

Abstract
Epidemiological studies and analysis of putative virulence genes have shown that Listeria monocytogenes has diverged into several phylogenetic divisions. We hypothesize that similar divergence has occurred for many genes that influence niche-specific fitness and virulence and that identifying these differences may offer new opportunities for the detection, treatment, and control of this important pathogen. To explore this issue further, we developed a microarray composed of fragmented DNA taken from 10 strains of L. monocytogenes. We then hybridized genomic DNA from 50 different strains to replicate arrays and analyzed the resulting hybridization patterns. A simple Euclidean distance metric permitted the reconstruction of previously described genetic relationships between serotypes, and only four microarray probes were needed to discriminate between the most important serotypes (1/2a, 1/2b, 1/2c, and 4). We calculated an index of linkage equilibrium from the microarray data and confirmed that L. monocytogenes has a strongly clonal population structure (IA = 3.85). Twenty-nine informative probes were retrieved from the library and sequenced. These included genes associated with repairing UV-damaged DNA, salt tolerance, biofilm formation, heavy metal transport, ferrous iron transport, and teichoic acid synthesis. Several membrane-bound lipoproteins and one internalin were identified, plus three phage sequences and six sequences with unknown function. Collectively, these data confirm that many genes have diverged between lineages of L. monocytogenes. Furthermore, these results demonstrate the value of mixed-genome microarrays as a tool for deriving biologically useful information and for identifying and screening genetic markers for clinically important microbes.
Listeria monocytogenes is considered a significant food-borne pathogen due to its potential for high mortality rates and because it is a difficult pathogen to control in production environments (29). There are 13 recognized serotypes of L. monocytogenes, and most human infections involve serotypes 1/2a, 1/2b, and 4b, with the latter being associated with the largest food-borne outbreaks (25, 34). Two major phylogenetic divisions clearly differentiate between the most commonly isolated L. monocytogenes serotypes (serotypes 1/2a and 1/2c versus 1/2b and 4b) (22, 25). Analysis of putative virulence genes suggests that the evolution of somatic and flagellar antigens has paralleled the evolution of genes associated with virulence (11, 33). We hypothesize that similar divergence has occurred for many other genes that influence niche-specific fitness and virulence. If this hypothesis is correct, then identifying these differences may offer new insights into the ecology of L. monocytogenes as well as new opportunities for the detection, treatment, and control of this important pathogen.
Differential fitness and virulence are likely to be controlled by either differential gene expression or the presence of lineage-specific genes or allelic variants. This paper focuses on the latter by identifying the genomic differences between strains of L. monocytogenes by using a comparative-genomics approach that we refer to as mixed-genome microarray (3). These arrays permit the rapid characterization of genetic differences at hundreds of loci in a format that is conducive to analyzing many strains of bacteria. Although we focus here on L. monocytogenes, this type of microarray is applicable to other bacteria and can serve as a means to both fingerprint isolates as well as identify genetic sequences unique to different phylogenetic divisions. In this study, the mixed-genome microarray clearly differentiated between well-recognized, phylogenetic divisions of L. monocytogenes and identified 29 probes that are polymorphic between strains. Only four of these probes are needed to genetically differentiate four major serotypes of L. monocytogenes. Polymorphic probe sequences include close matches for genes involved with environmental stress and virulence.
MATERIALS AND METHODS
Serotyping.
Listeria antiserum was obtained from Accurate Scientific (Westbury, N.Y.). Serotyping was performed according to the manufacturer's recommendations except that isolates were cultured in EB motility medium (0.3% beef extract, 0.1% peptone, 0.5% NaCl, 0.4% agar; pH 7.4) prior to H-antigen determination (12). Mismatch amplification mutation assay (MAMA)-PCR was used as described earlier to determine how our bacterial isolates were classified according to a previously published description of L. monocytogenes phylogeny (19).
Microarray fabrication.
Genomic DNA was extracted from 10 L. monocytogenes strains representing the 1/2a (n = 4), 1/2c (n = 1), 3a (n = 1), and 4b (n = 4) serotypes from human, milk, and veterinary isolations (3). Equal amounts of DNA from each strain were mixed and used to develop a shotgun library (Amplicon Express, Pullman, Wash.). Briefly, 10 μg of DNA was cut with the restriction enzyme CviJI (Chimerx, Milwaukee, Wis.) and fragments of approximately 600 bp were gel isolated, extracted, and ligated into pUC18. Ligation products were transformed into Escherichia coli, and recombinant clones were arrayed into 96-well plates. Clone inserts were amplified by PCR with M13 sequencing primers. PCR products of the correct size (500 to 1,000 bp) were purified by using a Montage PCR96 Cleanup kit (Millipore Corp, Bedford, Mass.) and stored at −20°C until ready for printing.
Purified PCR products were resuspended in print buffer (0.05% sodium dodecyl sulfate, 50 mM NaOH, pH 12.0) at approximately 350 ng/μl, followed by heat denaturing (95°C for 5 min) in 96-well trays. PCR products were allowed to cool and were then deposited as four subarrays on glass slides. Each set of subarrays was replicated on each slide. We used two slide formats with one being unmasked immunohistochemistry slides (Fisher Scientific Co., Pittsburgh, Pa.). The second format used 12-well, Teflon-masked slides (Erie Scientific, Portsmouth, N.H.) that were derivatized with 3-glycidoxypropyltrimethoxysilane (Sigma-Aldrich, Milwaukee, Wis.) (7). Because the physical dimensions of the full array exceeded a single well, each subarray was deposited within an independent well. Thus, each slide was composed of eight subarrays within eight wells to complete two full arrays.
Sample hybridization.
Genomic DNA was extracted from target strains by using a DNeasy tissue kit (Qiagen, Valencia, Calif.), and 1 μg was nick translated in the presence of biotin-dATP (BioNick labeling system; Invitrogen). Labeled DNA was then ethanol precipitated, resuspended in 210 μl of hybridization buffer consisting of 4× SSC (60 mM NaCl, 0.6 mM Na-citrate; pH 7.0) and 5× Denhardt's solution (0.1% Ficoll, 0.1% polyvinylpyrrolidone, 0.1% bovine serum albumin), and heat denatured before being added to the slide for overnight hybridizations at 55°C. Slides were preblocked at 23°C for 30 min with TNB buffer (100 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.5% blocking reagent [Tyramide Signal Amplification, {TSA} biotin system; Perkin-Elmer, Boston, Mass.]). Slides were washed according to the method described by Hegde et al. (17), and the remaining steps were done according to the manufacturer's instructions for the TSA kit. After biotinyl tyramide was washed from the slide, 2 μg streptavidin/ml conjugated to Alexa Fluor 546 (Molecular Probes, Eugene, Ore.) was added in 1× SSC and 5× Denhardt's solution. Slides were given a final wash and followed by drying and imaging with an Applied Precision (Issaquah, Wash.) ArrayWoRx scanner.
Image analysis.
Microarray images were quantified by using SPOT software (CSIRO Mathematical and Information Sciences, North Ryde, Australia). This program combines automatic spot finding with a seeded-region-growing algorithm (1) and morphological background subtraction suitable for spots that vary in shape and size. The final output included median pixel values that were imported into a relational database (MS Access; Microsoft Corp., Redmond, Wash.) for further processing. Because each array was printed as a group of four subarrays (eight subarrays per hybridization), signal intensity for each probe was normalized by dividing by the average signal for all probes within a subarray. Normalized signal intensity was averaged for replicate probes on each slide.
Probe sequencing.
Probes of interest were retrieved from the clone library and sequenced (Amplicon Express). One of these probe sequences (probe 302 [BH175018]) was selected to identify how sequence divergence was reflected by signal intensity on the microarray. PCR primers were selected to amplify a 500-bp region of the corresponding sequence from five L. monocytogenes isolates representing the two primary phylogenetic divisions (M12716E, M32771C, F3321, V8807, and M36467A). The resulting PCR products were sequenced, and the percent sequence similarity was calculated.
Data processing.
Three datasets were produced from three different batches of slides. The first data set resulted from optimization experiments with Teflon- masked slides where each subarray was printed in an independent well. Data from these experiments were used only as independent data for the validation of a classification function (see below). A second data set was compiled from Teflon-masked slides and used to describe the mixed-genome microarray concept and to highlight potential applications (3). The third data set resulted from arrays that were printed on unmasked slides. We were able to combine the last two data sets after normalization and use this larger data set for the analysis presented herein. We qualitatively identified several outlier experiments that yielded an unusually low hybridization signal. To objectively identify these and other outliers, we plotted a normal probability curve for the total signal intensity for all slides. Slides that deviated significantly from the normal plot were rejected from further analysis except that these slides were considered part of the independent data set used to test the classification function described below.
The majority of probes yielded a high signal intensity for most L. monocytogenes strains that were hybridized to the array. These probes provided no useful information and were rejected from further analysis. A subset of probes produced normalized signal intensities that were classified as either high (0.7 to 1.4) or low (<0.7) depending on the strain being hybridized. For our analysis, a probe was defined as bimodally distributed when at least 10% of bacterial isolates produced either low or high signal intensity for that probe. Our analysis was limited to the latter probes because they were expected to be the most discriminating with respect to phylogenetic divisions.
Data analysis.
Euclidean distances were calculated for every sample comparison by using the following formula:
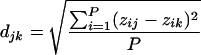 |
(1) |
where djk is the Euclidean distance between samples j and k, z is the normalized probe intensity, and P is the number of probes.
The resulting distance matrix was used to construct a dendrogram based on the Ward's minimum variance algorithm as implemented by NCSS 2001 (NCSS Statistical Software, Kaysville, Utah). This procedure grouped samples so that the pooled within-group sum of squares was minimized. Stepwise discriminant function analysis (DFA) was used to construct a classification function for sample serotypes. To implement this routine (SAS Institute, Carey, N.C.), the stepwise procedure was used to reduce the probe set to four probes suitable for classification. The classification function was assessed for accuracy by its ability to correctly classify the original data (training set) used to construct the function. More importantly, the accuracy was also assessed by using hybridization data from an independent set of microarray hybridizations that were excluded from development of the classification function.
Average linkage equilibrium was calculated by using an adaptation of the IA index (21, 35), where IA = (VO/VE − 1). The observed variance (VO) was calculated for all probes for all possible pairwise comparisons, and this value was divided by the expected value (VE):
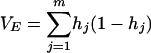 |
(2) |
where m is the number of loci and hj is the probability that two random individuals are different at the jth locus.
For microarray data, m is the number of probes considered in the analysis. Because we restricted the analysis to bimodal probes, we assumed that, on average, two randomly selected individuals could be characterized as having a high or low signal and, consequently, the probability that the individuals are different for a given probe was 0.5. Because only two states (high or low) were considered, VE simplified to m × 0.25. At linkage equilibrium, IA was expected to equal zero.
Replicate hybridizations were excluded from this analysis because these would artificially inflate the IA estimate. A bootstrap algorithm was used to construct an empirical confidence interval for IA. Fifty samples were randomly selected with replacement from the original pool of 50 samples. IA was then calculated, and a new set of 50 samples was randomly selected with replacement and calculations repeated. This process was continued for 1,000 iterations to construct an empirical probability distribution.
Nucleotide sequence accession numbers. The GenBank accession numbers for the probes sequenced in this study are BH175014 to BH175028 and BH861024 to BH861039.
RESULTS
We constructed a shotgun library after mixing equal-molar amounts of genomic DNA from 10 strains of L. monocytogenes (3). Probes (n = 585) were amplified from the clone library by PCR and printed onto glass slides. We then hybridized genomic DNA from 50 strains of L. monocytogenes (Table 1) to replicate arrays and assessed the signal intensity for each probe across all hybridizations. Twenty-nine probes were bimodally distributed, and subsequent analyses were limited to these probes to maximize the likelihood of identifying serotype or division-specific DNA sequences.
TABLE 1.
L. monocytogenes strains and associated characteristics
| Samplea | Sourceb | Serotype | Clusterc | MAMAd |
|---|---|---|---|---|
| E1163 | WADOH | 1/2a | IIa | 2 |
| E1165 | WADOH | 1/2a | IIa | 2 |
| E841 | WADOH | 1/2a | IIa | 2 |
| H1157 | WADOH | 1/2a | IIa | 2 |
| M32771C | USDA | 1/2a | IIa | 2 |
| M34058E | USDA | 1/2a | IIa | 2 |
| M36046A | USDA | 1/2a | IIa | 2 |
| M36509A | USDA | 1/2a | IIa | 2 |
| M36582B | USDA | 1/2a | IIa | 2 |
| M37952A | USDA | 1/2a | IIa | 2 |
| H1166 | WADOH | 1/2a | IIb | 2 |
| M35568A | USDA | 1/2a | IIb | 2 |
| M32490G | USDA | 1/2a | IIb | 2 |
| M32490E | USDA | 1/2a | IIb | 2 |
| M35568E | USDA | 1/2a | IIb | 2 |
| M37010C | USDA | 1/2a | IIb | 2 |
| E750 | WADOH | 1/2a | IIIc | 2 |
| H1162 | WADOH | 1/2a | IIIc | 2 |
| H1445 | WADOH | 1/2a | IIIc | 2 |
| M10867C | USDA | 1/2a | IIIc | 2 |
| M12716E | USDA | 1/2a | IIIc | 2 |
| F15C18 | FDA | 1/2b | Ia | 1 |
| F2492 | FDA | 1/2b | Ia | 1 |
| H1159 | WADOH | 1/2b | Ia | 1 |
| H1164 | WADOH | 1/2b | Ia | 1 |
| E9900101 | WADOH | 1/2b | Ib | 1 |
| H9900104 | WADOH | 1/2b | Ib | 1 |
| EH9066 | CDC | 1/2c | IIc | 2 |
| EH9067 | CDC | 1/2c | IIc | 2 |
| F3321 | CDC | 1/2c | IIc | 2 |
| H7973 | CDC | 1/2c | IIc | 2 |
| H900096 | WADOH | 1/2c | IIc | 2 |
| H9333 | CDC | 1/2c | IIc | 2 |
| M35584 | USDA | 4c | Ib | 3 |
| M36467A | USDA | 4c | Ib | 3 |
| M35402A | USDA | 4b | Ib | 1 |
| V013668A | USDA | 4b | Ib | 1 |
| H2365 | CDC | 4b | Ic | 1 |
| H1161 | WADOH | 4b | Ic | 1 |
| H1329 | USDA | 4b | Ic | 1 |
| H2172 | USDA | 4b | Ic | 1 |
| H9900094 | WADOH | 4b | Ic | 1 |
| M11056A | USDA | 4b | Ic | 1 |
| M13565A | USDA | 4b | Ic | 1 |
| M33027A | USDA | 4b | Ic | 1 |
| H5070 | CDC | 4b | Id | 1 |
| V8807 | USDA | 4b | Id | 1 |
| H2150 | WADOH | 4b | Id | 1 |
| H1092 | CDC | 4b | Id | 1 |
| H2140 | USDA | 4b | Id | 1 |
Sample identification prefix indicates where the strain was originally isolated: E, environment; H, human; M, bulk milk; and V, veterinary.
USDA, U.S. Department of Agriculture, Pullman, Wash.; WADOH, Washington State Department of Health, Shoreline; CDC, Centers for Disease Control and Prevention, Atlanta, Ga.; FDA, Food and Drug Administration.
Cluster analysis.
A dissimilarity matrix was generated based on the simple Euclidean distance across all probes, and this matrix was used with the Ward's minimum variance method to produce a dendrogram where the within-cluster variance was minimized (Fig. 1) (NCSS 2001). The algorithm clearly identified two previously described phylogenetic divisions for L. monocytogenes (25), and these findings were corroborated by the mismatch amplification mutation assay (19) (Fig. 1; Table 1). Clusters IIa and IIb were composed of serotype 1/2a, whereas cluster IIc included both serotypes 1/2a and 1/2c. Cluster Ia was composed of 1/2b strains, while clusters Ic and Id were composed of serotype 4b. Cluster Ib was composed of a mixture of the 1/2b, 4b, and 4c serotypes. Several replicated hybridizations were included in the dendrogram, and these grouped adjacent to their respective partners (M36509Ar1-2, V013668Ar1-2, M13565Ar1-3, and H1164r1-2). Epidemiologically linked isolates also grouped as adjacent neighbors on the dendrogram (E841 and E1163, H9900104 and E9900101, and H1092 and H5070). Strains associated with large food-borne outbreaks in Los Angeles, Calif. (H2365 [1985]), and Boston, Mass. (H1092 and H5070 [1983]), were grouped within clusters Ic and Id, respectively.
FIG. 1.

Euclidean distance between 55 microarray hybridization experiments. The dendrogram was constructed using the Ward's minimum variance algorithm. Division nomenclature follows that described by Piffaretti et al. (25).
Classification function.
The cluster analysis clearly separated the primary divisions of L. monocytogenes. We also used stepwise DFA to construct a classification function for the major serotypes (1/2a, 1/2b, 1/2c, and 4; 4b and 4c were pooled). The analysis resulted in the selection of four probes that produced divergent hybridization patterns for these serotypes (Fig. 2). The function correctly classified 53 of 55 sample hybridizations (96.4%) from the original data set. Two 1/2a samples were misclassified as 1/2c, which was not unexpected given the close genetic relationship between these two serotypes (Fig. 1, cluster IIc). A more meaningful validation procedure involved data from 28 hybridizations that were not used to develop the original classification function. In this case, one 1/2c isolate was misclassified as serotype 1/2a (96.4% correct classification; sample sizes for 1/2a, 1/2b, 1/2c, and 4b or 4c were 14, 4, 2, and 8, respectively).
FIG. 2.

Average signal intensity (error bar equals SEM) for the four probes used for serotype classification by DFA. Bars correspond to probe 1094 (open), probe 369 (black), probe 362 (grey), and probe 289 (lined).
Probe sequences.
We summarized probe intensity data for each of the clusters that were identified in Fig. 1 (Table 2). There were five probes (probes 317, 369, 791, 955, and 1069) that produced high signal intensities for all members of Division II and low signal intensities for Division I isolates. Only one probe (302) produced a consistently high signal intensity for all members of Division I while producing a low signal intensity for all members of Division II. All 29 probes identified in this analysis were sequenced, and the closest protein match was identified using BLASTx searches against GenBank sequences (Table 2). All probes had a significant match with the L. monocytogenes genome (e < 0.001), but some probe sequences produced better matches for genes from other species of bacteria. Only one probe sequence (297) spanned more than one open reading frame, and two of the probes appeared to be virtually identical (134 and 1072; 99% nucleotide identity). Probes included six sequences of unknown function, four phage-related genes, three membrane-associated lipoproteins and an associated transcription factor, several transport proteins, one gene related to the repair of UV-damaged DNA, two genes related to salt tolerance, one gene related to biofilm formation, and one internalin gene (Table 2).
TABLE 2.
Putative sequence identification for bimodally distributed microarray probes
| Probea | BLASTxb | Putative identity | Cluster and serotype profilec
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| IIa, 1/2a | IIb, 1/2a | IIc, 1/2a,c | Ia, 1/2b | Ib, mixed | Ic, 4b | Id, 4b | |||
| 362 | 8e-53 (92%) | Hypothetical protein Imo0068-L. monocytogenes | − | − | +/− | +/− | +/− | +/− | +/− |
| 1137 | 4e-10 (31%) | Bacteriophage capsid protein-Listeria innocua | − | − | +/− | +/− | − | − | − |
| 136 | 2e-89 (81%) | ATP-dependent Clp proteinase-Lactococcus lactis | − | − | +/− | + | − | − | − |
| 961 | 6e-89 (97%) | UV-damage repair protein-Listeria innocua | +/− | − | +/− | + | − | − | − |
| 1015 | 1e-76 (71%) | Tape measure protein-bacteriophage A118 | + | − | − | + | − | − | + |
| 1063 | e-105 (95%) | Portal protein (bacteriophage A118) | + | − | − | + | − | − | + |
| 118 | 3e-48 (78%) | Lmo2305-L. monocytogenes | + | − | − | +/− | − | − | + |
| 897 | 2e-78 (96%) | Coat protein-bacteriophage SPP1-L. monocytogenes | + | − | − | − | − | − | + |
| 297d | 9e-57 (89%) | Penicillin acylase and conjugated bile hydrolase-L. monocytogenes | + | +/− | + | +/− | + | − | − |
| 297d | 5e-25 (100%) | Glutamate decarboxylase-L. monocytogenes | + | +/− | + | +/− | + | − | − |
| 927 | 2e-64 (100%) | Conserved hypothetical protein-L. monocytogenes | + | +/− | + | +/− | + | − | − |
| 774 | 4e-50 (100%) | Gamma-glutamyl phosphate reductase-L. monocytogenes | + | + | +/− | − | − | − | − |
| 955f | 1e-66 (87%) | Phosphotransferase system, beta-glucoside-specific enzyme II L. monocytogenes | + | + | + | − | − | − | − |
| 369e | e-101 (97%) | 3-Isopropylmalate dehydrogenase-L. monocytogenes | + | + | + | − | − | − | − |
| 791e | e-115 (100%) | Hypothetical protein Imo0940-L. monocytogenes | + | + | + | − | − | − | − |
| 1069e | 2e-82 (92%) | Internalin proteins (LPXTG motif)-L. monocytogenes | + | + | + | − | − | − | − |
| 317e | 2e-49 (99%) | ABC transporter-L. monocytogenes | + | + | + | − | − | − | − |
| 380 | 2e-54 (100%) | Lmo0750-L. monocytogenes | + | + | + | +/− | +/− | − | +/− |
| 1094 | e-117 (93%) | Glycosyltransferase-L. monocytogenes | + | + | + | + | +/− | − | − |
| 312 | 2e-70 (100%) | Glycerol-3-phosphatecytidylyltransferase (gct), CDP-glycerol L. monocytogenes | + | + | + | + | +/− | − | − |
| 289 | e-114 (90%) | Heavy metal-transporting ATPase-L. monocytogenes | − | + | + | + | + | + | + |
| 134e,f | e-108 (99%) | Membrane-associated lipoprotein-L. monocytogenes | − | − | + | + | + | + | + |
| 1072e,f | e-108 (100%) | Membrane-associated lipoprotein-L. monocytogenes | − | − | + | + | + | + | + |
| 150e | 3e-86 (96%) | Transcription regulator (VirR from Streptococcus pyogenes) L. monocytogenes | − | − | + | + | + | + | + |
| 103e | 3e-94 (86%) | Membrane-associated lipoprotein-L. monocytogenes | − | − | + | + | + | + | + |
| 367e | 2e-91 (83%) | Membrane-associated lipoprotein-L. monocytogenes | − | − | + | + | + | + | + |
| 1092e | 5e-70 (85%) | Hypothetical protein Imo0461-L. monocytogenes | − | − | + | + | + | + | + |
| 302e | 1e-96 (91%) | Ferrous iron transport protein B-L. monocytogenes | − | − | − | + | + | + | + |
| 1085e | 2e-27 (96%) | Gamma-glutamyl kinase-L. monocytogenes | − | − | +/− | + | + | + | + |
| 343 | e-126 (100%) | 4b-4d-4e-serotype-specific gene cassette (gltA-gltB) L. monocytogenes | − | − | − | − | +/− | +/− | + |
Unique identifier for probes.
e-score from BLASTx search (% amino acid similarity).
Cluster designations are from Fig. 1. Symbols used in serotype profiles for each cluster indicate the following: −, all members of the cluster (column) have a low hybridization signal for that gene (row); +, all members of the cluster produced a strong hybridization signal; +/−, hybridization signal was mixed (high and low) for members of that cluster.
Probe 297 spanned two open reading frames.
Denotes probes originally sequenced by Borucki et al. (3).
Probes 134 and 1072 shared 99% sequence identity.
Linkage disequilibrium.
Genetic linkage can be a useful index to the degree of horizontal transfer occurring within the species L. monocytogenes. Our probes included several phage-related sequences that are consistent with horizontal gene flow and linkage equilibrium in bacteria. The well-defined clusters in Fig. 1, however, are indicative of clonality and linkage disequilibrium (21). We estimate that the average linkage equilibrium (IA) for the samples included in this study was 3.85 (Fig. 3). Several other published values for IA are included for reference in Fig. 3.
FIG. 3.

Bootstrap distribution of IA values for L. monocytogenes microarray data (bars). The point estimate for L. monocytogenes (d) is 3.85. Additional point estimates are provided for reference: (a) N. gonorrhoeae, (b) N. meningitidis, (c) Salmonella spp., and (e) Rhizobium spp. (21).
Sequence divergence and signal intensity.
Probe 302 has been identified earlier as a potential marker to discriminate between Divisions I and II of L. monocytogenes (3). For the present analysis, a primer set was designed to amplify a 500-bp region of this probe sequence from five isolates. Sequence similarity ranged between 98.6 and 99.4% for genetically related isolates (within Divisions I or II), whereas similarity ranged between 85.9 and 87.5% for comparisons between Divisions I and II. Thus, probe 302 probably originated from a serotype 4b strain that was included in the original reference library and the microarray was sensitive to a ca. 12 to 13% sequence divergence based on the hybridization conditions used in this study.
DISCUSSION
As illustrated in this study, mixed-genome microarrays present several unique advantages for characterizing clinically relevant microbes. First, these arrays are relatively simple to construct and use, making them very attractive tools for identifying and validating genetic markers. In this study, we identified 29 polymorphic probe sequences, several of which may be suitable for distinguishing between serotypes (Fig. 2) and a combination of which permits fingerprinting of bacterial isolates (Fig. 1). As discussed below, this approach to fingerprinting appears to retain information about phylogenetic relationships between isolates. Unlike conventional fingerprinting (e.g., pulsed-field gel electrophoresis or ribotyping), microarray fingerprinting offers the additional opportunity to identify unique DNA sequences that distinguish between the taxonomic and clinical isolates under study. These sequences can be used to construct smaller arrays, or they can be used to develop PCR-based detection assays. Unlike PCR-based fingerprinting methods, the arrays will be far less susceptible to target contamination (see below). Finally, as illustrated by this project, genetic markers identified by this technique may be relevant to differential survivorship and virulence and therefore offer new targets for pathogen detection or for studying the ecology, treatment, or control of important microbes.
The dendrogram (Fig. 1) illustrates two phylogenetic divisions that correspond to findings from multilocus enzyme electrophoresis (2, 25), pulsed-field gel electrophoresis (5), ribotyping (16), randomly amplified polymorphic DNA (22), and sequencing (11, 34). Classification of isolates into these two divisions is confirmed both by serotyping and by a PCR-based classification system (Table 1) (19). Samples with serotype 1/2a are distributed between three clusters (IIa, IIb, and IIc), although cluster IIc includes a mix of both serotypes 1/2a and 1/2c. There is a strong precedent for at least two 1/2a divisions in the literature. Using PCR with restriction enzyme analysis, Unnerstad et al. (31) showed that serotype 1/2a can be divided into two groups and Giovannacci et al. (14) showed a similar division by using ribotyping. Ericsson et al. (11) may have resolved two groups for 1/2a based on sequence variation for the inlB locus. Using multilocus enzyme electrophoresis, Bibb et al. (2) showed that serotype 1/2a has nearly twice as much genetic variation as other L. monocytogenes serotypes (1/2b, 3b, and 4b), a result consistent with the presence of well-delineated lineages for the 1/2a serotype. A recent analysis of a substracted genomic library demonstrated three unique hybridization patterns for serotype 1/2a (18). The findings in this latter report and a recent report by Mereghetti et al. (22) are consistent with our findings of three clusters for serotype 1/2a.
Five out of seven 1/2b isolates are placed in cluster Ia, which is consistent with the phylogenetic separation between the 1/2b and 4 serotypes (5, 11). Two epidemiologically linked 1/2b isolates, however, are grouped together in cluster Ib along with serotypes 4b and 4c. Furthermore, others describe a third lineage of L. monocytogenes that is composed of the 4a and 4c serotypes, but our cluster Ib is not consistent with this finding (19, 26, 36). The polyphyletic composition of cluster Ib may be an artifact of the original reference library. Due to logistical constraints at the time the library was constructed, serotypes 1/2b, 4a, and 4c are not represented and thus our present array design may be too limited to differentiate between some closely related strains. A larger, more representative library may resolve cluster Ib. Additionally, probes with fewer than five isolates on one side of the bimodal distribution were excluded from analysis and, consequently, probes that may have defined small subclusters (e.g., 4c isolates and 1/2b isolates 9900101 and 9900104) may have been omitted by this criterion. Serotype 4b is distributed between clusters Ic and Id. Ericsson et al. (10, 11) and Herd and Kocks (18) also presented evidence for two genetically distinct lineages with respect to serotype 4b. Our analysis places two epidemic strains (H2365; H1092 and H5070) into separate clusters (Ic and Id), which is consistent with the findings of Herd and Kocks (18). Thus, most of the genetic relationships shown in our dendrogram have prior precedence in the literature.
The DFA classification function demonstrates how a large number of probes can be distilled down to a small number that is suitable for classifying bacteria into predefined categories. In this case, only four probes were needed to distinguish major serotypes and the DFA was stable with data from independent hybridizations. These probes may be suitable targets for serotype-specific PCR assays (M. K. Borucki and D. R. Call, unpublished data), although the array format has the advantage of not being sensitive to template contamination. For example, the unique sensitivity of PCR means that this technique is vulnerable to minor contamination (on the order of 100 to 102 gene copies) whereas arrays are likely to require 104 to 106 copies for reliable detection. Sequence data from probe 302 and published findings by Wu et al. (37) suggest that when target identity diverges 10 to 15% from the probe sequence, one may expect a significantly lower hybridization signal than that with a perfect match. Clearly, this is a function of hybridization stringency and how the mismatched nucleotides are distributed throughout the target strand. Failure to detect a hybridized target, however, does not equate with a missing homologue, and in some cases, it is possible to detect the gene by PCR. When this occurs, the gene may be sequenced from a number of isolates to develop very specific mismatch PCR assays suitable for distinguishing between serotypes or phylogenetic lineages (3, 19).
Our estimate of linkage equilibrium indicates that L. monocytogenes is best characterized as having strong linkage disequilibrium (IA = 3.85). The empirically derived probability distribution shows that this index value is similar to the value reported for Salmonella spp. (IA = 3.11) (21). Salmonella has been described as having a clonal population structure characterized by a relatively high degree of linkage disequilibrium. Our estimated distribution for IA does not overlap with the values reported for Neisseria gonorrhoeae (IA = 0.04) and Neisseria meningitidis (IA = 1.96), both of which have population genetic structures approaching genetic equilibrium (Fig. 3).
The primary goal of this project was to assess the diversity of L. monocytogenes in a manner that identifies specific genetic differences between strains. Our relatively low-density microarray allowed the identification of several gene candidates that appear conserved within phylogenetic divisions and that might impact differential fitness and virulence. For example, L. monocytogenes is considered an environmentally ubiquitous organism but only a few serotypes are commonly found with food commodities (19, 29). One explanation for this distribution is that different strains of L. monocytogenes may be better adapted to grow or persist in areas conducive to product contamination. Several of our microarray probe sequences have close BLASTx matches for proteins that might confer differential fitness in different food processing or natural environments. These include a protein related to the repair of UV damage to the chromosome (probe 961), two proteins implicated in salt tolerance (probes 774 and 1085) (30), one gene related to exopolysaccharide synthesis (biofilm formation; probe 1094), and one heavy-metal transporter (probe 289). Additionally, probe 955 is most similar to beta-glucoside-specific enzyme II (bvrB) of the L. monocytogenes phosphotransferase system. BvrB is part of a locus that represses prfA-dependent genes in a soil environment (4, 32).
Epidemiology data suggest that not all strains of L. monocytogenes are equally virulent in humans and that there may be strains that are more virulent in other animals (22, 25, 36). A number of our polymorphic probes have high BLASTx matches for gene products that may allow enhanced survival and virulence in the mammalian host. For example, probe 302 is exclusive to Division I and it is most similar to a ferrous transport gene (3). Iron availability is limited in host tissues, and pathogenic bacteria characteristically evolve mechanisms to acquire iron. Additionally, iron is thought to have a role in L. monocytogenes virulence gene regulation (32). Probe 297 has amino acid sequence similarity to regions of two adjacent open reading frames, both of which encode proteins involved with gut colonization (8, 23). Probe 136 has 52% amino acid identity to L. monocytogenes ClpC ATPase, which is required for host cell adhesion and invasion and for stress tolerance (24, 27, 28). Additional polymorphic probes include close matches for four surface proteins similar to internalins. Probe 1069 includes an LPXTG motif that is found in all L. monocytogenes internalin proteins except InlB (9, 13), and probes 103, 134, and 1069 are most similar to membrane-bound lipoproteins. Both internalins and membrane-bound lipoproteins are implicated in the virulence potential of L. monocytogenes (reviewed in reference 6). In addition, probes 312 and 343 are related to the synthesis and expression of techoic acid-associated surface antigens. Probe 343 has 100% amino acid identity with a serotype 4b-4d-4e-specific gene cassette (20), and only serotype 4b (this study) had high intensity signals with this probe. Finally, four phage-related proteins and six polymorphic probe sequences with no putative functions were identified and these represent additional targets for investigation.
Library bias?
Two of the probe sequences from our array were virtually identical (probes 134 and 1072), and these and several related genes (150, 103, 367) are known to occur in close proximity on the L. monocytogenes genome (15). Furthermore, four of the probe sequences (13.8%) were phage related whereas only 1.4% of the L. monocytogenes genome encompasses phage-related sequences (15). The likelihood that even two identical sequences (or very closely linked sequences) would be found within the 585 probes (600-bp sequences) randomly selected for the array was very remote (2 × 10−3) for a genome of this size (3 Mbp). To have several of these unlikely combinations present on the array suggested that the method we used to make the original reference library biased the selection of insert DNA. The library was constructed by first fragmenting genomic DNA with a frequently cutting restriction enzyme (CviJI; 5-YGCR-3) followed by the size fractionation and purification of ca. 600-bp fragments for cloning. With the recent release of the Listeria genome sequence (NC_003210), we have now determined that this enzyme will cut the genome at approximately 34,119 sites, resulting in an average fragment length of 86 bp. In fact, there are only 486 predicted fragments between 400 and 800 bp, based on this restriction enzyme, which suggests that, if the restriction digest were 100% efficient, then we would have seen several replicate probe sequences.
Despite the potential limitations of our original clone library, we found a number of very useful genetic markers and identified several genes that might be related to differential fitness and virulence. Our dendrogram was not significantly affected by the library bias given that our findings are consistent with what has been reported in the literature. A new array constructed from truly random fragments (e.g., using a nebulizer or sonicator) may be much more informative with respect to more recent phylogenetic divisions and may provide a better estimate of genetic diversity while providing additional targets to investigate with respect to differential fitness and virulence.
Acknowledgments
We gratefully acknowledge the excellent technical assistance provided by M. Krug, W. Muraoka, and J. Reynolds. L. monocytogenes isolates were kindly provided by P. Hayes and L. Graves (CDC, Atlanta, Ga.), J. Hu (Washington State Department of Health, Olympia), and K. Jinneman (U.S. FDA, Bothel, Wash.). S. Mech and M. Evans provided statistical advice.
Funding was provided by U.S. Department of Agriculture-Agricultural Research Service CWU grant 5348-32000-017-00D and by the Agricultural Animal Health Program, College of Veterinary Medicine, Washington State University.
REFERENCES
- 1.Adam, R., and L. Bischof. 1994. Seeded region growing. IEEE Trans. Pattern Anal. Mach. Intell. 16:641-647. [Google Scholar]
- 2.Bibb, W. F., B. G. Gellin, R. Weaver, B. Schwartz, B. D. Plikaytis, M. W. Reeves, R. W. Pinner, and C. V. Broome. 1990. Analysis of clinical and food-borne isolates of Listeria monocytogenes in the United States by multilocus enzyme electrophoresis and application of the method to epidemiologic investigations. Appl. Environ. Microbiol. 56:2133-2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borucki, M., M. Krug, W. Muraoka, and D. Call. Discrimination among Listeria monocytogenes isolates using a mixed genome DNA microarray. Vet. Microbiol., in press. [DOI] [PubMed]
- 4.Brehm, K., M. T. Ripio, J. Kreft, and J. A. Vázquez-Boland. 1999. The bvr locus of Listeria monocytogenes mediates virulence gene repression by β-glucosides. J. Bacteriol. 181:5024-5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brosch, R., J. Chen, and J. B. Luchansky. 1994. Pulsed-field fingerprinting of listeriae: identification of genomic divisions for Listeria monocytogenes and their correlation with serovar. Appl. Environ. Microbiol. 60:2584-2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cabanes, D., P. Dehoux, O. Dussurget, L. Frangeul, and P. Cossart. 2002. Surface proteins and the pathogenic potential of Listeria monocytogenes. Trends Microbiol. 10:238-245. [DOI] [PubMed] [Google Scholar]
- 7.Call, D., D. Chandler, and F. Brockman. 2001. Fabrication of DNA microarrays using unmodified oligonucleotide probes. BioTechniques 30:368-379. [DOI] [PubMed] [Google Scholar]
- 8.Cotter, P., C. Gahan, and C. Hill. 2001. A glutamate decarboxylase system protects Listeria monocytogenes in gastric fluid. Mol. Microbiol. 40:465-475. [DOI] [PubMed] [Google Scholar]
- 9.Dramsi, S., P. Dehoux, M. Lebrun, P. L. Goossens, and P. Cossart. 1997. Identification of four new members of the internalin multigene family of Listeria monocytogenes EGD. Infect. Immun. 65:1615-1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ericsson, H., P. Stalhandske, M. L. Danielsson-Tham, E. Bannerman, J. Bille, C. Jacquet, J. Rocourt, and W. Tham. 1995. Division of Listeria monocytogenes serovar 4b strains into two groups by PCR and restriction enzyme analysis. Appl. Environ. Microbiol. 61:3872-3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ericsson, H., H. Unnerstad, J. G. Mattsson, M. L. Danielsson-Tham, and W. Tham. 2000. Molecular grouping of Listeria monocytogenes based on the sequence of the inIB gene. J. Med. Microbiol. 49:73-80. [DOI] [PubMed] [Google Scholar]
- 12.Food and Drug Administration. 1992. Bacteriological analytical manual. Center for Food Safety and Applied Nutrition, Washington, D.C.
- 13.Gaillard, J. L., P. Berche, C. Frehel, E. Gouin, and P. Cossart. 1991. Entry of L. monocytogenes into cells is mediated by internalin, a repeat protein reminiscent of surface antigens from gram-positive cocci. Cell 65:1127-1141. [DOI] [PubMed] [Google Scholar]
- 14.Giovannacci, I., C. Ragimbeau, S. Queguiner, G. Salvat, J. L. Vendeuvre, V. Carlier, and G. Ermel. 1999. Listeria monocytogenes in pork slaughtering and cutting plants. Use of RAPD, PFGE and PCR-REA for tracing and molecular epidemiology. Int. J. Food Microbiol. 53:127-140. [DOI] [PubMed] [Google Scholar]
- 15.Glaser, P., L. Frangeul, C. Buchrieser, C. Rusniok, A. Amend, F. Baquero, P. Berche, H. Bloecker, P. Brandt, T. Chakraborty, A. Charbit, F. Chetouani, E. Couve, A. de Daruvar, P. Dehoux, E. Domann, G. Dominguez-Bernal, E. Duchaud, L. Durant, O. Dussurget, K. D. Entian, H. Fsihi, F. G. Portillo, P. Garrido, L. Gautier, W. Goebel, N. Gomez-Lopez, T. Hain, J. Hauf, D. Jackson, L. M. Jones, U. Kaerst, J. Kreft, M. Kuhn, F. Kunst, G. Kurapkat, E. Madueno, A. Maitournam, J. M. Vicente, E. Ng, H. Nedjari, G. Nordsiek, S. Novella, B. de Pablos, J. C. Perez-Diaz, R. Purcell, B. Remmel, M. Rose, T. Schlueter, N. Simoes, A. Tierrez, J. A. Vazquez-Boland, H. Voss, J. Wehland, and P. Cossart. 2001. Comparative genomics of Listeria species. Science 294:849-852. [DOI] [PubMed] [Google Scholar]
- 16.Graves, L., B. Swaminathan, M. Reeves, S. Hunter, W. R. E., B. Plikaytis, and A. Schuchat. 1994. Comparison of ribotyping and multilocus enzyme electrophoresis for subtyping of Listeria monocytogenes isolates. J. Clin. Microbiol. 32:2936-2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hegde, P., R. Qi, K. Abernathy, C. Gay, S. Dharap, R. Gaspard, J. Hughes, E. Snesrud, N. Less, and J. Quackenbush. 2000. A concise guide to cDNA microarray analysis. BioTechniques 29:548-562. [DOI] [PubMed] [Google Scholar]
- 18.Herd, M., and C. Kocks. 2001. Gene fragments distinguishing an epidemic-associated strain from a virulent prototype strain of Listeria monocytogenes belong to a distinct functional subset of genes and partially cross-hybridize with other Listeria species. Infect. Immun. 69:3972-3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jinneman, K. C., and W. E. Hill. 2001. Listeria monocytogenes lineage group classification by MAMA-PCR of the listeriolysin gene. Curr. Microbiol. 43:129-133. [DOI] [PubMed] [Google Scholar]
- 20.Lei, X., F. Fiedler, Z. Lan, and S. Kathariou. 2001. A novel serotype-specific gene cassette (gltA-gltB) is required for expression of teichoic acid-associated surface antigens in Listeria monocytogenes of serotype 4b. J. Bacteriol. 183:1133-1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maynard Smith, J., N. Smith, M. O'Rourke, and B. Spratt. 1993. How clonal are bacteria? Proc. Natl. Acad. Sci. USA 90:4384-4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mereghetti, L., P. Lanotte, V. Savoye-Marczuk, N. Marquet-Van Der Mee, A. Audurier, and R. Quentin. 2002. Combined ribotyping and random multiprimer DNA analysis to probe the population structure of Listeria monocytogenes. Appl. Environ. Microbiol. 68:2849-2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moser, S., and D. Savage. 2001. Bile salt hydrolase activity and resistance to toxicity of conjugated bile salts are unrelated properties of lactobacilli. Appl. Environ. Microbiol. 67:3476-3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nair, S., E. Milohanic, and P. Berche. 2000. ClpC ATPase is required for cell adhesion and invasion of Listeria monocytogenes. Infect. Immun. 68:7061-7068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Piffaretti, J. C., H. Kressebuch, M. Aeschbacher, J. Bille, E. Bannerman, J. M. Musser, R. K. Selander, and J. Rocourt. 1989. Genetic characterization of clones of the bacterium Listeria monocytogenes causing epidemic disease. Proc. Natl. Acad. Sci. USA 86:3818-3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rasmussen, O. F., P. Skouboe, L. Dons, L. Rossen, and J. E. Olsen. 1995. Listeria monocytogenes exists in at least three evolutionary lines: evidence from flagellin, invasive associated protein and listeriolysin O genes. Microbiology 141:2053-2061. [DOI] [PubMed] [Google Scholar]
- 27.Rouquette, C., C. de Chastellier, S. Nair, and P. Berche. 1998. The ClpC ATPase of Listeria monocytogenes is a general stress protein required for virulence and promoting early bacterial escape from the phagosome of macrophages. Mol. Microbiol. 27:1235-1245. [DOI] [PubMed] [Google Scholar]
- 28.Rouquette, C., M. T. Ripio, E. Pellegrini, J. M. Bolla, R. I. Tascon, J. A. Vazquez-Boland, and P. Berche. 1996. Identification of a ClpC ATPase required for stress tolerance and in vivo survival of Listeria monocytogenes. Mol. Microbiol. 21:977-987. [DOI] [PubMed] [Google Scholar]
- 29.Ryser, E., and E. Marth (ed.). 1999. Listeria, listeriosis, and food safety, 2nd ed. Marcel Dekker, Inc., New York, N.Y.
- 30.Sleator, R., C. Gahan, and C. Hill. 2001. Identification and disruption of the proBA locus in Listeria monocytogenes: role of proline biosynthesis in salt tolerance and murine infection. Appl. Environ. Microbiol. 67:2571-2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Unnerstad, H., I. Nilsson, H. Ericsson, M. L. Danielsson-Tham, J. Bille, E. Bannerman, and W. Tham. 1999. Division of Listeria monocytogenes serovar 1/2a strains into two groups by PCR and restriction enzyme analysis. Appl. Environ. Microbiol. 65:2054-2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vázquez-Boland, J., M. Kuhn, P. Berche, T. Chakraborty, G. Domínguez-Bernal, W. Boebel, B. González-Zorn, J. Wehland, and J. Kreft. 2001. Listeria pathogenesis and molecular virulence determinants. Clin. Microbiol. Rev. 14:584-640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vines, A., M. W. Reeves, S. Hunter, and B. Swaminathan. 1992. Restriction fragment length polymorphism in four virulence-associated genes of Listeria monocytogenes. Res. Microbiol. 143:281-294. [DOI] [PubMed] [Google Scholar]
- 34.Vines, A., and B. Swaminathan. 1998. Identification and characterization of nucleotide sequence differences in three virulence-associated genes of Listeria monocytogenes strains representing clinically important serotypes. Curr. Microbiol. 36:309-318. [DOI] [PubMed] [Google Scholar]
- 35.Whittam, T., H. Ochman, and R. Selander. 1983. Multilocus genetic structure in natural populations of Escherichia coli. Proc. Natl. Acad. Sci. USA 80:1751-1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wiedmann, M., J. L. Bruce, C. Keating, A. E. Johnson, P. L. McDonough, and C. A. Batt. 1997. Ribotypes and virulence gene polymorphisms suggest three distinct Listeria monocytogenes lineages with differences in pathogenic potential. Infect. Immun. 65:2707-2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu, L., D. Thompson, G. Li, R. Hurt, J. Tiedge, and J. Zhou. 2001. Development and evaluation of functional gene arrays for detection of selected genes in the environment. Appl. Environ. Microbiol. 67:5780-5790. [DOI] [PMC free article] [PubMed] [Google Scholar]