

Abstract
The Rho family GTPases Cdc42 and Rac1 play fundamental roles in transformation and actin remodeling. Here, we demonstrate that the TRE17 oncogene encodes a component of a novel effector pathway for these GTPases. TRE17 coprecipitated specifically with the active forms of Cdc42 and Rac1 in vivo. Furthermore, the subcellular localization of TRE17 was dramatically regulated by these GTPases and mitogens. Under serum-starved conditions, TRE17 localized predominantly to filamentous structures within the cell. Epidermal growth factor (EGF) induced relocalization of TRE17 to the plasma membrane in a Cdc42-/Rac1-dependent manner. Coexpression of activated alleles of Cdc42 or Rac1 also caused complete redistribution of TRE17 to the plasma membrane, where it partially colocalized with the GTPases in filopodia and ruffles, respectively. Membrane recruitment of TRE17 by EGF or the GTPases was dependent on actin polymerization. Finally, we found that a C-terminal truncation mutant of TRE17 induced the accumulation of cortical actin, mimicking the effects of activated Cdc42. Together, these results identify TRE17 as part of a novel effector complex for Cdc42 and Rac1, potentially contributing to their effects on actin remodeling. The present study provides insights into the regulation and cellular function of this previously uncharacterized oncogene.
The Rho family GTPases are recognized for their role in malignant transformation. Members of this family, which includes Rho, Rac, and Cdc42, regulate diverse cellular processes, including cytoskeletal dynamics, cell cycle progression, and vesicular trafficking. It is likely that deregulation of all of these processes contributes to transformation induced by activated mutants of these GTPases. Indeed, cells transfomed by activated Cdc42 or Rac exhibit properties that reflect such aberrations. For example, these cells grow in a mitogen-independent manner (14, 19, 23, 37, 39, 58), indicating that signaling pathways that control cell cycle progression are constitutively activated. Cells transformed by Cdc42 or Rac also grow in an adhesion-independent manner, exhibit altered morphologies, and display increased motility and invasiveness, all of which reflect alterations in the actin cytoskeleton (16, 23, 38, 39, 56, 58). More recently, it has been shown that vesicular trafficking is also aberrantly regulated in cells expressing activated Cdc42 or Rac (1, 13, 18, 25, 57) contributing to reduced cell adhesion, altered receptor trafficking, and loss of polarity.
In recent years, much emphasis has been placed on understanding the molecular mechanisms by which Cdc42 and Rac exert their effects on the actin cytoskeleton. Early studies in mammalian cells revealed that expression of activated Cdc42 and Rac1 rapidly induces the formation of actin-based cell surface protrusions termed filopodia and lamellapodia (35, 45), respectively. These GTPases stimulate actin remodeling through multiple effectors (5). The best characterized are the WASP-related proteins, which include WASP and N-WASP, which are regulated directly by Cdc42, and WAVE, which is regulated by Rac via IRSp53. These proteins function by directly stimulating the actin nucleating activity of the Arp2/3 complex (30-32, 46, 49, 51). Another shared family of effectors are the p21-activated kinases (PAKs) (27). PAK1 phosphorylates and activates LIM kinase, which in turn phosphorylates and inhibits the actin depolymerizing activity of cofilin (11, 59). PAK1 has also been shown to phosphorylate myosin light chain, which stimulates actin/myosin-based contraction (43, 47). Another Cdc42-specific effector is MRCK, a kinase which also phosphorylates myosin light chain (21). Similarly, Rac uniquely targets several effectors, such as POR1, which functions through an unknown mechanism to promote actin-rich membrane protrusions (54). Another key effector of Rac is phosphatidylinositol-4-phosphate 5-kinase (52, 53), which catalyzes the production of phosphatidylinositol 4,5-bisphosphate, a potent regulator of various actin-binding proteins (50).
Studies in Saccharomyces cerevisiae have also provided fundamental insights into the functions of Cdc42. It was in yeast that Cdc42 was first shown to play a central role in actin organization and the establishment of cell polarity (15). Inactivation of Cdc42, through ablation of its guanine nucleotide exchange factor Cdc24, abolishes the polarization of actin cables that normally occurs during the budding process (4). Using a multicopy suppressor screen, Bi et al. identified proteins that could restore actin polarization in the absence of functional Cdc42. Two novel genes identified in this screen encoded the highly related proteins Msb3 and Msb4 (4). These proteins are functionally redundant, since deletion of either gene individually yielded no discernible phenotype. However, simultaneous disruption of MSB3 and MSB4 caused slowed growth and partial disorganization of the actin cytoskeleton (4). The mechanism by which Cdc42 signals to Msb3 and Msb4 remains undefined.
Sequence analysis of Msb3 and Msb4 revealed the presence of a domain that has alternatively been termed the PTM, TrH, Tbc, or Gyp domain (2, 3, 29, 44, 60). This domain is conserved throughout evolution, occurring in proteins from yeasts, Caenorhabditis elegans, Drosophila melanogaster, and mammals (34). Recent studies have revealed that these domains encode GTPase-activating proteins (GAPs) for Rab family G proteins (2, 9, 20, 42). Two PTM domain-containing proteins of particular interest were the human oncogenes, RN-tre and TRE17. RN-tre is a binding partner for the epidermal growth factor (EGF) receptor substrate, Eps8, which regulates trafficking of the receptor (20, 28). Overexpression of a truncated mutant of RN-tre causes anchorage-independent growth and enhances mitogenicity in response to EGF (28). The TRE17 gene was originally identified through a transformation-based assay using genomic DNA from a Ewing sarcoma (33). Specifically, expression of TRE17 in murine fibroblasts caused tumor formation in nude mice. However, its interaction with signaling pathways, regulation, and cellular function have remained completely unexplored.
In light of their homology to Msb3/Msb4, we explored whether TRE17 or RN-tre might function downstream of the Rho GTPases in mammalian cells. In the present study, we demonstrate that TRE17, but not RN-tre, is part of a novel effector pathway for Cdc42 and Rac1. Furthermore, we find that TRE17 is sufficient by itself to induce the accumulation of cortical actin and may therefore contribute to actin remodeling induced by Cdc42 and Rac1.
MATERIALS AND METHODS
Cell culture.
HeLa and COS cells were grown in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum, penicillin and streptomycin, and GlutaMax (Gibco-BRL). Cultures were maintained at 37°C in 6% CO2. EGF (Invitrogen) was used at a final concentration of 100 ng/ml. HeLa and COS cells were transfected by using FuGENE6 (Roche) and Lipofectamine (Invitrogen), respectively, as detailed below.
Plasmids.
The cDNAs encoding human Cdc42, Rac1, and RhoA and their mutant derivatives were subcloned into the mammalian expression vector pEBG to generate glutathione S-transferase (GST)-tagged fusion proteins (8). PBD/pEBG encodes the p21-binding domain (amino acids 70 to 117) of human PAK1.
The cDNA encoding TRE17 (clone 11-4 in pBluescript) was generously provided by Myriam Onno (33). The BamHI-SspI fragment encompassing the 5′-untranslated region and the TRE17 open reading frame was subcloned into a modified version of pcDNA3 containing a hemagglutinin (HA) epitope tag. The 5′-untranslated region was removed by digestion with BamHI-EcoRI and replaced with a PCR fragment restoring the normal TRE17 open reading frame. The sequence was confirmed by automated sequencing. HA-T17(447)/pcDNA3 encodes the first 447 amino acids of TRE17, HA-T17(ΔPTM)/pcDNA3 encodes a deletion of amino acids 80 to 303, and HA-T17(PTM)/pcDNA3 encompasses amino acids 80 to 327 of TRE17. In HA-T17(447/RK)/pCDNA, residue 149 was changed from arginine to lysine by PCR-mediated mutagenesis. In HA-T17(447/PA)/pCDNA, the proline-rich motifs P369RPVP373 and P389PGPP393 were mutated to ARAVA and PAGSA, respectively. Further details are available upon request.
β-Tubulin/pEGFP was kindly provided by Paul Janmey. The cDNA encoding RN-tre was generously provided by Pier Paolo di Fiore.
Antibodies and reagents.
For immunofluorescence of hemagglutinin (HA)-tagged proteins proteins, anti-HA (sc-805 [Santa Cruz] or monoclonal antibody 12CA5 [Roche]) was used; for immunoblotting, the former was used. Affinity-purified anti-GST antibody made against Escherichia coli-derived GST was used for all applications. Anti-ERK antibody was generously provided by John Blenis.
The secondary antibodies used were Cy3-conjugated donkey anti-mouse immunoglobulin G (IgG; heavy and light chain; Jackson Immunoresearch Laboratories), Alexa Fluor 633-conjugated goat anti-rabbit IgG (heavy and light chain; Molecular Probes), or FITC-conjugated donkey anti-rabbit IgG (heavy and light chain, Jackson Immunoresearch). F-actin was visualized with fluorescein isothiocyanate (FITC)-conjugated phalloidin (Molecular Probes).
Cytochalasin D (cytoD; 1 μM) and nocodazole (10 μg/ml; Sigma) were added for 1 or 2 h, respectively, where indicated.
GTPase pull-down assays.
COS cells were seeded at a density of 14.0 × 105 per 100-mm dish for TRE17 or at a density of 5.0 × 105 cells per 60-mm dish for T17(447) and RN-tre(466). The following day, cells were transfected by using either 16 μg of total DNA and 48 μl of Lipofectamine per plate (TRE17) or 6 μg of total DNA and 18 μl of Lipofectamine [T17(447) and RN-tre(466)]. Cells were incubated for 4 to 5 h, washed, and then allowed to recover overnight in growth medium. Cells were washed twice in ice-cold phosphate-buffered saline (PBS) and then lysed in either 750 μl (TRE17) or 500 μl [T17(447) and RN-tre(466)] of buffer I (PBS; 5 mM MgCl2; 0.1% Triton X-100; pepstatin, leupeptin, and aprotinin at 0.7, 2, and 1 μg/ml, respectively; 1 mM dithiothreitol [DTT]; 1 mM phenylmethylsulfonyl fluoride) Lysates were incubated on ice for 10 min and then centrifuged at 16,000 × g for 10 min at 4°C. An aliquot (30 μl) of the clarified supernatant was removed for immunoblotting analysis, and the remainder was incubated with glutathione-Sepharose beads (50-μl bead volume; Amersham Pharmacia Biotech) for 4 h at 4°C with constant mixing. The beads were washed twice in buffer II (PBS; 5 mM MgCl2; 0.01% Triton X-100; pepstatin, leupeptin, and aprotinin; 1 mM DTT) and then thrice in buffer III (PBS; 5 mM MgCl2; pepstatin, leupeptin, and aprotinin; 1 mM DTT). Proteins were eluted by boiling in sample buffer (125 mM Tris [pH 6.8], 2% sodium dodecyl sulfate, 5% β-mercaptoethanol, 7.5% glycerol, bromophenol blue). Expression of the GST-tagged GTPases and HA-tagged TRE17 or RN-tre constructs was detected by using enhanced chemiluminescence (Amersham Pharmacia Biotech).
Confocal immunofluorescence microscopy.
HeLa cells were seeded on 10-mm coverslips at a density of 2 × 105 to 2.4 × 105 per 35-mm dish. The following day, cells were transfected with 2 μg of total DNA and 6 μl of FuGENE6 per plate. The next day cells were either harvested for immunofluorescence or were serum starved for 24 h, followed by various treatments as indicated. Cells were fixed in (3.7% formaldehyde, 10 mM HEPES [pH 7.4], 100 mM NaCl) for 15 min and then chased with 50 mM glycine (pH 7.4) for 10 min. Cells were washed twice with PBS, permeabilized in TBST (10 mM Tris [pH 7.5], 0.1% Triton X-100, 150 mM NaCl) for 5 min, and then blocked in blocking buffer (2% bovine serum albumin in TBST) for 15 min. Coverslips were incubated with primary antibodies diluted in blocking buffer for 2 to 3 h at room temperature and then washed three times in TBST. Samples were incubated with fluorescently labeled secondary antibodies diluted in blocking buffer for 1 h and then washed two times in TBST and once in distilled water. Coverslips were mounted with SloFade (Molecular Probes) and viewed on a Zeiss confocal microscope with LSM510 software at excitation wavelengths of 488 nm (FITC), 546 nm (Cy3), or 633 nm (Alexa Fluor 633).
For quantitation of TRE17 localization to the plasma membrane versus filaments, 100 cells expressing low to medium levels of TRE17 were scored in the absence or presence of EGF. Cells were categorized into as having predominantly filamentous staining, predominantly plasma membrane staining, discernible staining at both locations, or diffuse staining of TRE17. For quantification of TRE17-induced cortical actin accumulation, a cell was scored as positive if the cortical actin was significantly stronger than the neighboring untransfected cells, as typified by the images shown in Fig. 7. The percentages given are the fraction of cells exhibiting cortical actin accumulation relative to the total number of cells expressing the indicated construct. All quantifications were done for at least three independent experiments, and the means and standard errors are depicted in the graphs.
FIG. 7.

T17(447) stimulates cortical actin accumulation downstream of Cdc42 and Rac1. (A) HA-T17(447) or GST-Cdc42V12 was transfected into HeLa cells. After serum starvation for 24 h, cells were fixed and T17(447), Cdc42V12, and F-actin were visualized. (B) HeLa cells were transfected with HA-TRE17, HA-T17(447), HA-T17(447/RK), or HA-T17(447/PA) and then quiesced for 24 h. The percentage of cells exhibiting an accumulation of cortical actin relative to the total number of cells expressing the indicated construct is given. The results are the average of seven independent experiments for untransfected cells, HA-TRE17, and HA-T17(447) or of four independent experiments for HA-T17(447/RK) and HA-T17(447/PA), with standard deviations indicated by the error bars. (C) HA-T17(447) was cotransfected with GST-Cdc42N17, -Rac1N17, or -PBD into HeLa cells. After 24 h of serum-starvation, HA-T17(447), F-actin, and the indicated GST-tagged construct were visualized. Scale bar, 10 μm.
Cell fractionation.
HeLa cells were seed at a density of 7.0 × 105 cells per 60-mm plate. The next day cells were transfected with 6 μg of DNA and 18 μl of FuGENE6 per sample. The following day, cells were washed twice in ice-cold PBS, and 1 ml of hypotonic lysis buffer (20 mM Tris [pH 7.4]; 10 mM KCl; 5 mM MgCl2; 5 mM β-glycerophosphate; 1 mM DTT; pepstatin, aprotinin, and leupeptin; 1 mM phenylmethylsulfonyl fluoride) was added. Cells were allowed to swell on ice for 10 min, scraped, and homogenized by 25 strokes in a Dounce homogenizer. The homogenate was centrifuged at 1,000 × g for 10 min at 4°C to remove nuclei and unbroken cells. The supernatant was collected, and NaCl was added to a final concentration of 150 mM. The supernatants were then centrifuged for an additional 10 min at 16,000 × g at 4°C to generate a pellet (containing membranes) and a soluble fraction (containing cytosolic proteins). The soluble fraction was boiled in sample buffer; the pellet was washed once in PBS and then resuspended in 200 μl of sample buffer. Portions of the soluble fraction (one twenty-fifth) and of the pellet (one-fifth) were analyzed by immunoblot analysis.
RESULTS
TRE17 associates with Cdc42 and Rac1 in a GTP-dependent manner in vivo.
Because the PTM domain-containing proteins Msb3 and Msb4 interact genetically with Cdc42 in S. cerevisiae, we sought to determine whether PTM proteins function downstream of Rho GTPases in mammalian cells. Effectors often form stable complexes with the active, GTP-bound form of G proteins. Therefore, we tested whether the human PTM-domain proteins TRE17 and RN-tre could associate with members of the Rho family of GTPases. HA-tagged TRE17 or RN-tre was cotransfected with GST-tagged alleles of Cdc42, Rac1, or RhoA into COS cells. The GTPases were purified by using glutathione-Sepharose beads, and association of TRE17 or RN-tre was monitored by anti-HA immunoblotting. As seen in Fig. 1A, TRE17 coprecipitated specifically with the activated alleles of Cdc42 (Cdc42V12) and Rac1 (Rac1V12) but not RhoA (RhoAV14). Dominant-negative mutants of Cdc42 (Cdc42N17) and Rac (Rac1N17), which are predominantly GDP bound, bound much more weakly. We also tested binding of TRE17 to activated Cdc42/Rac1 alleles bearing an additional mutation in the cysteine residue which is normally required for geranylation and membrane targeting (Cdc42V12/S189 and Rac1V12/S189). Interestingly, these mutants also failed to bind HA-TRE17. These results indicate that the in vivo interaction of TRE17 with Cdc42 and Rac1 is GTP dependent and requires membrane localization of the G proteins.
FIG. 1.

TRE17 associates with Cdc42 and Rac1 in a GTP-dependent manner. (A) COS cells were cotransfected with the indicated GST-tagged Cdc42, Rac1, or RhoA mutant and HA-tagged TRE17. The GTPases were pulled down with glutathione-Sepharose, and associated TRE17 was detected by anti-HA immunoblotting. Arrowhead, HA-TRE17. (B) The indicated mutant alleles of Cdc42 or Rac1 were cotransfected with HA-tagged T17(447), which encodes the first 447 amino acids of TRE17, and subjected to the same assay. (C) The indicated GTPases were cotransfected with HA-tagged RN-tre(466), which encodes the first 466 amino acids of RN-tre, or T17(447). Associated RN-tre(466) or T17(447) was detected as described above. PD, glutathione-Sepharose pull-down; WCL, whole-cell lysate.
To further delineate the requirements for coprecipitation of TRE17 with Cdc42 and Rac1, we tested binding of a C-terminally truncated mutant encoding the first 447 amino acids of TRE17 [T17(447)]. This mutant includes the PTM domain in addition to proline-rich sequences with potential for binding to SH3 domains (see Fig. 1B and 6A). Like full-length TRE17, T17(447) bound to Cdc42 and Rac1 in a GTP- and geranylation-dependent manner (Fig. 1B). We also examined TRE17's interaction with Cdc42V12 and Rac1V12 alleles bearing mutations in the effector domain. These mutants have been shown to selectively bind and activate distinct effectors (19, 22, 56). T17(447) bound to the effector mutants Cdc42V12/A37 and Cdc42V12/K40 as strongly as to Cdc42V12. In contrast, whereas Rac1V12/H40 bound to T17(447) as well as Rac1V12, Rac1V12/L37 bound poorly (Fig. 1B). Together, these results indicate that sequences within the N-terminal 447 amino acids of TRE17 are sufficient to mediate interaction with Cdc42 and Rac1. Furthermore, association requires contacts made with the effector domain of the GTPases. All of the above studies were performed in COS cells, where the high level of protein expression facilitated binding analysis. However, the specific interaction of TRE17 with the GTPases was also confirmed in HeLa cells (data not shown).
FIG. 6.
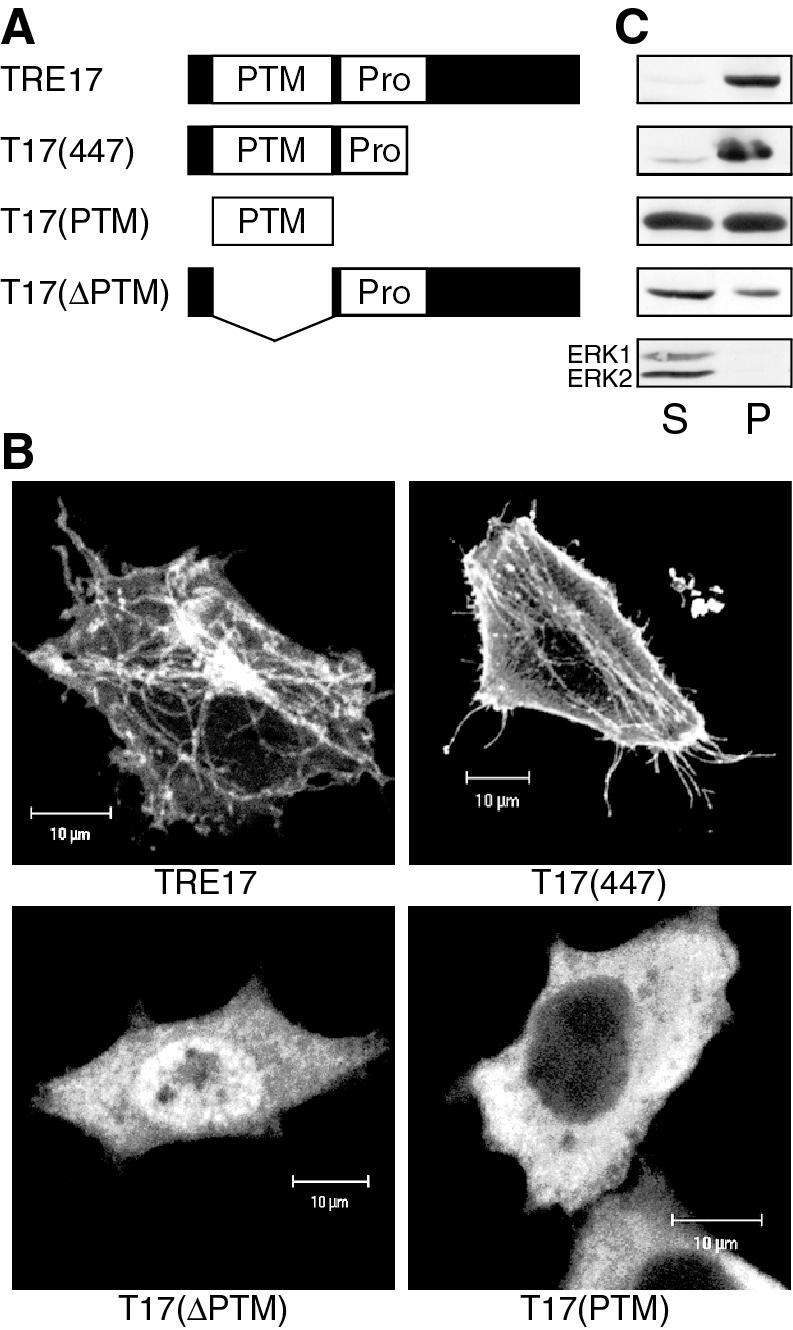
Structural determinants of TRE17 localization. (A) Schematic representation of full-length TRE17 and various mutants. PTM refers to the PTM/Gyp/Tbc domain, while Pro refers to the proline-rich region. (B) HeLa cells were transfected with the indicated constructs and then serum-starved for 24 h prior to visualization with anti-HA antibody. Scale bar, 10 μm. (C) TRE17 or the indicated mutants were transfected into HeLa cells. Cells were hypotonically lysed and fractionated as described in Materials and Methods. One twenty-fifth of the soluble fraction and one-fifth of the pellet were loaded into each lane. S, soluble fraction; P, pellet. Fractions were also probed for the presence of ERK (bottom panel), a cytoplasmic protein that should localize to the soluble fraction, to ensure proper fractionation.
TRE17 and RN-tre are highly related, sharing 35% identity and 55% similarity over their first 462 amino acids; however, their C termini are divergent in sequence (29). Given that this region of homology in TRE17 was sufficient to mediate interaction with Cdc42/Rac1, we tested whether the corresponding region of RN-tre [RN-tre(466)], encoding the N-terminal 466 amino acids, or full-length RN-tre could also bind. However, neither RN-tre(466) (Fig. 1C) nor full-length RN-tre (data not shown) associated with any of the Rho GTPases. These results suggest that among mammalian PTM-domain-containing proteins, TRE17 may be unique in its association with Rho GTPases.
To determine whether TRE17 binds directly to Cdc42 and Rac1, yeast two-hybrid analysis was performed. By this assay, no interaction was observed between TRE17 and Cdc42V12 or RacV12, either with the isoprenylation motif of the GTPases intact or mutated (data not shown). Similarly, no binding was seen by using overlay assays with [32P]GTP-labeled Cdc42V12 or Rac1V12 as a probe (data not shown). Thus, it appears that the interaction between TRE17 and Cdc42/Rac1 is indirect and that TRE17 is recruited to the GTPases as part of an effector complex.
TRE17's subcellular distribution is regulated by the actin cytoskeleton and microtubules.
In order to gain insight into the function of TRE17 and to explore how it might be regulated by Cdc42/Rac1, we examined its subcellular localization. Full-length HA-TRE17 was transiently transfected into HeLa cells, and its localization was analyzed by confocal microscopy. In all experiments, cells expressing low levels of TRE17 were analyzed to avoid artifacts that might arise from vast overexpression. In actively growing, unsynchronized HeLa cells, TRE17 exhibited a complex distribution, with localization at the plasma membrane, on filamentous structures, and punctate structures that were often aligned along the filaments (Fig. 2A).
FIG. 2.

TRE17 displays a complex subcellular distribution that is regulated by the actin cytoskeleton and microtubules. (A) HA-tagged TRE17 was transfected in HeLa cells, and its localization was determined by confocal microscopy with anti-HA antibody (left panel). F-actin in the same cell was visualized with FITC-phalloidin (right panel). (B) HA-TRE17-transfected HeLa cells were treated with 1 μM cytoD for 1 h, and TRE17 localization was analyzed as described in panel A. (C) HA-TRE17 (left panel) and GFP-tubulin (right panel) were cotransfected into HeLa cells, and their localization analyzed as in panel A. (D) HA-TRE17-transfected HeLa cells were treated with nocodazole (10 μg/ml) for 2 h. HA-TRE17 localization was analyzed as described above. Scale bar, 10 μm.
To define the filamentous structures which TRE17 decorates, we examined their colocalization with cytoskeletal markers and sensitivity to cytoskeleton-perturbing drugs. The TRE17-positive filaments were clearly distinct from F-actin labeled with phalloidin (Fig. 2A). Furthermore, in the presence of cytoD, which disrupted F-actin (data not shown), the TRE17-positive filaments persisted (Fig. 2B, 3C and F, and 4A). Interestingly, the punctate structures aligned along the filaments were enhanced by cytoD. In contrast, plasma membrane localization of TRE17 was completely abolished. Together, these results suggest that F-actin accumulation is required for plasma membrane localization but not for filamentous localization of TRE17.
FIG. 3.

Activated Rac1 and Cdc42 colocalize with TRE17 and stimulate its recruitment to the plasma membrane. (A to C) HA-TRE17 was cotransfected with GST-tagged WT Rac1 (A) or Rac1V12 (B and C) into HeLa cells. (C) Cells were treated with 1 μM cytoD for 1 h. TRE17 and Rac1V12 were detected as described above; F-actin was visualized with FITC-phalloidin. (D to F) HA-TRE17 was cotransfected with GST-tagged WT Cdc42 (D) or Cdc42V12 (E and F) and analyzed as described above. (F) Cells were treated with cytoD for 1 h and analyzed as in panel C. Scale bar, 10 μm.
FIG. 4.

EGF stimulates the recruitment of TRE17 to the plasma membrane in an F-actin-dependent manner. (A) HeLa cells were transfected with HA-tagged TRE17, and the following day cells were serum starved for 24 h. Cells were either left untreated (left panels), stimulated with 100 ng of EGF/ml for 10′ (middle panels), or pretreated with cytoD for 1 h prior to EGF stimulation (right panels). HA-TRE17 was visualized with anti-HA antibody (top row); F-actin was visualized with FITC-phalloidin (bottom row). Scale bar, 10 μm. (B) TRE17 localization was scored in either serum-starved cells (solid bars) or EGF-stimulated cells (open bars). The results are the average of three independent experiments, with the standard deviation indicated by the error bars.
We next compared the localization of TRE17 with microtubules, as detected by expression of green fluorescent protein-tagged tubulin. Although the TRE17-positive filaments did not precisely colocalize with microtubules, they were organized in an overlapping array, suggesting that these structures might be dependent on microtubules (Fig. 2C). Consistent with this, treatment with the microtubule depolymerizing drug nocodazole resulted in a complete loss of filamentous staining and TRE17 instead appeared in a perinuclear aggregate (Fig. 2D). Strikingly, plasma membrane localization of TRE17 persisted in the presence of nocodazole (Fig. 2D). Together, these results indicate that the localization of TRE17 is dynamically regulated by the cytoskeleton, such that plasma membrane localization is dependent on F-actin accumulation, and filamentous localization is dependent on microtubules.
Activated Cdc42 and Rac1 induce localization of TRE17 to the plasma membrane.
Since TRE17 associated with Cdc42 and Rac1, we sought to examine the effects of the GTPases on its subcellular localization. HA-TRE17 was cotransfected with the wild type (WT) or with constitutively active mutants of Rac1 or Cdc42, and their localization was analyzed by confocal microscopy. TRE17 colocalized strongly with WT Rac1 at the filamentous structures (Fig. 3A) and at the plasma membrane (data not shown). However, WT Rac1 had no effect on the subcellular distribution of TRE17. In contrast, coexpression with Rac1V12 led to a striking redistribution of TRE17, with a complete loss of filamentous staining and recruitment to the plasma membrane. TRE17 was recruited to both peripheral and dorsal ruffles, where it strongly colocalized with Rac1V12 (Fig. 3B). Treatment of cells with cytoD for 1 h reversed the effects of Rac1V12 and caused TRE17 to return to filamentous structures (Fig. 3C). Thus, Rac1V12-induced recruitment of TRE17 to the plasma membrane is dependent on integrity of the actin cytoskeleton.
Expression of Cdc42 mutants had distinct effects on TRE17 localization. WT Cdc42 induced recruitment of TRE17 to the plasma membrane, although a fraction of TRE17 remained cytosolic (Fig. 3D). Interestingly, in contrast to WT Rac1, Cdc42 did not localize to TRE17-positive filaments (Fig. 3D). More dramatic effects were seen with Cdc42V12, which induced essentially all of the TRE17 to relocalize to the plasma membrane. Furthermore, TRE17 colocalized with Cdc42V12 in filopodia (Fig. 3E). In distinct confocal sections, TRE17 did not colocalize with Cdc42 in the Golgi apparatus (data not shown). As seen with Rac1V12, Cdc42V12's ability to recruit TRE17 to the membrane was reversed by treatment with cytoD (Fig. 3F).
Together, these results indicate that activated Rac1 and Cdc42 can direct the relocalization of TRE17 to the plasma membrane in a manner that depends on integrity of the actin cytoskeleton.
TRE17 localization is regulated by growth factors.
Mitogenic growth factors, such as EGF, rapidly induce changes in the actin cytoskeleton and are known to activate Cdc42 and Rac1 (7, 17, 45). We therefore sought to determine whether such factors could also regulate the localization of TRE17. TRE17-transfected HeLa cells were serum starved for 24 h and then stimulated with EGF for 10 min. In serum-starved cells, TRE17 was found predominantly on filamentous structures (Fig. 4A, left panel). However, stimulation with EGF led to a dramatic relocalization of TRE17 to the plasma membrane, accompanied by strong accumulation of F-actin at the cell cortex (Fig. 4A, middle panels). Quantification of TRE17 localization in the absence or presence of EGF is presented in Fig. 4B. Pretreatment of cells with cytoD prior to EGF stimulation completely abolished plasma membrane recruitment of TRE17 (Fig. 4A, right panel). This result demonstrates that physiological agonists such as growth factors can also regulate the localization of TRE17, again in a manner that depends on cortical actin accumulation.
EGF-induced recruitment of TRE17 to the plasma membrane requires Rac1 and Cdc42.
Since Cdc42 and Rac1 are activated by EGF, we examined whether they might mediate EGF's effects on TRE17 relocalization. Coexpression of TRE17 with dominant-negative Rac1 (Rac1N17) or Cdc42 (Cdc42N17) led to an altered subcellular distribution of TRE17 under starved conditions. In Rac1N17 coexpressing cells, TRE17 was found predominantly on filamentous structures, but weak plasma membrane localization was also observed (data not shown). Stimulation of these cells with EGF resulted in no further increase in plasma membrane staining of TRE17, and a significant amount of TRE17 remained on filamentous structures (Fig. 5B). Thus, Rac1N17 partially blocked EGF-induced relocalization of TRE17. The effects of Cdc42N17 were more complicated, in part because many of the cells did not appear healthy. Under starved conditions, Cdc42N17 abolished the localization of TRE17 to filamentous structures and led to a weak recruitment to the plasma membrane (data not shown). A significant fraction of TRE17 was also found in intracellular aggregates (Fig. 5C). However, in the few healthy-looking cells coexpressing Cdc42N17 and TRE17, EGF failed to further enhance plasma membrane staining of TRE17 (Fig. 5C).
FIG. 5.
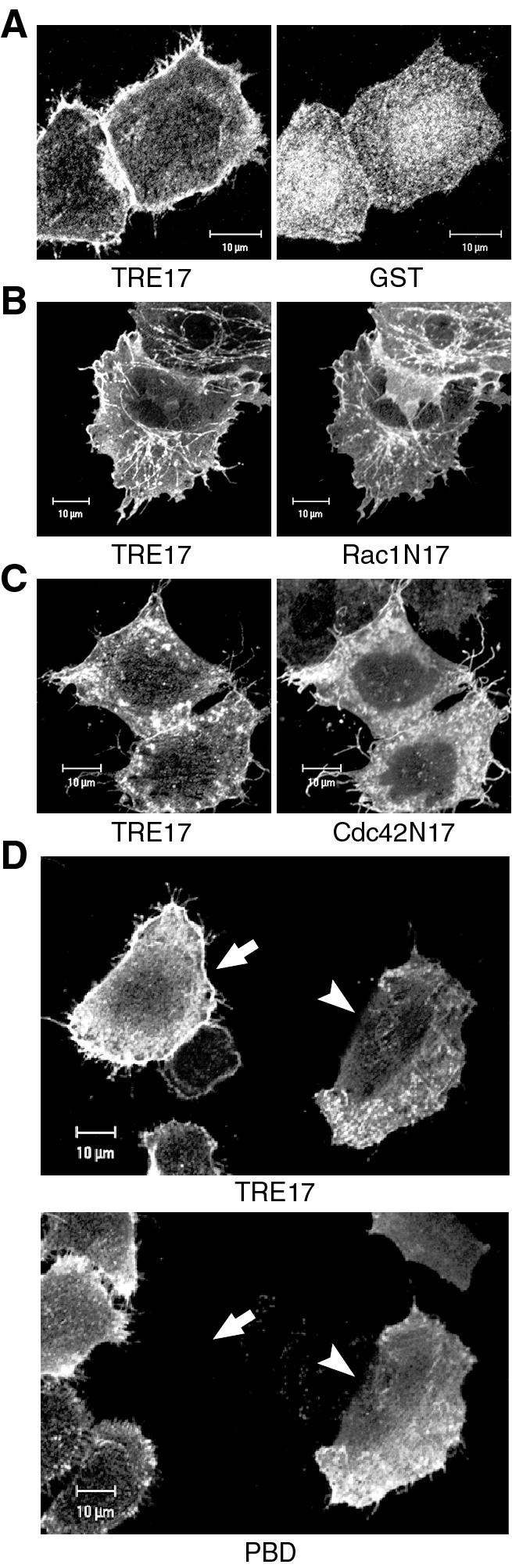
Rac1 and Cdc42 activity are required for EGF-stimulated recruitment of TRE17 to the plasma membrane. HA-tagged TRE17 was cotransfected with GST (A), GST-Rac1N17 (B), GST-Cdc42N17 (C), or GST-PBD (D), which encodes the p21-binding domain of PAK1. Cells were serum starved for 24 h and then stimulated with EGF (100 ng/ml) for 10 min. TRE17 localization was determined with anti-HA antibody, whereas localization of GST, Rac1N17, Cdc42N17, and PBD was determined with anti-GST antibody. In panel D, the upper panel represents the TRE17 localization, and the lower panel represents PBD localization. The arrow indicates a cell expressing TRE17 alone, whereas the arrowhead indicates a cell coexpressing PBD. Scale bar, 10 μm.
As an alternative means of blocking Cdc42 and Rac1 signaling simultaneously, we coexpressed TRE17 with the p21-binding domain (PBD) of PAK1. This peptide binds to Cdc42 and Rac1 specifically in their active GTP-bound state and prevents their interaction with downstream effectors. Coexpression of PBD essentially completely blocked the EGF-stimulated recruitment of TRE17 to the plasma membrane (Fig. 5D). In these cells, TRE17 was found on short filamentous and punctate structures. Together, these results suggest that perturbation of Cdc42- and Rac1-mediated signaling blocks EGF-induced recruitment of TRE17 to the plasma membrane.
Structural determinants of TRE17 localization.
To determine the requirements for its subcellular localization, structure-function analysis of TRE17 was performed. TRE17 contains a PTM domain near its N terminus, as well as a proline-rich domain that contains potential SH3 or WW domain binding sites (24). Three deletion constructs were generated as depicted in Fig. 6A. These mutants were transfected into HeLa cells, and their localization analyzed under serum-starved conditions. As noted above, full-length TRE17 was found predominantly on filamentous structures (Fig. 6B). However, a mutant expressing the first 447 amino acids [T17(447)] was found at the plasma membrane as well as on filamentous structures (Fig. 6B). Two additional constructs, encoding the PTM domain alone [T17(PTM)] or a deletion of the PTM domain [T17(ΔPTM)], were diffuse in the cytoplasm; no staining of filaments or the plasma membrane was discernible (Fig. 6B).
The distinct localization of the TRE17 mutants was confirmed by using biochemical cell fractionation. Cells were hypotonically lysed and then fractionated by centrifugation at low speed. As seen in Fig. 6C, a significant fraction of full-length TRE17 and T17(447) were found in the insoluble pellet fraction, whereas T17(PTM) and T17(ΔPTM) were largely in the soluble fraction. Together, these results indicate that the first 447 amino acids of TRE17 contain the determinants for localization at the plasma membrane and filamentous structures. Furthermore, they reveal that the PTM domain is necessary but not sufficient for normal localization of TRE17.
T17(447) stimulates cortical actin accumulation.
As shown above, T17(447) localized to the plasma membrane even in the absence of growth factor stimulation. This led us to speculate that it might represent a constitutively active allele. Surprisingly, we found that expression of T17(447) in serum-starved cells induced strong accumulation of F-actin at the cell cortex, a finding similar to what was observed in some cells expressing Cdc42V12 (Fig. 7A). Quantification revealed that ca. 65% of T17(447)-expressing cells exhibited strong cortical actin staining compared to 18% of the untransfected cells. Full-length TRE17 was also able to induce cortical actin accumulation, albeit at a lower frequency than T17(447) (Fig. 7B).
In order to gain further insights into the mechanism of TRE17-induced actin remodeling, we analyzed the phenotype induced by two additional mutants. As mentioned above, TRE17 contains a Rab GAP homology domain at its N terminus. All Rab GAPs characterized to date contain a conserved arginine residue that is essential for catalytic activity; mutation of this residue to lysine completely abolishes GAP function (2, 3, 20, 42). We found that mutation of the corresponding residue in TRE17 (arginine 149) had no effect on the ability of T17(447) [mutant T17(447/RK)] to induce cortical actin accumulation (Fig. 7B). Another notable feature of T17(447) is the presence of two proline-rich motifs with potential for binding to WW or SH3 domains (24). However, we found that mutation of these motifs also left T17(447)'s actin remodeling activity intact [mutant T17(447/PA); Fig. 7B]. Both T17(447/RK) and T17(447/PA) exhibited localization patterns identical to T17(447) (data not shown). Together, these results indicate that TRE17's ability to localize to the plasma membrane and induce cortical actin accumulation is independent of its putative RabGAP activity and of interactions mediated by the proline-rich motifs.
We next tested whether T17(447)'s membrane localization or ability to induce cortical actin accumulation required Cdc42 and Rac1. Surprisingly, localization of T17(447) to the plasma membrane was not blocked by coexpression with Cdc42N17, Rac1N17, or PBD (Fig. 7C). Similarly, the accumulation of cortical actin induced by T17(447) persisted in the presence of these inhibitory molecules (Fig. 7C). These data suggest that T17(447) functions downstream of Cdc42 and Rac1 and that it may serve to mediate some of their effects on reorganization of the actin cytoskeleton. Our data further suggest a dynamic relationship between TRE17 and the actin cytoskeleton, such that cortical actin polymerization is required for stable plasma membrane recruitment of TRE17; once recruited, TRE17 may stabilize or further stimulate the accumulation of cortical actin.
DISCUSSION
In the present study, we establish TRE17 as part of a novel effector pathway for the Rho family GTPases Cdc42 and Rac1. TRE17 associates with Cdc42 and Rac1 in a manner that requires activation and membrane localization of the G proteins. Consistent with this interaction, Cdc42 and Rac1 regulate the subcellular localization of TRE17. Constitutively active mutants of Cdc42 and Rac1 induce its recruitment to the plasma membrane, where the GTPases colocalize with TRE17 in filopodia and lamellapodia, respectively. In addition, WT Rac1 colocalizes with TRE17 on filamentous structures. Membrane recruitment of TRE17 is also induced by EGF in a manner that is dependent on activation of the GTPases and actin polymerization. Finally, we find that a TRE17 mutant encoding the N-terminal 447 amino acids is constitutively localized to the plasma membrane. Surprisingly, this mutant was able to stimulate the accumulation of F-actin at the cell cortex under serum-starved conditions, recapitulating some of the effects of Cdc42V12. Together, these data suggest that TRE17 may contribute to Cdc42's and Rac1's effects on reorganization of the actin cytoskeleton. Our work thus links this previously uncharacterized oncogene to a well-studied signaling pathway.
Taken together, our data lead to the following model for the regulation of TRE17 (Fig. 8). In quiescent cells, TRE17 is localized to intracellular filamentous and punctate structures in the cytoplasm, folded in an inactive conformation. Upon growth factor addition, Cdc42 and Rac1 become activated and recruit TRE17 to the plasma membrane. Stable membrane localization of TRE17 also requires polymerized actin. This recruitment process leads to a conformational change in TRE17, such that the N-terminal portion of the molecule further stimulates the accumulation of cortical actin. Consistent with this model, deletion of the C terminus results in constitutive activation of TRE17, such that it localizes to the plasma membrane and stimulates cortical actin accumulation in a mitogen- and Cdc42/Rac1-independent manner.
FIG. 8.

Model of TRE17 regulation and function. In quiescent cells, TRE17 is localized to filamentous structures in the cytoplasm in an inactive conformation. EGF stimulates the activation of Cdc42 and Rac1, leading to cortical actin accumulation, as well as recruitment of TRE17 to the plasma membrane. This process leads to a conformational change in TRE17, such that it now stimulates further cortical actin accumulation via its N-terminal 447 amino acids. See the text for further details.
Presently, the precise mechanism by which polymerized actin contributes to plasma membrane recruitment of TRE17 remains unknown. TRE17 does not bind directly to G- or F-actin (data not shown). It is possible that F-actin (or actin-binding proteins) may not actively promote recruitment but instead may be required passively for translocation of TRE17 from the filamentous structures to the plasma membrane. It also remains unclear how TRE17 induces cortical actin remodeling. TRE17 does not bind N-WASP or Arp2 or recruit them to sites of cortical actin accumulation (data not shown). Finally, TRE17 has no effect on PAK activity (data not shown). Thus, TRE17 appears to be a component of a novel pathway leading to actin remodeling. Consistent with this notion, the binding profile of TRE17 with the effector domain mutants of Cdc42 and Rac1 is distinct from that of previously characterized effectors (19, 22, 56). Of note, the effector domain mutants that do associate with TRE17 retain the ability to induce actin remodeling (19, 56).
Given that the role of Cdc42 and Rac1 in cellular transformation and actin remodeling is well established, their identification as upstream regulators of TRE17 was highly illuminating. These GTPases regulate cytoskeletal dynamics through multiple effector pathways. As discussed above, Cdc42's targets include WASP, the PAK kinases, and MRCK. Effectors of Rac1 include IRSp53/WAVE, the PAK kinases, POR1, and phosphatidylinositol-4-phosphate 5-kinase. Our results suggest that TRE17 may be an effector for both Cdc42 and Rac1 in actin remodeling. In HeLa cells, RacV12 induced peripheral and dorsal ruffles, whereas Cdc42V12 induced filopodia in ca. 75% of transfected cells and cortical actin accumulation (in the absence of pronounced filopodia) in the remaining 25%. It is interesting that the pattern of F-actin accumulation induced by T17(447) mimicked only the latter Cdc42 phenotype. How can this be reconciled with a role for TRE17 in signaling downstream of both Cdc42 and Rac1? One likely contributing factor is that T17(447) is deregulated, as demonstrated by its mitogen- and Cdc42-/Rac1-independent localization at the plasma membrane. This persistent localization may drive the accumulation of F-actin at the cell cortex in a manner distinct from the full-length protein, which is regulated more dynamically by the GTPases. Another probable factor is that both Rac1 and Cdc42 utilize multiple effectors to regulate the cytoskeleton, as discussed above. The cytoskeletal alterations induced by these GTPases represent a composite of the coordinated activities of all of these effectors. Therefore, it is not surprising that a single effector is not sufficient to recapitulate the full range of their effects. This hypothesis is supported by the literature, where overexpression of a single effector, such as WASP (49), PAK, MRCKα (21), ROCK, or mDia1 (55), is unable to phenocopy the precise actin rearrangements induced by activated Rho GTPases. Indeed, overexpression of activated PAK, which is believed to be an effector for both Cdc42 and Rac1, only induces lamellipodia and not filopodia in multiple cell types (10-12, 47, 48). Furthermore, in HeLa cells it was found that activated PAK alleles do not induce either lamellipodia or filopodia but rather inhibit stress fiber and focal adhesion formation (26).
Another possibility is that TRE17 may not function as an effector for both GTPases in actin remodeling but may instead link them to other cellular processes, such as vesicular trafficking. In this context, it is important to highlight the colocalization of TRE17 and Rac1 on filamentous structures (Fig. 3A). In HeLa cells, endogenous Rac1 has been shown to localize to similar structures which, like the TRE17-positive filaments, are enhanced in the presence of cytoD (41). These structures in fact represent a tubular endosomal compartment of a plasma membrane recycling system that is regulated by the Arf6 GTPase (6, 40, 41). Nocodazole has been shown to disrupt this compartment, leading to its collapse into a perinuclear aggregrate (40), a finding that is again similar to what was observed with TRE17. The potential localization of TRE17 to a trafficking compartment is particularly exciting since it contains a PTM/Tbc/Gyp domain. As mentioned above, the PTM/Tbc/Gyp domains of other proteins have been shown to function as GAPs for Rab family GTPases (2, 9, 20, 42), which regulate both the endocytic and secretory pathways (36).
In summary, we provide here the first insights into the cellular functions and molecular interactions of this previously uncharacterized oncogene. Future efforts will be aimed at elucidating the mechanism by which TRE17 induces actin remodeling and assessing its role in vesicular trafficking. Furthermore, its role in Cdc42- and Rac-mediated transformation will be investigated. Through these combined studies, we hope to ultimately gain insights into how TRE17 causes cellular transformation.
Acknowledgments
We thank Miriam Onno for providing the cDNA encoding TRE17. We thank Gerd A. Blobel, Sally Zigmond, Erfei Bi, and Judy Meinkoth for insightful comments and suggestions on the manuscript. We epecially thank Sally Zigmond for assistance on the actin-related studies.
This work was supported by the NIH (grant 1 RO1 CA81415-01A1).
REFERENCES
- 1.Akhtar, N., and N. A. Hotchin. 2001. RAC1 regulates adherens junctions through endocytosis of E-cadherin. Mol. Biol. Cell 12:847-862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Albert, S., and D. Gallwitz. 1999. Two new members of a family of Ypt/Rab GTPase activating proteins: promiscuity of substrate recognition. J. Biol. Chem. 274:33186-33189. [DOI] [PubMed] [Google Scholar]
- 3.Albert, S., E. Will, and D. Gallwitz. 1999. Identification of the catalytic domains and their functionally critical arginine residues of two yeast GTPase-activating proteins specific for Ypt/Rab transport GTPases. EMBO J. 18:5216-5225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bi, E., J. B. Chiavetta, H. Chen, G. C. Chen, C. S. Chan, and J. R. Pringle. 2000. Identification of novel, evolutionarily conserved Cdc42p-interacting proteins and of redundant pathways linking Cdc24p and Cdc42p to actin polarization in yeast. Mol. Biol. Cell 11:773-793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bishop, A. L., and A. Hall. 2000. Rho GTPases and their effector proteins. Biochem. J. 348(Pt. 2):241-255. [PMC free article] [PubMed] [Google Scholar]
- 6.Brown, F. D. 2001. Phosphatidylinositol 4,5-bisphosphate and Arf6-regulated membrane traffic. J. Cell Biol. 154:1007-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chinkers, M., J. A. McKanna, and S. Cohen. 1979. Rapid induction of morphological changes in human carcinoma cells A-431 by epidermal growth factors. J. Cell Biol. 83:260-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chou, M. M., and J. Blenis. 1996. The 70-kDa S6 kinase complexes with and is activated by the Rho family G proteins Cdc42 and Rac1. Cell 85:573-583. [DOI] [PubMed] [Google Scholar]
- 9.Cuif, M. H., F. Possmayer, H. Zander, N. Bordes, F. Jollivet, A. Couedel-Courteille, I. Janoueix-Lerosey, G. Langsley, M. Bornens, and B. Goud. 1999. Characterization of GAPCenA, a GTPase activating protein for Rab6, part of which associates with the centrosome. EMBO J. 18:1772-1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dharmawardhane, S., L. C. Sanders, S. S. Martin, R. H. Daniels, and G. M. Bokoch. 1997. Localization of p21-activated kinase 1 (PAK1) to pinocytic vesicles and cortical actin structures in stimulated cells. J. Cell Biol. 138:1265-1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edwards, D. C., L. C. Sanders, G. M. Bokoch, and G. N. Gill. 1999. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat. Cell Biol. 1:253-259. [DOI] [PubMed] [Google Scholar]
- 12.Frost, J. A., A. Khokhlatchev, S. Stippec, M. A. White, and M. H. Cobb. 1998. Differential effects of PAK1-activating mutations reveal activity-dependent and -independent effects on cytoskeletal regulation. J. Biol. Chem. 273:28191-28198. [DOI] [PubMed] [Google Scholar]
- 13.Garrett, W. S., L. M. Chen, R. Kroschewski, M. Ebersold, S. Turley, S. Trombetta, J. E. Galan, and I. Mellman. 2000. Developmental control of endocytosis in dendritic cells by Cdc42. Cell 102:325-334. [DOI] [PubMed] [Google Scholar]
- 14.Gjoerup, O., J. Lukas, J. Bartek, and B. M. Willumsen. 1998. Rac and Cdc42 are potent stimulators of E2F-dependent transcription capable of promoting retinoblastoma susceptibility gene product hyperphosphorylation. J. Biol. Chem. 273:18812-18818. [DOI] [PubMed] [Google Scholar]
- 15.Johnson, D. I., and J. R. Pringle. 1990. Molecular characterization of CDC42, a Saccharomyces cerevisiae gene involved in the development of cell polarity. J. Cell Biol. 111:143-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keely, P. J., J. K. Westwick, I. P. Whitehead, C. J. Der, and L. V. Parise. 1997. Cdc42 and Rac1 induce integrin-mediated cell motility and invasiveness through PI(3)K. Nature 390:632-636. [DOI] [PubMed] [Google Scholar]
- 17.Kozma, R., S. Ahmed, A. Best, and L. Lim. 1995. The Ras-related protein Cdc42Hs and bradykinin promote formation of peripheral actin microspikes and filopodia in Swiss 3T3 fibroblasts. Mol. Cell. Biol. 15:1942-1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kroschewski, R., A. Hall, and I. Mellman. 1999. Cdc42 controls secretory and endocytic transport to the basolateral plasma membrane of MDCK cells. Nat. Cell Biol. 1:8-13. [DOI] [PubMed] [Google Scholar]
- 19.Lamarche, N., N. Tapon, L. Stowers, P. D. Burbelo, P. Aspenstrom, T. Bridges, J. Chant, and A. Hall. 1996. Rac and Cdc42 induce actin polymerization and G1 cell cycle progression independently of p65PAK and the JNK/SAPK MAP kinase cascade. Cell 87:519-529. [DOI] [PubMed] [Google Scholar]
- 20.Lanzetti, L., V. Rybin, M. G. Malabarba, S. Christoforidis, G. Scita, M. Zerial, and P. P. Di Fiore. 2000. The Eps8 protein coordinates EGF receptor signalling through Rac and trafficking through Rab5. Nature 408:374-377. [DOI] [PubMed] [Google Scholar]
- 21.Leung, T., X. Q. Chen, I. Tan, E. Manser, and L. Lim. 1998. Myotonic dystrophy kinase-related Cdc42-binding kinase acts as a Cdc42 effector in promoting cytoskeletal reorganization. Mol. Cell. Biol. 18:130-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li, R., B. Debreceni, B. Jia, Y. Gao, G. Tigyi, and Y. Zheng. 1999. Localization of the PAK1-, WASP-, and IQGAP1-specifying regions of Cdc42. J. Biol. Chem. 274:29648-29654. [DOI] [PubMed] [Google Scholar]
- 23.Lin, R., S. Bagrodia, R. Cerione, and D. Manor. 1997. A novel Cdc42Hs mutant induces cellular transformation. Curr. Biol. 7:794-797. [DOI] [PubMed] [Google Scholar]
- 24.Macias, M. J., S. Wiesner, and M. Sudol. 2002. WW and SH3 domains, two different scaffolds to recognize proline-rich ligands. FEBS Lett. 513:30-37. [DOI] [PubMed] [Google Scholar]
- 25.Malecz, N., P. C. McCabe, C. Spaargaren, R. Qiu, Y. Chuang, and M. Symons. 2000. Synaptojanin 2, a novel Rac1 effector that regulates clathrin-mediated endocytosis. Curr. Biol. 10:1383-1386. [DOI] [PubMed] [Google Scholar]
- 26.Manser, E., H. Y. Huang, T. H. Loo, X. Q. Chen, J. M. Dong, T. Leung, and L. Lim. 1997. Expression of constitutively active alpha-PAK reveals effects of the kinase on actin and focal complexes. Mol. Cell. Biol. 17:1129-1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Manser, E., and L. Lim. 1999. Roles of PAK family kinases. Prog. Mol. Subcell. Biol. 22:115-133. [DOI] [PubMed] [Google Scholar]
- 28.Matoskova, B., W. T. Wong, N. Nomura, K. C. Robbins, and P. P. Di Fiore. 1996. RN-tre specifically binds to the SH3 domain of eps8 with high affinity and confers growth advantage to NIH 3T3 upon carboxy-terminal truncation. Oncogene 12:2679-2688. [PubMed] [Google Scholar]
- 29.Matoskova, B., W. T. Wong, N. Seki, T. Nagase, N. Nomura, K. C. Robbins, and P. P. Di Fiore. 1996. RN-tre identifies a family of tre-related proteins displaying a novel potential protein binding domain. Oncogene 12:2563-2571. [PubMed] [Google Scholar]
- 30.Miki, H., T. Sasaki, Y. Takai, and T. Takenawa. 1998. Induction of filopodium formation by a WASP-related actin-depolymerizing protein N-WASP. Nature 391:93-96. [DOI] [PubMed] [Google Scholar]
- 31.Miki, H., S. Suetsugu, and T. Takenawa. 1998. WAVE, a novel WASP-family protein involved in actin reorganization induced by Rac. EMBO J. 17:6932-6941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miki, H., H. Yamaguchi, S. Suetsugu, and T. Takenawa. 2000. IRSp53 is an essential intermediate between Rac and WAVE in the regulation of membrane ruffling. Nature 408:732-735. [DOI] [PubMed] [Google Scholar]
- 33.Nakamura, T., J. Hillova, R. Mariage-Samson, M. Onno, K. Huebner, L. A. Cannizzaro, L. Boghosian-Sell, C. M. Croce, and M. Hill. 1992. A novel transcriptional unit of the tre oncogene widely expressed in human cancer cells. Oncogene 7:733-741. [PubMed] [Google Scholar]
- 34.Neuwald, A. F. 1997. A shared domain between a spindle assembly checkpoint protein and Ypt/Rab-specific GTPase-activators. Trends Biochem. Sci. 22:243-244. [DOI] [PubMed] [Google Scholar]
- 35.Nobes, C. D., and A. Hall. 1995. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81:53-62. [DOI] [PubMed] [Google Scholar]
- 36.Novick, P., and M. Zerial. 1997. The diversity of Rab proteins in vesicle transport. Curr. Opin. Cell Biol. 9:496-504. [DOI] [PubMed] [Google Scholar]
- 37.Olson, M. F., A. Ashworth, and A. Hall. 1995. An essential role for Rho, Rac, and Cdc42 GTPases in cell cycle progression through G1. Science 269:1270-1272. [DOI] [PubMed] [Google Scholar]
- 38.Qiu, R. G., A. Abo, F. McCormick, and M. Symons. 1997. Cdc42 regulates anchorage-independent growth and is necessary for Ras transformation. Mol. Cell. Biol. 17:3449-3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qiu, R. G., J. Chen, D. Kirn, F. McCormick, and M. Symons. 1995. An essential role for Rac in Ras transformation. Nature 374:457-459. [DOI] [PubMed] [Google Scholar]
- 40.Radhakrishna, H. 1997. ADP-ribosylation factor 6 regulates a novel plasma membrane recycling pathway. J. Cell Biol. 139:49-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Radhakrishna, H., O. Al-Awar, Z. Khachikian, and J. G. Donaldson. 1999. ARF6 requirement for Rac ruffling suggests a role for membrane trafficking in cortical actin rearrangements. J. Cell Sci. 112:855-866. [DOI] [PubMed] [Google Scholar]
- 42.Rak, A., R. Fedorov, K. Alexandrov, S. Albert, R. S. Goody, D. Gallwitz, and A. J. Scheidig. 2000. Crystal structure of the GAP domain of Gyp1p: first insights into interaction with Ypt/Rab proteins. EMBO J. 19:5105-5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ramos, E., R. B. Wysolmerski, and R. A. Masaracchia. 1997. Myosin phosphorylation by human cdc42-dependent S6/H4 kinase/gammaPAK from placenta and lymphoid cells. Recept. Signal Transduct. 7:99-110. [PubMed] [Google Scholar]
- 44.Richardson, P. M., and L. I. Zon. 1995. Molecular cloning of a cDNA with a novel domain present in the tre-2 oncogene and the yeast cell cycle regulators BUB2 and cdc16. Oncogene 11:1139-1148. [PubMed] [Google Scholar]
- 45.Ridley, A. J., H. F. Paterson, C. L. Johnston, D. Diekmann, and A. Hall. 1992. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 70:401-410. [DOI] [PubMed] [Google Scholar]
- 46.Rohatgi, R., L. Ma, H. Miki, M. Lopez, T. Kirchhausen, T. Takenawa, and M. W. Kirschner. 1999. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell 97:221-231. [DOI] [PubMed] [Google Scholar]
- 47.Sells, M. A., J. T. Boyd, and J. Chernoff. 1999. p21-activated kinase 1 (Pak1) regulates cell motility in mammalian fibroblasts. J. Cell Biol. 145:837-849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sells, M. A., U. G. Knaus, S. Bagrodia, D. M. Ambrose, G. M. Bokoch, and J. Chernoff. 1997. Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr. Biol. 7:202-210. [DOI] [PubMed] [Google Scholar]
- 49.Symons, M., J. M. Derry, B. Karlak, S. Jiang, V. Lemahieu, F. McCormick, U. Francke, and A. Abo. 1996. Wiskott-Aldrich syndrome protein, a novel effector for the GTPase CDC42Hs, is implicated in actin polymerization. Cell 84:723-734. [DOI] [PubMed] [Google Scholar]
- 50.Takenawa, T., T. Itoh, and K. Fukami. 1999. Regulation of phosphatidylinositol 4,5-bisphosphate levels and its roles in cytoskeletal re-organization and malignant transformation. Chem. Phys. Lipids 98:13-22. [DOI] [PubMed] [Google Scholar]
- 51.Takenawa, T., and H. Miki. 2001. WASP and WAVE family proteins: key molecules for rapid rearrangement of cortical actin filaments and cell movement. J. Cell Sci. 114:1801-1809. [DOI] [PubMed] [Google Scholar]
- 52.Tolias, K. F., L. C. Cantley, and C. L. Carpenter. 1995. Rho family GTPases bind to phosphoinositide kinases. J. Biol. Chem. 270:17656-17659. [DOI] [PubMed] [Google Scholar]
- 53.Tolias, K. F., J. H. Hartwig, H. Ishihara, Y. Shibasaki, L. C. Cantley, and C. L. Carpenter. 2000. Type Iα phosphatidylinositol-4-phosphate 5-kinase mediates Rac-dependent actin assembly. Curr. Biol. 10:153-156. [DOI] [PubMed] [Google Scholar]
- 54.Van Aelst, L., T. Joneson, and D. Bar-Sagi. 1996. Identification of a novel Rac1-interacting protein involved in membrane ruffling. EMBO J. 15:3778-3786. [PMC free article] [PubMed] [Google Scholar]
- 55.Watanabe, N., T. Kato, A. Fujita, T. Ishizaki, and S. Narumiya. 1999. Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nat. Cell Biol. 1:136-143. [DOI] [PubMed] [Google Scholar]
- 56.Westwick, J. K., Q. T. Lambert, G. J. Clark, M. Symons, L. Van Aelst, R. G. Pestell, and C. J. Der. 1997. Rac regulation of transformation, gene expression, and actin organization by multiple, PAK-independent pathways. Mol. Cell. Biol. 17:1324-1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu, W. J., J. W. Erickson, R. Lin, and R. A. Cerione. 2000. The gamma-subunit of the coatomer complex binds Cdc42 to mediate transformation. Nature 405:800-804. [DOI] [PubMed] [Google Scholar]
- 58.Wu, W. J., R. Lin, R. A. Cerione, and D. Manor. 1998. Transformation activity of Cdc42 requires a region unique to Rho-related proteins. J. Biol. Chem. 273:16655-16658. [DOI] [PubMed] [Google Scholar]
- 59.Yang, N., O. Higuchi, K. Ohashi, K. Nagata, A. Wada, K. Kangawa, E. Nishida, and K. Mizuno. 1998. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature 393:809-812. [DOI] [PubMed] [Google Scholar]
- 60.Zhang, S. D., J. Kassis, B. Olde, D. M. Mellerick, and W. F. Odenwald. 1996. Pollux, a novel Drosophila adhesion molecule, belongs to a family of proteins expressed in plants, yeast, nematodes, and man. Genes Dev. 10:1108-1119. [DOI] [PubMed] [Google Scholar]