

Abstract
Dmrt1 is a recently described gene that is specifically expressed in the gonads and is required for postnatal testis differentiation. Here, we describe the transcriptional mechanisms regulating the Dmrt1 proximal promoter in testicular Sertoli cells. A genomic clone containing exon 1 of the rat Dmrt1 gene and more than 9 kilobases of 5′ flanking sequence was isolated and characterized. Several prominent transcriptional start sites were identified, with the major site located 102 bases from the translational start. The Dmrt1 5′ flanking region from −5000 to + 74 was transcriptionally active in primary Sertoli cells, and deletion analysis of this fragment identified 2 major regions needed for full Dmrt1 promoter function. These regions were located between −3200 and −2000 base pairs (bp) and downstream of −150 bp relative to the major transcriptional start site. DNase I footprint analysis of the region downstream of −150 bp revealed 3 regions that are bound by proteins from Sertoli cell nuclear extracts. Site-directed mutagenesis of these regions identified 2 elements that activate the Dmrt1 promoter and 2 that repress it. The positive elements bind the transcription factors Sp1, Sp3, and Egr1, suggesting that these transcription factors play a critical role in Dmrt1 regulation in the testis.
Keywords: developmental biology, gene regulation, Sertoli cells, spermatogenesis, testis
INTRODUCTION
In mammals, sex determination occurs in the embryo when the indifferent gonad is instructed to differentiate along either the male or female pathway. This differentiation choice is ultimately determined by the nature of the sex chromosomes, whereby the presence of a Y chromosome results in testis formation and its absence results in ovary formation. Subsequently, the endocrine environment provided by the testis or ovary directs further sexual differentiation, resulting in the characteristic male or female phenotypes. The genetic components of sex determination and differentiation have, in part, been described and include the genes for Sry, SF-1, Dax1, WT1, Lim1, Emx2, Lhx9, and SOX9 [1–8]. More recently, a gene encoding a DM domain (a novel DNA-binding motif) protein, Dmrt1, has emerged as an important player in the male sexual differentiation pathway [9–13]. This gene was first recognized for its likeness to mab-3 and doublesex, known regulators of male differentiation in nematodes and flies, respectively [12]. The similarity between these proteins resides within their DNA binding or DM domains, which are now thought to represent a common structural feature found in proteins important to male sexual differentiation in organisms of different phylogenetic groups [12]. Prior to the discovery of the DM domain proteins, genetic or molecular similarities that traverse different phyla had not been described for the sexual differentiation pathways.
A role for Dmrt1 in male sexual differentiation has been implicated not only by its evolutionary conservation but also by its chromosome location, which maps to a region on human chromosome 9 associated with XY sex reversal, and by its mRNA expression profile, which is male specific and found only in the testis [9, 11–14]. In mice, Dmrt1 mRNA is present only in the genital ridge of early XX and XY embryos and becomes testis specific as development proceeds [9]. Within the developing and the postnatal testis, Dmrt1 expression is restricted to cells of the seminiferous epithelium, the site where male germ cells develop into spermatozoa [9, 10, 15]. Studies on Dmrt1 in the testis of postnatal rats and mice have shown that its mRNA and protein is expressed in Sertoli cells and germ cells of the seminiferous epithelium and that the levels of both change as a function of the age of the animal [10, 16].
A role for Dmrt1 in testis differentiation was recently secured by studies demonstrating severe testicular defects upon ablation of the gene from the mouse genome [10]. As in humans missing portions of chromosome 9, mice lacking Dmrt1 had a failure in testis differentiation accompanied by germ cell death. In addition, the defects observed from the Dmrt1 deletion were male specific and only apparent in postnatal animals. Further analysis of the Dmrt1−/− animals revealed that the germ cells failed to enter meiosis and were eventually lost from the epithelium and that the Sertoli cells overproliferated, failed to adopt a differentiated phenotype, and eventually died [10]. In the testis, Sertoli cells and germ cells are found in very close association and greatly influence the function and development of each other. Thus, defects in the Sertoli cells can have a profound impact on germ cell differentiation and function and vice versa. The functional implications of the gene knockout studies are, therefore, confounded by these interactions, making it difficult to attribute the phenotype to any one cell type. However, in Dmrt1−/− testes, Sertoli cell morphology and gene expression patterns were significantly abnormal and the phenotype differed from that of other mutants that only lack germ cells, indicating a direct impact on Sertoli cell differentiation [10].
The Sertoli cells play a critical role in testis development and function after birth [17–20]. In the embryo, Sertoli cells, under the direction of Sry, are the first somatic cells to differentiate in the gonad and are thought to orchestrate subsequent events in testis formation and sex determination [21–26]. After birth, Sertoli cells support the differentiation of germ cells into viable sperm. During early postnatal and pubertal development, Sertoli cells exhibit remarkable structural and functional changes that are required for the initiation and maintenance of spermatogenesis [27]. Many of these changes are regulated by hormones and growth factors, 1 of which is the pituitary glycoprotein hormone FSH. Consistent with its role in postnatal testis function and differentiation, Dmrt1 expression has recently been shown to be regulated by FSH [16].
Given the recognized importance of Dmrt1 in the testis and its highly restricted pattern of expression, identification of the molecular events required for its transcription will significantly extend our understanding of the factors necessary for spermatogenesis and fertility and cell-specific gene expression in the testis. Here, we present studies on the functional characterization of the Dmrt1 promoter in primary cultures of Sertoli cells. Deletion and site-directed mutagenesis in conjunction with DNase I footprint analysis were used to delineate regions of the promoter that are required for full transcriptional activity. The studies revealed 2 main regions necessary for Dmrt1 expression, and we identified the transcription factors Sp1, Sp3, and Egr1 as important contributors to Dmrt1 promoter function.
MATERIALS AND METHODS
Cell Preparation and Transfection Analysis
Preparation and transient transfection of Day 15 primary rat Sertoli cells were as previously described [28–30]. Sertoli cells were seeded onto 12-well plates in Ham/F12 medium and 1.5 mM Hepes (pH 7.1) in the presence of 5% fetal bovine serum (FBS). After 40 h in culture, the cells were treated with a hypotonic solution of 10 mM Tris (pH 7.4) for 2 min to remove germ cells, and Ham/F12 medium supplemented with 1.5 mM Hepes and 1% penicillin/streptomycin was added. Approximately 12–16 h later, the medium was changed to Dulbecco modified Eagle medium (DMEM) containing 110 mg/L sodium pyruvate. Two to 4 h later, the cells were transfected with 0.2 μg of firefly luciferase reporter and 40 ng of pRL-TK using 1 μl lipofectamine reagent and 2 μl PLUS reagent according to the manufacturer’s recommendation (Promega, Madison, WI). pRL-TK expresses Renilla luciferase from the herpes simplex virus thymidine kinase promoter and was included to control for transfection efficiency (Promega). Three hours later, an additional 300 μl of DMEM was added into the transfected medium. Thus, serum and FSH were absent from the medium during the course of the transfection. Cells were harvested approximately 48 h later and assayed for firefly and Renilla luciferase activities using the dualluciferase reporter assay system as described previously [30]. The purity of the primary cultures was assessed by morphological characterization to identify Sertoli cells and examination of alkaline phosphatase activity to detect myoid cell contamination. Typically, cultures are greater than 85–95% Sertoli cells.
For transient transfection, TM4 cells were plated onto 24-well dishes at a density of 26 000 cells/well. Approximately 24 h later, cells were transfected with 200 ng of firefly luciferase reporter construct and 10 ng of pRL-TK using 1 μl of lipofectamine. After 16–24 h, the transfection medium was removed, and TM4 medium (50% DMEM, 50% Ham/F12 medium, 1.2 g/L sodium bicarbonate, 5% horse serum, 2% FBS, and 15 mM Hepes, pH 7.1) was added. Cells were harvested and assayed as above. All plasmid DNAs were prepared from overnight bacterial cultures using Qiagen DNA plasmid columns according to the supplier’s protocol (Qiagen, Valencia, CA). All investigations involving animals were conducted in accordance with the Guide for the Care and Use of Laboratory Animals.
Promoter Clones
A rat genomic library (Clontech Laboratories, Palo Alto, CA) was screened using a radiolabeled probe corresponding to the 5′ end of the rat Dmrt1 cDNA [16]. Library screening was done according to the manufacturer’s recommendations. Lambda clone 56.1611 was isolated and found to contain approximately 16 kilobases (kb) of 5′ flanking sequence, exon 1, and 1 kb of the first intron as determined by DNA sequence analysis and restriction endonuclease mapping. Dmrt1(−5000/+74)Luc was generated by subcloning a region of 56.1611 spanning from an NheI site approximately 5000 base pairs (bp) upstream of the transcriptional start site to a BamHI site 74 bp 3′ to the transcriptional start site into the respective sites in mpGL3-Basic. mpGL3-Basic is pGL3-basic (Promega) modified to remove the BamHI and SalI sites. Dmrt1(−5000/+74)Luc was used to create a series of deletion mutants using restriction endonuclease digestion followed by a Klenow fill-in reaction and ligation. A restriction endonuclease site within the upstream polylinker was used together with the following specific restriction endonuclease sites within the Dmrt1 promoter: Dmrt1(−3200/+74)Luc (SpeI site), Dmrt1(−2000/+74)Luc (HindIII site), Dmrt1(−1200/+74)Luc (BamHI site), Dmrt1(−400/+74)Luc (SalI site), and Dmrt1(−300/+74)Luc (SstI site). Smaller deletion mutants were generated by amplification of the promoter sequence using unique upstream oligodeoxynucleotide primers, a downstream primer that binds within the luciferase gene (Luc1 5′-TTTGGCGTCTTCCATTTTACCAACAGTACC-3′), and Dmrt1(−5000/+74)Luc as template. DNA amplification was performed using Bio-X-Act according to the manufacturer’s recommendations (Bioline USA, Springfield, NJ). The upstream primers contained an SstI site near the 5′ end. After amplification, the DNA fragments were digested with SstI and cloned into the SstI site of mpGL3 containing the Dmrt1 region from the SstI site at −8 bp relative to the BamHI site at +74 bp. The upstream primers and the clones they generated are as follows: DM1 (5′-GCGCGAGCTCCAGCAACGCTGTTGGCG-3′), Dmrt1(−221/+74)Luc; DM2 (5′-GCGCGAGCTCCCTGCCATCAGAAGAAGCG-3′), Dmrt1(−189/+74)Luc; DM3 (5′-GCGCGAGCTCACACCAGTGGGCGTGGC-3′), Dmrt1(−150/+74)Luc; DM4 (5′-GCGCGAGCTCGGAAGGTTCTGAAAAGTATC-3′), Dmrt1(−98/+74) Luc; and DM5(5′-GCGCGAGCTCAAAAGTCCTGCGCTAGGC-3′), Dmrt1-(−74/+74)Luc.
Block replacements m1–m3 were generated by amplification of the promoter region from −150 to +74 bp using Dmrt1-specific oligodeoxynucleotide primers containing the intended mutations, Dmrt1(−300/+74)Luc as template, and Luc1 as the downstream primer. DNA amplification was performed using Bio-X-Act as described above. The upstream primers for each of the block mutants were BM1 (5′-GC GCGCTAGCACACACTGTCTATGTTAGCATACGCGTAGCGCCCTCTC-3′), BM2 (5′-GCGCGCTAGCACACCAGTGGGCGTGGCCACGCATACGCTACAAAGATCCCGCCCACGCAATC-3′), and BM3 (5′-GCGCGCTAGCACACCAGTGGGCGTGGCCACGCCGCGTAGCGCCCTCGAAGTAAGCATACCGATTCGGAAGGTTCTGAAAAG-3′). The upstream primers contained an NheI site near the 5′ end that was used as the 5′ cloning site. The amplified fragments were digested by NheI and HindIII and subcloned into mpGL3 digested with NheI and HindIII.
Block replacement mutants m4–m6 and m456 were generated by amplification of the region between −150 and −45 bp with an upstream primer Rvprimer3 (5′-CTAGCAAAATAGGCTGTCCC-3′; Promega), downstream primers containing the intended mutation, and Dmrt1(−150/+74)Luc as template. The downstream primer for each mutant is as follows: BM4 (5′-CCTTTCGGCGCGCCTAGCGCAGGACTTTTGGATACACACTCTCTCCTTCCGAGGATTG-3′), BM5 (5′-CCTTTCGGCGCGCCTAGCGCAGGACTTGCTTGCCTTTTTCAGAACCTTC-3′), BM6 (5′-CCTTTCGGCGCGCCTAGCGTCTTGTCCTTGGATACTTTTC-3′), and BM456 (5′-CTTTCGGCGCGCCTAGCGCAGTGAGCCGTTCGCACGGCACTAACCTTCCGAGGATTG-3′). The amplified products were digested by SstI and AscI and subcloned into Dmrt1(−150/+74)Luc digested with SstI and AscI.
Block replacement mutants m7–m9 and m7′ were generated by amplification of the region between −65 and +74 bp with a downstream primer Luc1 and the upstream primers listed below that contained the intended mutation. Amplification reactions were done using Dmrt1(−150/+74)Luc as template. The upstream primer for each mutant is as follows: BM7 (5′-GCGCTAGGCGCGCCGAAAGGCAGGCCGGGCCCGGTACAACCCCCCACC-3′), BM8 (5′-GCGCTAGGCGCGCCGAAAGGCAGAAAAAAAAATAGCACGACCCCCACCCACTTCTC-3′), BM9 (5′-GCGCTAGGCGCGCCGAAAGGCAGAAAAAAAAAGGTACAACTTATGGTTCACTTCTCGAGCTC-3′), and BM7′ (5′-GCGCTAGGCGCGCCGAAAGGCAGGGTTCGGTCGGTACAACCCCCCACC-3′). The amplified products were digested by AscI and HindIII and subcloned into Dmrt1(−150/+74)Luc digested with AscI and HindIII.
The mutation m10 was generated by amplification of the region between −17 and +74 bp with downstream primer Luc1 and an upstream primer containing the intended mutation (BM10, 5′-CACTTCTCGAGCTCTGGCCGGGGACTACAGGTCC-3′), with Dmrt1(−150/+74)Luc as template. The amplified product was digested by XhoI and HindIII and subcloned into Dmrt1(−150/+74)Luc digested with XhoI and HindIII.
The smaller site-directed mutants RM, sm1–sm4, and sm7–sm10 were generated by amplification of the region between −141 and +74 bp (RM, sm2, sm3, sm7–sm10) or between −150 and +74 bp (sm1 and sm4), using downstream primer Luc1 and upstream primers containing the intended mutation. The template was Dmrt1(−150/+74)Luc. The upstream primers were as follows: RM (5′-GGGCGTGGCCACGCCGCGTAGCGCCCTCTCCCTCCCAGGCAGTCCCGGGAATGTTCTGAAAAGTATC-3′), Sm1 (5′-GCGCGAGCTCACACCAGTGGAGATGGCCACGCCGCG-3′), Sm2 (5′-GGCGTGGCCACGCCGCGTAGACTCCTCTCCCGCCCAC-3′), Sm3 (5′-GGCGTGGCCACGCCGCGTAGCGCCCTCTCCCGAAAGAGCAATCCTCGGAAG-3′), Sm4 (5′-GCGCGAGCTCACACCAGTGGGCGTGGCCAACACGCGTAGCGCCC-3′), Sm7 (5′-GGCGTGGCCACGCCGCTCCTCGCCCTCTCCCGCCCACG-3′), Sm8 (5′-GGC G T G G C C A C G C C G C G T A G C G C A T G A T C C C G C C C A CGCAATCCTC-3′), Sm9 (5′-GGCGTGGCCACGCCGCGTAGCGCCCTCGAAATCCCACGCAATCCTC3′), and Sm10 (5′-GGCGTGGCCACGCCGCGTAGCGCCCTCTCCCGCCCAC TACCGAATCGGAAGGTTCTG-3′). The amplified products were digested by Msc1 and HindIII (RM, sm2, sm3, sm7–sm10) or SstI alone (sm1 and sm4) and subcloned into Dmrt1(−150/+74)Luc digested with Msc1 and HindIII (RM, sm2, sm3, sm7–sm10) or SstI (sm1 and sm4). All clones were confirmed by sequencing using the ABI Prism dRhodamine Terminatior Cycle Sequence Ready Reaction Kit (PE Applied Biosystems, Foster City, CA).
Transcriptional Start Site Mapping
Transcriptional start site mapping was performed using total RNA isolated from primary cultures of rat Sertoli cells using TRIZOL reagent (Life Technologies, Rockville, MD). For RNase protection, a region of the rat Dmrt1 gene corresponding to 325 bp 5′ to the putative translational start codon and 55 bp 3′ of this site was amplified using the Dmrt1 genomic clone 56.1611 as template, upstream primer Dmrt1.18 (5′-AGCAACGCTGTTGGCGTG-3′), and downstream primer Dmrt1.16B (5′-GGGC T C TA ATA C G A C T C A C TATA G G G A G G C G T G A G G A A C C TCCGTCGG-3′). DNA amplification was performed as above. The downstream primer contained a T7 polymerase recognition sequence that was used to generate an antisense RNA probe from the amplified DNA product using T7 polymerase according to the manufacturer’s recommendations (Promega). The conditions for the RNase protection assay were as described previously [16]. Protected fragments were resolved by denaturing PAGE together with a DNA sequencing ladder generated with Sequenase 2.0 (USB, Cleveland, OH), a cloned region of the Dmrt1 gene as template, and a primer identical to the downstream amplification primer but lacking the T7 sequences.
5′ Rapid amplification of cDNA ends (RACE) was performed as described previously, using a set of nested 5′ RACE primers generated against the rat Dmrt1 cDNA [16, 30–32]. Complementary DNA was made from total Sertoli cell RNA using the most 3′ primer Dmrt1.7 (5′-CCTGAGGAGGACTCAGCAG-3′) and was used as the initial template for DNA amplification. DNA products from 2 rounds of amplification with Dmrt1-specific primers Dmrt1.8 (5′-GACAAGGCCATGGAGTACTG-3′) and Dmrt1.9 (5′-AGTAAGGAAACAGAGACGGCTG-3′) were sub-cloned into pGem4Z (Promega), and individual clones were sequenced. Transcriptional start sites were assigned based on DNA sequence analysis of the RACE clones, and the size of protected RNA fragments was adjusted for differences in migration between RNA and DNA.
DNase I Footprint Analysis
Nuclear extracts were prepared as described previously [30]. End-labeled DNA probes were generated using a DNA amplification reaction in which one of the oligodeoxynucleotide primers was radiolabeled with 32P using T4 kinase. After amplification, the DNA probes were resolved by agarose gel electrophoresis and isolated from the gel using the Qiaex II gel extraction kit (Qiagen). For the sense probe, the upstream primer dm1 (5′-CAGCAACGCTGTTGGCG-3′) was radiolabeled and used with downstream primer Dmrt1.19 (5′-CTCACGGATCCAGCCAGCAGGC-3′). These primers amplified the region between −221 and +74 bp of the Dmrt1 promoter. For the antisense probe, the downstream primer Dmrt1.19 was radiolabeled and used with upstream primer Dmrt1.25 (5′-CGCTTGAGCCCGTGACC-3′). These primers amplified the region between −526 and +74 bp. The DNase I footprint procedure was described previously [33]. Nuclear extracts (32 μg) from cultured Sertoli cells were incubated in 79 μl of incubation buffer (10 mM Hepes, pH 7.8, 40 mM KCl, 4 mM MgCl2, 5% glycerol, 0.25 mM dithiothreitol, and 1 μg poly dI-dC) for 15 min at 37°C. One microliter of radiolabeled double-stranded probe (20 000 cpm/μl) was then added to the reaction and incubated for an additional 15 min at room temperature. Next, DNase I (Worthington Biochemical Corp., Lakewood, NJ) was added and incubated for 1 min at room temperature (0.01 units DNase I for reactions without nuclear extracts; 0.15 units for reactions with nuclear extracts). The DNase I reaction was stopped immediately following the incubation period by the addition of 200 μl of Tris-EDTA–saturated phenol, 120 μl H2O, and 10 μg tRNA (10 mg/ml). Following phenol-chloroform extraction and precipitation, the DNA samples were resolved by denaturing PAGE and visualized by autoradiography.
Electrophoretic Mobility Shift Assays
Electrophoretic mobility shift assays (EMSAs) were performed as described previously [29, 30]. Nuclear extracts (10 μg protein) were incubated with 25 fmol of radiolabeled double-stranded oligodeoxynucleotide in the presence of 10 mM Hepes (pH 7.9), 3 mM MgCl2, 30 mM KCl, 1.0 mM dithiothreitol, 12% glycerol, 1.0 mM EDTA, 0.2 mM PMSF, 100 ng salmon sperm DNA, 1 μg dIdC, 10 μM ZnCl2, and 1 μg/ml BSA in a 20-μl reaction volume. Competitors and antibodies, when used, were added to the reaction immediately before the addition of the nuclear extract. Reactions were incubated on ice for 10 min prior to addition of probe, for an additional 20 min at room temperature, and then for 15 min on ice after the probe was added. Competitors were added at a concentration 100 times that of the probe unless otherwise indicated. Protein/DNA complexes were resolved on a 4% polyacrylamide gel (acrylamide:bisacrylamide 40:1) run in 25 mM Tris (pH 8.5), 190 mM glycine, 0.5 mM EDTA at 240 volts for 1.5 h at 4°C. Gels were dried and then analyzed by autoradiography. Antibodies for Sp1, Sp2, Sp3, Sp4, Egr1, Egr2, Egr3, and WT1 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies were added to the binding reactions at a concentration of 1 μg per binding reaction.
RESULTS
Cloning and Characterization of the Rat Dmrt1 Promoter
A genomic clone containing exon 1 of the rat Dmrt1 gene and more than 9 kb of 5′ flanking sequence was isolated and characterized by restriction endonuclease site mapping (Fig. 1A). DNA sequence analysis was performed, and the sequence of the rat proximal promoter region was compared with that of the human DMRT1 gene (GenBank accession number AL136365), revealing 3 regions of significant sequence identity in the Dmrt1 promoter regions of these 2 species (regions 1–3, Fig. 1B). Also apparent in the sequence of the Dmrt1 promoter was the lack of a TATA box consensus sequence and a high number of guanine and cytosine residues, which are often found associated with promoters lacking TATA elements.
FIG. 1.
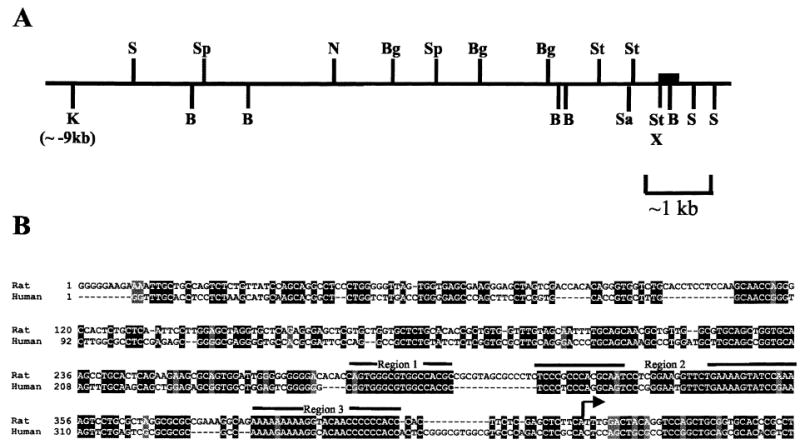
Dmrt1 5′ flanking region. A) Approximately 9 kb of 5′ flanking sequence, exon 1, and a portion of the first intron residing in rat Dmrt1 lambda clone 56.1611 were mapped using a variety of restriction endonucleases: NheI (N), KpnI (K), SmaI (S), BamHI (B), BglII (Bg), SstI (St), and XhoI (X). The filled box depicts the position of exon 1. B) The region between −426 and +35 of the rat Dmrt1 promoter was aligned with the corresponding region of the human Dmrt1 gene using the ClustalW multiple sequence alignment program followed by Boxshade. Shaded regions represent areas of identity (solid) or conserved changes (shaded). The arrow depicts the major transcriptional start site identified in rat.
Transcriptional start site mapping was performed on RNA isolated from primary cultures of rat Sertoli cells using 2 independent techniques: RNase protection analysis (RPA) and 5′ RACE. RPA, using a probe that extended from 325 bp 5′ to the putative translational start codon to 55 bp 3′ of this site, identified multiple transcriptional start sites, with the major ones residing within the first 200 bp upstream of the translational start codon (arrows a–d, Fig. 2, A and B). Similarly, 5′ RACE analysis revealed several 5′ cDNA ends, with the most frequent terminations occurring in a region of significant sequence similarity to an initiator element (Fig. 2B, numbers with downward arrows refer to the number of terminations observed at the indicated base). Under the conditions used for denaturing PAGE in the RPA, the protected RNA bands (Fig. 2A) were estimated to run approximately 10% slower than the corresponding DNA size fragments in the accompanying sequencing ladder [34]. Taking into consideration this migration difference, the major transcriptional start site revealed by RPA (arrow b, ~170 bp, Fig. 2A) had an actual protected size of about 158 bases. Thus, both methods identified an adenine residue located 102 bases upstream of the initial methionine codon as the major transcriptional start site. Although multiple transcriptional start sites were apparent, this major site was adopted as the +1 position (Fig. 2B).
FIG. 2.
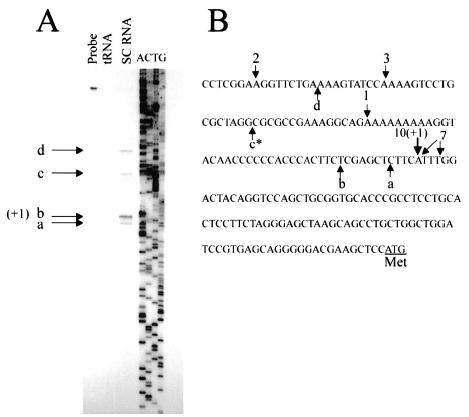
Identification of Dmrt1 transcriptional start sites. A) RNase protection analysis of Dmrt1 mRNA in Sertoli cells. An in vitro transcribed RNA probe was generated from the Dmrt1 genomic sequence to span the putative transcriptional start site (325 bp 5′ to the putative translational start codon and 55 bp 3′ of this site). RNA from cultured Sertoli cells (SC RNA) or tRNA was used in the protection analysis. The protected fragments were resolved by denaturing PAGE with a sequencing ladder corresponding to the region used to generate the RNA probe. The ladder was generated by the Sanger dideoxysequencing method, and A, C, G, T refers to the dideoxynucleotide in the termination reaction. Probe refers to undigested probe. The major protected sites are indicated as a–d. The +1 indicates the major transcriptional start site. B) Positions of identified transcriptional start sites on the corresponding sequence of the Dmrt1 gene. Upward arrows with the letters a–d represent the sites shown in A. Downward arrows show the results of 5′ RACE analysis. The accompanying number for the RACE arrows gives the number of clones that terminated at the indicated position. The major transcriptional start site identified by both RPA (taking into consideration differences in DNA and RNA migration) and 5′ RACE is marked as +1. The asterisk indicates that identification of the exact base for band c in A was obscured because of sequence compression. The double arrow associated with the number 7 reflects the inability to definitively name the termination base in the sequenced RACE products because of the polyadenosine tract added during RACE analysis.
Two Regions Within the 5′ Flanking Sequence of Dmrt1 Are Needed for Full Promoter Activity
To evaluate the transcriptional mechanisms regulating Dmrt1, a portion of the gene from +74 bp to the NheI site within the 5′ flanking region (N in Fig. 1A) was cloned upstream of the firefly luciferase reporter gene to generate Dmrt1(−5000/+74)Luc. Basal activity of this promoter construct was evaluated in primary cultures of rat Sertoli cells; previous studies have confirmed Dmrt1 expression in these cells [10, 16]. The −5000/+74 Dmrt1 promoter was functional in primary Sertoli cells, where its activity was approximately 20-fold higher than that of the promoterless control, pGL3-Basic (Fig. 3). The activity of a vector containing the SV40 enhancer/promoter sequences driving luciferase (pGL3-control) was approximately 93-fold greater than that of pGL3-Basic (Fig. 3, Control).
FIG. 3.
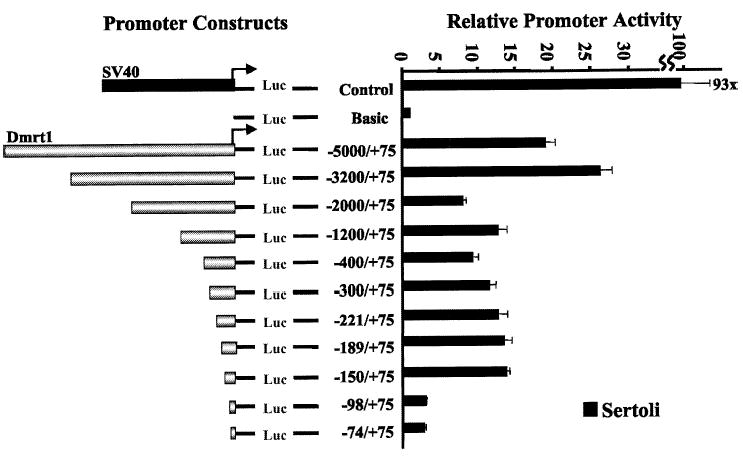
Transient transfection analysis of Dmrt1 promoter deletion mutants in primary rat Sertoli cell cultures and in mouse TM4 cells. The rat Dmrt1 gene from −5000 to +74 was cloned upstream of the firefly luciferase reporter gene in pGL3-Basic, and various 5′ deletion mutants were generated. The promoter constructs were transfected into primary Sertoli cell cultures together with pRL-TK, which expresses Renilla luciferase from the thymidine kinase promoter and was used as control for transfection efficiency. The data represent the firefly/Renilla luciferase activity of each promoter construct made relative to the firefly/Renilla luciferase activity of the promoterless control vector pGL3-Basic (Basic). Transfections were done a minimum of 3 times. Error bars represent the SEM. Control represents the pGL3-Control vector that contains the SV40 promoter and enhancer sequences.
To help identify regions of the Dmrt1 promoter that contain important cis-acting elements, sequentially larger deletions from the 5′ end of Dmrt1(−5000/+74)Luc were generated and tested for promoter activity by transient transfection analysis (Fig. 3). Deletions from −3200 to −2000 and from −150 to −98 substantially (~70–80%) decreased transcriptional activity of the Dmrt1 promoter. In contrast, only modest changes in promoter activity were observed with the intervening deletions between −2000 and −150, and an increase in promoter activity was seen with a deletion from −5000 to −3200 bp. These results indicate that important positive regulatory elements reside between −3200 and −2000 bp and downstream of −150 bp, whereas negative regulatory elements lie upstream of − 3200 bp.
The region downstream of −150 bp was further characterized using DNase I footprint analysis. The analysis was performed using nuclear extracts isolated from primary cultures of Sertoli cells and end-labeled probes that spanned the Dmrt1 promoter from −221 to +74 (sense probe) and −526 to +74 (antisense probe). With both probes, 2 regions were protected from DNase I digestion in the presence of Sertoli cell nuclear extracts (regions I and II, Fig. 4, A and B). Region I resides just 3′ to position −150, and region II resides to the 3′ side of position −98. A more modestly protected region (III) was observed between regions I and II, but only when the antisense probe was employed. Regions revealed by footprint analysis corresponded to regions identified as having high sequence conservation between the human and rat promoters (Figs. 1B and 4B). Furthermore, results of the functional analysis of the deletion mutants (Fig. 3) supported those of the DNase I footprint analysis; both indicated the presence of important response elements between −150 and −98. Thus, these complementary approaches support a role for regions I and III (Fig. 4B) in Dmrt1 transcriptional regulation. Although the transient transfection data did not directly support a role for region II, the ability to reveal this site might have been masked by the already compromised nature of the promoter.
FIG. 4.
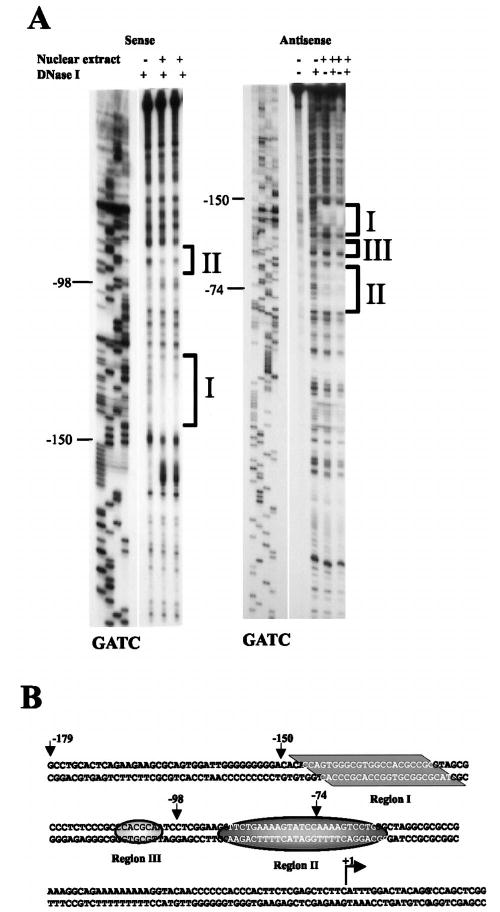
In vitro DNase I footprint analysis of the Dmrt1 proximal promoter region. A) End-labeled DNA probes spanning the region from either −221 to +74 (labeled on sense strand, sense probe) or −526 to +74 (labeled on antisense strand, antisense probe) were generated. The probes were incubated either in the presence (+) or absence (−) of primary rat Sertoli cell nuclear extracts and then digested with DNase I. A dideoxynucleotide sequencing ladder was generated using a Dmrt1 promoter clone as template and the same primer that was end labeled to generate each of the probes. Regions protected from DNase I digestion in the presence of Sertoli cell nuclear proteins are labeled I, II, and III. B) Sequence of the Dmrt1 promoter in the region protected from DNase I digestion in the presence of Sertoli cell proteins. The bases protected from digestions are shaded and indicated as regions I–III. The major transcriptional start site is marked with an arrow.
Both Positive and Negative Elements Regulate the Dmrt1 Proximal Promoter
The DNase I footprint and sequence alignment data were used as a guide to generate specific mutations to help identify the nature of the response elements residing downstream of −150 and of the proteins that bind them. Eleven different block-replacement mutations (m1–m10, m456) and a mutation that converts the bases within footprinted region III (Fig. 4) to those found within the human promoter (RM) were generated and placed into the context of the Dmrt1(−150/+74)Luc construct and evaluated for their effect on promoter activity by transient transfection analysis in primary rat Sertoli cells and the mouse Sertoli cell line TM4 (Fig. 5). In both Sertoli and TM4 cells, mutations m1 and m3 significantly diminished promoter activity, but mutations m2, m4, and m6 had little influence (Fig. 5B). Within the region mutated by m3, conversion of the rat sequence to that observed in the human gene had only a modest impact on promoter activity, where slightly elevated activity was observed in Sertoli cells and slightly decreased activity was observed in TM4 cells (RM mutant, Fig. 5). Mutation m5 specifically increased promoter activity in Sertoli cells, as did the larger block-replacement mutant m456. However, only the larger mutant increased promoter activity in TM4 cells (Fig. 5B). Several mutations were also generated within conserved region III of the Dmrt1 promoter (Fig. 1, m7–m9 in Fig. 5A). Within this region, m7 significantly increased promoter activity, m8 modestly decreased promoter activity, and m9 had no substantial impact (Fig. 5B). A mutation spanning the transcriptional start site (m10) diminished promoter activity approximately 30–50% (Fig. 5B). These data indicate that important transcriptional activators bind within the promoter regions mutated by m1 and m3, whereas proteins that repress promoter activity bind within regions mutated by m4, m5, m6, and m7. The functional characteristics of the proximal promoter region are similar in primary rat Sertoli cells and in the mouse Sertoli cell line TM4.
FIG. 5.
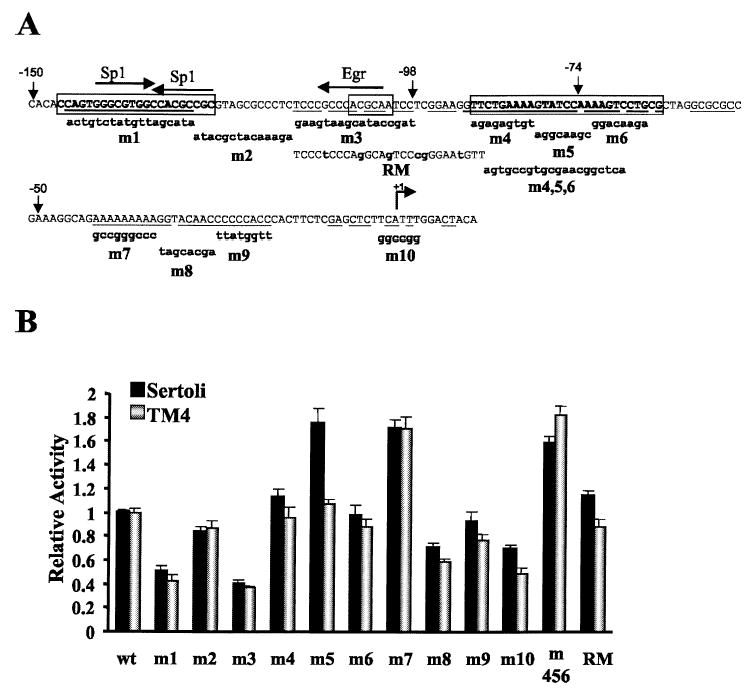
Transient transfection analysis of Dmrt1 promoter mutants in primary rat Sertoli cell cultures and mouse TM4 cells. A) Twelve different block-replacement mutants (bm1–bm10, bm456, RM) were generated and placed into the context of the Dmrt1(−150/+74)Luc construct. Wild-type sequence of the Dmrt1 promoter from −150 to +12 is shown with the sequence of each of the mutations below. Uppercase letters reflect identity to the wild type, and lowercase letters indicate bases that have been changed. Potential transcription factor binding sites identified through search of the Transfac database [35] are indicated above the promoter sequence. The arrows indicate the orientation of the putative element. Bases within the box correspond to those within regions I–III identified by in vitro DNase I footprinting. The underlined bases within these regions mark the conserved bases within the promoters of rats and humans. B) Promoters containing the mutations in A were transfected together with pRL-TK into primary cultures of rat Sertoli cells or mouse TM4 cells, and the cells were later lysed and assayed for fire-fly and Renilla luciferase activities. The data represent the firefly/Renilla luciferase activity of each promoter construct made relative to the firefly/Renilla luciferase activity of the wild-type Dmrt1(−150/+74)Luc. Transfections were done a minimum of 3 times, and the error bars represent the SEM.
Computer-assisted sequence analysis of the positive regulatory regions (i.e., those mutated by m1 and m3) revealed putative bindings sites for the transcriptional regulators Sp1, Egr1, Egr2, and Egr3 (Fig. 5A) [35]. To further resolve the bases important for promoter activity, smaller mutations (sm1–sm4 and sm7–sm10, Fig. 6) were generated within the promoter regions implicated by the m1–m3 mutations and evaluated by transient transfection. Two of the mutations, sm1 and sm4, were designed to disrupt core bases of the 2 putative Sp1 sites within the region mutated by m1. Evaluation of these mutations revealed that only the most 5′ Sp1 site significantly contributed to Dmrt1 promoter function (Fig. 6). Thus mutation sm1 diminished promoter activity to a similar extent as the larger mutation m1, but sm4 had no impact on Dmrt1 promoter function. A second mutation, sm3, which disrupts the core sequence of the putative Egr binding site, also had a significant impact on Dmrt1 promoter activity (Fig. 6). With the exception of a slight increase in promoter activity with the sm10 mutant, the remaining mutations spanning this region had little impact on promoter function. These studies better define the functional elements within the Dmrt1 promoter and implicate Sp1 and Egr in control of Dmrt1 transcription.
FIG. 6.
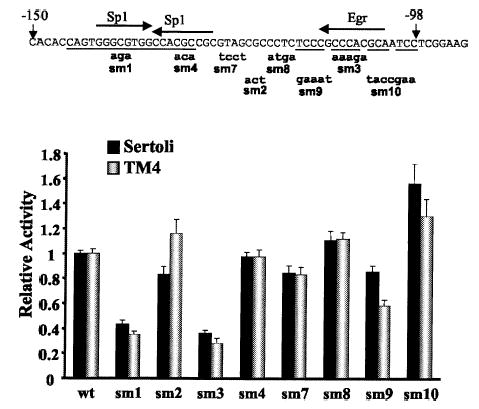
Transient transfection analysis of smaller mutants in the Sp1 and Egr sites of the Dmrt1 promoter. A) Eight different block-replacement mutants (sm1–sm4 and sm7–sm10) were generated and placed into the context of the Dmrt1(−150/+74)Luc construct. Wild-type sequence of the Dmrt1 promoter from −150 to +12 is shown with the sequence of each of the mutations below. Uppercase letters reflect identity to the wild type, and lowercase letters indicate bases that have been changed. Potential binding sites for the transcription factors Sp1 and Egr (includes Egr1, Egr2, and Egr3) were identified through search of the Transfac database [35] and are indicated above the promoter sequence. The arrows indicate the orientation of the putative element. The underlined bases within the promoter indicate those that are conserved between rats and humans. Promoters containing the mutations were transfected together with pRL-TK into primary cultures of rat Sertoli cells or mouse TM4 cells, and the cells were later lysed and assayed for firefly and Renilla luciferase activities. The data represent the firefly/Renilla luciferase activity of each promoter construct made relative to the firefly/Renilla luciferase activity of the wild-type Dmrt1(−150/+74)Luc. Transfections were done a minimum of 3 times, and the error bars represent the SEM.
Sp1, Sp3, and Egr1 Bind Important Regulatory Elements Within the Dmrt1 Promoter
To examine proteins binding important regulatory elements, EMSAs were performed using radiolabeled oligodeoxynucleotide probes that span regions contributing to promoter activity. A probe spanning from −150 to −118, which includes the regions mutated by m1, sm1, and sm4, bound 3 major complexes when incubated with nuclear extracts isolated from primary rat Sertoli cells (−150/−118wt, Table 1 and Fig. 7A). These complexes were effectively competed by unlabeled oligodeoxynucleotides containing sequences corresponding to that of the probe (wt) or a consensus Sp1 binding site (Sp1, Table 1 and Fig. 7A). In contrast, competitors containing sequences for an AP2 element or a nonspecific scrambled sequence (NS) failed to compete for binding of these complexes (Table 1 and Fig. 7A). The competition profile demonstrated that these complexes bound specifically to the Dmrt1 promoter element. Inclusion of antibodies generated against Sp1, Sp2, Sp3, and Sp4 in the binding reactions revealed that the most slowly migrating complex cross-reacted with the Sp1 antibody, and the 2 more quickly migrating complexes cross-reacted with both the Sp1 and Sp3 antibodies (Fig. 7A). Complexes that cross-reacted with Sp2 or Sp4 antibodies were not detected. Similar results were observed when nuclear proteins isolated from TM4 cells were used, except some cross-reactivity was observed with the Sp2 antibody (Fig. 7B). These data indicate that Sp1 and Sp3 bind an important functional element of the Dmrt1 promoter.
TABLE 1.
Sequences of oligodeoxynucleotides used in EMSA.
| Oligodeoxy-nucleotide | Sequence* |
|---|---|
| −150/−118 wt | CACACCAGTGGGCGTGGCCACGCCGCGTAGCG |
| −150/−118 sm1 | CACACCAGTGGagaTGGCCACGCCGCGTAGCG |
| −150/−118 sm4 | CACACCAGTGGGCGTGGCCACacaGCGTAGCG |
| −125/−94 wt | GTAGCGCCCTCTCCCGCCCACGCAATCCTCGG |
| −125/−94 sm3 | GTAGCGCCCTCTCCCGaaagaGCAATCCTCGG |
| −125/−94 sm9 | GTAGCGCCCTCgaaatCCCACGCAATCCTCGG |
| −125/−94 sm10 | GTAGCGCCCTCTCCCGCCCACtaccgaaTCGG |
| RM | GTAGCGCCCTCTCCCtCCCAgGCAgTCCcgGGAAtGTT |
| Egr | GGGAGTCAGTCTTGCGTGGGCGTTAGTCAGTCGGG |
| Sp1 | ATTCGATCGGGGCGGGGCGAGC |
| NS | CTAGAGTCGACCTGCAGGCATGCAAGCTTGGCATTC |
| AP2 | GATCGAACTGACCGCCCGCGGCCCGT |
Lowercase letters indicate bases that have been changed.
FIG. 7.
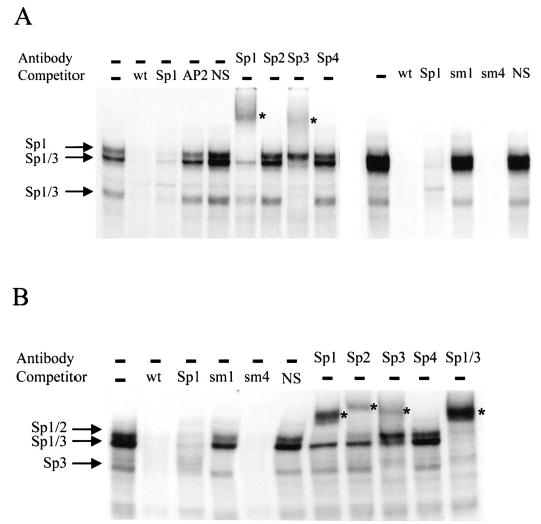
Sp1 binds region I in the Dmrt1 promoter. A double-stranded radiolabeled oligodeoxynucleotide probe containing the region from −150 to −118 of the Dmrt1 promoter (−150/−118 wt, Table 1) was used in a EMSA with nuclear extracts from either primary rat Sertoli cells (A) or TM4 cells (B). The radiolabeled wt probe (25 fmole) was incubated with 10 μg of nuclear extract and resolved on a 4% polyacrylamide gel. Where indicated, competitor oligodeoxynucleotides were added to the reaction at a concentration equal to 100-fold that of the labeled probe. Oligodeoxynucleotide sequences are given in Table 1. Antibodies against the transcription factors Sp1, Sp2, Sp3, and Sp4 were added to the reactions where indicated. The arrows mark the major binding complexes, and the antibodies to which they cross-reacted are indicated next to the respective arrows. Asterisks mark the supershifted complexes.
To correlate promoter function with the binding of Sp1 and Sp3 transcription factors, additional EMSAs were performed using competitors containing the sm1 and sm4 mutations. The binding studies showed that an oligodeoxynucleotide containing the sm1 mutation (Table 1) failed to compete for the Sp1 and Sp3 proteins, indicating that the mutation disrupted binding of these factors (Fig. 7, A and B). In contrast, an oligodeoxynucleotide containing the sm4 mutation was able to compete and therefore bound the Sp1 and Sp3 complexes (Table 1 and Fig. 7, A and B). Thus, the bases required for Sp1 and Sp3 binding were strongly correlated with those needed for promoter function, suggesting that Sp1 and/or Sp3 regulate transcription of the Dmrt1 gene.
The region spanning from −125 to −94, which includes the sites mutated by the m3 and sm3 mutations, was similarly examined for binding complexes (Table 1). EMSA revealed multiple complexes from primary rat Sertoli cell extracts that bound this region of the Dmrt1 promoter (Fig. 8A). The protein complexes were effectively competed with unlabeled oligodeoxynucleotides containing either homologous sequence (wt) or an Egr1 consensus binding site (Table 1 and Fig. 8A). An oligodeoxynucleotide containing the functionally disruptive sm3 mutation failed to compete and thus did not bind any of the observed complexes (Fig. 8A). A similar pattern was observed when nuclear extracts from TM4 cells were used (Fig. 8B). Oligodeoxynucleotides containing either the sm9 or sm10 mutations were used as competitors in the assay and revealed differences in the binding requirements for the different complexes in both Sertoli cell and TM4 extracts (Fig. 8, A and B). Thus the sm9 mutant failed to compete for the upper complexes but had full affinity for the middle complex and partial affinity for the lower complex. In contrast, sm10 was able to fully compete for the upper complex but had lower binding affinity for the lower complexes. The lower affinity for the 2 complexes was more apparent with the TM4 extracts than with the Sertoli cell extracts (Fig. 8). An oligodeoxynucleotide-containing sequence corresponding to a consensus Sp1 site competed only for the most slowly migrating complex (Fig. 8B), whereas competitors containing a non-specific scrambled sequence (NS) failed to compete for complex binding (Fig. 8). Thus, the complexes are specific for the element, bind differentially to Egr1 and Sp1 consensus elements and the sm9 and sm10 mutations, and fail to bind an oligodeoxynucleotide containing the sm3 mutation. An oligodeoxynucleotide containing the sequence of the human gene within this region (Table 1, RM) was able to compete partially for each of the complexes but had significantly lower affinity for the more quickly migrating proteins (Fig. 8). Inclusion of antibodies generated against Egr1, Egr2, Egr3, or Sp1 revealed that the most slowly migrating complex cross-reacted with the Sp1 antibody, whereas the middle complex cross-reacted with the Egr1 antibody (Fig. 8, A and B). With respect to the most quickly migrating complex, additional EMSAs revealed very modest cross-reactivity with an antibody generated against Sp3 but no cross-reactivity with antibodies generated against Sp1, Sp2, Sp4, Egr1, Egr2, Egr3, and Wilms tumor WT1 (Fig. 8 and data not shown). This finding suggests that a minor portion of the complex contained Sp3 or a protein that cross-reacted with its antibody, but other proteins within the complex remain unknown.
FIG. 8.
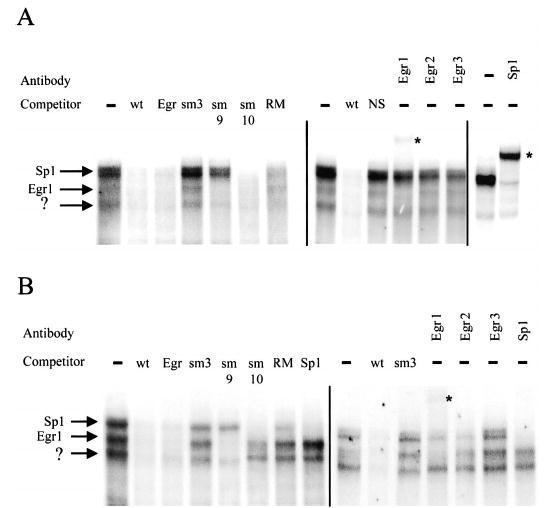
Egr1 and Sp1 bind within region III of the Dmrt1 promoter. A double-stranded radiolabeled oligodeoxynucleotide probe containing the region from −125 to −94 of the Dmrt1 promoter (−125/−94 wt, Table 1) was used in a EMSA with nuclear extracts from either primary rat Sertoli cells (A) or TM4 cells (B). The radiolabeled wt probe (25 fmole) was incubated with 10 μg of nuclear extract and resolved on a 4% polyacrylamide gel. Where indicated, competitor oligodeoxynucleotides were added to the reaction at a concentration equal to 100-fold that of the labeled probe. Oligodeoxynucleotide sequences are given in Table 1. Antibodies against the transcription factors Sp1, Egr1, Egr2, and Egr3 were added to the reactions where indicated. The arrows mark the major binding complexes, and the antibodies to which they cross-reacted are indicated next to the respective arrows. Asterisks mark the supershifted complexes.
DISCUSSION
The recognized role of Dmrt1 in testis differentiation and function underscores the need to understand the transcriptional mechanisms regulating the Dmrt1 gene in the testis. Characterization of the 5′ flanking region of the rat Dmrt1 gene revealed that transcription initiation of Dmrt1 occurs at multiple sites that cluster around a primary initiation site located 102 bases upstream of the translational start codon. Sequence analysis of this region revealed that the major initiation site resides within a sequence that corresponds to a consensus sequence of an initiator element (Inr, Py- PyA+1N T/A PyPy), an element that acts to direct accurate basal transcription and is important for core promoter activity in TATA-containing as well as TATA-less genes [36–39]. Notably, the Dmrt1 promoter lacks a discernible TATA motif within the core promoter sequence. A block replacement mutation through the putative Inr (m10, Fig. 5) decreased promoter activity by 30% and 50% in rat Sertoli and TM4 cells, respectively, suggesting that Dmrt1 transcription partially depends on the Inr. Inrs frequently work in conjunction with upstream elements that bind the transcription factor Sp1, which was also implicated in the regulation of the Dmrt1 promoter [37]. A strict requirement for the Inr in Dmrt1 transcriptional initiation was not suggested by the transient transfection studies, indicating that other initiation sites contribute to Dmrt1 transcription or that in the absence of the Inr sequence alternative sites were used.
From the data obtained in this investigation, we know that the Dmrt1 promoter is regulated by both negative and positive elements, and we have identified several of the proteins important for positive regulation of the gene. The transcription factors Sp1, Sp3, Egr1, and an as yet unidentified factor bound important positive regulatory elements and are thus implicated in the regulation of Dmrt1 transcription. Sp1 and Sp3 are members of a family of Sp1-like proteins that are zinc-finger transcription factors expressed in a variety of cells and tissues [40]. Studies on mice lacking Sp1 suggested that this transcription factor plays a critical role in the maintenance of differentiated cells [41]. Although ubiquitous expression patterns for Sp1 and Sp3 indicate they are not directly responsible for restricting Dmrt1 to the testis, the functional implications from the Sp1 gene knockout studies suggest they are important for sustained Dmrt1 expression, which in turn regulates events necessary for initiation and maintenance of Sertoli cell differentiation [40]. Some members of the Sp1-like family, such as EKLF, LKLF, and Sp4, are expressed in a tissue-enriched manner, raising the possibility that other members of this family play a more cell-specific role in Dmrt1 expression at different times during development [40]. To this end, Sp1-like family members BTEB2 and GKLF are preferentially expressed in the testis [42, 43].
There are 4 closely related transcription factors in the Egr family: Egr1 (NGFI-A, krox24, and zif-268), Egr2 (krox20), Egr3, and Egr4 (NGFI-C and pAT133). These proteins are also zinc-finger transcription factors and have nearly identical DNA binding domains [44]. The Egr proteins are expressed in a variety of tissues and are thought to regulate critical genetic programs involved in cellular growth and differentiation [45–48]. Studies on mice lacking Egr1 and Egr4 have revealed sexually dimorphic functions in male and female fertility [45, 47, 49]. Of 2 different studies describing Egr1-deficient mice, in only 1 was a defect in male fertility observed, implying that mouse strain differences influence male reproductive capabilities in the absence of Egr1. In these male Egr1-deficient mice, the infertility defect was at least partially due to the loss of LH; a single LH injection restored fertility in about half of the animals. However, because Egr1 is also expressed in the testis, it is not certain that the fertility defect is entirely due to LH loss or whether it has some direct influence on testis function. Thus, the fertility defect may in part reflect mis-regulation of Dmrt1 in the absence of Egr1.
Egr4-deficient mice have fertility defects that are male specific [47]. In wild-type mice, Egr4 was observed at low levels in male meiotic germ cells, and Egr4−/− mice had obvious defects in germ cell maturation during the early to middle pachytene stage. Thus, Egr4 is implicated in the regulation of genes important to germ cell function at some point early in meiosis. Dmrt1−/− mice also had male-specific germ cell defects that occurred during meiosis. Although Egr4 appears to be absent from Sertoli cells and thus not relevant to the current investigation, these observations suggest it may be an important transcriptional regulator of Dmrt1 in male germ cells [47]. Evaluation of Dmrt1 expression in the Egr4-deficient mice will help determine whether such a relationship exists and may further our understanding of the mechanism involved in male fertility.
Transcriptional repression was also implicated in the control of Dmrt1 gene expression. DNase I footprinting showed that conserved region II bound nuclear proteins from rat Sertoli cells, and mutations in this region (m5, m456) significantly increased promoter activity (Figs. 4 and 5). Because mutations may inadvertently introduce a binding site for a transactivator, 2 different mutations (m5, m456) were introduced across this region, both of which caused a marked increase in promoter activity. This observation, together with the highly conserved nature of the region and the presence of a binding site for Sertoli cell nuclear proteins, indicates that a transcriptional repressor binds this site of the promoter. However, an unexplained difference between the primary Sertoli cells and TM4 cells was revealed by examination of mutations in this promoter region. In primary rat Sertoli cells, the m5 mutant resulted in significant promoter activation, whereas there was no activation in TM4 cells. However, the larger block mutant (m456) increased promoter activity in both cell types, which suggests that there are differences in the binding factors interacting within this region in the 2 cell types. Evaluation of the sequence of the repressor element revealed the presence of a potential binding site for the transcription factor C/EBPalpha, a protein expressed in Sertoli cells that is known to act as a transcriptional repressor [50, 51]. A repressor-binding site was also implicated within conserved region III; mutation m7 resulted in a 1.8-fold increase in promoter activity. A second mutation (m7′) in this region (5′-AAAAAAAAA-3′ to 5′-ggttcggtc-3′) had a similar impact on promoter function (data not shown). Within this region, there was no indication of protein binding by DNase I footprint analysis (Fig. 4). However, a binding site within this region may have been overlooked because of the poor sensitivity of the region to DNase I digestion. Further analysis of these 2 negative regulatory elements is necessary to better understand the role of transcriptional repression in Dmrt1 expression.
The studies presented in this report represent the first characterization of the rat Dmrt1 promoter and focused predominantly on characterization of the proximal promoter region (downstream of −150). In these studies, Dmrt1 expression was examined in postnatal immature rat Sertoli cells, where a role for Dmrt1 was revealed through gene knockout studies. Currently, it is unknown whether the identified proteins and regions important for Dmrt1 expression in immature Sertoli cells are involved in gene regulation during development or in male germ cells. Further investigation in this area and an examination of the mechanisms responsible for hormone response and cell-specific expression are needed to fully understand the regulation of Dmrt1 and its physiological role in the testis.
Dmrt1 mRNA is highly induced by the pituitary hormone FSH and its downstream signaling molecule cAMP. Within the proximal promoter, sequence analysis failed to reveal any obvious cAMP response elements (CREs), suggesting that hormone response requires elements outside this region. Upstream of −150, there were also no obvious consensus CREs, but several AP1-like elements have been identified (data not shown). Studies to evaluate FSH-stimulated induction of Dmrt1 transcription are ongoing and will provide important insight into the mechanism of FSH response in Sertoli cells. Dmrt1 expression is highly restricted to the gonads and is testis specific in the postnatal animal. The mechanism that drives testis-specific expression is currently not understood. The ubiquitous nature of the factors regulating the proximal promoter suggests that elements outside this region are required for cell-specific expression. Analysis of the various deletion mutants identified a region located between −3200 and −2000 bp that contains important positive regulatory elements. Studies to further evaluate this and other regions of the gene are needed and will likely provide important insight into the mechanisms driving cell-specific expression of Dmrt1.
Acknowledgments
We thank Jiangkai Chen for cell culture preparation and technical assistance and Dr. Michael Wolfe for his generosity and helpful suggestions.
Footnotes
This work was supported by a Madison and Lila Self Graduate Fellowship.
References
- 1.Sinclair AH, Berta P, Palmer MS, Hawkins JR, Griffiths BL, Smith MJ, Foster JW, Frischauf AM, Lovell-Badge R, Goodfellow PN. A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature. 1990;346:240–244. doi: 10.1038/346240a0. [DOI] [PubMed] [Google Scholar]
- 2.Birk OS, Casiano DE, Wassif CA, Cogliati T, Zhao L, Zhao Y, Grinberg A, Huang S, Kreidberg JA, Parker KL, Porter FD, Westphal H. The LIM homeobox gene Lhx9 is essential for mouse gonad formation. Nature. 2000;403:909–913. doi: 10.1038/35002622. [DOI] [PubMed] [Google Scholar]
- 3.Kreidberg JA, Sariola H, Loring JM, Maeda M, Pelletier J, Housman D, Jaenisch R. WT-1 is required for early kidney development. Cell. 1993;74:679–691. doi: 10.1016/0092-8674(93)90515-r. [DOI] [PubMed] [Google Scholar]
- 4.Kent J, Wheatley SC, Andrews JE, Sinclair AH, Koopman PA. Male-specific role for SOX9 in vertebrate sex determination. Development. 1996;122:2813–2822. doi: 10.1242/dev.122.9.2813. [DOI] [PubMed] [Google Scholar]
- 5.Morais-da-Silva S, Hacker A, Harley V, Goodfellow P, Swain A, Lovell-Badge R. Sox9 expression during gonadal development implies a conserved role for the gene in testis differentiation in mammals and birds. Nat Genet. 1996;14:62–68. doi: 10.1038/ng0996-62. [DOI] [PubMed] [Google Scholar]
- 6.Swain A, Narvaez V, Burgoyne P, Camerino G, Lovell-Badge R. Dax1 antagonizes Sry action in mammalian sex determination. Nature. 1998;391:761–767. doi: 10.1038/35799. [DOI] [PubMed] [Google Scholar]
- 7.Shawlot W, Behringer RR. Requirement for Lim1 in head-organizer function. Nature. 1995;374:425–430. doi: 10.1038/374425a0. [DOI] [PubMed] [Google Scholar]
- 8.Miyamoto N, Yoshida M, Kuratani S, Matsuo I, Aizawa S. Defects of urogenital development in mice lacking Emx2. Development. 1997;124:1653–1664. doi: 10.1242/dev.124.9.1653. [DOI] [PubMed] [Google Scholar]
- 9.Raymond CS, Kettlewell JR, Hirsch B, Bardwell VJ, Zarkower D. Expression of Dmrt1 in the genital ridge of mouse and chicken embryos suggests a role in vertebrate sexual development. Dev Biol. 1999;215:208–220. doi: 10.1006/dbio.1999.9461. [DOI] [PubMed] [Google Scholar]
- 10.Raymond CS, Murphy MW, O’Sullivan MG, Bardwell VJ, Zarkower D. Dmrt1, a gene related to worm and fly sexual regulators, is required for mammalian testis differentiation. Genes Dev. 2000;14:2587–2595. doi: 10.1101/gad.834100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raymond CS, Parker ED, Kettlewell JR, Brown LG, Page DC, Kusz K, Jaruzelska J, Reinberg Y, Flejter WL, Bardwell VJ, Hirsch B, Zarkower D. A region of human chromosome 9p required for testis development contains two genes related to known sexual regulators. Hum Mol Genet. 1999;8:989–996. doi: 10.1093/hmg/8.6.989. [DOI] [PubMed] [Google Scholar]
- 12.Raymond CS, Shamu CE, Shen MM, Seifert KJ, Hirsch B, Hodgkin J, Zarkower D. Evidence for evolutionary conservation of sex-determining genes. Nature. 1998;391:691–695. doi: 10.1038/35618. [DOI] [PubMed] [Google Scholar]
- 13.Smith CA, McClive PJ, Western PS, Reed KJ, Sinclair AH. Conservation of a sex-determining gene. Nature. 1999;402:601–602. doi: 10.1038/45130. [DOI] [PubMed] [Google Scholar]
- 14.Kettlewell JR, Raymond CS, Zarkower D. Temperature-dependent expression of turtle Dmrt1 prior to sexual differentiation. Genesis. 2000;26:174–178. [PubMed] [Google Scholar]
- 15.De Grandi A, Calvari V, Bertini V, Bulfone A, Peverali G, Camerino G, Borsani G, Guioli S. The expression pattern of a mouse doublesex-related gene is consistent with a role in gonadal differentiation. Mech Dev. 2000;90:323–326. doi: 10.1016/s0925-4773(99)00282-8. [DOI] [PubMed] [Google Scholar]
- 16.Chen J, Heckert LL. Dmrt1 expression is regulated by follicle-stimulating hormone and phorbol esters in postnatal Sertoli cells. Endocrinology. 2001;142:1167–1178. doi: 10.1210/endo.142.3.8021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bardin CW, Cheng CY, Mustow NA, Gunsalus GL. The Sertoli cell. In: Knobil E, Neill JD (eds.), The Physiology of Reproduction, 2nd ed. New York: Raven Press; 1994: 1291–1333.
- 18.Clermont Y. Introduction to the Sertoli cell. In: Russell LD, Griswold MD (eds.), The Sertoli Cell. Clearwater: Cache River Press; 1993: xxi–xxv.
- 19.Griswold MD. Actions of FSH on mammalian Sertoli cells. In: Russell LD, Griswold MD (eds.), The Sertoli Cell. Clearwater: Cache River Press; 1993: 493–508.
- 20.Pelliniemi LJ, Frojdman K, Paranko J. Embryological and prenatal development and function of Sertoli cells. In: Russell LD, Griswold MD (eds.), The Sertoli Cell. Clearwater: Cache River Press; 1993: 87–113.
- 21.Magre S, Jost A. The initial phases of testicular organogenesis in the rat. An electron microscopy study. Arch Anat Microsc Morphol Exp. 1980;69:297–318. [PubMed] [Google Scholar]
- 22.McLaren A. Development of the mammalian gonad: the fate of the supporting cell lineage. Bioessays. 1991;13:151–156. doi: 10.1002/bies.950130402. [DOI] [PubMed] [Google Scholar]
- 23.McLaren A. Gonad development: assembling the mammalian testis. Curr Biol. 1998;8:R175–R177. doi: 10.1016/s0960-9822(98)70104-6. [DOI] [PubMed] [Google Scholar]
- 24.Karl J, Capel B. Sertoli cells of the mouse testis originate from the coelomic epithelium. Dev Biol. 1998;203:323–333. doi: 10.1006/dbio.1998.9068. [DOI] [PubMed] [Google Scholar]
- 25.Capel B. Sex in the 90s: SRY and the switch to the male pathway. Annu Rev Physiol. 1998;60:497–523. doi: 10.1146/annurev.physiol.60.1.497. [DOI] [PubMed] [Google Scholar]
- 26.Capel B. The battle of the sexes. Mech Dev. 2000;92:89–103. doi: 10.1016/s0925-4773(99)00327-5. [DOI] [PubMed] [Google Scholar]
- 27.Gondos B, Berndston WE. Postnatal and pubertal development. In: Knobil E, Neill JD (eds.), The Physiology of Reproduction, 2nd ed. New York: Raven Press; 1994: 116–154.
- 28.Karl AF, Griswold MD. Sertoli cells of the testis: preparation of cell cultures and effects of retinoids. Methods Enzymol. 1990;190:71–75. doi: 10.1016/0076-6879(90)90010-x. [DOI] [PubMed] [Google Scholar]
- 29.Heckert LL, Daggett MA, Chen J. Multiple promoter elements contribute to activity of the follicle-stimulating hormone receptor (FSHR) gene in testicular Sertoli cells. Mol Endocrinol. 1998;12:1499–1512. doi: 10.1210/mend.12.10.0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Daggett MA, Rice DA, Heckert LL. Expression of steroidogenic factor 1 in the testis requires an E box and CCAAT box in its promoter proximal region. Biol Reprod. 2000;62:670–679. doi: 10.1095/biolreprod62.3.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frohman MA. RACE: rapid amplification of cDNA ends. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds.), PCR Protocols: A Guide to Methods and Applications. San Diego: Academic Press; 1990: 28–38.
- 32.Frohman MA. Rapid amplification of complementary DNA ends for generation of full-length complementary DNAs: thermal RACE. Methods Enzymol. 1993;218:340–356. doi: 10.1016/0076-6879(93)18026-9. [DOI] [PubMed] [Google Scholar]
- 33.Iannello RC. DNase I footprinting using PCR-generated end-labeled DNA probes. Methods Mol Biol. 1995;37:379–391. doi: 10.1385/0-89603-288-4:379. [DOI] [PubMed] [Google Scholar]
- 34.Sambrook J, Fritz IB, Maniatis T. Extraction, purification, and analysis of messenger RNA from eukaryotic cells. In: Nolan C (ed.), Molecular Cloning: A Laboratory Manual Plainview, NY: Cold Spring Harbor Laboratory Press; 1989: 7:76.
- 35.Heinemeyer T, Chen X, Karas H, Kel AE, Kel OV, Liebich I, Meinhardt T, Reuter I, Schacherer F, Wingender E. Expanding the TRANSFAC database towards an expert system of regulatory molecular mechanisms. Nucleic Acids Res. 1999;27:318–322. doi: 10.1093/nar/27.1.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smale ST. Transcription initiation from TATA-less promoters within eukaryotic protein-coding genes. Biochim Biophys Acta. 1997;1351:73–88. doi: 10.1016/s0167-4781(96)00206-0. [DOI] [PubMed] [Google Scholar]
- 37.Weis L, Reinberg D. Transcription by RNA polymerase II: initiator-directed formation of transcription-competent complexes. FASEB J. 1992;6:3300–3309. doi: 10.1096/fasebj.6.14.1426767. [DOI] [PubMed] [Google Scholar]
- 38.Emami KH, Navarre WW, Smale ST. Core promoter specificities of the Sp1 and VP16 transcriptional activation domains. Mol Cell Biol. 1995;15:5906–5916. doi: 10.1128/mcb.15.11.5906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smale ST, Baltimore D. The ‘‘initiator’’ as a transcription control element. Cell. 1989;57:103–113. doi: 10.1016/0092-8674(89)90176-1. [DOI] [PubMed] [Google Scholar]
- 40.Cook T, Gebelein B, Urrutia R. Sp1 and its likes: biochemical and functional predictions for a growing family of zinc finger transcription factors. Ann NY Acad Sci. 1999;880:94–102. doi: 10.1111/j.1749-6632.1999.tb09513.x. [DOI] [PubMed] [Google Scholar]
- 41.Marin M, Karis A, Visser P, Grosveld F, Philipsen S. Transcription factor Sp1 is essential for early embryonic development but dispensable for cell growth and differentiation. Cell. 1997;89:619–628. doi: 10.1016/s0092-8674(00)80243-3. [DOI] [PubMed] [Google Scholar]
- 42.Sogawa K, Imataka H, Yamasaki Y, Kusume H, Abe H, Fujii-Kuriyama Y. cDNA cloning and transcriptional properties of a novel GC box-binding protein, BTEB2. Nucleic Acids Res. 1993;21:1527–1532. doi: 10.1093/nar/21.7.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shields JM, Christy RJ, Yang VW. Identification and characterization of a gene encoding a gut-enriched Kruppel-like factor expressed during growth arrest. J Biol Chem. 1996;271:20009–20017. doi: 10.1074/jbc.271.33.20009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gashler A, Sukhatme VP. Early growth response protein 1 (Egr-1): prototype of a zinc-finger family of transcription factors. Prog Nucleic Acid Res Mol Biol. 1995;50:191–224. doi: 10.1016/s0079-6603(08)60815-6. [DOI] [PubMed] [Google Scholar]
- 45.Lee SL, Sadovsky Y, Swirnoff AH, Polish JA, Goda P, Gavrilina G, Milbrandt J. Luteinizing hormone deficiency and female infertility in mice lacking the transcription factor NGFI-A (Egr-1) Science. 1996;273:1219–1221. doi: 10.1126/science.273.5279.1219. [DOI] [PubMed] [Google Scholar]
- 46.Sadovsky Y, Crawford PA, Woodson KG, Polish JA, Clements MA, Tourtellotte LM, Simburger K, Milbrandt J. Mice deficient in the orphan receptor steroidogenic factor 1 lack adrenal glands and gonads but express P450 side-chain-cleavage enzyme in the placenta and have normal embryonic serum levels of corticosteroids. Proc Natl Acad Sci U S A. 1995;92:10939–10943. doi: 10.1073/pnas.92.24.10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tourtellotte WG, Nagarajan R, Auyeung A, Mueller C, Milbrandt J. Infertility associated with incomplete spermatogenic arrest and oligozoospermia in Egr4-deficient mice. Development. 1999;126:5061–5071. doi: 10.1242/dev.126.22.5061. [DOI] [PubMed] [Google Scholar]
- 48.Tourtellotte WG, Nagarajan R, Bartke A, Milbrandt J. Functional compensation by Egr4 in Egr1-dependent luteinizing hormone regulation and Leydig cell steroidogenesis. Mol Cell Biol. 2000;20:5261–5268. doi: 10.1128/mcb.20.14.5261-5268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Topilko P, Schneider-Maunoury S, Levi G, Trembleau A, Gourdji D, Driancourt MA, Rao CV, Charnay P. Multiple pituitary and ovarian defects in Krox-24 (NGFI-A, Egr-1)-targeted mice. Mol Endocrinol. 1998;12:107–122. doi: 10.1210/mend.12.1.0049. [DOI] [PubMed] [Google Scholar]
- 50.Slomiany BA, D’Arigo KL, Kelly MM, Kurtz DT. C/EBPalpha inhibits cell growth via direct repression of E2F-DP-mediated transcription. Mol Cell Biol. 2000;20:5986–5997. doi: 10.1128/mcb.20.16.5986-5997.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gronning LM, Dahle MK, Tasken KA, Enerback S, Hedin L, Tasken K, Knutsen HK. Isoform-specific regulation of the CCAAT/enhancer-binding protein family of transcription factors by 3′,5′-cyclic adenosine monophosphate in Sertoli cells. Endocrinology. 1999;140:835–843. doi: 10.1210/endo.140.2.6526. [DOI] [PubMed] [Google Scholar]