

Abstract
Secretory carrier membrane proteins (SCAMPs) are integral membrane proteins found in secretory and endocytic carriers implicated to function in membrane trafficking. Using expressed sequence tag database and library screens and DNA sequencing, we have characterized several new SCAMPs spanning the plant and animal kingdoms and have defined a broadly conserved protein family. No obvious fungal homologue has been identified, however. We have found that SCAMPs share several structural motifs. These include NPF repeats, a leucine heptad repeat enriched in charged residues, and a proline-rich SH3-like and/or WW domain–binding site in the N-terminal domain, which is followed by a membrane core containing four putative transmembrane spans and three amphiphilic segments that are the most highly conserved structural elements. All SCAMPs are 32–38 kDa except mammalian SCAMP4, which is ∼25 kDa and lacks most of the N-terminal hydrophilic domain of other SCAMPs. SCAMP4 is authentic as determined by Northern and Western blotting, suggesting that this portion of the larger SCAMPs encodes the functional domain. Focusing on SCAMP1, we have characterized its structure further by limited proteolysis and Western blotting with the use of isolated secretory granules as a uniformly oriented source of antigen and by topology mapping through expression of alkaline phosphatase gene fusions in Escherichia coli. Results show that SCAMP1 is degraded sequentially from the N terminus and then the C terminus, yielding an ∼20-kDa membrane core that contains four transmembrane spans. Using synthetic peptides corresponding to the three conserved amphiphilic segments of the membrane core, we have demonstrated their binding to phospholipid membranes and shown by circular dichroism spectroscopy that the central amphiphilic segment linking transmembrane spans 2 and 3 is α-helical. In the intact protein, these segments are likely to reside in the cytoplasm-facing membrane interface. The current model of SCAMP1 suggests that the N and C termini form the cytoplasmic surface of the protein overlying a membrane core, which contains a functional domain located at the cytoplasmic interface with little exposure of the protein on the ectodomain.
INTRODUCTION
Secretory carrier membrane proteins (SCAMPs) are a family of membrane proteins that were initially discovered as components of regulated secretory carriers in exocrine, neural, and endocrine cells (Brand et al., 1991). Further characterization has indicated that the distribution extends more broadly to include most, if not all, membranes that recycle between the cell surface and internal compartments, including early and late endosomes, Golgi-derived vesicles, membranes that circulate various transporters, and secretory granules and vesicles in hematopoietic cells (Brand and Castle, 1993; Laurie et al., 1993; Brumell et al., 1995; Haass et al., 1996; Singleton et al., 1997; Wu and Castle, 1997, 1998; Guo et al., 1998). Although the function of SCAMPs has not yet been identified, their concentration in recycling carriers suggests that they serve a role in membrane trafficking or its regulation. This possibility is supported by recent reports that genetic ablation of SCAMP1 affects exocytosis and possibly endocytosis (Fernandez-Chacon et al., 1999, 2000) and that conserved, SCAMP-derived peptides are potent inhibitors of exocytosis (Guo et al., 1998).
SCAMPs have been classified as integral membrane proteins on the basis that they are not extracted from membranes by alkaline sodium carbonate and that they bind Triton X-114 detergent micelles and sediment during phase separation (Brand et al., 1991). Furthermore, hydropathy analysis has suggested that the SCAMPs may have four closely spaced transmembrane spans that are located centrally in the sequence (Brand and Castle, 1993; Singleton et al., 1997). Finally, the mAb originally used to identify the SCAMPs, SG7C12, has a cytoplasmically oriented epitope that is cleaved from purified intact secretory granules by trypsin and also is quite effective for organelle isolation by immunoadsorption (Brand et al., 1991; Laurie et al., 1993).
In view of the growing interest in SCAMPs as prospective components of the membrane trafficking machinery, we have conducted a number of studies to define SCAMP organization within membranes in more detail and to identify what portions of the structure deserve closest scrutiny as prospective function-encoding domains. We have deduced the primary structures of several new SCAMPs, documenting their phylogenetic conservation across the plant and animal kingdoms and identifying highly conserved sequences that are hallmarks of the family. Among these new SCAMPs, we have demonstrated the existence of mammalian SCAMP4, a shorter (∼25 kDa) SCAMP that is presumed to share the function of the longer (32–38 kDa) SCAMPs. Focusing on mammalian SCAMP1 as the prototype, we have used limited proteolysis and Western blotting to demonstrate the cytoplasmic orientation of its N- and C-terminal segments, the sequential cleavage within these segments that probably relates to protein folding, and the presence of a protease-resistant membrane core. We have gone on to map four transmembrane spans within the membrane core and have used synthetic peptides to evaluate the membrane binding of three cytoplasmically oriented amphiphilic segments that are also within the membrane core: one immediately preceding the first transmembrane span, one within the linker between transmembrane spans 2 and 3, and one immediately after the last transmembrane span. In combination, our data led to a model of the SCAMPs in which the N and C termini, particularly the extended N termini of longer SCAMPs, form a cytoplasmic surface overlying the membrane core with little exposure on the ectodomain. Because the membrane core alone is present in all family members, we infer that it is the functional domain and that its highly conserved amphiphilic segments may carry out an interfacial activity.
MATERIALS AND METHODS
Expressed Sequence Tags from Genome Sequencing Projects and Full-Length cDNAs
Genomic sequencing projects have resulted in the submission of several SCAMP homologues from a variety of eukaryotes. Novel SCAMP homologues, in addition to the paralogues that we identified and characterized previously (Singleton et al., 1997), include SCAMPs in pig (SCAMP1; Wen et al., 1998) and pea (Krajinski et al., 1998). Homologues in other species were identified by using the rat SCAMP1 protein sequence as a query for BLAST homology searches against the nonredundant nucleotide and expressed sequence tag (EST) divisions of GenBank. A possible SCAMP homologue was identified in chromosome I in Caenorhabditis elegans, and a full-length cDNA was isolated from a C. elegans early embryo cDNA library constructed in λGT11. Additional cDNAs encoding potential SCAMP homologues were identified as EST sequences in Arabidopsis thaliana, Oryza sativa (rice), Drosophila melanogaster, and Mus musculus. These were sequenced in their entirety to identify complete ORFs and are summarized in Table 1. Also, partial homologues were identified by sequence similarity from flounder, zebrafish, Xenopus, and various plant EST databases (our unpublished observations). Two additional SCAMP homologues were also identified from the A. thaliana genome sequencing project. The predicted reading frames identified by computer analysis of the BAC clones for the GenBank entries indicated in Table 1 differ from those presented in this work. The genomic nucleotide sequence was compared with the cDNA sequence for the available Arabidopsis clone to identify intron/exon boundaries resulting in an ORF more similar to the previously obtained SCAMP sequences. The deduced protein sequences from these sources were used for multiple sequence alignments by the Pileup and Lineup programs in the GCG package (Genetics Computer Group, Madison, WI).
Table 1.
Summary of SCAMP sequences
| Sequence | GenBank entry | Predicted size |
|---|---|---|
| atSC1 | AC006234 (G) | 289 aa; 31.8 kDa |
| atSC2 | AC002560 (G) | 281 aa; 31.8 kDa |
| atSC3 | AC002294 (G) | 289 aa; 32.6 kDa |
| T42180 (C) | ||
| riSC | C98157 (C) | 286 aa; 32.2 kDa |
| ceSC | AF003739 (G) | 335 aa; 36.8 kDa |
| T00564 (C) | ||
| dmSC | AA392499 (C) | 344 aa; 38.4 kDa |
| mSC4 | W71023 (C) | 230 aa; 25.4 kDa |
GenBank accession numbers for newly characterized full-length cDNAs are: atSC3, AF225920; riSC, AF225922; ceSC, AF225921; dmSC, AF224720; and mSC4, AF224721.
SC, SCAMP; at, Arabidopsis thaliana; ri, rice; ce, Caenorhabditis elegans; dm, Drosophila melanogaster; m, mouse; (G), gene; (C), cDNA; aa, amino acids.
Antibodies
mAb 7C12 was generated and characterized as described previously (Brand et al., 1991; Brand and Castle, 1993). Preparation and characterization of rabbit polyclonal anti-SCAMP1 antibodies against specific peptide sequences 1α (SDFDSNPFADPDLNN-NorLeu(C)), 1ς (KKVHGLYRTTGASFEK), and 1ω ((C)TSAAQNAFKGNQM) were described previously (Wu and Castle, 1997, 1998). Rabbit polyclonal antibodies 1γ, 1ε, and 4ω against the peptide sequences (C)KPTEEHPAYTQITK, CFYQDFSVDIPVEFQKTVK, and (C)LPE-YPTVPTVPSYS, respectively, were prepared by the same procedures as for 1α and 1ω. The peptides were conjugated to maleimide-activated keyhole limpet hemocyanin (Imject, Pierce Chemical, Rockford, IL); injection and antiserum collection were performed by Covance (Denver, PA); and antibodies were affinity purified from sera on columns containing the cysteinyl peptides coupled to Sulfolink resin (Pierce Chemical). In the case of the epitope peptide of 1ς, which did not include a cysteinyl residue, the peptide was immobilized by succinimide ester–mediated coupling to Affigel (Bio-Rad, Richmond, CA). Standard low-pH elution procedures were used in all cases. A rabbit antibody against bacterial alkaline phosphatase was purchased from Eppendorf-5′ (Boulder, CO).
RNA Preparation and Northern Analysis
Total RNA was prepared from NIH 3T3 cells with the use of Trizol (Life Technologies/BRL, Grand Island, NY). The RNA was electrophoresed on formaldehyde-impregnated agarose, transferred to reinforced nylon, and probed with the use of 32P-labeled cDNA in aqueous hybridization buffer and washed under normal stringency.
N-terminal Truncations of SCAMP1 Fused to GST and Epitope Analysis
Nested deletions were constructed by PCR with the use of an N-terminal EcoRI/HindIII fragment encoding amino acids 1–150 of the full-length rat SCAMP1 cDNA. The following sense strand oligonucleotides were used to generate N-terminally truncated versions of SCAMP1: Δ1, GGGAATTCTAAGAAATGTGCCACCG; Δ2, GGGAATTCTAAGAACGCCTCCACCA; Δ3, GGGAATTCTAAA-AATGCCTAATGTA; and Δ4, GGGAATTCTAAAGCCGACCGAGGAG. In each case, the oligonucleotide was designed to introduce an EcoRI site at the 5′ end of the product; the antisense oligonucleotide for PCR was in the polylinker of the vector. PCR products encoding SCAMP truncations beginning with residues R28, R44, K52, and K65 were subcloned downstream of GST in the fusion protein vector pGEX-KG (Guo and Dixon, 1991), and the resulting plasmids were used to transform a protease-deficient strain of Escherichia coli (BL21 DE3). Bacteria expressing the N-terminal domain of SCAMP1 and the four truncations fused to GST were prepared by standard procedures, which included a 2-h induction with 1 mM isopropylthio-β-galactoside followed by sonication in Tris buffer, pH 8, containing proteinase inhibitors (1 mM PMSF, 1 mM 4-(2-aminoethyl)benzenesulfonylfluoride, 100 μM leupeptin, and 2 mM EDTA). Equal fractions of the purified fusion proteins were subjected to SDS-PAGE and Western blotting on nitrocellulose (Singleton et al., 1997; Wu and Castle, 1997).
ELISA of 7C12; Competition by SCAMP Peptides
Ninety-six–well plates were coated with 100 ng of SCAMP1 N-terminal domain and washed with PBS, 0.05% Tween 20. Stock solutions (1.1 mM) and serial dilutions of each SCAMP-derived peptide were prepared in blocking buffer (PBS, 0.05% Tween 20, 0.5 mg/ml gelatin, 0.2 mg/ml BSA, goat serum [final dilution, 12.5×], and 0.25 mM PMSF). Aliquots of the serial dilutions were mixed with 1 ng of 7C12 antibody, transferred to coated wells, and incubated for 2 h at room temperature. After washing, goat anti-mouse immunoglobulin G–peroxidase was used as secondary antibody, and peroxidase activity with the use of 2,2′-azino-bis(3-ethyl)benzthiazoline-6-sulfonic acid substrate was read at an absorbance of 414 nm.
Subcellular Fractionation and Proteolysis of Purified Secretory Granules
Secretory granules from rat parotid glands (8–12 glands from four to six overnight-fasted rats per experiment) were purified with the use of isoosmotic self-forming Percoll gradients as described previously (Zastrow and Castle, 1987; Wu and Castle, 1997). After collecting the granules from the bottom of the gradients, the samples were diluted with three volumes of 0.3 M sucrose containing 2 mM MOPS, 1 mM EDTA, pH 6.7, pelleted (2000 × g, 25 min), and resuspended in the same sucrose solution. An aliquot was immediately assayed for protein content with the use of the bicinchoninic acid procedure (Pierce Chemical) to standardize the amount of proteolytic enzyme to be added to each digest. Aliquots of the granule suspension were incubated with serial dilutions of proteolytic enzymes (trypsin or proteinase K) for 2 h on ice. At the end of the incubation, proteinase inhibitors (0.2 mg/ml lima bean trypsin inhibitor and 0.25 mM PMSF, final concentrations) were added and the granules were pelleted by centrifugation. Protein in the resulting supernatants and resuspended pellets was assayed to calculate granule intactness after digestion. Where appropriate, the protein values were corrected for added trypsin inhibitor. Granules in the resuspended pellets were lysed by dilution in hypoosmotic medium (50 mM NaHCO3, 1 mM EDTA, 0.25 mM PMSF), and the membranes were pelleted, resuspended, and solubilized in sample buffer for electrophoresis. An equal fraction of the total of each sample was subjected to SDS-PAGE and Western blotting on nitrocellulose. Bound antibody was detected with the use of peroxidase-conjugated secondary antibodies and enhanced chemiluminescence.
Generation and Analysis of SCAMP–PhoA Fusions
PCR-generated truncations of rat SCAMP1 coding sequence were subcloned into an expression vector encoding bacterial alkaline phosphatase (AP) as a C-terminal fusion partner. Briefly, pSM969 (a gift of Dr. S. Michaelis, Johns Hopkins University Medical School, Baltimore, MD) contains a lac promoter immediately upstream of the coding sequence for the Saccharomyces cerevisiae membrane protein STE6p, which is flanked by unique 5′ BamHI and 3′ NheI restriction sites. The coding sequence for AP (minus its signal sequence) lies ∼10 codons downstream and is in frame with the NheI junction. Native SCAMP1 cDNA has unique BamHI and NheI sites 27 and 877 nucleotides downstream, respectively, from the start codon. Previously, we doubly mutagenized the cDNA to abrogate the endogenous BamHI site and to add a new one immediately upstream of the start codon (our unpublished results). This modified DNA was used as a template for PCR-based generation of 11 truncations of the coding sequence as well as one product encoding the full-length protein (truncations are identified in RESULTS). All products included the upstream BamHI site afforded by a common forward primer. The 10 shortest truncations were generated by reverse primers (all primers were from Operon Technologies, Alameda, CA), which placed an NheI site immediately downstream of the last codon. The eleventh truncation (residues 1–290) and full-length SCAMP1 (minus the stop codon) were engineered to contain an XbaI site downstream of the final codon, allowing eventual subcloning into the NheI site of pSM969. PCR products were ligated into either pCR-II TOPO (TOPO-TA) or pCR-II (TA) vectors (Invitrogen, Carlsbad, CA). All products were sequenced with the use of PCR/dye terminator chemistry followed by automated electrophoresis and analysis on an ABI PRISM TM 377 DNA Sequencer at the University of Virginia Biomolecular Research Facility (Charlottesville, VA). Upon confirmation of correct nucleotide sequences, PCR products were subcloned into the BamHI–NheI remnant of pSM969.
For evaluation of AP activity, all constructs were expressed in E. coli strain UT5600 (number 7092, Yale E. coli Genetic Stock Center, New Haven, CT), which is deficient in the membrane-associated protease ompT. Our method was adapted from previously described procedures (Manoil, 1991; Geller et al., 1996). Cultures of transformants expressing the 12 SCAMP1/AP constructs, as well as untransformed UT5600, were grown overnight at 37°C (16–18 h). Aliquots of each were diluted 50 times into fresh medium, grown at 30°C until OD600 ≅ 0.6 (∼2.5 h), and then grown for another 2 h at 30°C in the presence of isopropylthio-β-galactoside (1 mM final concentration). Cultures were then chilled and maintained on ice. Aliquots were withdrawn for measurement of final OD600 and for SDS-PAGE/Western blotting. For AP assay, 1-ml samples of each culture were pelleted (three min at 2800 × g), washed once with 1 ml of 10 mM Tris, pH 8.0, 10 mM MgSO4, 1 mM iodoacetic acid (IAA), washed twice with 1 ml of 1 M Tris, pH 8.0, 1 mM IAA, and then resuspended in 1 ml of the same buffer. Aliquots made up to 1 ml and containing 1 M Tris, pH 8.0, 0.1 mM ZnCl2, 1 mM IAA were mixed with 50 μl of 1% SDS, incubated for 5 min at 37°C (to permeabilize cell walls), chilled to add 100 μl of 0.4% p-nitrophenylphosphate, 1 M Tris, pH 8.0, and then incubated at 37°C until sufficient color development was observed (usually 30 min). Reactions were stopped by chilling and adding 120 μl of 1 M K2HPO4 pH 8.0, 0.1 M EDTA, and the samples were spun for 5 min at 12,000 × g before reading OD420 on the supernatants. Relative AP activities were calculated according to Brickman and Beckwith (1975), normalizing for assay time. The relative AP values were further normalized for differences in protein expression with the use of quantitative densitometric data obtained from the Western blots with the use of 125I-labeled secondary antibody, phosphorimaging, and analysis with ImageQuant software (Molecular Dynamics, Sunnyvale, CA).
Peptide Synthesis
A series of peptides based on the three amphiphilic
cytoplasmically localized segments that belong to the membrane core of
SCAMP were synthesized and purified by the Biomolecular Research
Facility at the University of Virginia, and the identity of each
peptide was confirmed by mass spectrometry. The sequences of these
peptides are listed in Table 2. Peptides
corresponding to the cytoplasmic segments preceding the first and
succeeding the last transmembrane helices were also synthesized with a
C-terminal cysteine residue. This facilitated the derivatization of
each peptide segment with the cysteine-specific methanethiosulfonate
spin-label (MTSSL) shown in Scheme
1 to produce a series of
peptides with the labeled side chain R1.

Table 2.
SCAMP-derived peptides
| Peptide | Sequence |
|---|---|
| D | Ac-QDFSVDIPVEFQKTVK-NH |
| E | Ac-CWYRPIYKAFR-NH |
| F | Ac-KKVHGLYRTTGASFEK |
| D-SLa | Ac-R1-FYQDFSVDIPVEFQKTVK-NH |
| E-SL | Ac-R1-WYRPIKAFR-NH |
| F-SL | Ac-R1-KKVHGLYRTTGASFEK-NH |
For spin-labeled (SL) peptides, the R1 side chain was derivatized onto a free cysteine residue located at the peptide N terminus with the use of the MTSSL label as described in the text.
Lipid Vesicle Preparation
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine (PC) and 1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphatidylserine (PS) were obtained from Avanti Polar Lipids (Alabaster, AL). Lipid mixtures containing the desired molar ratio of PC and PS were produced by mixing the appropriate lipid solutions in chloroform, removal of the solvent by vacuum desiccation overnight, and hydration of the lipid film in a buffer solution containing 100 mM KCl, 10 mM MOPS, pH 7. For electron paramagnetic resonance (EPR) binding measurements, unilamellar vesicles were then formed by freeze-thawing the suspension five times followed by extrusion of the mixture through polycarbonate filters with a 0.1-μm pore diameter (Poretics, Livermore, CA) with the use of a hand-held extruder (Avestine, Ottawa, Canada). For circular dichroism (CD) measurements, the mixture was sonicated with the use of a probe sonicator at 4°C for ∼30 min to produce small 300-Å unilamellar vesicles as described previously (Castle and Hubbell, 1976).
Partition Coefficients Determined by EPR
The membrane binding of the SCAMP-derived peptides was determined by EPR with the use of a procedure similar to that described previously (Archer et al., 1991). Briefly, membrane-bound and aqueous EPR spectra of a spin-labeled macromolecule exhibit dramatically different line shapes because of the altered rotational rates for the macromolecule in the two environments. If the spin-labeled macromolecule partitions between the aqueous and membrane phases, the resulting EPR spectrum is a sum arising from the spectra of membrane-bound and aqueous spin populations. As a result, the partitioning can then be determined accurately by quantitating the aqueous and membrane populations from the composite EPR spectrum. This method has been shown numerous times to be an accurate and reliable approach to determine the membrane binding of peptides (Lewis and Cafiso, 1999; Victor et al., 1999).
EPR spectra were obtained with the use of a Varian (Palo Alto, CA) E-line Century series spectrometer fitted with a standard rectangular TE102 X-band cavity with a microwave power of 10 mW and modulation amplitude of 1 gauss. Samples (100 μL) contained identical peptide concentrations (typically 20 μM) with varied concentrations of unilamellar lipid vesicles in buffer (100 mM KCl, 10 mM MOPS, pH 7.0). The peak-to-peak amplitude of the mI=−1 resonance was recorded, and for each spectrum the fraction of membrane-bound peptide was determined as described previously (Cafiso and Hubbell, 1981). The molar partition coefficient K (units of M−1) was then determined by fitting the data to the relationship:
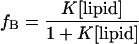 |
1 |
where fB is fraction bound and [lipid] is the molar concentration of accessible lipid. Because the peptides are unlikely to be membrane permeable in these samples, this concentration is simply taken as half the total lipid concentration.
CD Spectroscopy
CD spectra of the three SCAMP peptides were obtained with the use of a Jasco (Easton, MD) J-720 spectropolarimeter with a 1-mm path length sample cell. Unless indicated otherwise, the following instrumental parameters were used: bandwidth, 2.0 nm; sensitivity, 100 mdeg; response time, 0.25 s; scan speed, 50 nm/s. Spectra were obtained over the range of 190–300 nm and were typically the sum of 10–20 scans. Sonicated lipid samples were used for these measurements to minimize the effects of light scattering, and peptides were added to a concentration of 20–40 μM.
RESULTS
Conserved Primary Structure of SCAMPs
Previously, we reported the amino acid sequences that were conceptually translated from cDNAs for rat SCAMP1 and human SCAMPs 1–3 (Brand and Castle, 1993; Singleton et al., 1997). These efforts provided the first indication that there are multiple paralogues of SCAMPs within individual mammalian species, each deriving from separate but related genes, and that sequence identity/similarity is >90% for particular paralogues compared between different mammals but ∼60% between distinct paralogues of the same species. The multiple genome sequencing projects and databases of ESTs have now made it possible to search for SCAMPs in several different organisms spanning the plant and animal kingdoms. A representative set of those we have characterized is presented in Figure 1. We have obtained a variety of cDNAs from the EST sources and have used them either directly for sequencing or for library screening to obtain cDNAs encoding full-length polypeptides for sequencing. Pairwise multiple sequence alignment produces a dendrogram that recapitulates the evolutionary relationship between the individual species in which SCAMP homologues have been identified (Figure 1A). Multiple SCAMP paralogues have been found in vertebrate species, and the A. thaliana genome and various plant sequencing projects suggest that individual types of plants also have multiple SCAMPs. However, invertebrate genome sequencing projects (C. elegans and D. melanogaster) have indicated only a single SCAMP locus in each species. Significantly, no homologous sequences have been identified so far in fungi from the completed S. cerevisiae genome and the essentially completed Schizosaccharomyces pombe and Candida albicans genomes.
Figure 1.

Deduced SCAMP protein sequences and their relationships. (A) Dendrogram constructed by pairwise comparison of sequences with the use of the GCG module Pileup illustrating the relative similarities between the homologues characterized here and previously (Singleton et al., 1997). Grouping within the plant and animal kingdoms is evident. (B) Alignment of selected plant and animal SCAMP sequences relative to rat SCAMP1, the prototype SCAMP (Brand and Castle, 1993), for which residue numbers are provided. Putative transmembrane spans are boxed in black and along with gray-shaded regions identify structural similarities among the multiply aligned sequences. rSC1, rat SCAMP1; mSC4, mouse SCAMP4; dmSC, D. melanogaster SCAMP; ceSC, C. elegans SCAMP; atSC1, -2, and -3, A. thaliana SCAMP forms; riSC, rice SCAMP. (C) Structural modules representing regions of similarity between the aligned SCAMPs (modules alsoidentified between the rows of sequences in B). Box A represents the NPF repeats located near the N terminus, with the number indicating how many repeats are found in each isoform. Box B is the leucine heptad repeat, which is predicted to have a helical configuration with alternating rows of positively and negatively charged residues on its surface. Box C is a proline-rich segment that is similar to ligands for SH3 domains and is located just past the N terminus of SCAMP4. Box D is the final segment before the putative first transmembrane span. Boxes TM1–TM4 are the four predicted transmembrane spans. Box E, connecting transmembrane spans 2 and 3, is the most highly conserved segment of the protein. Box F immediately follows the fourth transmembrane span and is predicted to be a helix. Box G is an alanine/serine-rich segment that is near or at the C terminus of all SCAMPs except SCAMP4.
Aligned sequences demonstrate the highly conserved domain structure of the SCAMP family (Figure 1, B and C). All SCAMP family members share a common central core domain that includes four predicted transmembrane spans of similar length and conserved amino acid sequence (Figure 1C, blocks TM1–TM4). Loops between the transmembrane spans are also conserved in length, and the amino acid sequence linking spans 2 and 3 (Figure 1C, block E, sequence [199]FVCWYRPLYGAFRSDSS[215] in rSC1) is especially conserved in all SCAMP homologues examined. In addition, sequences preceding and succeeding the first and last transmembrane spans (Figure 1C, blocks D and F, respectively) are quite similar across the different SCAMPs, particularly in their amphiphilic character, and contribute to the conserved core domain. Preceding block D is a proline-rich segment having a resemblance to both an SH3-binding motif (PXXP) and a WW domain–binding motif (PPXY) (Sudol, 1996) (Figure 1C, block C) that is again highly conserved in all SCAMPs. With the exception of the shorter mSC4 (and a newly identified rodent SCAMP5 [GenBank accession numbers AAF64491 and AAF64466] and a zebrafish EST), all homologues have two other types of conserved segments (Figure 1C, blocks A and B) that are located in the N-terminal domain upstream from the conserved core. Block A indicates that longer SCAMPs all have either two or three NPF repeats that begin near the N terminus and are potential binding sites for EH domain–containing proteins (Salcini et al., 1997; Paoluzi et al., 1998). Block B identifies a predicted helical segment that is characterized by a heptad repeat of aliphatic amino acids and intervening charged residues that appear organized in positive and negative rows along the surface (Brand and Castle, 1993). The C-terminal domain succeeding the conserved core of the SCAMPs is shorter than the N-terminal domain preceding the core. Beyond block F (Figure 1C), the primary structure is more variable (as for the segment between blocks A and B), although several of the homologues (human, rodent [except mSC4], fly, nematode, and plant) have an alanine-rich segment (Figure 1C, block G) either immediately before (animal) or at (plant) the C terminus.
Authenticity of SCAMP4
The attenuated N-terminal domain of mSC4 that was deduced by conceptual translation of the cognate cDNA differed from that of the other SCAMPs that we have characterized more thoroughly. Therefore, we sought to confirm that this SCAMP was both authentic and expressed. As shown in Figure 2A, we detected an ∼2.1-kilobase (kb) RNA by Northern blotting of total RNA from mouse NIH 3T3 cells. The cDNA used to probe the Northern blot was ∼1.8 kb, consistent with the possibility that it encodes a full-length SCAMP polypeptide. Using an antibody raised to a peptide that corresponds to a unique segment near the C terminus of the deduced sequence of SCAMP4, we identified a polypeptide of Mr ∼ 25,000 by Western blotting (Figure 2B). The polypeptide is widely expressed, and its size matches that deduced by conceptual translation of the cDNA.
Figure 2.

Expression of SCAMP4. (A) Northern blot of total RNA from mouse NIH 3T3 cells showing the presence of an ∼2.1-kb band. (B) Western blot of lysates of NRK and AtT-20 cells and a postnuclear supernatant fraction from rat pancreas showing a single ∼25-kDa band as detected with the use of a rabbit antibody raised against a unique peptide located near the C terminus of SCAMP4.
Location of the Epitope for mAb 7C12
Trypsinization of intact secretory granules abolished detection of SCAMPs 1 and 2 by Western blotting with the use of mAb 7C12, suggesting the presence of the epitope in one of the two extended hydrophilic domains forming N- and C-terminal segments of the full-length polypeptide (Brand and Castle, 1993). To locate the epitope, we inserted portions of the SCAMP1 cDNA corresponding to the first 149 amino acids and the final 53 amino acids into the pGEX-KG vector and expressed the GST fusion proteins in E. coli (BL21 DE3). Western blotting of bacterial lysates indicated that the epitope was present in the N-terminal segment. We then prepared a series of N-terminal truncations of SCAMP1 in the GST chimeras, which initiated the SCAMP1 sequence at residues R28, R44, K52, and K65 (Figure 1). Western blots of bacterial lysates showed that the 7C12 epitope was detected in the chimeras beginning at the authentic N terminus and at R28 but not in the shorter residues (Figure 3A). The level of expression of each of the chimeras was normalized by blotting with an anti-peptide antibody (1γ) whose epitope is made up of residues 65–78 (KPTEEHPAYTQITK) of SCAMP1. Use of 125I-labeled secondary antibodies and quantitation of antibody binding by phosphorimaging demonstrated that the ratio of 7C12/1γ binding decreased by 65–70% as a result of the first N-terminal deletion. Thus, we concluded that the epitope of the mAb was likely to include portions of the sequence between residues 1–28 and 28–43 but nothing closer to the C terminus.
Figure 3.

Characterization of the epitope of mAb 7C12 within the primary structure of SCAMP1. (A) Comparative Western blots of the N-terminal cytoplasmic domains of SCAMP1 and successive truncations fused to GST and expressed in E. coli. Blots were probed with 7C12 and with antibody 1γ, whose epitope is retained in all deletions and thus enables normalization for different levels of expression. Bound antibodies were detected with the use of 125I-labeled secondary antibodies followed by autoradiography and phosphorimaging (for quantitation with the use of ImageQuant). The autoradiograms indicate that 7C12 binding substantially decreases with the first deletion and is fully lost with the second deletion. Quantitation shows that the first deletion reduces binding by 65–70% (binding of 1γ was used to normalize for differing amounts of fusion protein among the samples). (B) Use of synthetic peptides to compete the binding of 7C12 to the recombinant N-terminal cytoplasmic domain of SCAMP1 in an ELISA. Peptide 1 (residues 2–16 of SCAMP1) competes about one-fifth as well as peptides 2 and 3 (residues 30–44 and 34–44, respectively). Peptide 4 (residues 65–78) does not compete binding.
To characterize the epitope further, we examined the ability of selected synthetic peptides to compete the binding of 7C12 antibody to the N-terminal (149-residue) segment of SCAMP1 as detected by ELISA. Four peptides proved to be useful for this purpose. Peptide 1 (SDFDSNPFADPDLNN) contains residues 2–16 at the N terminus of SCAMP1. Peptide 2 (VPPGLDEYNPFSDSR) corresponds to residues 30–44, the peptide determined above to contain the most distal portion of the epitope. Peptide 3 (LDEYNPFSDSR) is a truncation of peptide 2 lacking the N-terminal VPPG residues. Peptide 4, residues 65–78 of SCAMP1, is the epitope of anti-peptide antibody 1γ (identified above) and served as a control. As shown in Figure 3B, peptides 1, 2, and 3 all competed antibody binding to immobilized antigen with half-maximal decreases in peroxidase activity at concentrations of 8, 3.5, and 2 μM, respectively, whereas the fourth peptide did not compete. Comparison of the sequences indicates that NPFXD is the common element in peptides 1–3 and is likely to be largely responsible for competing antibody binding. This possibility would also explain the results shown in Figure 3A in that the removal of two of the three NPFXD motifs would reduce antibody binding by two-thirds. However, the epitope may be more complex than the simple linear sequence for at least two reasons. First, 7C12 does not bind to nonmammalian SCAMPs (e.g., C. elegans SCAMP) that contain NPFXD motifs (our unpublished results). Second, the original antigen used to generate 7C12 was a preparation of purified granule membranes that were treated with sodium carbonate but were not otherwise denatured (Brand et al., 1991). Thus, the epitope may involve higher-order structure within the N-terminal NPFXD-containing region. For the purpose of these studies, however, the important observation is that the peptide segment between residues 30 and 44 of SCAMP1 defines the C-terminal limit of the antibody's epitope.
Organization of SCAMP1 as Examined by Limited Proteolysis and Western Blotting
In addition to mAb 7C12, which binds mammalian SCAMPs 1–3 (Singleton et al., 1997), we have generated a series of five antibodies against peptides that correspond to sequences in SCAMP1. The antibodies are identified as 1α, 1γ, 1ε, 1ς, and 1ω; the epitopes of the first three are located in the N-terminal hydrophilic domain of SCAMP1 preceding the first putative transmembrane domain, and those for the last two are located in the C-terminal hydrophilic domain succeeding the last putative transmembrane domain. Their positions and the deduced location of the 7C12 epitope are indicated in the cartoon shown in Figure 4 (bottom panel). To determine how the hydrophilic terminal domains of SCAMP1 are positioned in relation to the membrane, we conducted a limited proteolysis study on purified parotid secretory granules, a source of SCAMP1 that is expected to be uniformly oriented with respect to the membrane. The purified secretory granules were resuspended in buffered isoosmotic sucrose, assayed for protein, and incubated with enzymes as specified in MATERIALS AND METHODS. After digestion and subsequent addition of proteinase inhibitors, the samples were processed further by pelleting granules to determine intactness and by lysis to prepare membranes for SDS-PAGE and Western blotting. Blotting with the antibodies 1α, 7C12, 1γ, 1ς, and 1ω was performed on separate blots of the digestion series for each antibody. Antibody 1ε worked poorly for Western blotting and was used only for immunoadsorption to confirm the presence of the epitope in the trypsin-insensitive domain of membrane-associated SCAMP1.
Figure 4.

Organization of SCAMP1 as deduced from combined proteolysis and Western blotting of purified secretion granules from rat parotid gland. Six different antibodies were used in the study, 1α, 7C12, 1γ, 1ε, 1ς, and 1ω, and the positions of their epitopes within the primary structure of SCAMP1 are indicated in the bottom panel. All antibodies except for 7C12 are specific for SCAMP1 (Mr 37,000); 7C12 also detects SCAMP2 (Mr 39,000). Enzyme digestions were carried out at 0°C on suspensions of intact granules with the use of a series of trypsin concentrations and selected concentrations of proteinase K (PK). Amounts of enzyme used are expressed as the log of the ratio mg enzyme:mg total protein in each sample (−5 and −2 are the minimum and maximum ratios, respectively, used for trypsin). After digestion, enzyme was inhibited and granules were pelleted. Protein in supernatants and pellets was assayed to estimate granule intactness after proteolysis, and the granules werelysed and membranes sedimented for Western blotting. Decreased molecular weight and loss of epitopes in the blots (A–E) are used as indices of epitope accessibility in the native and partially digested forms of the protein. The profiles indicate that the 1ς epitope (D) is resistant to trypsinization and is still detected in an Mr ∼ 20,000 fragment after digestion with the highest level of enzyme used. This epitope is lost from intact granules when digested with 10−3 mg PK/mg protein (D, right). As seen in A–E, trypsin digestion proceeds stepwise from the N terminus (epitopes 1α, 7C12, and 1γ are cleaved in order) and then at the C terminus (1ω epitope is lost). Six successive sites of trypsinization (T1–T6, bottom panel) were deduced by comparing the locations and sizes of predicted tryptic fragments within the primary sequence of SCAMP1 with the changes in apparent Mr and detection of the bands observed in the Western blots. Antibody 1ε, which is not satisfactory for Western blotting, was used to immunoprecipitate the smallest tryptic fragment shown in D, and the resultant immunoprecipitate was blotted with 1ς to confirm that the epitope of 1ε is retained in the trypsin-resistant core of SCAMP 1 (F). HC and LC in F refer to heavy and light chains, respectively, of immunoglobulin G. Low levels of uncleaved SCAMP1 observed in A and E (1α and 1ω are the strongest antibodies) identify a small amount of inaccessible antigen, possibly derived from inside-out membrane vesicles in the granule fraction.
The essential results of the enzymatic digestion study are shown in Figure 4 and were reproduced in four experiments conducted with different preparations of granules. Digestion of SCAMP1 with trypsin appeared to proceed stepwise beginning with the N terminus. As the amount of enzyme was increased in orders of magnitude from 10−5 to 10−2 mg/mg granule protein, the epitopes of 1α, 7C12, and 1γ (Figure 4, A–C, respectively) were cleaved in order, generating a descending ladder of lower Mr fragments. The epitope of 1ω at the C terminus of SCAMP1 mostly persisted on an Mr ∼21,000 fragment after almost complete cleavage of the N terminus with 10−3 mg trypsin/mg protein (Figure 4E). However, this epitope was removed by 10−2 mg trypsin/mg protein, leaving an Mr 19,000–20,000 fragment detected by 1ς that appears refractory to further trypsin cleavage (Figure 4D). This trypsin-insensitive portion of SCAMP1 could be immunoadsorbed by 1ε after solubilization from the digested sample in RIPA buffer and was detected by immunoblotting with 1ς (Figure 4F). Thus, the trypsin-resistant core of SCAMP1 corresponded to the putative transmembrane domains and flanking peptide segments. Using the primary sequence of SCAMP1 and the apparent molecular weight changes for each cleavage product, we have deduced six successive sites of trypsinization, which are identified as T1–T6 in the bottom panel of Figure 4. The probable cleavage sites are: T1 at R28, T2 at either K52 or K65, T3 at KR89/90, T4 at K117, T5 at K311, and T6 at K298. The trypsin-resistant core, residues 118–298, has a calculated Mr of 20,946, in agreement with the final trypsin-resistant band observed in Figure 4D. Two other observations are noted from the trypsin digestion series. First, SCAMP2 (apparent Mr 39,000), which is also present in secretion granule membranes, was detected only by 7C12 (Figure 4B). The relative resistance of the N terminus of SCAMP2 to trypsinization compared with SCAMP1 is likely to reflect the absence of appropriate K and R residues in the N-terminal domain in SCAMP2, which is more acidic than SCAMP1 (Singleton et al., 1997). Second, in the 1α and 1ω trypsin series (Figure 4, A and E), a small fraction of SCAMP1 appeared to be refractory to digestion, as detected by our strongest antibodies. We assume that this antigen is associated with vesicular contaminants in the granule fraction (possibly having inside-out orientation, as discussed below) in which the SCAMP is inaccessible.
Digestions with proteinase K (PK) supported and extended the results obtained with trypsin. At moderate levels of PK (0.4 μg/mg granule protein), the same-sized cleavage product (Mr 19,000–20,000) containing the 1ς epitope but not the 1α, 1γ, and 1ω epitopes, as with the largest amount of trypsin tested, was observed (Figure 4, A and D). More significantly, 2.5-fold more PK (10−3 mg/mg protein) abolished binding to the 1ς epitope (Figure 4D). For all of the digested samples except for 10−3 mg PK/mg protein, granule intactness after digestion ranged from 90 to 94%, the same as in control samples that were incubated without enzymes. At 10−3 mg PK/mg protein, granule integrity decreased to 84–85%, which, although lower, still suggested that to a good approximation all cleavages occurred from the cytoplasmic side of the granule membrane. This deduction was also supported indirectly by other experiments in which efforts to digest SCAMPs on the ectoplasmic surface during cycling to the plasma membrane of insulin-stimulated adipocytes were uniformly unsuccessful (T. Wu and D. Castle, unpublished results). Together, the results obtained with the use of proteolytic digestion suggest that the N-terminal hydrophilic domain of SCAMP1 forms the cytoplasmic surface of the folded protein, extending over much of the C-terminal hydrophilic domain, which is cytoplasmically oriented, and the protease-resistant core, which includes the putative transmembrane spans and flanking sequences.
Presence of Four Transmembrane Spans in SCAMP1
The identification of cytoplasmically oriented N and C termini for SCAMP1 by limited proteolysis indicated that as an integral membrane component, the polypeptide must have an even number of transmembrane spans. To explore the topology further, we assumed that the original hydropathy plot (Brand and Castle, 1993) correctly predicted four transmembrane spans, and we attempted to tag the putative ectodomain loops between transmembrane spans 1/2 and 3/4 of SCAMP1 by myc epitope insertion (Borjigin and Nathans, 1994). However, in both cases, the SCAMP expressed from the recombinant DNAs appeared to accumulate in the endoplasmic reticulum and nuclear envelope as judged by immunofluorescent staining, suggesting that tagging at these internal sites in the primary structure induced misfolding. As an alternative approach, we chose to map the transmembrane topology with the use of AP gene (PhoA) fusions expressed in E. coli (Manoil, 1991). Prokaryotic AP correctly folds and is active only when exported to the periplasm, and in chimeras with integral membrane proteins, AP activity is observed with odd numbers of transmembrane spans. This approach has been used widely to map the topology of prokaryotic and eukaryotic membrane proteins that span the bilayer multiple times (reviewed by Traxler et al., 1993), and it reproduces the topologies determined in eukaryotic cells (Geller et al., 1996).
We constructed 12 chimeras in which cDNAs encoding N-terminal segments of SCAMP1 of increasing length were fused upstream of PhoA in place of sequence encoding the AP signal sequence. The C-terminal residue of the SCAMP1 portion of each chimera is identified in Figure 5A, illustrating its position in relation to the putative transmembrane spans. The chimeras include fusion sites near the C-terminal end of each hydrophilic domain (Boyd et al., 1993) of SCAMP1 as well as more proximal fusion sites within each hydrophilic segment. After transformation and induction of expression, the relative steady-state levels of each chimera were assessed by quantitative Western blotting with the use of 125I-labeled secondary antibody. As shown in Figure 5B, chimeric polypeptides of the appropriate size were detected in each sample, and steady-state levels of expression in almost all cases differed by a factor of 4 or less. The exceptions are chimeras 241 and 250, in which the levels were 13-fold and 7-fold less, respectively, than the maximum (chimera 338). Comparable relative levels of expression (all within a factor of 5) were quantitated from a separate Western blot with the use of anti-AP antibody in place of anti-SCAMP. The results of the AP assays, after subtracting the background activity of nontransformed cells (1% of maximum activity observed) and normalizing for level of expression, are shown in Figure 5C. Enzyme activity is either very high or essentially background. Because of this clear outcome and the relatively similar steady-state levels observed for all of the chimeras, we believe that our results are not likely to be affected by any differences in stability among the chimeras in E. coli. Therefore, we have normalized activity to the steady-state level and not to the rate of biosynthesis (Calamia and Manoil, 1990; Geller et al., 1996) in Figure 5C.
Figure 5.

Membrane topology of SCAMP1 as deduced from PhoA gene fusion analysis in E. coli. (A) Cartoon of SCAMP1 identifying the residues that terminate the SCAMP portion of each chimera and their positions in relation to the four putative transmembrane spans. (B) Western blot of SCAMP1-AP chimeras with the use of antibody 1α to detect the SCAMP epitope with detection of bound primary antibody with the use of 125I-labeled secondary antibody. (C) Plot showing AP activity for each chimera normalized for the level of expression of the chimera as determined by Western blotting (from B). The asterisk identifies an unexpectedly high activity that is discussed in the text. The results shown in B and C are from one of three independent experiments giving the same results.
The two peaks of enzyme activity flanked by three background levels observed in Figure 5C indicate a four-transmembrane topology for SCAMP1 in which the N- and C-terminal segments and the loop between transmembrane spans 2 and 3 are protoplasmic (cytoplasmic) and the loops between spans 1 and 2 and between spans 3 and 4 are periplasmic (ectoplasmic). Only one chimera (286) resulted in an orientation of AP that was opposite what was predicted. We believe that this outcome occurred because the SCAMP1 portion of this chimera terminated after a hydrophobic segment and thereby lacked the basic amino acids needed to create a strong topogenic signal that would maintain the subsequent AP in the protoplasm. Indeed, chimera 290, four residues more distal, gives the expected orientation, which is in agreement with the cytoplasmic orientation of the antibody 1ς epitope (which includes residues 286 and 290) that was observed by limited proteolysis (Figure 4). Also, there is precedence for the anomalous orientation of AP fusions when chimeras are made immediately C terminal to hydrophobic segments (Calamia and Manoil, 1990). Therefore, our data strongly support a four-transmembrane-span topology of SCAMP1, as diagrammed in Figure 5A.
Membrane Binding and Secondary Structure of Synthetic Peptides Corresponding to Conserved Amphiphilic Segments within SCAMP1's Membrane Core
The three cytoplasmically oriented segments—just preceding the first transmembrane span, linking transmembrane spans 2 and 3, and just succeeding the last transmembrane span—within the conserved core of SCAMP have sequences that include nonpolar and polar residues and are consistent with forming amphiphilic structures that associate with the membrane interface. To test whether these segments have an affinity for membranes independent of their adjacent transmembrane tethers, we examined membrane binding of the corresponding synthetic peptides (Table 2) that were derivatized by spin-labeled side chain R1 (see MATERIALS AND METHODS) by EPR spectroscopy. Shown in Figure 6A are plots of the fraction of peptide bound as a function of the accessible lipid concentration, along with fits of the data to Equation 1 (above). Because the peptides are unlikely to be membrane permeable, the accessible lipid concentration is taken as the external vesicle lipid concentration (or one-half the total lipid concentration of the extruded vesicles). A summary of the binding constants obtained is given in Table 3. Although all three peptides bind to lipid bilayers, peptide E, which forms the linker between the second and third transmembrane segments, shows the greatest membrane affinity. In the presence of PC alone, the peptide has a modest membrane affinity that increases by approximately 2 orders of magnitude in the presence of PS. The binding of peptide E is estimated to be at least 104 M−1 in the presence of 25 mol% PS, and >90% of the peptide is bound at lipid concentrations of ∼1 mM. This increase in affinity in the presence of PS is consistent with an electrostatic attraction of the basic peptide to the acidic phospholipid surface, and similar binding increases have been seen for other basic peptides that interact with membranes containing acidic lipid (see, for example, Buser et al., 1994).
Figure 6.

(A) Membrane binding of SCAMP-derived peptides determined by EPR. Peptide concentration was ∼10–20 μM, and the fraction of peptide bound was determined as described in the text. Data are shown for E peptide in the presence of PC (▴) or PC/PS (3:1) (●), F peptide in the presence of PC (○) or PC/PS (3:1) (▵), and D peptide in the presence of PC/PS (3:1) (♦). The accessible lipid concentration is taken as the concentration of lipid in the exterior vesicle monolayer. (B) CD spectra for 35 μM E peptide in the presence of buffer (dashed line), 1 mM PC (dotted line), or PC/PS (3:1) vesicles (solid line). The D, E, and F peptides are presumed to reside at the cytoplasmic membrane interface and are part of the trypsin-resistant core of the polypeptide. The peptides include the segments that flank the first and last transmembrane spans and the segment that links transmembrane spans 2 and 3 (boxes D, G, and F, respectively, in Figure 1C).
Table 3.
Membrane-binding constants for SCAMP-derived peptides
| Peptide | PC (K, M−1) | PC/PS (3∶1) (K, M−1) |
|---|---|---|
| D-SL | 20 | 15 |
| E-SL | 328 | >104 |
| F-SL | 65 | 53 |
Membrane-binding constants determined for these peptides have an error of 10–20%. We were not able to make an accurate prediction of the binding of peptide E in PC/PS with EPR because the concentration of peptide needed for the assay significantly alters the membrane charge density at low lipid concentrations.
Compared with peptide E, peptides D and F show a more modest membrane affinity, with peptide F achieving 50% binding at ∼20 mM lipid and peptide D reaching 50% binding at ∼50 mM lipid in PC/PS mixtures. Unlike peptide E, neither peptide D nor peptide F shows a strong preference for PC/PS- versus PC-containing membranes. This is consistent with the low net charge on peptide D, but it is somewhat surprising for peptide F, which has a net valence of +4 and should have a significant electrostatic attraction in the presence of PS.
The CD spectrum for peptide E was examined in buffer as well as in the presence of PC and PC/PS. Although peptide E assumes a largely random configuration in buffer, binding of the peptide to lipid vesicles is accompanied by an increase in helical content (Figure 6B). In the presence of PC/PS under conditions in which the peptide is almost completely membrane-associated, peptide E is estimated to have between 20 and 30% helical content based on the molar ellipticity at 222 nm (Luo and Baldwin, 1997). Peptides D and F are also in a random configuration in buffer, but they exhibit CD spectra that are indicative of β-structure in the presence of lipid bilayers (our unpublished observations). However, the decreased membrane affinity of these peptides makes a structural assessment by CD difficult, because the lipid concentration required to achieve substantial peptide binding results in severe light scattering.
Given the high membrane affinity of peptide E and its attachment to two transmembrane spans in the full-length polypeptide, it is almost certainly an interfacial segment in intact SCAMP. When modeled as a helix, the segment is highly amphipathic, and the isolated peptide has significant helical content when membrane bound. Thus, we believe that the behavior of the isolated peptide provides strong evidence that this segment of SCAMP has an interfacial location and helical configuration. For peptides D and F, our information is not adequate to make structural predictions with regard to intact SCAMP; however, these segments are also likely to have an interfacial location. Because they are tethered to the membrane interface in the intact protein, they experience a greater effective membrane concentration in the intact protein than they do as isolated peptide fragments. As a result, the modest membrane affinities for the isolated peptides are significant in the context of the intact protein. With the use of a simple “ball-and-chain” model (Kim et al., 1994), it is easy to show that these tethered segments will experience a membrane surface-to-aqueous volume ratio that is several orders of magnitude greater than the vesicle surface-to-aqueous volume ratio at the highest lipid concentrations used in these experiments. Thus, peptides D and F will be bound within the interface if they are not complexed within the protein. Such interfacial association is consistent with our trypsin digestion study (Figure 4), in which peptides D and F were not cleaved from the membrane core despite containing lysine residues at their junctions with the transmembrane spans.
DISCUSSION
Our studies have substantially extended current knowledge about the broad distribution of SCAMPs in the animal and plant kingdoms and have identified the structural features within the polypeptide that serve as signatures for the family. SCAMPs are expressed in a range of metazoans including nematodes, insects, fish, frogs, and mammals, and in both monocot and dicot plants. Furthermore, our studies mostly in mammals have indicated that SCAMPs are detected in every cell type examined except mature erythrocytes. Conservation of this magnitude raises the possibility that SCAMPs perform a ubiquitous role in vesicular trafficking in post-Golgi compartments, where they are mostly concentrated. Interestingly, intersectin and γ-synergin, proteins thought to function in endocytosis and trafficking from the trans-Golgi network, respectively (Hussain et al., 1999; Page et al., 1999), have been identified recently as prospective binding partners for SCAMP1 (Fernandez-Chacon et al., 2000). Although it is tempting to view these interactions, which appear to involve binding to the NPF repeats of SCAMP1, as a possible reflection of a general role in trafficking, we are reminded that this interaction might relate more to the binding and relocation of selected SCAMPs, in analogy with the role of NPFXD as a trafficking ligand for selected receptors in yeast (Tan et al., 1996). At present, we are hesitant to suggest that SCAMP function is essential for two reasons. First, unlike other proteins that are currently regarded as essential for the operation of vesicular trafficking, we have been unable to identify a prospective SCAMP homologue from the genome-sequencing projects in three different types of yeast. The absence of an obvious yeast homologue may suggest that SCAMPs serve a regulatory function that is observed only in multicellular eukaryotes (Sugita et al., 1999). On the other hand, there are well-known cases in which proteins that have the same function in vesicular trafficking in yeast and mammals have no detectable primary sequence identity in the two organisms (e.g., phosphatidylinositol transfer protein [Phillips et al., 1999]). Thus, a functional homologue of SCAMPs may still be identified in yeast. Second, genetic ablation of SCAMP1 is not lethal (Fernandez-Chacon et al., 1999), even though this SCAMP is by far the most prevalent SCAMP detected in brain tissue (Singleton et al., 1997). Again, however, it is possible that a yet to be characterized SCAMP (e.g., SCAMP4, SCAMP5) or some other functional homologue may cover for the loss of SCAMP1, and neither of the negative findings rule out the possibility that SCAMPs may perform a key role in trafficking.
As a part of this study, we have now confirmed the original prediction (Brand and Castle, 1993) that SCAMP1 is an integral protein that spans membranes four times with cytoplasmically oriented N- and C-terminal domains. This topology is the same as that of at least three other mammalian membrane protein families: the tetraspanin superfamily (Maeker et al., 1997), the physins (synaptophysin, synaptoporin, pantophysin, and synaptogyrin), and connexins (Kumar and Gilula, 1986; Knaus et al., 1990; Kanter et al., 1994; Leube, 1994; Stenius et al., 1995). However, at several levels, the structural organization of the SCAMPs sets them apart from each of these other families. First, the transmembrane spans are spaced very differently. In SCAMPs, they are quite closely bunched with interconnecting segments that are all less than 20 residues long. Tetraspanins have one and physins and connexins each have two ectodomain loops connecting the transmembrane spans that are ≥30 residues long. Although the single endodomain loop between transmembrane spans 2 and 3 in tetraspanins and physins is short, as it is in SCAMPs, the loop in connexins is very extended. Second, the closely linked transmembrane spans in SCAMPs are situated between extended N- and C-terminal domains. All three of the other families have much shorter N-terminal domains, and the C-terminal domains of tetraspanins and one of the physins, pantophysin, also are shorter. Although the C-terminal domains of synaptophysin, synaptoporin, synaptogyrin, and the connexins are extended like those of SCAMPs, the primary structures are not similar, especially in the case of the physins, which have several repeats rich in D, P, and Y residues. Third, SCAMPs are not glycosylated and do not contain disulfide bonds in ectodomains. Tetraspanins and physins are predicted to contain both of these features (Johnston et al., 1989; Johnston and Sudhof, 1990; Maeker et al., 1997), and both ectodomain loops of connexins contain multiple cysteines, which would be compatible with the formation of disulfide bonds. Additionally, the two ectodomain loops of the SCAMPs are likely to be situated close to or within the membrane interface, the first loop being extremely short and the second being amphiphilic.
Although the structural organization of SCAMPs contrasts substantially with that of tetraspanins, physins, and connexins, it appears much more similar to the organization of the yeast proteins Sft2p and Got1p (Conchon et al., 1999). Both yeast proteins have the same membrane topology as the SCAMPs, and like SCAMPs, their transmembrane spans are concentrated within <130 residues of the entire sequence and encompass the most conserved portion of the primary structure. Furthermore, the length and composition of the transmembrane spans of SCAMPs and Sft2p and Got1p are fairly similar. Although the segments connecting the transmembrane spans in both Sft2p and Got1p are predicted to be even shorter than in the SCAMPs, the cytoplasmic segment connecting spans 2 and 3 shares a similar positively charged and amphiphilic character with a segment that is highly conserved in the SCAMPs. The close succession of transmembrane spans within the primary structure implies that the spans, which by their length are probably helices, are closely situated to one another within the membrane interior. This organization would result in a rather compact structure for the membrane core (as envisioned in our model of SCAMP1; Figure 7). Interactions among transmembrane spans or the flanking segments may contribute to the oligomerization of SCAMPs (Wu and Castle, 1997), including SCAMP4 (our unpublished observations). The possible resemblance of SCAMPs to Sft2p and Got1p leads us to suggest that the function of SCAMPs may be more similar to that of these yeast proteins than to the functions of tetraspanins, physins, and connexins. Both Sft2p and Got1p interact genetically with syntaxin family proteins (Banfield et al., 1995; Conchon et al., 1999), and they are thought to promote fusion in endosome/Golgi and ER/Golgi transport, respectively (Conchon et al., 1999). Quite strikingly, a similar role has been proposed for SCAMP1 based on the results of a gene knockout in mice in which a potential defect in achieving a stable fusion pore during exocytosis was identified (Fernandez-Chacon et al., 1999). Thus, although both Got1p and Sft2p are similar to each other and to homologues of higher eukaryotic (including mammalian) polypeptides (Conchon et al., 1999), the SCAMPs, which may be more distantly related, may either substitute or collaborate in the same process.
Figure 7.

Model of the organization of SCAMP1 incorporating the information gained about its membrane topology, the nature of cytoplasmic interfacial segments, and the protease sensitivity of the extended cytoplasmic N- and C-terminal domains. The N terminus is folded such that the most accessible trypsin cleavage sites are exposed on the surface, with the NPF repeats being prospective ligands for EH domain–like interactions. Interactions with the N-terminal domain are envisioned to regulate the functions of the longer (Mr 38,000) SCAMPs. The leucine heptad repeat with alternating rows of charge has strong potential for forming coiled coils; it may also function in protein–protein interactions, potentially including intramolecular interactions with the interfacial segments in the folded protein. The SH3-like binding site is drawn as a proline helix, similar to the three-dimensional structure of other SH3-binding do-mains (Yu et al., 1994). The protease-resistant core of the protein includes the transmembrane spans and flanking segments, in accord with data presented in Figures 4–6. Because this core encompasses most of the structure of SCAMP4, we envision that it represents the functional domain throughout the SCAMP family. This view is consistent with the presence of highly conserved polypeptide segments and their spacing (Figure 1) within this region.
We have used the results of the present studies to construct a speculative model of the structure of SCAMPs (Figure 7). The N-terminal segment, which is readily and progressively proteolyzed by trypsin (Figure 4), constitutes the most accessible portion of the cytoplasmic surface of the protein. The C-terminal cytoplasmic segment, although exposed on the surface, contains tryptic sites that may be occluded by the more proteolytically accessible N terminus. The clustered transmembrane spans, their linkers, and the interfacial segments that flank the spans on the cytoplasmic surface together make up most of the protease-resistant core of SCAMP (Figure 4). We regard this core as SCAMP's functional domain based on the extensive conservation of sequence and spacing within this region (Figure 1) and especially on the existence of SCAMP4, an authentic SCAMP (Figure 2) that is mainly made up of the core (Figure 1). If SCAMP's core is indeed the functional domain of the protein, it seems quite reasonable to regard the N-terminal domain found on most SCAMPs as a regulatory domain. The conserved NPF repeats at its surface are prospective ligands for an EH domain–containing protein (Salcini et al., 1997; Paoluzi et al., 1998), and binding at these sites, e.g., by intersectin, γ-synergin, or other EH domain–containing proteins, may be required to activate the function of SCAMPs. In the inactive state, we envision that SCAMPs may be tightly folded such that the conserved and highly charged leucine heptad repeat interacts with the conserved amphiphilic helices located at the membrane interface. Thus, activation would entail binding to NPF repeats and unfolding the compact structure, thereby freeing the functional domain. Because SCAMP4 lacks most of the N-terminal domain, we speculate that it may be a constitutively active SCAMP. Alternatively, its activity (as well as that of the other SCAMPs) may be controlled by protein interactions at the conserved proline-rich SH3-like (and/or WW domain) binding site that precedes the SCAMP core. This model serves as a basis for several structural and functional predictions that need to be tested in future studies, particularly with respect to interactions with membrane fusion machinery and facilitation of interactions and reorganization of the lipid bilayers during the fusion event.
ACKNOWLEDGMENTS
We are grateful to Erin Code for generating and initially characterizing the GST-SCAMP chimera encoding the full N terminus of SCAMP1. We are also grateful to Dr. Susan Michaelis for providing E. coli strain UT5600, the phoA-encoding plasmid, and for helpful advice in conducting the topology analysis by means of the gene fusion approach. We thank Dr. Anna Castle for helpful advice and assistance with experiments, Dr. Yongde Bao (University of Virginia Biomolecular Research Facility) for exceptional efforts in DNA sequencing, and Amy Huang for help in preparing illustrations. These studies were supported by grant DE09655 from the National Institutes of Health (NIH), and D.S. gratefully acknowledges postdoctoral support initially from NIH training grant T32 DK07646 and subsequently from an a National Research Service award (F32 DE05680) from the NIH.
REFERENCES
- Archer SJ, Ellena JF, Cafiso DS. Dynamics and aggregation of the peptide ion channel alamethicin: measurements using spin-labeled peptides. Biophys J. 1991;60:389–398. doi: 10.1016/S0006-3495(91)82064-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banfield PD, Lewis MJ, Pelham HRB. A SNARE-like protein required for traffic through the Golgi complex. Nature. 1995;375:806–809. doi: 10.1038/375806a0. [DOI] [PubMed] [Google Scholar]
- Borjigin J, Nathans J. Insertional mutagenesis as a probe of rhodopsin's topography, stability, and activity. J Biol Chem. 1994;269:14715–14722. [PubMed] [Google Scholar]
- Boyd D, Traxler B, Beckwith J. Analysis of the topology of a membrane protein by using a minimum number of alkaline phosphatase fusions. J Bacteriol. 1993;175:553–556. doi: 10.1128/jb.175.2.553-556.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand SH, Castle JD. SCAMP 37, a new marker within the general cell surface recycling system. EMBO J. 1993;12:3753–3761. doi: 10.1002/j.1460-2075.1993.tb06053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand SH, Laurie SM, Mixon MB, Castle JD. Secretory carrier membrane proteins 31–35 define a common protein composition among secretory carrier membranes. J Biol Chem. 1991;266:18949–18957. [PubMed] [Google Scholar]
- Brickman E, Beckwith J. Analysis of the regulation of Escherichia coli alkaline phosphatase synthesis using deletions and phi80 transducing phages. J Mol Biol. 1975;96:307–316. doi: 10.1016/0022-2836(75)90350-2. [DOI] [PubMed] [Google Scholar]
- Brumell JH, Volchuk A, Sengelov H, Borregaard N, Cieutat AM, Bainton DF, Grinstein S, Klip A. Subcellular distribution of docking/fusion proteins in neutrophils, secretory cells with multiple exocytic compartments. J Immunol. 1995;155:5750–5759. [PubMed] [Google Scholar]
- Buser CA, Sigal CT, Resh MD, McLaughlin SA. Membrane binding of myristylated peptides corresponding to the NH2-terminus of src. Biochemistry. 1994;22:13092–13101. doi: 10.1021/bi00248a019. [DOI] [PubMed] [Google Scholar]
- Cafiso DS, Hubbell WL. EPR determination of membrane potentials. Annu Rev Biophys Bioeng. 1981;10:217–244. doi: 10.1146/annurev.bb.10.060181.001245. [DOI] [PubMed] [Google Scholar]
- Calamia J, Manoil C. Lac permease of Escherichia coli: topology and sequence elements promoting membrane insertion. Proc Natl Acad Sci USA. 1990;87:4937–4941. doi: 10.1073/pnas.87.13.4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castle JD, Hubbell WL. Estimation of membrane surface potential and charge density from the phase equilibrium of a paramagnetic amphiphile. Biochemistry. 1976;15:4818–4831. doi: 10.1021/bi00667a011. [DOI] [PubMed] [Google Scholar]
- Conchon S, Cao X, Barlowe C, Pelham HRB. Got1p and Sft2p: membrane proteins involved in traffic to the Golgi complex. EMBO J. 1999;18:3934–3946. doi: 10.1093/emboj/18.14.3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Chacon R, Achiriloaie M, Janz R, Albanesi JP, Sudhof TC. SCAMP1 function in endocytosis. J Biol Chem. 2000;274:12752–12756. doi: 10.1074/jbc.275.17.12752. [DOI] [PubMed] [Google Scholar]
- Fernandez-Chacon R, Toledo GA, Hammer RE, Sudhof TC. Analysis of SCAMP1 function in secretory vesicle exocytosis by means of gene targeting in mice. J Biol Chem. 1999;274:32551–32554. doi: 10.1074/jbc.274.46.32551. [DOI] [PubMed] [Google Scholar]
- Geller D, Taglicht D, Edgar R, Tam A, Pines O, Michaelis S, Bibi E. Comparative topology studies in Saccharomyces cerevisiae and in Escherichia coli. J Biol Chem. 1996;271:13746–13753. doi: 10.1074/jbc.271.23.13746. [DOI] [PubMed] [Google Scholar]
- Guo KL, Dixon JE. Eukaryotic proteins expressed in Escherichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione transferase. Anal Biochem. 1991;192:262–267. doi: 10.1016/0003-2697(91)90534-z. [DOI] [PubMed] [Google Scholar]
- Guo Z, Turner C, Castle D. Relocation of the t-SNARE SNAP-23 from lamellipodia-like cell surface projections regulates compound exocytosis in mast cells. Cell. 1998;94:537–548. doi: 10.1016/s0092-8674(00)81594-9. [DOI] [PubMed] [Google Scholar]
- Haass NK, Kartenbeck MA, Leube RE. Pantophysin is a ubiquitously expressed synaptophysin homologue and defines constitutive transport vesicles. J Cell Biol. 1996;134:731–746. doi: 10.1083/jcb.134.3.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain NK, Yamabhai M, Ramjaun AR, Guy AM, Baranes D, O'Bryan JP, Der CJ, Kay BK, McPherson PS. Splice variants of intersectin are components of the endocytic machinery in neurons and nonneuronal cells. J Biol Chem. 1999;274:15671–15677. doi: 10.1074/jbc.274.22.15671. [DOI] [PubMed] [Google Scholar]
- Johnston PA, Jahn R, Sudhof TC. Transmembrane topology and evolutionary conservation of synaptophysin. J Biol Chem. 1989;264:1268–1273. [PubMed] [Google Scholar]
- Johnston PA, Sudhof TC. The multisubunit structure of synaptophysin: the relationship between disulphide bonding and homo-oligomerization. J Biol Chem. 1990;265:7849–7852. [PubMed] [Google Scholar]
- Kanter HL, Saffitz JE, Beyer EC. Molecular cloning of two human cardiac gap junction proteins, connexin 40 and connexin 45. J Mol Cell Cardiol. 1994;26:861–868. doi: 10.1006/jmcc.1994.1103. [DOI] [PubMed] [Google Scholar]
- Kim JK, Blackshear PJ, Johnson DJ, McLaughlin SA. Phosphorylation reverses the membrane association of peptides that correspond to the calmodulin-binding domains of MARCKS and neuromodulin. Biophys J. 1994;67:227–237. doi: 10.1016/S0006-3495(94)80473-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaus P, Marqueze-Pouey B, Schaer H, Betz H. Synaptoporin, a novel putative channel protein of synaptic vesicles. Neuron. 1990;5:453–462. doi: 10.1016/0896-6273(90)90084-s. [DOI] [PubMed] [Google Scholar]
- Krajinski F, Martin-Laurent F, Gianinazzi S, Gianinazzi-Pearson V, Franken P. Cloning and analysis of psam 2, a gene from Pisum sativum L. regulated in symbiotic arbuscular mycorrhiza and pathogenic root-fungus interactions. Physiol Mol Plant Pathol. 1998;52:297–307. [Google Scholar]
- Kumar NM, Gilula NB. Cloning and characterization of human and rat liver cDNAs coding for a gap junction protein. J Cell Biol. 1986;103:767–776. doi: 10.1083/jcb.103.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurie SM, Cain CC, Lienhard GE, Castle JD. The glucose transporter GluT4 and secretory carrier membrane proteins (SCAMPs) colocalize in rat adipocytes and partially segregate during insulin stimulation. J Biol Chem. 1993;268:19110–19117. [PubMed] [Google Scholar]
- Leube RE. Expression of the synaptophysin gene family is not restricted to neuronal and neuroendocrine differentiation in rat and human. Differentiation. 1994;56:163–171. doi: 10.1046/j.1432-0436.1994.5630163.x. [DOI] [PubMed] [Google Scholar]
- Lewis JR, Cafiso DS. Membrane spontaneous curvature modulates the binding energy of a channel forming voltage-gated peptide. Biochemistry. 1999;38:5932–5938. doi: 10.1021/bi9828167. [DOI] [PubMed] [Google Scholar]
- Luo P, Baldwin RL. Mechanism of helix induction by trifluoroethanol: a framework for extrapolating the helix-forming properties of peptides from trifluoroethanol/water mixtures back to water. Biochemistry. 1997;36:8413–8421. doi: 10.1021/bi9707133. [DOI] [PubMed] [Google Scholar]
- Maeker HT, Todd SC, Levy S. The tetraspanin superfamily: molecular facilitators. FASEB J. 1997;11:428–442. [PubMed] [Google Scholar]
- Manoil C. Analysis of membrane protein topology using alkaline phosphatase and β-galactosidase gene fusions. Methods Cell Biol. 1991;34:61–75. doi: 10.1016/s0091-679x(08)61676-3. [DOI] [PubMed] [Google Scholar]
- Page LJ, Sowerby PJ, Lui WW, Robinson MS. γ-Synergin: an EH domain-containing protein that interacts with γ-adaptin. J Cell Biol. 1999;146:993–1004. doi: 10.1083/jcb.146.5.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoluzi S, Castagnoli L, Lauro I, Salcini AE, Coda L, Fre S, Calfonieri S, Pelicci PG, DiFiore P-P, Cesareni G. Recognition specificity of individual EH domains of mammals and yeast. EMBO J. 1998;17:6541–6550. doi: 10.1093/emboj/17.22.6541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips SE, et al. Yeast Sec14p deficient in phosphatidylinositol transfer activity is functional in vivo. Mol Cell. 1999;4:187–197. doi: 10.1016/s1097-2765(00)80366-4. [DOI] [PubMed] [Google Scholar]
- Salcini AE, Confalonieri S, Doria M, Santolini E, Tassi E, Minenkova O, Cesareni G, Pelicci PG, DiFiore PP. Binding specificity and in vivo targets of the EH domain, a novel protein-protein interaction module. Genes Dev. 1997;11:2239–2249. doi: 10.1101/gad.11.17.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton DR, Wu TT, Castle JD. Three mammalian SCAMPs (secretory carrier membrane proteins) are highly related products of distinct genes having similar subcellular distributions. J Cell Sci. 1997;110:2099–2107. doi: 10.1242/jcs.110.17.2099. [DOI] [PubMed] [Google Scholar]
- Stenius K, Janz R, Sudhof TC, Jahn R. Structure of synaptogyrin (p29) defines novel synaptic vesicle protein. J Cell Biol. 1995;131:1801–1809. doi: 10.1083/jcb.131.6.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudol M. Structure and function of the WW domain. Prog Biophys Mol Biol. 1996;65:113–132. doi: 10.1016/s0079-6107(96)00008-9. [DOI] [PubMed] [Google Scholar]
- Sugita S, Janz R, Sudhof TC. Synaptogyrins regulate Ca-dependent exocytosis in PC12 cells. J Biol Chem. 1999;274:18893–18901. doi: 10.1074/jbc.274.27.18893. [DOI] [PubMed] [Google Scholar]
- Tan PK, Howard JP, Payne GS. The sequence NPFXD defines a new class of endocytosis signal in Saccharomyces cerevisiae. J Cell Biol. 1996;135:1789–1800. doi: 10.1083/jcb.135.6.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traxler B, Boyd D, Beckwith J. The topological analysis of integral cytoplasmic membrane proteins. J Membr Biol. 1993;132:1–11. doi: 10.1007/BF00233047. [DOI] [PubMed] [Google Scholar]
- Victor K, Jacob J, Cafiso DS. Interactions controlling the membrane binding of basic protein domains. Biochemistry. 1999;38:12527–12536. doi: 10.1021/bi990847b. [DOI] [PubMed] [Google Scholar]
- Wen G, Leeb T, Hui D, Baumgartner BG, Robic A, Hameister H, Brenig P. Mammal. Genome. 1998;9:536–539. doi: 10.1007/s003359900814. [DOI] [PubMed] [Google Scholar]
- Wu TT, Castle JD. Evidence for colocalization and interaction between 37 and 39 kDa isoforms of secretory carrier membrane proteins (SCAMPs) J Cell Sci. 1997;110:1533–1541. doi: 10.1242/jcs.110.13.1533. [DOI] [PubMed] [Google Scholar]
- Wu TT, Castle JD. Tyrosine phosphorylation of selected secretory carrier membrane proteins, SCAMP1 and SCAMP3, and association with the EGF receptor. Mol Biol Cell. 1998;9:1661–1674. doi: 10.1091/mbc.9.7.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Chen JK, Feng S, Dalgarno DC, Brauer AW, Schreiber SL. Structural basis for the binding of proline-rich peptides to SH3 domains. Cell. 1994;75:933–945. doi: 10.1016/0092-8674(94)90367-0. [DOI] [PubMed] [Google Scholar]
- Zastrow M, Castle JD. Protein sorting among two distinct export pathways occurs from the content of maturing exocrine storage granules. J Cell Biol. 1987;105:2675–2684. doi: 10.1083/jcb.105.6.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]