

Abstract
The IκB kinase (IKK) complex mediates activation of transcription factor NF-κB by phosphorylation of IκB proteins. Its catalytic subunits, IKKα and IKKβ, require association with the regulatory IKKγ (NEMO) component to gain full basal and inducible kinase activity. However, the oligomeric composition of the IKK complex and its regulation by IKKγ are poorly understood. We show here that IKKγ predominantly forms tetramers and interacts with IKKα or IKKβ in this state. We propose that tetramerization is accomplished by a prerequisite dimerization through a C-terminal coiled-coil minimal oligomerization domain (MOD). This is followed by dimerization of the dimers with their N-terminal sequences. Tetrameric IKKγ sequesters four kinase molecules, yielding a γ4(α/β)4 stoichiometry. Deletion of the MOD leads to loss of tetramerization and of phosphorylation of IKKβ and IKKγ, although the kinase can still interact with the resultant IKKγ monomers and dimers. Likewise, MOD-mediated IKKγ tetramerization is required to enhance IKKβ kinase activity when overexpressed in 293 cells and to reconstitute a lipopolysaccharide-responsive IKK complex in pre-B cells. These data thus suggest that IKKγ tetramerization enforces a spatial positioning of two kinase dimers to facilitate transautophosphorylation and activation.
A large number of physiological stimuli activate NF-κB transcription factors to regulate gene expression in innate or adaptive immune responses in development and cellular growth control. In unstimulated cells, NF-κB proteins are sequestered by the small cytosolic IκB molecules α, β, and ɛ or by the precursor proteins NF-κB1/p105 and NF-κB2/p100 (24, 36). Upon cellular stimulation with proinflammatory agents, including tumor necrosis factor alpha (TNF-α), interleukin 1 (IL-1), and bacterial lipopolysaccharide (LPS), or with antigens, viral pathogens, growth factors, or morphogens, these IκBs are phosphorylated by an IκB kinase (IKK) complex. Phosphorylated IκB proteins are ubiquitinated by the SCFβTRCP ubiquitin ligase complex and subject to proteolysis by the proteasome, resulting in liberation and nuclear translocation of NF-κB (11, 32).
The prevalent cellular IKK complex contains three components, two kinases, IKKα (also called IKK1) and IKKβ (also called IKK2) and the noncatalytic IKKγ (also called NEMO) protein. The kinases share 52% identity and contain an N-terminal catalytic domain, a central leucine zipper, and a C-terminal helix-loop-helix motif. The kinases form homo- or heterodimers via their leucine zippers, and their activity depends on their ability to dimerize (16, 27, 35, 45). Direct interaction with IKKγ is mediated by a short motif at the extreme C terminus of IKKα and IKKβ (23). Deletion of this motif or introduction of a short peptide containing this sequence into cells impairs binding of both kinases to IKKγ and activation of IKK kinase activity and of NF-κB (23).
Murine IKKγ was identified by complementation cloning using NF-κB activation-deficient rodent cells (39). The human homologue was obtained by biochemical purification of the IKK complex and microsequencing (26, 31) and cloned as an adenovirus (Ad E3-14.7K)-interacting protein (21). ΙKKγ is highly conserved between species, and structural predictions indicate a high α-helical content, with extended coiled-coil regions, a leucine zipper, and a C-terminal zinc finger. The binding site of IKKγ for the kinases has been confined to a region within the first half of the molecules, although different results have been reported for the more precise localization of the IKKα-IKKβ interaction region (23, 26, 29, 31, 41). IKKγ is essential for activation of IKKα and IKKβ by all physiological stimuli analyzed, and C-terminal sequences of IKKγ are specifically required for responsiveness of the complex to proinflammatory stimuli (10, 31, 39).
At present, it is not known by which mechanism the IKK complex is activated. A critical step is phosphorylation of the kinases at activation loop residues. Stimulation of IKK kinase activity by TNF-α coincides with serine phosphorylation of IKKα and IKKβ and with serine/threonine phosphorylation of IKKγ. For IKKβ, two of the sites were mapped in the activation loop to residues 177 and 181, and mutation of these sites eliminates kinase activation (7). However, IKKα and IKKβ are biochemically not equivalent, and IKKα activation loop phosphorylation is not required for IKK activation by TNF-α or IL-1 (7). Gene ablation experiments have established that IKKβ and IKKγ are required for NF-κB activation by proinflammatory stimuli, while IKKα is essential for morphogenic signals (11, 15, 16). The activation loop serines of IKKα are required for RANK-induced cyclin D1 expression and proliferation of mammary epithelial cells (4). The function of IKKα in keratinocyte differentiation in the epidermis, however, is independent of its kinase activity and of NF-κB (13). It is not known how distinct functions are mediated by a canonical heteromeric IKKα-IKKβ-IKKγ complex and whether separate IKKα complexes exist.
Activation loop phosphorylation of IKKβ, induced by proinflammatory signals, could be caused by transautophosphorylation or by upstream acting kinases that perhaps are sequestered by the C-terminal Zn finger region of IKKγ. However, no direct and functional interaction between this domain and any known component of the TNF-α or IL-1 signaling pathways has yet been demonstrated. The kinases RIP1 and IRAK1, which are functionally essential constituents of the ligand-induced TNF receptor and IL-1 receptor complexes, respectively, do not need their kinase activity to stimulate IKK (8, 20). Furthermore, none of the MAP3K-like kinases which regulate IKK upon overexpression, such as NIK, MEKK1, and Cot/Tpl-2 (11), have been confirmed as physiological, proinflammatory IKK activators by genetic evidence (9, 37, 42, 43). In contrast, MEKK3 appears to be required for TNF-α-induced IKK activation in fibroblasts and functions downstream of RIP and TRAF2. However, how this kinase activates IKK is not understood (40).
Engagement of TNF receptor 1 has been shown to result in recruitment of the IKK complex into the receptor complex along with TRAF2 and RIP1 (8, 46). RIP is essential for TNF-α induction of IKK activity (8, 17) and interacts with IKKγ (46). Since the enzymatic activity of RIP1 is dispensable (8), activation of IKK following recruitment could be due to an induced proximity mechanism. In fact, enforced oligomerization of an N-terminal portion of IKKγ or of the IKKα or IKKβ kinase domains can induce IKK and NF-κB activation (29). Furthermore, oligomerization of IKKγ was proposed to be induced by TNF-α stimulation or by RIP overexpression (29).
On the basis of stoichiometric analyses of recombinant components in vitro and of reconstituted IKK complexes in Saccharomyces cerevisiae, an equal stoichiometric amount of IKKα, IKKβ, and IKKγ in the heteromeric complex was proposed (19, 28). Given that IKKα and IKKβ form homo- and heterodimers and IKKγ was proposed to form dimers (31, 39), it was assumed that an IKKα-IKKβ dimer is bound by a dimer of IKKγ (28, 30). However, a recent report claimed that IKKγ predominantly forms trimers (1). It has not been established in these studies whether oligomerization is essential for a functional, inducible IKK complex.
We have determined the oligomerization state of IKKγ by hydrodynamic analysis, protein-protein interaction assays, and chemical cross-linking in intact cells and in vitro and found that IKKγ forms tetramers. We identified a C-terminal oligomerization domain, which is a prerequisite for IKKγ tetramerization. Our data suggest that IKKγ tetramerization is mediated by antiparallel dimerization of two dimers and allows docking of two kinase dimers. Functional analysis demonstrates that the oligomeric assembly is requisite to activation of the holocomplex, likely by providing a spatial arrangement of kinase dimers.
MATERIALS AND METHODS
Cell culture.
HeLa and 293 cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 1 mM sodium pyruvate, and 100 U of penicillin and streptomycin per ml. Pre-murine B-cell lines 70Z/3 and 1.3E2 were maintained in RPMI 1640 medium supplemented with 7.5% fetal calf serum, 2 mM l-glutamine, 100 U of penicillin and streptomycin per ml, and 50 μM β-mercaptoethanol. Stable 1.3E2 IKKγ clones were generated as described previously (12), and cells were maintained with 1 μg of G418 per ml of medium.
Expression plasmids and purification of recombinant proteins.
Human IKKγ cDNA was cloned as described elsewhere (19). Using standard PCR techniques, IKKγ and IKKγ mutants were cloned into pcDNA3/FLAG. IKKγ expressed in bacteria was purified for analytical ultracentrifugation under native conditions using the pGEX system according to the manufacturer's protocol (Amersham). Briefly, in Escherichia coli BL21/pLysS, expressed GST-IKKγ was bound to glutathione-Sepharose 4B, and glutathione S-transferase (GST) was cleaved by PreScission protease (Amersham). Eluted IKKγ was further purified by MonoQ 10/10 chromatography, concentrated by step elution from MonoQ 5/5 concentration step, and finally separated on a Superose 6 column. T7-tagged IKKγ was bacterially expressed and purified with nickel-agarose using pRSET vector (Invitrogen). Cell pellets were lysed in a solution containing 10 mM Tris (pH 8), 100 mM NaH2PO4, and 8 M urea. Expressed proteins were bound to Ni2+-agarose (Qiagen) for 3 h at room temperature (RT), washed three times with lysis buffer, and eluted for 1 h at RT with 300 mM imidazol in phosphate-buffered saline (PBS). Purified proteins were renatured by stepwise dialysis.
GST pull-down.
GST and GST-IKKγ246-365 (IKKγ containing amino acids 246 to 365) were expressed in E. coli BL21/pLysS, bound to glutathione Sepharose 4B under native conditions, and eluted with 20 mM glutathione. GST or GST-IKKγ246-365 (4 μg) was incubated with 4 μl of in vitro-translated, [35S]methionine-labeled IKKγ in a total volume of 50 μl of binding buffer (PBS, 0.2% Nonidet P-40 [NP-40], 0.5 mg of bovine serum albumin per ml) for 1 h at RT. Binding buffer (500 μl) containing 30 μl of glutathione Sepharose 4B (50% slurry) was added. After incubation for 2 h at 4°C on a turning wheel, beads were pelleted and washed three times with a solution containing 20 mM Tris-HCl (pH 7.9), 150 mM NaCl, and 0.2% NP-40. Proteins were eluted in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer and resolved by SDS-12% PAGE.
Antibodies.
IKKγ (FL-419) and HA (Y-11) antibodies (Santa Cruz Company) and FLAG M5 antibody (Sigma) were used. A specific IKKγ antibody was raised against a peptide comprising amino acids 57 to 72 of IKKγ. Peptide synthesis and immunization were done by Eurogentec.
Luciferase assay.
For reporter assays, 293 cells were seeded on 60-mm-diameter dishes and transfected by the calcium phosphate precipitation method (with a total of 5 μg of DNA). The following DNA amounts were transfected in 293 cells: 100 ng of 6xNF-κBluc as a reporter, 50 ng of pRL-TKluc (Promega) as an internal control, and the indicated amounts of IKKγ constructs. 70Z/3 and 1.3E2 cells were electroporated using a total amount of 30 μg in a Bio-Rad gene pulser at 950 μF and 220 V. For these cell lines, 4 μg of 6xNF-κBluc, 2 μg of pRL-TKluc, and 20 μg of IKKγ constructs were used. Cells were stimulated 36 h posttransfection for 6 h as indicated, and luciferase activity was determined using the dual-luciferase kit (Promega).
Chemical cross-linking of metabolically labeled cell lysates.
HeLa cells grown to 80% confluence were incubated in Dulbecco's modified Eagle's medium without methionine for 45 min and then labeled with 100 μCi of [35S]methionine per ml for 5 h. Labeling of 107 1.3E2 or 1.3E2 cells stably expressing IKKγ was done in RPMI 1640 without methionine as described above for HeLa cells. After the cells were washed twice with PBS, they were lysed in a solution containing 20 mM HEPES (pH 8.4), 150 mM NaCl, 0.5% NP-40, 1 mM EDTA, 1 mM dithiothreitol (DTT), 10 mM NaF, 8 mM β-glycerophosphate, 0.1 mM orthovanadate, 10% glycerol, and protease inhibitors (0.4 mM Pefabloc, 1 μg of leupeptin per ml, 1 μg of aprotinin per ml, and 1 μg of pepstatin A per ml). Lysates were rotated on a turning wheel for 15 min at 4°C and then clarified by high-speed centrifugation. Extracts were incubated with the homobifunctional cross-linking reagent ethyleneglycol-bis-succinimidylsuccinate (EGS) (Pierce) at a final concentration of 5 mM for 30 min at RT. Reactions were stopped by addition of 1 M Tris (pH 8) to a final concentration of 20 mM. After incubation for 30 min at RT, lysates were precleared with protein A-Sepharose for 1 h at 4°C, and immunoprecipitation was performed overnight at 4°C. Precipitates were washed three times with lysis buffer. Supernatants were analyzed by SDS-PAGE and autoradiography.
Chemical cross-linking of labeled proteins.
For chemical cross-linking of [35S]methionine-labeled proteins, coupled in vitro transcription-translation reactions were performed according to the manufacturer's protocol (Promega). Proteins were cross-linked in PBS-0.5% NP-40 with EGS at a final concentration of 2 mM in a volume of 15 μl. After incubation at RT for 30 min, 500 μl of a solution containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.5% NP-40, 1 mM EDTA, 1 mM DTT, 10 mM NaF, 8 mM β-glycerophosphate, 0.1 mM orthovanadate, 10% glycerol, and protease inhibitors (0.4 mM Pefabloc, 1 μg of leupeptin per ml, 1 μg of aprotinin per ml, and 1 μg of pepstatin A per ml) was added prior to immunoprecipitation.
Cleavage of immunoprecipitated EGS cross-linked proteins.
Cross-link with EGS and immunoprecipitation of proteins from labeled cell lysates were controlled by SDS-PAGE and autoradiography. To elute cross-linked proteins from polyacrylamide gels, fivefold-more input was applied to an SDS-polyacrylamide gel, the band of interest was excised, and proteins were recovered by electroelution (Bio-Rad Electro-Eluter 422) for 5 h at 9 mA. Cleavage of EGS was achieved by addition of an equal volume of 2 M hydroxylamine-HCl in PBS (pH 8.5) and incubation for 4 h at 37°C. The solution was dialyzed against PBS prior to immunoprecipitation with IKKγ antibody (FL-419). Precipitates were separated by SDS-PAGE and detected with a phosphorimager.
Coimmunoprecipitation.
293 cells grown on 60-mm-diameter plates were cotransfected with hemagglutinin-tagged full-length IKKγ and indicated FLAG-tagged IKKγ deletion mutants. Cells were lysed 36 h after transfection in a solution containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.5% NP-40, 1 mM EDTA, 1 mM DTT, 10 mM NaF, 8 mM β-glycerophosphate, 0.1 mM orthovanadate, 10% glycerol, and protease inhibitors (0.4 mM Pefabloc, 1 μg of leupeptin per ml, 1 μg of aprotinin per ml, and 1 μg of pepstatin A per ml). Precleared extracts were used for precipitation of HA-tagged IKKγ. Precipitates were washed four times with lysis buffer, resuspended in SDS loading buffer, boiled for 5 min, applied to an SDS-15% polyacrylamide gel, and analyzed by Western blotting.
In vitro kinase assays.
293 cells were cotransfected with HA-tagged IKKβ and indicated IKKγ constructs. Thirty-six hours later, the cells were lysed and immunoprecipitated in a solution containing 50 mM HEPES (pH 7.5), 150 mM NaCl, 1.5 mM MgCl2, 1 mM EDTA, 1% Triton X-100, 10% glycerol, 1 mM DTT, 10 mM NaF, 8 mM β-glycerophosphate, 0.1 mM orthovanadate, 0.4 mM Pefabloc, 1 μg of leupeptin per ml, 1 μg of aprotinin per ml, and 1 μg of pepstatin A per ml. The precipitates were washed twice in lysis buffer and once in kinase reaction buffer containing 20 mM HEPES (pH 7.5), 10 mM MgCl2, 20 mM β-glycerophosphate, 50 μM orthovanadate, 1 mM DTT, and 20 μM ATP. Kinase reactions were performed with 5 μCi of [γ-32P]ATP and GST-IκBα1-53 as the substrate in a 15-μl reaction mixture volume for 20 min at 37°C. After the addition of SDS loading buffer, reaction mixtures were boiled for 5 min and applied to an SDS-polyacrylamide gel. The dried gel was analyzed by autoradiography.
Analytical ultracentrifugation.
IKKγ was expressed in E. coli using the pGEX system and purified as described above. Molecular mass studies on IKKγ were performed with an XL-A type analytical ultracentrifuge (Beckman Instruments, Palo Alto, Calif.) equipped with UV absorbance optics. Sedimentation equilibrium distribution of IKKγ was analyzed using externally loaded six-channel centerpieces with a 12-mm-long optical path with the capacity to handle three solvent-solution pairs of 75 μl of liquid. Seven of these cells were used to simultaneously analyze different samples or fractions of IKKγ and the same run. Sedimentation equilibrium was reached after 2 h of centrifugation at 14,000 rpm followed by centrifugation at an equilibrium speed of 10,000 rpm at 10°C for about 30 h. The radial absorbencies of each compartment were recorded at three different wavelengths between 220 and 280 nm according to the concentration in the different fractions of IKKγ. Molecular mass calculations were performed by simultaneously fitting the sets of three radial absorbance distribution curves described by equation 1 with equation 2
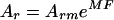 |
(1) |
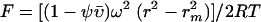 |
(2) |
using the POLYMOLE program (3). In these equations, Ar is the radial absorbance, Arm is the radial absorbance at the meniscus position, ρ is the solvent density, ῡ is the partial specific volume, ω is the angular velocity, R is the gas constant, T is the absolute temperature, M is the molecular mass, and rm is the radius at meniscus point.
RESULTS
IKKγ forms tetramers in vitro and in intact cells.
Inducible kinase activity of IKKα and IKKβ depends on their association with IKKγ. An important basis for understanding the activation mechanism is the knowledge of the stoichiometric composition of the IKK complex. To analyze the oligomeric state of IKKγ in intact cells, we have used the homobifunctional cross-link agent EGS. Lysates of metabolically labeled 1.3E2 cells, which are devoid of IKKγ, or 1.3E2 cells stably transfected with IKKγ were treated with EGS. Immunoprecipitated IKKγ revealed the presence predominantly of tetramers and minor amounts of monomers, dimers, and trimers (Fig. 1A). To confirm that the precipitated 200-kDa protein complex is an IKKγ tetramer and not a result of cross-linked kinase and IKKγ, we applied the fivefold amount of cross-linked IKKγ precipitates to a polyacrylamide gel and eluted the 200-kDa protein complex from the gel. The eluted proteins were treated with hydroxylamine-HCl, which leads to cleavage of the EGS-mediated cross-link, and analysis of IKKγ-associated proteins revealed only one protein species with a molecular mass of 50 kDa (IKKγ) (Fig. 1A, right panel). We next tested metabolically labeled HeLa cells (Fig. 1B). EGS cross-linking yielded exclusively tetramers of the endogenous protein, the immunoprecipitation of which could be inhibited with a specific peptide. Prior TNF-α stimulation did not affect the oligomeric state (not shown). The observed sizes of the products (∼50, ∼150, and ∼200 kDa [Fig. 1A] and ∼200 kDa [Fig. 1B]) are consistent with oligomeric γ subunits and cannot be explained by cross-linked IKKα or IKKβ (around 85 kDa each). Thus, EGS appears to cross-link preferentially the IKKγ subunits in the IKKα-IKKβ-IKKγ holocomplex.
FIG. 1.

Tetrameric oligomerization of IKKγ. (A) IKKγ-deficient 1.3E2 cells and 1.3E2 cells stably expressing IKKγ were metabolically labeled with [35S]methionine. Lysates were treated with EGS, IKKγ was immunoprecipitated (IP: IKKγ), and pellets were washed and analyzed by SDS-PAGE and autoradiography. To confirm that the 200-kDa band resulting from the EGS cross-link consists exclusively of IKKγ, the band was excised from a polyacrylamide gel, and as indicated, proteins were electroeluted and treated with hydroxylamine-HCl to cleave EGS. IKKγ complexes were precipitated and separated by SDS-PAGE, and components were detected with a phosphorimager (top gels). The lack of immunoprecipitated labeled proteins in 1.3E2 cells confirms the specificity of the antibody. IKKγ expression before cross-linking was determined by Western blotting (WB). (B) Metabolically labeled HeLa cells were lysed, and proteins were cross-linked with sulfo-EGS. Immunoprecipitation with an IKKγ antibody (IP: IKKγ) was performed with (+) or without (−) preblocking with a specific peptide, as indicated above the gel. Products were analyzed by SDS-PAGE and autoradiography. The arrows in panels A and B indicate the position of IKKγ tetramers. (C) [35S]methionine-labeled, N-terminally FLAG-tagged IKKγ (FLAG-IKKγ) and C-terminally HA-tagged IKKγ (IKKγ-HA) were prepared by coupled in vitro transcription and translation and cross-linked with EGS. The reaction was stopped by the addition of Tris, and immunoprecipitations (IP) with the indicated antibodies were performed. The positions of molecular mass markers (in kilodaltons) are shown to the left of the gel. (D) (Left) Recombinant IKKγ was purified from E. coli and analyzed on a Coomassie blue-stained SDS-polyacrylamide gel along with protein molecular mass markers, as indicated. (Right) Analytical ultracentrifugation analysis of purified recombinant IKKγ (Superose 6 peak fractions). Radial concentration distribution curves of IKKγ dissolved in 20 mM Tris-HCl (pH 7.5) containing 100 mM NaCl are shown. The profiles were recorded at 10,000 rpm and wavelengths of 220 (○), 225 (•), or 230 (□) nm. The three curves were fitted globally using the polymole program, resulting in a molecular mass of 192.6 ± 3.6 kDa.
Predominantly tetrameric oligomerization was further observed for cross-linked, in vitro-translated IKKγ, irrespective of the HA or FLAG tag sequences, which were fused to the N or C termini, respectively (Fig. 1C).
The EGS cross-linking reagent may have affected the oligomeric state by chemical modification, or it may not have been able to link some oligomeric forms due to the lack of lysine residues in suitable positions. To rule out this possibility, we determined the oligomeric state of recombinant IKKγ by equilibrium sedimentation. This method allows determination of the molecular mass of the native protein in solution. The concentration distribution at sedimentation equilibrium depends only on molecular mass and is independent of the shape. Highly purified bacterially expressed IKKγ (gel in Fig. 1D) was used for analysis at three different wavelengths (graph in Fig. 1D). Radial concentration distributions were recorded, which are typical for a more or less monodisperse system. The optimal fit using the POLYMOLE program (3) yielded a molecular mass of about 193 kDa, indicating that purified IKKγ forms a tetramer with a theoretical molecular mass of 192.8 kDa.
A carboxy-terminal coiled-coil domain in IKKγ mediates oligomerization.
To determine the domain in IKKγ, which is minimally required for self association, various FLAG-tagged deletion mutants (Fig. 2A) were cotransfected along with HA-tagged full-length IKKγ into 293 cells (Fig. 2B). HA immunoprecipitation revealed that only those IKKγ mutants that contained sequences within amino acids 246 to 365 were recovered efficiently. This sequence is localized in the C-terminal half of IKKγ and contains two coiled-coil (CC) domains with leucine zipper motifs (Fig. 2A, CC2, amino acids 254 to 298, and CC3, amino acids 308 to 349).
FIG. 2.

Oligomerization of IKKγ is mediated by a C-terminal domain between amino acids 246 to 365. (A) Conserved motifs and structural predictions of IKKγ by PredictProtein and MultiCoil are shown at the top. The positions of alpha-helical regions (open boxes), coiled-coil domains (black bars), and zinc fingers are shown. Below the IKKγ map, a schematic summary of IKKγ deletion mutants is shown. The numbers are amino acids. IKKγ oligomerization is shown to the right as follows: +, IKKγ oligomerizes; −, IKKγ does not oligomerize; ++, IKKγ oligomerizes strongly; (+), IKKγ oligomerizes weakly; n.d., not determined. (B) Interaction of FLAG-tagged IKKγ and IKKγ deletion mutants with HA-tagged full-length IKKγ coexpressed in 293 cells. Lysates were analyzed for expression with anti-FLAG antibody (top gel). Immunoprecipitation (IP) was performed with an anti-HA antibody. Coimmunoprecipitated proteins were detected in a Western blot (WB) with anti-FLAG antibody (middle gel), and precipitated HA-tagged IKKγ (HA-IKKγ) was detected with anti-HA antibody (bottom gel). The asterisk marks a signal caused by the HA antibody heavy chain. (C) GST or GST-tagged IKKγ246-365 and [35S]methionine-labeled full-length IKKγ (input [middle and bottom gels]) were used for an interaction analysis (top gel), as indicated.
Internal deletion of this sequence (in IKKγΔ246-365 [IKKγ in which amino acids 246 to 365 were deleted]) likewise resulted in complete loss of interaction (Fig. 2B, lane 14). In contrast, internal deletion of its first or second half (in IKKγΔ246-302 and IKKγΔ302-365, respectively) still allowed some, albeit strongly reduced binding (Fig. 2B, lanes 12 and 13, note the overlapping nonspecific signal), indicating an equivalent, redundant function of the two subregions containing CC2 and CC3, respectively. However, this was depending on the context, since IKKγ1-302 retained some binding activity, while IKKγ302-419 or IKKγ65-302 did not (lanes 3, 6, and 9). Some very weak interaction was seen with IKKγ1-196 (lane 3), containing CC1.
A GST pull-down assay revealed that the sequences within amino acids 246 to 365 are sufficient to bind to full-length IKKγ efficiently (Fig. 2C). Thus, strong self-association of IKKγ is conferred by a minimal oligomerization domain (MOD) spanning amino acids 246 to 365.
C-terminal dimerization is a prerequisite for IKKγ tetramerization.
To define sequences of IKKγ which are required for tetramerization, in vitro-translated deletion mutants were subjected to chemical cross-linking. To exclude overreaction of the cross-linking reagent, again conditions were chosen such that residual amounts of monomeric IKKγ were still detected. Consistent with the previous results (Fig. 1), full-length IKKγ yielded tetramers (Fig. 3A). The MOD was in fact required for tetramer formation, since upon its internal deletion (IKKγΔ246-365), only monomers were observed (Fig. 3A). Deletion of subregions of the MOD revealed that its first half (amino acids 246 to 302) was critical for tetramerization, while the second half (residues 302 to 365) was not (Fig. 3A). However, while necessary for tetramerization, the MOD was not sufficient, as indicated by analysis of C- and N-terminal deletion mutants (Fig. 3B). For these mutants, tetramers were observed only with IKKγ1-365, which is devoid of the C-terminal Zn finger motif but contains the complete N-terminal sequence (lane 4). Upon further deletion of the first 65 amino acids (IKKγ65-365), trimers and dimers were formed (lane 11), which also were the predominant species of cross-linked IKKγ65-419 (lane 5). The shortest mutants containing the MOD (IKKγ246-419 and IKKγ196-365) yielded only dimers (lanes 8 and 10). The N-terminal half of IKKγ (residues 1 to 196), devoid of the MOD, formed only trace amounts of dimers (lane 2). Thus, no discrete subdomain alone could confer tetramerization, while the shortest N- and C-terminal deletion mutants (retaining amino acids 1 to 196 and 246 or 302 to 419) formed dimers. Taken together, these results suggest that tetramerization of IKKγ is achieved by two subsequent oligomerization steps, a prerequisite dimerization through the MOD, followed by dimerization of these dimers engaging an N-terminal region of IKKγ. This N-terminal interaction site apparently has an affinity that is too low to be effective in the absence of prior MOD-mediated dimerization.
FIG. 3.

C-terminal dimerization is a prerequisite for IKKγ tetramerization. Labeled FLAG-tagged IKKγ mutants were prepared by in vitro translation. After EGS cross-linking, products were precipitated with anti-FLAG antibodies and analyzed by SDS-PAGE and autoradiography. The positions of molecular mass markers are indicated. (A) Analysis of full-length IKKγ and internal deletion mutants IKKγΔ246-302, IKKγΔ302-365, and IKKγΔ246-365. Migration of monomers and tetramers is indicated. (B) (Top) Analysis of N- and C-terminal IKKγ deletion mutants. Major and minor multimeric forms are indicated below the autoradiogram (minor forms shown in parentheses). (Bottom) Schematic presentation of major oligomerization states of IKKγ subregions. The positions of alpha-helical regions (open boxes), coiled-coil domains (black bars), and zinc finger are shown.
Since most of the IKKγ molecule is predicted to assume an alpha-helical conformation with three extended coiled-coil regions (Fig. 2A and 3B, bottom), it is possible that in the sequence context of some deletion mutants, these motifs may account for the formation of not only dimers but also of trimers. Trimers were not significantly formed by endogenous or recombinant full-length IKKγ (Fig. 1 and 3).
IKKγ interacts with IκB kinases as a tetramer, but oligomerization is not required for binding of IKKα or IKKβ.
A tetrameric oligomerization state of IKKγ was also determined for the endogenous protein in HeLa cells (Fig. 1B), which is bound to IKKα and IKKβ. To ensure that preformed IKKγ tetramers can interact with the kinases, in vitro-translated IKKγ tetramers were covalently fixed with EGS, immunoprecipitated, and tested for IKKβ interaction. Indeed, IKKβ bound efficiently to fixed IKKγ tetramers (Fig. 4). IKKβ was also sequestered by IKKγ added without prior cross-linking, and the recovery was the same as for fixed tetramers. Thus, we conclude that IKKγ tetramers, whether covalently stabilized or not, interact with IKKβ. The near stoichiometric recovery of IKKγ and kinase also reflects that recombinant IKKγ is in an active state and correctly folded.
FIG. 4.

IKKγ binds as a tetramer to IKKβ. [35S]methionine-labeled FLAG-tagged IKKγ (FLAG-IKKγ) and HA-tagged IKKβ (HA-IKKβ) were prepared by in vitro translation (input [left gel]). Equal aliquots of FLAG-tagged IKKγ were not treated or were cross-linked with EGS, followed by inactivation of EGS. Both samples were immunoprecipitated with anti-FLAG antibodies (IP: FLAG). Precipitates were washed and incubated with HA-tagged IKKβ on ice for 30 min. After an additional washing step, proteins were separated by SDS-PAGE and analyzed by autoradiography.
We next asked whether IKKγ tetramerization would be a requirement for IKKγ interaction with IKKα or IKKβ or whether it would influence interaction affinity. In vitro-translated IKKγ mutants were incubated with lysates of 293 cells expressing HA-tagged IKKα or HA-tagged IKKβ and analyzed for kinase interaction by anti-HA immunoprecipitation (Fig. 5A). The experiment revealed that tetrameric oligomerization is not required for binding of IKKγ to IKKα or IKKβ. IKKγΔ246-302 or IKKγΔ246-365, which are monomeric (Fig. 3A), were sequestered efficiently by both kinases compared to tetramer-forming IKKγΔ302-365 or full-length IKKγ (Fig. 5A, lanes 1 to 4).
FIG. 5.

Physical interaction of IKKα and IKKβ with IKKγ involves overlapping but distinct N-terminal domains of IKKγ and does not require IKKγ tetramerization. (A) [35S]methionine-labeled IKKγ mutants were prepared by coupled in vitro transcription and translation. Labeling efficiency was controlled by SDS-PAGE or autoradiography (top gel). 293 cells were transiently transfected with HA-tagged IKKα (HA-IKKα) or HA-tagged IKKβ (HA-IKKβ). Lysates were checked for equal expression of transfected proteins by Western blotting (WB) with anti-HA antibodies. Labeled IKKγ mutants were added to lysates with either overexpressed HA-tagged IKKα or HA-tagged IKKβ and incubated for 30 min on ice. After immunoprecipitation (IP) with anti-HA antibodies, precipitated proteins were analyzed by SDS-PAGE and autoradiography. (B) (Left) In vitro-translated, [35S]methionine-labeled HA-tagged IKKβ, left untreated (−) or incubated with EGS (+), was analyzed for cross-linked IKKβ dimers after anti-HA immunoprecipitation (IP:HA), PAGE, and autoradiography. (Right) In vitro-translated, [35S]methionine-labeled, FLAG-tagged IKKγ1-196 was incubated without or with baculovirus-expressed, purified recombinant IKKβ and treated with EGS, as indicated. Cross-linked species were visualized after anti-FLAG immunoprecipitation (IP: FLAG) by PAGE and autoradiography. The positions of the monomeric and dimeric proteins are indicated by the arrows.
The N- and C-terminal deletion mutants allowed delineation of the domain of IKKγ which interacts with both kinases (Fig. 5A). Surprisingly, the boundaries of the kinase interaction domain for IKKα and IKKβ overlapped but were not identical. While interaction with IKKβ was conferred minimally by amino acids 65 to 196 of IKKγ, binding of IKKα additionally involved further N-terminal sequences and was within residues 1 to 196 of IKKγ (compare lanes 5, 10, 11, and 13). However, the relative binding efficiencies of IKKα and IKKβ to full-length IKKγ, internal and C-terminal deletion mutants (lanes 1 to 9 and 12) were indistinguishable, indicating that quantification and folding of IKKα and IKKβ were equivalent.
On the basis of the roughly equal stoichiometric amounts of IKKγ and kinase subunits in reconstituted IKK complexes (19, 28), tetrameric IKKγ would bind two kinase dimers. It was not possible to covalently link IKKγ and IKKβ with EGS (data not shown), perhaps because the IKKγ binding domain of the kinase (23) does not contain lysines. In contrast, IKKβ dimers could be readily cross-linked (Fig. 5B, left gel), consistent with lysines present in its dimerization region or central leucine zipper region. In an IKKγ4-IKKβ4 complex, one IKKβ dimer could interact with two IKKγ molecules. We analyzed IKKγ1-196, which contains the kinase binding region (Fig. 5A) and dimerizes only poorly (Fig. 3), for enhanced dimerization upon IKKβ binding. In fact, while IKKγ1-196 alone formed only trace amounts of dimers, dimerization was strongly increased in the presence of IKKβ (Fig. 5B, right gel), suggesting that IKKβ dimers bind to two IKKγ moieties in an IKKγ4-IKKβ4 complex.
Formation of an active IKK complex and IKK autophosphorylation require IKKγ oligomerization.
The inducible kinase activity of IKKα and IKKβ critically depends on their association with IKKγ. To analyze if the catalytic activity of the IKK complex requires IKKγ oligomerization, 293 cells were cotransfected with FLAG-tagged IKKγ mutants and HA-tagged IKKβ. IKK complexes were retrieved by immunoprecipitation and tested for in vitro kinase activity (Fig. 6A). Only full-length IKKγ and IKKγ1-365 stimulated the kinase activity of IKKβ (compare lane 1 to lanes 2 and 5). However, mutants which do not form tetramers (IKKγ1-196, IKKγΔ246-365, and IKKγ1-302 [lanes 3, 4, and 7]) or lack the kinase binding domain (IKKγ196-419 [lane 6]) did not stimulate IKKβ kinase activity. These data thus suggest that MOD-mediated oligomerization of IKKγ is a requirement for the formation of an active IKK complex.
FIG. 6.

Activity and inducibility of the IKK complex require IKKγ tetramerization. (A) 293 cells were cotransfected with HA-tagged IKKβ (HA-IKKβ) and FLAG-tagged IKKγ wild-type or mutant proteins, as indicated. Complexes were immunoprecipitated with HA antibody (IP: HA) and tested in in vitro kinase assays (KA) with GST-IκBα1-53 as the substrate (top gel). Aliquots of immunoprecipitated IKKβ were analyzed by Western blotting (WB) using HA antibody (middle gel), and expression of IKKγ proteins was controlled in whole-cell extracts with FLAG antibody (bottom gel). (B) 70Z/3, 1.3E2, and 1.3E2 cells stably expressing the indicated constructs were tested for LPS-induced NF-κB activation by an electrophoretic mobility shift assay (EMSA). The presence (+) and absence (−) of LPS is shown above the gel. Expression control of endogenous and ectopic IKKγ proteins and of endogenous IKKβ analyzed by Western blotting (WB) of whole-cell extracts is shown at the bottom. Immunoprecipitation (IP) of endogenous and ectopic IKKγ proteins was performed to confirm equal interaction of IKKβ with mutant and wild-type IKKγ. (C) 1.3E2 cells were cotransfected with the NF-κB reporter plasmid 6xNF-κBluc, internal control plasmid, and IKKγ or IKKγΔ246-365 or empty expression vector (mock), as indicated. Cells were stimulated with LPS (+) for 6 h or not treated with LPS (−), and luciferase activity was determined. (D) IKKβ autophosphorylation and phosphorylation of IKKγ. 293 cells were transfected with HA-tagged IKKβ (HA-IKKβ) and FLAG-tagged IKKγ (FLAG-IKKγ) constructs, as indicated. HA-immunoprecipitated complexes were incubated with [γ-32P]ATP in kinase assays (KA), and phosphorylated proteins were detected by SDS-PAGE and autoradiography (top gel). Expression of IKKβ and IKKγ proteins and IKKβ precipitation were controlled by anti-HA or anti-FLAG immunoblotting of extracts (middle gel) and pellets (bottom gel), respectively. WB, Western blotting; IP, immunoprecipitation.
To examine the requirement of IKKγ oligomerization for LPS-induced NF-κB activity in intact cells, IKKγ-deficient 1.3E2 pre-B cells were stably transfected either with wild-type IKKγ or IKKγΔ246-365, devoid of the MOD (Fig. 6B). NF-κB DNA binding activity could be induced by LPS in 70Z/3 cells, but not in 1.3E2 cells, unless the cells were complemented with full-length IKKγ (lanes 2, 4, and 6), as was expected. Internal deletion of the MOD, however, completely eliminated the capacity to functionally complement 1.3E2 cells (lanes 7 and 8). The lack of NF-κB activation in 1.3E2 cells stably expressing oligomerization-deficient IKKγ is not due to loss of interaction between IKKβ and the deletion mutant, as shown by coimmunoprecipitation analysis (Fig. 6B, lower blots). Similarly, NF-κB transcriptional activity could be induced only by LPS in transiently transfected 1.3E2 cells, when wild-type IKKγ was complemented (Fig. 6C). Again, oligomerization-deficient IKKγ did not reconstitute LPS-induced NF-κB transcriptional activity, indicating that IKKγ tetramerization is a prerequisite for LPS inducibility of the IKK complex.
One possibility is that IKKγ tetramerization could serve to juxtapose bound IKKα-IKKβ dimers for transphosphorylation. Kinase assays (Fig. 6D) revealed that cotransfection of IKKγ and IKKβ indeed resulted in IKKβ phosphorylation only when full-length IKKγ (lane 1) or IKKγ1-365 (lane 3), containing MOD and kinase binding regions, were present. IKKβ phosphorylation was not observed when IKKγ1-302 or IKKγΔ246-365, retaining kinase binding but lacking the MOD (lanes 2 and 4), were expressed. Not only IKKβ but also IKKγ and IKKγ1-365 were phosphorylated in the same reactions (lanes 1 and 3). However, IKKγ mutants lacking the MOD were not phosphorylated, despite their full ability to interact with IKKβ (Fig. 5A). These results indicate that MOD-mediated IKKγ oligomerization affects spatial positioning of the bound kinase molecules.
Since short IKKγ mutants containing the MOD region efficiently bind to full-length IKKγ (Fig. 2), overexpression of these molecules should result in formation of heteromers with endogenous IKKγ and interfere with normal tetramerization and with IKK kinase activity. In fact, overexpression of the MOD alone (IKKγ246-365) inhibited TNF-α-induced NF-κB activation in a dose-dependent fashion (Fig. 7). Overexpression of IKKγ1-196, which is expected to titrate out IKKα and IKKβ, similarly inhibited TNF-α-dependent NF-κB activity in a dose-dependent manner using equivalent amounts of expression vector. In a similar dose-response range, overexpression of full-length IKKγ, which is likely to dilute out the limited amounts of kinases bound to endogenous IKKγ, inhibited TNF-α-induced NF-κB activation (Fig. 7). These data further support the conclusion that an oligomeric state of the IKKγ-kinase complex is required for its inducibility.
FIG. 7.

Dose-dependent inhibition of NF-κB activation by the MOD. 293 cells were cotransfected with the indicated amounts of expression vectors for wild-type IKKγ, IKKγ1-196, or IKKγ246-365, NF-κB reporter plasmid 6xNF-κBluc, and an internal control plasmid. After stimulation of the cells with TNF-α for 6 h, luciferase activity was measured.
DISCUSSION
Knowledge of the molecular composition of the IκB kinase complex and of the oligomeric state of its constituents is an essential basis for understanding its mechanism of activation. By chemical cross-linking of endogenous or in vitro-translated IKKγ and by sedimentation analysis of purified recombinant IKKγ, we show in this study that the IKKγ subunit forms tetramers. Tetramer formation was observed for IKKγ when associated with endogenous IKK components, including IKKα and IKKβ, or when present alone, and tetrameric oligomerization did not grossly affect the kinase binding affinity. On the basis of binary protein-protein interaction analysis with IKKγ mutants, we showed that a MOD is confined to a region between amino acids 246 and 365 in the C-terminal half of the molecule and that this region is sufficient to bind to full-length IKKγ. By chemical cross-linking, this region was shown to be required for tetramerization, although by itself it forms only dimers. Similarly, the N-terminal half of IKKγ was able to dimerize weakly. We therefore propose that IKKγ forms a dimer of dimers by utilizing the C-terminal MOD for primary dimerization, followed by subsequent dimerization of dimers through the N-terminal domains (Fig. 8). The dimerization of dimers possibly occurs by antiparallel interaction, since in a parallel interaction mode, dimerization of the N terminal domain should result in molecules dimerized both N- and C-terminally, finally resulting in dimers instead of tetramers. The weak N-terminal dimerization region overlaps with the sequences bound by IKKα and IKKβ, and binding of IKKβ dimers enhances dimerization of this region, indicating that a kinase dimer may attract two IKKγ moieties simultaneously (Fig. 8).
FIG. 8.

Hypothetical model for tetrameric assembly of IKKγ by head-to-head dimerization of two dimers and interaction with two kinase dimers. The positions of zinc fingers, MODs, and kinase binding domains (KBD) are shown.
We have delineated the kinase binding domain of IKKγ to amino acids 1 to 196 for IKKα and to amino acids 65 to 196 for IKKβ. The IKKγ interaction site for IKKβ has been mapped previously to amino acids 44 to 86 (23). This could indicate that a relatively short α-helical region between residues 65 and 86 is contacted by IKKβ. However, Rothwarf et al. (31) observed IKKβ binding to residues 134 to 419 and Poyet et al. (29) described binding to residues 105 to 200 of IKKγ. These discrepancies might by due to different oligomeric states of the various IKKγ deletion mutants that have been used. Kinase interaction with IKKγ is perhaps more complex than assumed for the short conserved IKKγ-binding peptide of IKKα and IKKβ, as reflected by our surprising observation that IKKα binding, but not IKKβ binding, requires a contribution of the N-terminal 65 amino acids of IKKγ. In line with this, it has been shown recently that the IKKγ (NEMO)-binding peptides of IKKα and IKKβ do in fact differ (25). Nonidentical contacts of both kinases with IKKγ could be important for the assembly of two IKKα-IKKβ heterodimers with tetrameric IKKγ in a sterically selective way. This could ensure that the α and β subunits in the two IKKγ-bound kinase dimers are juxtaposed to each other in a symmetrical way (Fig. 8).
Our findings that N- or C-terminally truncated fragments of IKKγ form mainly dimers but also form trimers is also in accordance with coiled-coil predictions made by MultiCoil (34). Mostly dimeric coiled coils, and with much lower probability, trimeric coiled coils were predicted for the three separate coiled-coil regions (CC1 [amino acids 98 to 196], CC2 [amino acids 254 to 298], and CC3 including the leucine zipper [amino acids 308 to 349]). For Drosophila IKKγ, a much weaker prediction for less-extended N- and C-terminal coiled-coils are obtained compared to human, murine, or bovine IKKγ (not shown), indicating that structural organization of the Drosophila IKK complex may differ. This is intriguing, given the fact that for Drosophila, only one kinase (most highly related to mammalian IKKβ) has been reported (33).
Agou and coworkers recently proposed that NEMO/IKKγ forms trimers and claimed that the majority of IKKγ is present in a monomeric state (1). Several reasons may account for the discrepancy between their study and our results. Agou et al. used detergent to achieve solubility of bacterially expressed, His-tagged murine IKKγ, which under their conditions copurified with DnaK (E. coli Hsp70) protein. However, the determination of the oligomeric state of a protein from a complex with another one (DnaK), which itself can form an association equilibrium of monomers, dimers, and trimers (18) by the sedimentation equilibrium technique is questionable. For the latter analysis, a heterogeneous preparation containing IKKγ and DnaK was used (1). The diffusion coefficient determination is critical, since the analytical solution of the Lamm equation, used by these researchers, is valid only for monodisperse systems (2). Furthermore, a different, Cys-directed, reagent was used for cross-linking of IKKγ in intact cells, and only truncated IKKγ was analyzed by sedimentation velocity analysis (1). Comparable deletion constructs also yielded trimers in our study, as shown by cross-linking. Dimers and trimers were also reported by Rothwarf et al. (31) for IKKγ deletion mutants, using EGS and six-His-tagged, bacterially expressed proteins, but with full-length IKKγ, larger species were also detected.
In gel filtration experiments, the apparent molecular mass of the cellular IKK complex relative to globular marker proteins is 800 to 900 kDa (19, 27, 44), which shifts to 400 to 500 kDa in the absence of IKKγ (39). Gel filtration analysis of complexes reconstituted with recombinant IKKβ and IKKγ or MOD-deficient IKKγ revealed a pronounced shift to a lower apparent molecular mass in the absence of the MOD, as was expected (data not shown). Recently, it was shown that the 800- to 900-kDa cellular heterocomplex is assembled by direct interactions of the chaperones Cdc37 and Hsp90 which bind to the kinase domains of IKKα and IKKβ and the Hsp90 inhibitor geldanamycin disrupts this heterocomplex (5). Cdc37 or Hsp90 and IKKα, IKKβ, or IKKγ have been estimated to be present in the heterocomplex at a stoichiometric ratio of 1:1 (5). Thus, an IKKα2-IKKβ2-IKKγ4 × (Cdc37/Hsp90)2 complex would have a predicted mass between 800 and 900 kDa, consistent with the observed apparent molecular weight.
It has been proposed that both activation and recruitment to TNF receptor 1 of the IKK complex requires the presence of Cdc37 and Hsp90 (5). Although it is not understood whether these chaperones are involved in the activation process per se, as opposed to the more general role for folding, conformational state, and stability of their substrates, it is feasible that these molecules participate in the assembly of a heteromeric cellular complex containing tetrameric IKKγ.
We showed that the MOD of IKKγ is required for activation of IKKβ kinase activity in transfected 293 cells and for reconstitution of an LPS-inducible IKK complex in pre-B cells. Furthermore, transautophosphorylation of IKKβ and IKKγ required the presence of the MOD. These data suggest that MOD-mediated tetramerization is required to assemble a functional IKK complex. IKKγ is already tetrameric in the endogenous unstimulated IKK complex (Fig. 1), and TNF-α or LPS stimulation has no effect on its oligomerization state (data not shown). Since IKKβ is activated by phosphorylation and the activity of the kinase depends on its phosphorylation state, tetramerization could be necessary to bring two kinase dimers in spatial proximity, allowing activation by mutual phosphorylation. In the endogenous situation, where the IKK complex is bound by chaperones, conformational changes in the tetrameric IKKγ scaffold, induced by interaction with activating cellular or viral factors, could be needed to facilitate the transphosphorylation reaction. Such a mechanism could also explain why IKK-activating proteins do not need to be kinases. Examples are viral Tax (38) or enzymatically inactive RIP1 and IRAK1 (8, 20).
Defective IKK activation upon deletion of the MOD could also indicate that the MOD region itself is required for binding of adapter molecules and that tetramerization is functionally not required. However, our observations that upon overexpression, where endogenous factors are limiting, IKKγ-dependent activity of IKKβ depends on an intact MOD and the MOD conferred autophosphorylation argues against this possibility.
Ectopic expression of IKKγ at low levels is known to activate NF-κB, whereas higher doses efficiently diminish induced NF-κB activation (19). We observed the same effect of dose-dependent loss of TNF-α-induced NF-κB activation, when only the kinase binding domain or the MOD of IKKγ was expressed. This could be explained by titrating out kinases or by formation of heteromeric IKKγ, which cannot tetramerize, respectively, in each case reducing the amounts of functional complexes containing tetrameric IKKγ bound to two kinase dimers.
Previously, C-terminal sequences of IKKγ, containing the MOD and the Zn finger domain, were proposed to mediate functional interaction with unknown activating components of proinflammatory pathways (31). In fact, when C-terminal fragments containing both the MOD and zinc finger act as dominant negatives, these molecules could titrate out tetramers, rather than any postulated, but unidentified adapter proteins. It has recently been shown that mutations of leucines in CC3 inactivate the ability of IKKγ to functionally complement IκB kinases (22). These mutations and some IKKγ mutations described for HED-ID, which reside in the coiled-coil regions (6), may in fact result in functional defects by disrupting the tetrameric structure of IKKγ. It has been proposed that the zinc finger module is largely dispensable for NF-κB activation by the rapid and strong inducers LPS and TNF-α, while being essential for UV induction (14). The exact mechanisms by which C-terminal IKKγ sequences mediate IKK activation by the various stimuli will be addressed in future studies. An attractive possibility is that protein interactions with these tetramerized structures induce allosteric structural alterations within the IKK complex.
Acknowledgments
We thank Daniel Krappmann for helpful suggestions and critical comments on the manuscript. We also thank Rudolf Dettmer for providing purified proteins.
REFERENCES
- 1.Agou, F., F. Ye, S. Goffinont, G. Courtois, S. Yamaoka, A. Israel, and M. Veron. 2002. NEMO trimerizes through its coiled-coil C-terminal domain. J. Biol. Chem. 277:17464-17475. [DOI] [PubMed] [Google Scholar]
- 2.Behlke, J., and O. Ristau. 2002. A new approximate whole boundary solution of the Lamm differential equation for the analysis of sedimentation velocity experiments. Biophys. Chem. 95:59-68. [DOI] [PubMed] [Google Scholar]
- 3.Behlke, J., O. Ristau, and H. J. Schonfeld. 1997. Nucleotide-dependent complex formation between the Escherichia coli chaperonins GroEL and GroES studied under equilibrium conditions. Biochemistry 36:5149-5156. [DOI] [PubMed] [Google Scholar]
- 4.Cao, Y., G. Bonizzi, T. N. Seagroves, F. R. Greten, R. Johnson, E. V. Schmidt, and M. Karin. 2001. IKKα provides an essential link between RANK signaling and cyclin D1 expression during mammary gland development. Cell 107:763-775. [DOI] [PubMed] [Google Scholar]
- 5.Chen, G., P. Cao, and D. V. Goeddel. 2002. TNF-induced recruitment and activation of the IKK complex require Cdc37 and Hsp90. Mol. Cell 9:401-410. [DOI] [PubMed] [Google Scholar]
- 6.Courtois, G., A. Smahi, and A. Israel. 2001. NEMO/IKKγ: linking NF-κB to human disease. Trends Mol. Med. 7:427-430. [DOI] [PubMed] [Google Scholar]
- 7.Delhase, M., M. Hayakawa, Y. Chen, and M. Karin. 1999. Positive and negative regulation of IκB kinase activity through IKKβ subunit phosphorylation. Science 284:309-313. [DOI] [PubMed] [Google Scholar]
- 8.Devin, A., A. Cook, Y. Lin, Y. Rodriguez, M. Kelliher, and Z. Liu. 2000. The distinct roles of TRAF2 and RIP in IKK activation by TNF-R1: TRAF2 recruits IKK to TNF-R1 while RIP mediates IKK activation. Immunity 12:419-429. [DOI] [PubMed] [Google Scholar]
- 9.Dumitru, C. D., J. D. Ceci, C. Tsatsanis, D. Kontoyiannis, K. Stamatakis, J. H. Lin, C. Patriotis, N. A. Jenkins, N. G. Copeland, G. Kollias, and P. N. Tsichlis. 2000. TNF-α induction by LPS is regulated posttranscriptionally via a Tpl2/ERK-dependent pathway. Cell 103:1071-1083. [DOI] [PubMed] [Google Scholar]
- 10.Ghosh, S., and M. Karin. 2002. Missing pieces in the NF-κB puzzle. Cell 109:S81-S96. [DOI] [PubMed]
- 11.Hatada, E. N., D. Krappmann, and C. Scheidereit. 2000. NF-κB and the innate immune response. Curr. Opin. Immunol. 12:52-58. [DOI] [PubMed] [Google Scholar]
- 12.Heissmeyer, V., D. Krappmann, E. N. Hatada, and C. Scheidereit. 2001. Shared pathways of IκB kinase-induced SCFβTrCP-mediated ubiquitination and degradation for the NF-κB precursor p105 and IκBα. Mol. Cell. Biol. 21:1024-1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu, Y., V. Baud, T. Oga, K. I. Kim, K. Yoshida, and M. Karin. 2001. IKKα controls formation of the epidermis independently of NF-κB. Nature 410:710-714. [DOI] [PubMed] [Google Scholar]
- 14.Huang, T. T., S. L. Feinberg, S. Suryanarayanan, and S. Miyamoto. 2002. The zinc finger domain of NEMO is selectively required for NF-κB activation by UV radiation and topoisomerase inhibitors. Mol. Cell. Biol. 22:5813-5825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Israel, A. 2000. The IKK complex: an integrator of all signals that activate NF-κB? Trends Cell Biol. 10:129-133. [DOI] [PubMed] [Google Scholar]
- 16.Karin, M. 1999. How NF-κB is activated: the role of the IκB kinase (IKK) complex. Oncogene 18:6867-6874. [DOI] [PubMed] [Google Scholar]
- 17.Kelliher, M. A., S. Grimm, Y. Ishida, F. Kuo, B. Z. Stanger, and P. Leder. 1998. The death domain kinase RIP mediates the TNF-induced NF-κB signal. Immunity 8:297-303. [DOI] [PubMed] [Google Scholar]
- 18.Kim, D., Y. J. Lee, and P. M. Corry. 1992. Constitutive HSP70: oligomerization and its dependence on ATP binding. J. Cell. Physiol. 153:353-361. [DOI] [PubMed] [Google Scholar]
- 19.Krappmann, D., E. N. Hatada, S. Tegethoff, J. Li, A. Klippel, K. Giese, P. A. Baeuerle, and C. Scheidereit. 2000. The IκB kinase (IKK) complex is tripartite and contains IKKγ but not IKAP as a regular component. J. Biol. Chem. 275:29779-29787. [DOI] [PubMed] [Google Scholar]
- 20.Li, X., M. Commane, C. Burns, K. Vithalani, Z. Cao, and G. R. Stark. 1999. Mutant cells that do not respond to interleukin-1 (IL-1) reveal a novel role for IL-1 receptor-associated kinase. Mol. Cell. Biol. 19:4643-4652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li, Y., J. Kang, J. Friedman, L. Tarassishin, J. Ye, A. Kovalenko, D. Wallach, and M. S. Horwitz. 1999. Identification of a cell protein (FIP-3) as a modulator of NF-κB activity and as a target of an adenovirus inhibitor of tumor necrosis factor alpha-induced apoptosis. Proc. Natl. Acad. Sci. USA 96:1042-1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Makris, C., J. L. Roberts, and M. Karin. 2002. The carboxyl-terminal region of IκB kinase γ (IKKγ) is required for full IKK activation. Mol. Cell. Biol. 22:6573-6581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.May, M. J., F. D'Acquisto, L. A. Madge, J. Glockner, J. S. Pober, and S. Ghosh. 2000. Selective inhibition of NF-κB activation by a peptide that blocks the interaction of NEMO with the IκB kinase complex. Science 289:1550-1554. [DOI] [PubMed] [Google Scholar]
- 24.May, M. J., and S. Ghosh. 1997. Rel/NF-κB and IκB proteins: an overview. Semin. Cancer Biol. 8:63-73. [DOI] [PubMed] [Google Scholar]
- 25.May, M. J., R. B. Marienfeld, and S. Ghosh. 2002. Characterization of the IκB-kinase NEMO binding domain. J. Biol. Chem. 277:45992-46000. [DOI] [PubMed] [Google Scholar]
- 26.Mercurio, F., B. W. Murray, A. Shevchenko, B. L. Bennett, D. B. Young, J. W. Li, G. Pascual, A. Motiwala, H. Zhu, M. Mann, and A. M. Manning. 1999. IκB kinase (IKK)-associated protein 1, a common component of the heterogeneous IKK complex. Mol. Cell. Biol. 19:1526-1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mercurio, F., H. Zhu, B. W. Murray, A. Shevchenko, B. L. Bennett, J. Li, D. B. Young, M. Barbosa, M. Mann, A. Manning, and A. Rao. 1997. IKK-1 and IKK-2: cytokine-activated IκB kinases essential for NF-κB activation. Science 278:860-866. [DOI] [PubMed] [Google Scholar]
- 28.Miller, B. S., and E. Zandi. 2001. Complete reconstitution of human IκB kinase (IKK) complex in yeast. Assessment of its stoichiometry and the role of IKKγ on the complex activity in the absence of stimulation. J. Biol. Chem. 276:36320-36326. [DOI] [PubMed] [Google Scholar]
- 29.Poyet, J. L., S. M. Srinivasula, J. H. Lin, T. Fernandes-Alnemri, S. Yamaoka, P. N. Tsichlis, and E. S. Alnemri. 2000. Activation of the IκB kinases by RIP via IKKγ/NEMO-mediated oligomerization. J. Biol. Chem. 275:37966-37977. [DOI] [PubMed] [Google Scholar]
- 30.Rothwarf, D. M., and M. Karin. 1999. The NF-κB activation pathway: a paradigm in information transfer from membrane to nucleus. Sci. Signal Transduction Knowledge Environ. 5:1.. [DOI] [PubMed]
- 31.Rothwarf, D. M., E. Zandi, G. Natoli, and M. Karin. 1998. IKK-γ is an essential regulatory subunit of the IκB kinase complex. Nature 395:297-300. [DOI] [PubMed] [Google Scholar]
- 32.Silverman, N., and T. Maniatis. 2001. NF-κB signaling pathways in mammalian and insect innate immunity. Genes Dev. 15:2321-2342. [DOI] [PubMed] [Google Scholar]
- 33.Silverman, N., R. Zhou, S. Stoven, N. Pandey, D. Hultmark, and T. Maniatis. 2000. A Drosophila IκB kinase complex required for Relish cleavage and antibacterial immunity. Genes Dev. 14:2461-2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wolf, E., P. S. Kim, and B. Berger. 1997. MultiCoil: a program for predicting two- and three-stranded coiled coils. Protein Sci. 6:1179-1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woronicz, J. D., X. Gao, Z. Cao, M. Rothe, and D. V. Goeddel. 1997. IκB kinase-β: NF-κB activation and complex formation with IκB kinase-α and NIK. Science 278:866-869. [DOI] [PubMed] [Google Scholar]
- 36.Wulczyn, F. G., D. Krappmann, and C. Scheidereit. 1996. The NF-κB/Rel and IκB gene families: mediators of immune response and inflammation. J. Mol. Med. 74:749-769. [DOI] [PubMed] [Google Scholar]
- 37.Xia, Y., C. Makris, B. Su, E. Li, J. Yang, G. R. Nemerow, and M. Karin. 2000. MEK kinase 1 is critically required for c-Jun N-terminal kinase activation by proinflammatory stimuli and growth factor-induced cell migration. Proc. Natl. Acad. Sci. USA 97:5243-5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiao, G., and S. C. Sun. 2000. Activation of IKKα and IKKβ through their fusion with HTLV-I tax protein. Oncogene 19:5198-5203. [DOI] [PubMed] [Google Scholar]
- 39.Yamaoka, S., G. Courtois, C. Bessia, S. T. Whiteside, R. Weil, F. Agou, H. E. Kirk, R. J. Kay, and A. Israel. 1998. Complementation cloning of NEMO, a component of the IκB kinase complex essential for NF-κB activation. Cell 93:1231-1240. [DOI] [PubMed] [Google Scholar]
- 40.Yang, J., Y. Lin, Z. Guo, J. Cheng, J. Huang, L. Deng, W. Liao, Z. Chen, Z. Liu, and B. Su. 2001. The essential role of MEKK3 in TNF-induced NF-κB activation. Nat. Immunol. 2:620-624. [DOI] [PubMed] [Google Scholar]
- 41.Ye, J., X. Xie, L. Tarassishin, and M. S. Horwitz. 2000. Regulation of the NF-κB activation pathway by isolated domains of FIP3/IKKγ, a component of the IκB-α kinase complex. J. Biol. Chem. 275:9882-9889. [DOI] [PubMed] [Google Scholar]
- 42.Yin, L., L. Wu, H. Wesche, C. D. Arthur, J. M. White, D. V. Goeddel, and R. D. Schreiber. 2001. Defective lymphotoxin-beta receptor-induced NF-κB transcriptional activity in NIK-deficient mice. Science 291:2162-2165. [DOI] [PubMed] [Google Scholar]
- 43.Yujiri, T., M. Ware, C. Widmann, R. Oyer, D. Russell, E. Chan, Y. Zaitsu, P. Clarke, K. Tyler, Y. Oka, G. R. Fanger, P. Henson, and G. L. Johnson. 2000. MEK kinase 1 gene disruption alters cell migration and c-Jun NH2-terminal kinase regulation but does not cause a measurable defect in NF-κB activation. Proc. Natl. Acad. Sci. USA 97:7272-7277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zandi, E., and M. Karin. 1999. Bridging the gap: composition, regulation, and physiological function of the IκB kinase complex. Mol. Cell. Biol. 19:4547-4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zandi, E., D. M. Rothwarf, M. Delhase, M. Hayakawa, and M. Karin. 1997. The IκB kinase complex (IKK) contains two kinase subunits, IKKα and IKKβ, necessary for IκB phosphorylation and NF-κB activation. Cell 91:243-252. [DOI] [PubMed] [Google Scholar]
- 46.Zhang, S. Q., A. Kovalenko, G. Cantarella, and D. Wallach. 2000. Recruitment of the IKK signalosome to the p55 TNF receptor: RIP and A20 bind to NEMO (IKKγ) upon receptor stimulation. Immunity 12:301-311. [DOI] [PubMed] [Google Scholar]