC at the single cell level. Traditional methods using HPLC [16] can assess the total amount of DNA damage per genomes whereas LSC indicates DNA damage at each individual cell. The detection of 8-oxo-dG by fluorescence in a flow cytometer [30] or by LSC might be useful tools for estimating DNA damage at the single cell level. Consistent with Dizdaroglu et al. [26], we have shown that 8-oxo-dG accounts for 5% of the total oxidized bases known to occur in DNA, and that even untreated cells showed 8-oxo-dG positivity (2.1%; Fig. 2B). In addition, we have demonstrated that H2O2 induced 8-oxo-dG-positive cells appear in a concentration-dependent manner.
The Comet assay is a sensitive method for the detection of DNA strand-breaks and alkali-labile (e.g. apurinic) sites in cells exposed to genotoxic carcinogens [15]. In this study, we did not use a modified comet assay with a purified DNA repair enzyme, formamidopyrimidine DNA-glycosylase (FPG), even though this method is more sensitive than the assay for detecting FPG-sensitive sites than our classic comet assay [31,32]. However, FPG is one of the multifunctional DNA repair enzymes participating in base excision repair [33]. We used the classic comet assay to detect repair phase of DNA breaks. To evaluate DNA fragmentation as a consequence of oxidative stress, cells treated with H2O2 showed an initial concentration-dependent increase in strand-breaks up to 1 h before a subsequent decrease in DNA fragmentation, which may reflect the mechanism involved in the repair of damaged DNA.
The increasing amounts of 8-oxo-dG and tail moments following H2O2 exposure suggest that 8-oxo-dG might give an estimate of the levels of DNA fragmentation. This finding is disagrees with previous reports [34,35], which demonstrated significant increases in fragmented DNA in cells treated with H2O2 but no concomitant increase in 8-oxo-dG. Glutathione (GSH) is a major non-protein antioxidant that protects cells from oxidative stress. The airway epithelium is a target for ROIs, either inhaled or released during lung inflammation, and is resistant to oxidant injury due to its abundant level of intracellular GSH [36]. Another explanation is that lung epithelial cells may be susceptible to repair DNA damage by FPG as well as 8-oxo-dG DNA glycosylase (OGG-1) [33]. In airway epithelial cells where DNA repair occurs, we see parallel induction of both DNA strand-breaks and DNA modifications, albeit both at low levels.
In this study, the possibility exits that resistance of the SV-40 transformed cell line to DNA damage may not be the same as that of human primary lung epithelium, because SV-40 large T antigen simultaneously disables the retinoblastoma (pRB) and p53 tumor suppressor pathways [37]. More experiments on other types of lung epithelial cells transformed by different means need to be analysed to establish the mechanism of DNA repair.
Recent progress has shown the central role of mitochondria in death signaling [38]. In certain situations, death stimuli induce an increase in the permeability of the outer mitochondrial membrane that releases apoptosis inducers, such as cytochrome c [39] and apoptosis-inducing factor (AIF) [40]. In our study, the ΔΨm was quantified by the flow cytometric analysis of JC-1-stained cells. This potential is the driving force for crucial cellular functions, ranging from ATP synthesis to the participation of mitochondria in Ca2+ signaling. We have shown that H2O2 causes a concentration-dependent dissipation of ΔΨm (Fig. 4). Under conditions where extensive growth-arrest and moderate apoptosis were observed, others have reported a general degradation of mitochondrial, but not nuclear polynucleotides during the early stage response to oxidative stress [41]. Mitochondria are not only the major site for the generation of reactive oxygen species, but one of the main targets of oxidative damage. One of the major products of DNA oxidation, 8-oxo-dG, accumulates in mitochondrial DNA (mtDNA) to three times the level in nuclear DNA [42]. Mitochondrial DNA is particularly susceptible to damage by ROIs because of its close proximity to the electron transport chain and its lack of protective histones. Prolonged dissipation of ΔΨm might be attributed to this susceptibility of mitochondrial DNA damage.
The involvement of peroxynitrite (ONOO-) in inflammatory diseases has been implicated by the detection of 3-nitrotyrosine, an allegedly characteristic protein oxidation product, in various inflamed tissues. The formation of reactive nitrogen species (RNS), including peroxynitrite and nitrogen dioxide (NO2), may be the prime reason in many cases why NO· can contribute to the etiology of inflammatory lung diseases, such as asthma [43]. Detection of 3-nitrotyrosine at inflammatory sites serves as a biochemical marker for peroxynitrite formation [18,44]. Our results indicate that H2O2 and the SIN-1 might produce peroxynitrite, which was detected as 3-nitrotyrosine immunoreactivity. We suggest that H2O2, as well as peroxynitrite, might induce DNA damage and mitochondrial injury.
In conclusion, DNA modifications, detected as 8-oxo-dG positive cells, and DNA fragmentation, assessed by the comet tail length, showed a similar pattern and were increased in lung epithelial cell lines exposed to H2O2. H2O2 induced the cells to undergo apoptosis and/or G1 arrest, which is the cell-cycle phase for repairing DNA damage. The nucleus and mitochondria of the SV-40 transformed lung epithelial cells exhibited different susceptibilities to oxidative stress injury. This may be the mechanism of apoptosis, which is a programmed event, and is irreversible in terms of DNA cleavage. The mechanism of lung resistance to oxidative stress may be in the dissociation of mitochondria damage from the cascade that induces apoptosis, thus increasing the threshold for DNA repair.
Methods
Cell culture
BEAS-2B cells, which are SV-40 immortalized cells derived from normal human bronchial epithelium [45], were obtained from American Type Culture Collection (Manassas, VA). The cells (passages 32 to 70) were cultured in serum-free keratinocyte basal medium (Sigma; St. Louis, MO), which was supplemented with 10 ng/ml of epidermal growth factor (Gibco BRL; Grand Island, NY) and 30 μg/ml of bovine pituitary extract (Gibco BRL) in the presence of 5% CO2 at 37°C. The human lung cell line WI-26VA4 was kindly provided by Human Science Research Resource Bank (Osaka, Japan). Cells (passages 162 to 180) were cultured in Dulbecco's modified Eagle's medium (DMEM; IWAKI, Chiba, Japan) supplemented with 10% fetal calf serum, amino acids (L-arginine HCl 252.8 mg/l, L-cystine 48.0 mg/l, L-histidine HCl 83.8 mg/l, L-isoleucine 105.0 mg/l, L-leucine 105.0 mg/l, L-lysine HCl 146.2 mg/l, L-methionine 29.8 mg/l, L-phenylalanine 66.0 mg/l, L-threonine 95.2 mg/l, L-tryptophan 20.4 mg/l, L-tyrosine 72.4 mg/l, and L-valine 93.8 mg/l), mixed vitamins (folic acid 1.0 mg/ml, nicotinamide 1.0 mg/ml, pyridoxal HCl 1.0 mg/ml, riboflavin 0.1 mg/ml, and thiamin HCl 1.0 mg/ml), penicillin (100 U/ml), and streptomycin (100 μg/ml) in the presence of 5% CO2 at 37°C.
Cell cycle analysis using BrdU incorporation
BEAS-2B cells exposed to H2O2 were pulse-labeled with 50 μM 5-bromo-2'-deoxyuridine (BrdU) (Sigma Aldrich; Tokyo, Japan) for 30 min one day after exposure, and harvested by trypsinization, pelleted, and gently resuspended in phosphate buffered saline (PBS; OXOID, Hampshire, England). The cells were fixed with ice-cold 70% ethanol, centrifuged and resuspended in 1 ml 2 N HCl containing 0.05% Triton X-100 (v/v). The cells were incubated at room temperature for 30 min followed by centrifugation, rinsed in 0.1 M borate buffer, pH 8.5, and PBS/bovine serum albumin (BSA; Sigma-Aldrich) (1 mg/ml BSA in PBS), after washing with PBS/0.5% Tween 20/1% BSA. Appropriately diluted anti-BrdU antibody (clone BU-1; Amersham, Buckinghamshire, UK) was added to the cell pellet. After incubation for 1 h at room temperature, the cells were rinsed twice in PBS/BSA. For visualization, fluorescein isothiocyanate (FITC)-conjugated Fab2 fragments of rabbit anti-mouse Ig (DAKO, Glostrup, Denmark) antibody were added in a 1:30 dilution. After incubation for 45 min at room temperature, the samples were rinsed twice in PBS/BSA and the cells were resuspended in 0.5 ml cold PBS supplemented with 100 μg/ml RNase (Sigma-Aldrich) and 20 μg/ml propidium iodide (PI; Sigma-Aldrich). The samples were allowed to stand for 15 min on ice in the dark before flow cytometric analysis. For the negative control, the primary antibody was omitted. The stained cells were analyzed by FACScan (Becton-Dickinson, Franklin Lakes, NJ), using the CELL QUEST program.
Measurement of 8-oxo-2'-deoxyguanosine (8-oxo-dG) levels in DNA by laser scanning cytometer
The extent of cell DNA damage was determined using a commercial kit (Calbiochem-Novabiochem Corporation; Darmstadt, Germany) on WI-26VA4 cells at 50 to 70% confluence on coverslips in a 6-well culture plate. The cells were fixed with fresh 4% paraformaldehyde (Wako Life Science, Osaka, Japan) in PBS and dehydrated in 70% followed by 95% methanol at -20°C. Cells were permeabilized in 99% methanol at -20°C, and incubated for 30 min on ice, rehydrated with 95% and 70% methanol at -20°C. The rehydrated cells were washed with PBS/Tween 20, containing thimerosal. To block non-specific binding sites, a blocking solution was added to each well and incubated for 1 h at 37°C. The blocking solution was removed and the cells washed with PBS/Tween 20 before being incubated with FITC-conjugated substrate for 1 h at 37°C, and washed 5 times with PBS/Tween 20 with 3 min of gentle shaking per wash. Next, the cells were incubated with PI-RNase for 10 min at 37°C, washed 5 times with PBS/Tween 20 with 3 min of gentle shaking per wash, and rinsed once with distilled water. The coverslips were freed, mounting solution was added, and placed on slides to be observed under fluorescent microscopy with FITC filters.
Cells were subsequently analyzed with an LSC (LSC101; Olympus, Tokyo, Japan) as previously described [46], using a krypton/argon laser operating at 488 nm and 568 nm. Dual-channel imaging was performed with two photomultipliers, using a band-pass filter specifically designed to detect FITC fluorescence and a long-pass (590 nm) filter for detecting PI fluorescence. Images were generated using integral computational and display software. To assess mean fluorescence intensity (MFI) of 8-oxo-dG, each nucleus was identified by areas counterstained with PI. We analyzed a 10-mm2 scan area, occupied by approximately ~1 × 104 cells.
Comet assay
The alkaline version of the Comet assay was performed according to the method of Singh et al. (1988) with minor modifications [15]. Briefly, fully-frosted microscope slides were pre-coated with 1 ml of 0.75% normal melting point agarose (NMA; Promega, Madison, WI) and stored at 4°C. This layer was removed before use and 120 μl of 0.75% NMA was pipetted onto the slides, which were then covered with coverslips. Cell suspensions of BEAS-2B cells (1 × 104 cells/10 μl) were mixed with 70 μl of 1% low melting point agarose (LMA) and pipetted over the first layer of agarose. NMA (80 μl) was used as a final protective layer. After each step, the slides were incubated at 4°C for 10 min to allow the agarose to gel. Slides were placed in cold lysing solution (2.5 M NaCl, 100 mM Na2EDTA, 10 mM Tris, pH 10, and 1% sodium N-lauroyl sarcosinate to which 10% dimethyl sulfoxide and 1% Triton X-100 had been added immediately prior to use) for 1 h. After lysis, the slides were placed in electrophoresis buffer (300 mM NaOH and 1 mM Na2EDTA, pH 13) for 20 min to allow unwinding of the DNA. Electrophoresis was conducted in the same buffer by applying an electric current of 0.8 V/cm (300 mA) for 20 min, using an electrophoresis power supply (Pharmacia LKB, Sweden). Finally, the slides were washed in neutralization buffer (0.4 M Tris, pH 7.5) 3 times for 5 min each, dried and stained with Hoechst 33342 (10 μg/ml).
Image analysis involved a microscope equipped with an LSC. Nuclei were excited with green light and the emitted red spectrum was captured by a 40 × dry objective. Images of 50 randomly selected nuclei were analyzed per duplicate slide (100 nuclei in total). The tail moment (TM) was determined to assess the extent of DNA damage. The TM value was defined as the product of the percentage of DNA in the comet tail and the tail length.
Determination of mitochondrial membrane potential (ΔΨm)
BEAS-2B cells were stained with 1 μg/ml of 5,5',6,6'-tetrachloro-1,1',3,3'-tetraethylbenzimidazolylcarbocyanine (JC-1; Molecular Probes, Eugene, OR) for 30 min at 37°C, trypsinized, resuspended in medium at a density of approximately 1 × 107/ml, and transferred on ice to the flow cytometer. JC-1 was excited at 488 nm and the monomer signal (green) was analyzed at 525 nm (FL1) on a FACScan (Becton-Dickinson, Mountain-View, CA) flow cytometer. Simultaneously, the aggregate signal (red) was analyzed at 590 nm (FL2). In BEAS-2B cells, the percentage of PI-negative cells (1 × 104) was analyzed and the mean fluorescence was calculated for each sample.
Immunohistochemistry for nitrotyrosine
BEAS-2B cells were cultured in 6-well plates on coverslips and were untreated or treated with H2O2 alone or with a peroxynitrite generator 3-morpholinosyndonimine (SIN-1), which is the precursor of peroxynitrite, for 1 h. The coverslips were then washed with PBS, prefixed in 2% formalin at room temperature for 10 min, and fixed in methanol/acetone (1:1) at -20°C for 10 min. The coverslips were washed with PBS and then were blocked in PBS containing 20% normal swine serum (Dako), 0.4% Triton X and 0.1% BSA. The fixed cells were stained with a primary antibody against nitrotyrosine (Upstate Biotechnology, Lake Placid, NY) diluted at 1:50 in PBS containing 0.4% Triton X and 0.1% BSA for 1 h. After removing the first antibody by washing, the cells were incubated with biotinylated goat anti-rabbit Ig (Dako) in PBS containing 0.4% Triton X and 0.1% BSA for 45 min. After removing the second antibody, the cells were incubated with FITC-conjugated streptavidin (Dako) in PBS containing 0.4% Triton X and 0.1% BSA for 45 min. Finally, the cells were counter-stained with 20% hematoxylin and the coverslips were mounted on slides with mounting medium (DPX). The stained cells were visualized and photographed using a confocal laser scanning microscope (Leica, Heidelberg, Germany).
Data analysis
Group data are expressed as the mean (± SD). Comparisons between data were tested using the Mann-Whitney U test. A p value < 0.05 was considered significant.
Contributor Information
Yoshihiro Fujii, Email: yoshihir@grape.med.tottori-u.ac.jp.
Katsuyuki Tomita, Email: tom0223@grape.med.tottori-u.ac.jp.
Hiroyuki Sano, Email: hsano@grape.med.tottori-u.ac.jp.
Akira Yamasaki, Email: yamasaki@grape.med.tottori-u.ac.jp.
Yutaka Hitsuda, Email: hitsuda@grape.med.tottori-u.ac.jp.
Ian M Adcock, Email: ian.Adcock@ic.ac.uk.
Eiji Shimizu, Email: eiji@grape.med.tottori-u.ac.jp.
References
- Cross CE, Frei B, Louie S. The adult respiratory distress syndrome (ARDS) and oxidative stress: therapeutic implications. Adv Exp Med Biol. 1990;264:435–438. doi: 10.1007/978-1-4684-5730-8_69. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Cross CE. Reactive oxygen species, antioxidants, and acquired immunodeficiency syndrome. Sense or speculation? Arch Intern Med. 1991;151:29–31. doi: 10.1001/archinte.151.1.29. [DOI] [PubMed] [Google Scholar]
- Hoidal JR, Niewoehner DE. Lung phagocyte recruitment and metabolic alterations induced by cigarette smoke in humans and in hamsters. Am Rev Respir Dis. 1982;126:548–552. doi: 10.1164/arrd.1982.126.3.548. [DOI] [PubMed] [Google Scholar]
- Cosio MG, Guerassimov A. Chronic obstructive pulmonary disease. Inflammation of small airways and lung parenchyma. Am J Respir Crit Care Med. 1999;160:S21–25. doi: 10.1164/ajrccm.160.supplement_1.7. [DOI] [PubMed] [Google Scholar]
- Asami S, Manabe H, Miyake J, Tsurudome Y, Hirano T, Yamaguchi R, Itoh H, Kasai H. Cigarette smoking induces an increase in oxidative DNA damage, 8-hydroxydeoxyguanosine, in a central site of the human lung. Carcinogenesis. 1997;18:1763–1766. doi: 10.1093/carcin/18.9.1763. [DOI] [PubMed] [Google Scholar]
- Barnes PJ. Potential novel therapies for chronic obstructive pulmonary disease. Novartis Found Symp. 2001;234:255–267. [PubMed] [Google Scholar]
- Ichinose M, Sugiura H, Yamagata S, Koarai A, Shirato K. Increase in reactive nitrogen species production in chronic obstructive pulmonary disease airways. Am J Respir Crit Care Med. 2000;162:701–706. doi: 10.1164/ajrccm.162.2.9908132. [DOI] [PubMed] [Google Scholar]
- Hanazawa T, Kharitonov SA, Barnes PJ. Increased nitrotyrosine in exhaled breath condensate of patients with asthma. Am J Respir Crit Care Med. 2000;162:1273–1276. doi: 10.1164/ajrccm.162.4.9912064. [DOI] [PubMed] [Google Scholar]
- Wattenberg LW. Inhibition of chemical carcinogenesis by antioxidants. Carcinog Compr Surv. 1980;5:85–98. [PubMed] [Google Scholar]
- van Delft JH, Baan RA, Roza L. Biological effect markers for exposure to carcinogenic compound and their relevance for risk assessment. Crit Rev Toxicol. 1998;28:477–510. doi: 10.1080/10408449891344254. [DOI] [PubMed] [Google Scholar]
- Pryor WA. Cigarette smoke radicals and the role of free radicals in chemical carcinogenicity. Environ Health Perspect. 1997;105:875–882. doi: 10.1289/ehp.97105s4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward JF. The complexity of DNA damage: relevance to biological consequences. Int J Radiat Biol. 1994;66:427–432. doi: 10.1080/09553009414551401. [DOI] [PubMed] [Google Scholar]
- Helbock HJ, Beckman KB, Ames BN. 8-Hydroxydeoxyguanosine and 8-hydroxyguanine as biomarkers of oxidative DNA damage. Methods Enzymol. 1999;300:156–166. doi: 10.1016/s0076-6879(99)00123-8. [DOI] [PubMed] [Google Scholar]
- Li X, Darzynkiewicz Z. The Schrodinger's cat quandary in cell biology: integration of live cell functional assays with measurements of fixed cells in analysis of apoptosis. Exp Cell Res. 1999;249:404–412. doi: 10.1006/excr.1999.4525. [DOI] [PubMed] [Google Scholar]
- Singh NP, McCoy MT, Tice RR, Schneider EL. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res. 1988;175:184–191. doi: 10.1016/0014-4827(88)90265-0. [DOI] [PubMed] [Google Scholar]
- Chen SK, Tsai MH, Hwang JJ, Chan WP. Determination of 8-oxoguanine in individual cell nucleus of gamma-irradiated mammalian cells. Radiat Res. 2001;155:832–836. doi: 10.1667/0033-7587(2001)155[0832:dooiic]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Cossarizza A, Kalashnikova G, Grassilli E, Chiappelli F, Salvioli S, Capri M, Barbieri D, Troiano L, Monti D, Franceschi C. Mitochondrial modifications during rat thymocyte apoptosis: a study at the single cell level. Exp Cell Res. 1994;214:323–330. doi: 10.1006/excr.1994.1264. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271:C1424–1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- Knaapen AM, Seiler F, Schilderman PA, Nehls P, Bruch J, Schins RP, Borm PJ. Neutrophils cause oxidative DNA damage in alveolar epithelial cells. Free Radic Biol Med. 1999;27:234–240. doi: 10.1016/S0891-5849(98)00285-8. [DOI] [PubMed] [Google Scholar]
- Yoneda K, Peck K, Chang MM, Chmiel K, Sher YP, Chen J, Yang PC, Chen Y, Wu R. Development of high-density DNA microarray membrane for profiling smoke- and hydrogen peroxide-induced genes in a human bronchial epithelial cell line. Am J Respir Crit Care Med. 2001;164:S85–89. doi: 10.1164/ajrccm.164.supplement_2.2106062. [DOI] [PubMed] [Google Scholar]
- Goldkorn T, Balaban N, Matsukuma K, Chea V, Gould R, Last J, Chan C, Chavez C. EGF-Receptor phosphorylation and signaling are targeted by H2O2 redox stress. Am J Respir Cell Mol Biol. 1998;19:786–798. doi: 10.1165/ajrcmb.19.5.3249. [DOI] [PubMed] [Google Scholar]
- Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- Rao KS. Genomic damage and its repair in young and aging brain. Mol Neurobiol. 1993;7:23–48. doi: 10.1007/BF02780607. [DOI] [PubMed] [Google Scholar]
- Dizdaroglu M. Facts about the artifacts in the measurement of oxidative DNA base damage by gas chromatography-mass spectrometry. Free Radic Res. 1998;29:551–563. doi: 10.1080/10715769800300591. [DOI] [PubMed] [Google Scholar]
- Kaur H, Halliwell B. Measurement of oxidized and methylated DNA bases by HPLC with electrochemical detection. Biochem J. 1996;318:21–23. doi: 10.1042/bj3180021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dizdaroglu M. Oxidative damage to DNA in mammalian chromatin. Mutat Res. 1992;275:331–342. doi: 10.1016/0921-8734(92)90036-O. [DOI] [PubMed] [Google Scholar]
- Yarborough A, Zhang YJ, Hsu TM, Santella RM. Immunoperoxidase detection of 8-hydroxydeoxyguanosine in aflatoxin B1-treated rat liver and human oral mucosal cells. Cancer Res. 1996;56:683–688. [PubMed] [Google Scholar]
- Nehls P, Seiler F, Rehn B, Greferath R, Bruch J. Formation and persistence of 8-oxoguanine in rat lung cellsas an important determinant for tumor formation following particle exposure. Environ Health Perspect. 1997;105S:1291–1296. doi: 10.1289/ehp.97105s51291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyokuni S, Tanaka T, Hattori Y, Nishiyama Y, Yoshida A, Uchida K, Hiai H, Ochi H, Osawa T. Quantitative immunohistochemical determination of 8-hydroxy-2'-deoxyguanosine by a monoclonal antibody N45.1: its application to ferric nitrilotriacetate-induced renal carcinogenesis model. Lab Invest. 1997;76:365–374. [PubMed] [Google Scholar]
- Futaki M, Igarashi T, Watanabe S, Kajigaya S, Tatsuguchi A, Wang J, Liu JM. The FANCG Fanconi anemia protein interacts with CYP2E1: possible role in protection against oxidative DNA damage. Carcinogenesis. 2002;23:67–72. doi: 10.1093/carcin/23.1.67. [DOI] [PubMed] [Google Scholar]
- Slamenova D, Kuboskova K, Horvathova E, Robichova S. Rosemary-stimulated reduction of DNA strand breaks and FPG-sensitive sites in mammalian cells treated with H2O2 or visible light-excited methylene blue. Cancer Lett. 2002;177:145–153. doi: 10.1016/S0304-3835(01)00784-4. [DOI] [PubMed] [Google Scholar]
- Collins AR, Duthie SJ, Dobson VL. Direct enzymic detection of endogenous oxidative base damage in human lymphocyte DNA. Carcinogenesis. 1993;14:1733–1735. doi: 10.1093/carcin/14.9.1733. [DOI] [PubMed] [Google Scholar]
- He YH, Xu Y, Kobune M, Wu M, Kelley MR, Martin WJ 2nd. Escherichia coli FPG and human OGG1 reduce DNA damage and cytotoxicity by BCNU in human lung cells. Am J Physiol Lung Cell Mol Physiol. 2002;282:L50–55. doi: 10.1152/ajplung.00316.2001. [DOI] [PubMed] [Google Scholar]
- Jornot L, Petersen H, Junod AF. Hydrogen peroxide-induced DNA damage is independent of nuclear calcium but dependent on redox-active ions. Biochem J. 1998;335:85–94. doi: 10.1042/bj3350085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi Y, Linn S. Purification of all forms of HeLa cell mitochondrial DNA and assessment of damage to it caused by hydrogen peroxide treatment of mitochondria or cells. J Biol Chem. 1995;270:7950–7956. doi: 10.1074/jbc.270.14.7950. [DOI] [PubMed] [Google Scholar]
- Rahman I, Mulier B, Gilmour PS, Watchorn T, Donaldson K, Jeffery PK, MacNee W. Oxidant-mediated lung epithelial cell tolerance: the role of intracellular glutathione and nuclear factor-kappaB. Biochem Pharmacol. 2001;62:787–794. doi: 10.1016/S0006-2952(01)00702-X. [DOI] [PubMed] [Google Scholar]
- Hahn WC, Dessain SK, Brooks MW, King JE, Elenbaas B, Sabatini DM, DeCaprio JA, Weinberg RA. Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol Cell Biol. 2002;22:2111–2123. doi: 10.1128/MCB.22.7.2111-2123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M. et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Abramova NE, Davies KJ, Crawford DR. Polynucleotide degradation during early stage response to oxidative stress is specific to mitochondria. Free Radic Biol Med. 2000;28:281–288. doi: 10.1016/S0891-5849(99)00239-7. [DOI] [PubMed] [Google Scholar]
- de Souza-Pinto NC, Eide L, Hogue BA, Thybo T, Stevnsner T, Seeberg E, Klungland A, Bohr VA. Repair of 8-oxodeoxyguanosine lesions in mitochondrial dna depends on the oxoguanine dna glycosylase (OGG1) gene and 8-oxoguanine accumulates in the mitochondrial dna of OGG1-defective mice. Cancer Res. 2001;61:5378–5381. [PubMed] [Google Scholar]
- Grisham MB, Jourd'Heuil D, Wink DA. Nitric oxide. I. Physiological chemistry of nitric oxide and its metabolites: implications in inflammation. Am J Physiol. 1999;276:G315–321. doi: 10.1152/ajpgi.1999.276.2.G315. [DOI] [PubMed] [Google Scholar]
- Cross AH, Manning PT, Keeling RM, Schmidt RE, Misko TP. Peroxynitrite formation within the central nervous system in active multiple sclerosis. J Neuroimmunol. 1998;88:45–56. doi: 10.1016/S0165-5728(98)00078-2. [DOI] [PubMed] [Google Scholar]
- Reddel RR, Ke Y, Gerwin BI, McMenamin MG, Lechner JF, Su RT, Brash DE, Park JB, Rhim JS, Harris CC. Transformation of human bronchial epithelial cells by infection with SV40 or adenovirus-12 SV40 hybrid virus, or transfection via strontium phosphate coprecipitation with a plasmid containing SV40 early region genes. Cancer Res. 1988;48:1904–1909. [PubMed] [Google Scholar]
- Tomita K, Chikumi H, Tokuyasu H, Yajima H, Hitsuda Y, Matsumoto Y, Sasaki T. Functional assay of NF-kappaB translocation into nuclei by laser scanning cytometry: inhibitory effect by dexamethasone or theophylline. Naunyn Schmiedebergs Arch Pharmacol. 1999;359:249–255. doi: 10.1007/pl00005349. [DOI] [PubMed] [Google Scholar]